

Abstract
Recent studies describe a novel role of fibroblast growth factor 23 (Fgf23)-klotho activity in the systemic regulation of calcium and phosphate homeostasis. Both Fgf23 and klotho ablated mice develop extensive vascular and soft tissue calcification. Inability to clear the required amount of phosphate by the kidney, due to the absence of Fgf23-klotho activity, leads to increased serum accumulation of phosphate in these genetically modified mice, causing extensive calcification. Serum calcium levels are also elevated in both Fgf23 and klotho ablated mice. Moreover, increased sodium-phosphate co-transporter activity in both Fgf23 and klotho ablated mice increases renal phosphate reabsorption which in turn can facilitate calcification. Collectively, these observations bring new insights into our understanding of the roles of the Fgf23-klotho axis in the development of vascular and soft tissue calcification.
Keywords: Fgf23, klotho, calcification, hyperphosphatemia, NaPi2a
Fibroblast growth factor 23
FGF23 is an approximately 30 kDa secreted protein that is mostly synthesized by the osteocytes in the bone 1,2. FGF23 is a master in vivo regulator of phosphate homeostasis. Under physiological conditions, it controls renal phosphate excretion according to the need of the body through the regulation of the renal sodium-dependent phosphate cotransporter NaPi2a and NaPi2c 3,4. Genetic defects in FGF23 gene can produce distinct human diseases. For instance, gain-of-function mutations of FGF23 are responsible for the clinical symptoms observed in patients suffering from autosomal dominant hypophosphatemic rickets (ADHR) 5. These mutations prevent the proteolytic cleavage of the FGF23 protein, leading to its increased biological activity and resulting in severe renal phosphate wasting. Similarly, increased serum levels of FGF23 in the patients with oncogenic osteomalacia (OOM) are found to be the causative factor for tumor-induced renal phosphate wasting 6. Patients affected by X-linked hypophosphatemia (XLH), a dominant disorder caused by inactivating mutations of the gene encoding PHEX (the phosphate-regulating gene with homologies to endopeptidases on the X chromosome), exhibit increased serum FGF23 levels, phosphaturia and osteomalacia 7. A similar phosphate wasting effect, due to increased FGF23 serum level, has been detected in patients with autosomal recessive hypophosphatemia (ARHP) – a rare genetic disorder with essentially similar clinical features as those seen in the patients with OOM, XLH and ADHR 8,9. Recent studies using wild-type and ADHR mutant proteins have identified key FGF23-specific receptor-mediated signaling 10,11.
FGF-23 signaling
FGF23 exerts its bioactivity on selected target tissues by interacting with its cognate FGF receptors (FGFRs) in the presence the cofactor klotho 10–12. The klotho gene encodes a single-pass transmembrane protein with an extracellular domain consisting of two homologous domains that share sequence homology with the [beta]-glucosidase of bacteria and plants. Klotho facilitates the binding of FGF23 to FGFR1c, -3c, and -4 11,12. FGFRs contain a signal-transducing extracellular ligand-binding domain and an intracellular tyrosine kinase domain. The restricted expression of klotho determines the tissue specificity of FGF23 function 12,13. Klotho is mostly expressed in the renal distal tubular epithelial cells, the parathyroid gland, and the pituitary gland 13,14.
FGF23, in the presence of klotho can activate downstream signaling molecules, as determined by activation or phosphorylation of FGFR substrate-2a, extracellular signal-regulated kinase (ERK), and early growth response element-1 (Egr-1) 10,11. Only in presence of klotho, cells exposed to FGF23 underwent ERK phosphorylation and increased the expression of Egr-1 protein. Klotho also enhances FGF23 binding to its receptor since FGF23 has a greater affinity to the Klotho/FGFR complex with than to the FGFR alone, underscoring the important role of klotho as a cofactor in the FGF23 FGFR interaction and subsequent signaling 11. Our understanding of FGF23 and its receptor interactions, along with the downstream signaling events helps us focus on its biological functions. Recent animal genetic studies, generating Fgf23 and klotho ablated mice, have shown that altered mineral ion metabolism in the mutant mice is associated with extensive vascular and soft tissue calcification 15–17.
Vascular calcification
Vascular calcification is a complex, regulated process that involves the molecular interplay between calcification stimulators and inhibitors. Although numerous individual molecules and/or factors have been identified as stimulators of calcification, including inorganic phosphate, calcium, sodium-phosphate cotransporters, Runx2, tissue non-specific alkaline phosphase (TNAP), glucose, acetylated LDL, tumor necrosis factor-alpha (TNF-α), and bone morphogenetic protein 2 (BMP-2) 18–20, their exact mechanism to induce vascular calcification and their interaction with the calcification inhibitors is not yet clearly understood.
Recent studies have shed some light on vascular calcification and how a disrupted balance between calcification inhibiting and promoting factors can lead to calcification. As mentioned, there are several key factors that have been shown to directly regulate the induction and progression of vascular calcification; these include but are not limited to circulating factors (i.e., phosphate, calcium, pyrophosphate, parathyroid hormone: PTH) and their signaling components, matrix molecules (i.e., Matrix Gla Protein: MGP), and catalyzing enzymes (i.e., TNAP).
Serum phosphate and calcium levels are important determinants of vascular calcification, as inadequate regulation of these minerals can lead to spontaneous deposition of calcium-phosphate in the blood vessels and soft tissues. Hyperphosphatemia in dialysis patient correlates with vascular calcification and effective phosphate control with noncalcium phosphate binders is correlated with attenuated progression of vascular calcification in these patients 21. In addition, in vitro studies have shown that smooth muscle cells grown in the in the presence of elevated inorganic phosphorus undergo a dramatic phenotypic change characterized by the downregulation of smooth muscle cell lineage genes and the upregulationof the osteochondrogenic lineage genes 22. Similar to phosphorus, a positive calcium balance is linked to vascular calcification in humans 23. In vitro, calcium promotes mineralization in vascular smooth muscle cell and the calcium-induced mineralization upregulates the expression of the major sodium-dependent phosphate cotransporters in these cells 24.
Inorganic pyrophosphate inhibits vascular calcification by restricting hydroxyapatite formation and propagation through its biophysical chelator-like role, as well as stabilizing the aortic phenotype by acting as a paracrine regulator 19. Reduced plasma pyrophosphate levels are reported in hemodialysis patients and are exacerbated as a result of pyrophosphate clearance 25. It is therefore likely that restoring pyrophosphate levels may help in limiting vascular calcification. Another factor, TNAP, an enzyme produced in several tissues including bones, serves as a functional phenotypic marker of osteoblasts and is often used as a molecular marker for vascular calcification. Since pyrophosphate is a substrate for TNAP and phosphorus is the product of its catalytic activity, one can theoretically anticipate that upregulated TNAP expression acts as a precursor to vascular calcification 26. PTH can also influence vascular calcification. Uncontrolled secretion of PTH can release excessive amount of calcium form bone, which can precipitate as calcifying foci in blood vessels and soft tissue 27.
BMP-2 plays a role in calcification by exerting osteogenic effects on blood vessels and soft tissues 28. Studies have shown a positive correlation between BMP2 and vascular calcification. Furthermore, matrix proteins, like MGP can inhibit vascular calcification. A positive correlation exists between the local expression of MGP and calcification in arteries. In MGP knockout mice and human Keutel Syndrome, the deficiency of MGP, are associated with ectopic calcification 29. MGP is able to control vascular calcification partly through its Gla residues, which have a calcium/hydroxyl apatite chelating capacity.
Vascular calcification is histologically divided into four main types: 1) atherosclerotic intimal calcificaiton, 2) medial artery calcification (Monckeberg sclerosis), 3) cardiac valve calcificaiton, and 4) arteriole calcification in the form of calciphylaxis. Although systemic factors have great importance in inducing calcification, the interplay between the resident cells of the vasculature usually determines the extent of the damage; cross talks and phenotypic alteration of endothelial cells, smooth muscle cells, pericytes and perhaps mesenchymal stem cells, in response to systemic dysregulation of mineral balance can significantly influence the calcification process. In general, there are significant similarities between skeletal mineralization and vascular and soft tissue calcification 28. The reader is referred to recent reviews for an in-depth overview of the general aspects of bone and vascular calcification 18,28. The purpose of this review is to briefly discuss the potential effects of FGF23-klotho activity on the development of vascular and soft tissue calcification.
Does FGF23-klotho activity influence calcification?
Both human and animal studies have shown that reduced FGF23 or klotho activities are closely associated with vascular and soft tissue calcification.
Animal studies
Extensive vascular and soft tissue calcification is observed in Fgf23 knockout mice by 6 weeks of age; small and medium sized arteries and the proximal tubules in the kidneys are the most extensively affected sites, in addition to the aorta. Interestingly, the expression of the sodium-phosphate co-transporter, NaPi2a of the proximal tubular epithelial cells is also upregulated in Fgf23 knockout mice 1. Increased tubular sodium-phosphate co-transporter expression (Figure 1) in these mice may translocate extracellular phosphate within the cells to facilitate calcification. In addition to kidney and blood vessels (Figure 2), Fgf23 knockout mice also exhibit widespread soft tissue calcification in the lungs, skeletal muscle, skin, urinary bladder, testes, and cardiac muscle. Vascular and soft tissue calcification appear as early as 6 weeks postnatally and progress with age in Fgf23 knockout mice 15,16. Such widespread calcification in Fgf23 knockout mice is associated with osteopenia. Autoradiographic studies of both fore- and hind limbs show that the bone mineral density (BMD) is strikingly reduced in Fgf23 knockout mice, compared to their control littermates 1. Interestingly, despite reduced BMD, the total body mineral content (BMC) in Fgf23 knockout mice is higher due to their extensive vascular and soft tissue calcification 1,15. The Fgf23 knockout mouse phenotype has clinical relevance, as human studies have also shown an association between reduced BMD and vascular calcification 30; low BMD is suggested to independently predict coronary artery disease in women, with a higher odds ratio than traditional risk factors 31. Lower BMD, yet higher BMC in Fgf23 knockout mice, therefore, provides a unique model to study molecular regulation of osteoporosis and vascular calcification 1,15.
Figure 1.
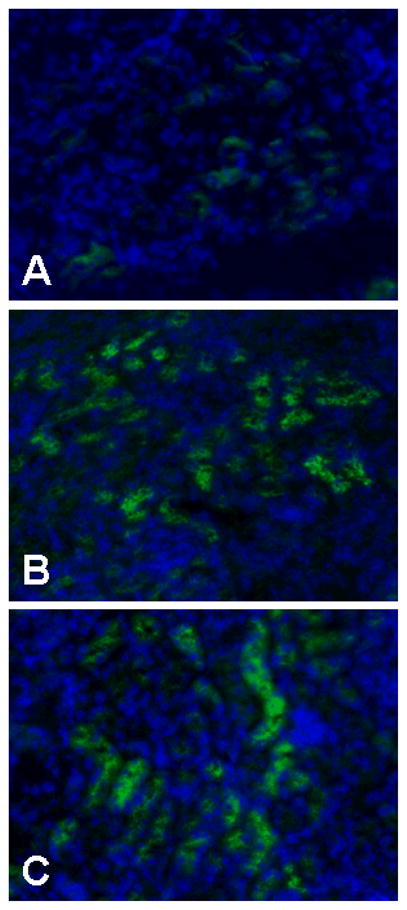
Immunostaining of NaPi2a protein in kidneys obtained from control (A), Fgf23−/− (B) and klotho−/− (C) mice. An increased expression of NaPi2a protein is detected in Fgf23−/− and klotho−/− mice, compared to wild-type mice. Please note that NaPi2a protein is exclusively present in the lumnal side of the proximal tubular epithelial cells.
Figure 2. Soft tissue and vascular calcification in Fgf-23−/− mice.
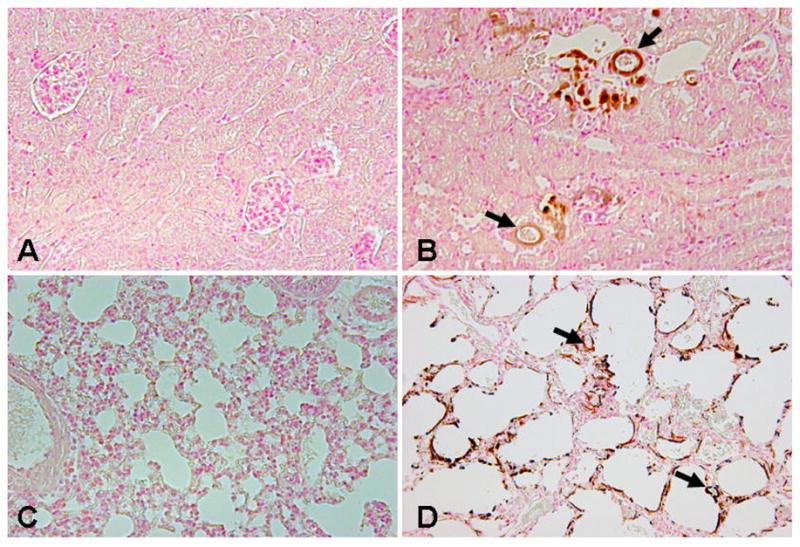
von Kossa staining on paraffin sections of the kidney (A, B) and lung (C, D), showing widespread renal (B) and pulmonary (D) calcifications in Fgf-23−/− mice. No such calcification is noted in the wild-type littermates (A, C). Arrows depict the calcified vessels in the kidney and lung of the Fgf-23−/− mice. (Magnification: kidney ×20; lung ×10).
Similar to the Fgf23 knockout animals, mice homozygous for the hypomorphic alleles of the klotho gene show increased expression of NaPi2a and NaPi2c co-transporters in the proximal tubular epithelial cells (Figure 1). Furthermore, extensive calcification in both vascular and soft tissues, including lungs, skin, testis, and heart is noted in klotho ablated mice. Taking into consideration the phenotypes of both Fgf23 and klotho ablated mice, it seems likely that in vivo dysregulation of the FGF23-klotho axis can lead to vascular calcification, possibly by affecting mineral ion metabolism 4,32–34. Needless to mention that extensive vascular and soft tissue calcification in both Fgf23 and klotho ablated mice is associated with severe hyperphosphatemia, and increased serum level of 1,25 hydroxyvitamin D 15–17. The experimental relevance of both Fgf23 and klotho ablated mice has significantly increased due to the fact that mutations in either human FGF23 or klotho genes are also associated with ectopic calcification.
Human studies
In accord with the animal studies, human diseases associated with inactivating mutations in either FGF23 or Klotho gene express severe ectopic calcification. For instance, familial tumoral calcinosis (FTC) is an autosomal recessive disorder characterized by hyperphosphatemia and ectopic calcifications; and is associated with diaphysitis, hyperostosis, arterial aneurysms, dental abnormalities, and angioid streaksof the retina. Genetic studies have shown evidence that missense mutations in the human FGF23 gene or GALNT3 gene (UDP – N – acetyl – alpha – D - galactosamine: polypeptide N-acetylgalactosaminyltransferase 3) cause the disease 35. Mutations in GLANT3 prevent its ability to selectively O-glycosylate a furin-like convertase recognition sequence in FGF23, thus preventing the proteolytic processing of FGF23 and the secretion of intact FGF23 protein 36. Hence, mutations in either FGF23 or GALNT3 genes reduce FGF23 activities, which lead to hyperphosphatemia and eventually to tumoral calcinosis in patients with FTC.
Klotho is a recently identified co-factor in FGF23 signaling. Experimental studies have demonstrated convincingly that FGF23, in the absence of klotho, cannot exert its bioactivities. For instance, despite extremely high serum levels of Fgf23 (about 2000-fold higher) in klotho ablated mice, Fgf23 is unable to exert its phosphaturic effects in these mice 12. The lack of phosphaturic activity despite extremely high levels of Fgf23 in klotho ablated mice signifies that Fgf23 is incapable to perform its physiological functions in the absence of klotho 32. Recently, a point mutation in the human Klotho gene was reported in a 13 year old patient with severe vascular and soft-tissuecalcification despite significantly high serum level of FGF23. Lack of function of the Klotho gene in this patient can attenuate the ability of FGF23 to exert its phosphate lowering effects, which can eventually lead to the severe vascular and soft tissuecalcifications 37.
Can FGF23 suppress calcification?
Since reduced FGF23 activity is associated with vascular and soft tissue calcification in both experimental and human studies, the clinically relevant question would be: does FGF23 have any calcification inhibitory effects. Current observations suggest that the manipulation of FGF23 activity can delay calcification through the lowering of serum calcium and phosphate levels. Also, Shimada and coworkers demonstrated that FGF23 can suppress the renal expression of 1a-hydroxylase, the rate limiting enzyme that converts the inactive vitamin-D metabolite to its active form 38. It is possible that FGF23 can reduce calcification by inhibiting vitamin-D activity. Inaba et al. recently reported that FGF23 is an independent factor that is negatively associated with hand-artery, but not aortic calcification in haemodialysis patients, and proposed plasma FGF23 levels as a reliable marker for medial peripheral artery calcification in these patients 39. Nevertheless, extensive cardiovascular calcification is the leading cause of death in chronic kidney disease patients undergoing dialysis, despite their significantly high serum FGF23 levels 40–42. Variability in the degree of failing kidneys can account for this apparent contradiction: diminishing renal function interferes to various extents with the ability of FGF23 to exert its inhibitory effects due to potential defects in klotho and/or FGFR expression, leading to both elevated serum FGF23 levels and vascular calcification. Finally, a recent study on subjects with normal kidney function found no correlation between serum intact FGF23 and/or fetuin-A levels, and coronary artery score 43. These findings suggest that under normal renal function, where the kidneys are effectively maintaining a normal phosphate balance, FGF23 is not a suitable marker for coronary artery calcification. Further studies are needed to better understand the role of FGF23 in vascular and soft tissue calcification under various pathological conditions.
Concluding remarks
The recent understanding of the systemic regulation of mineral ion homeostasis and vitamin D metabolism by FGF23-klotho signaling leads us to revisit the mechanistic aspect of calcification 44–47. Whether the FGF23-klotho duo can directly inhibit calcification or the effect is indirect due to reduced availability of calcification promoting mineral ions, needs additional study. Hypothetically, enhanced FGF23-klotho activity can delay the process of calcification by negatively impacting the serum phosphate balance. Further studies will determine if the pharmacological manipulation of FGF23 activity can be beneficial in fine-tuning existing treatments of vascular and/or soft tissue calcification 48. Such studies will also expand our understanding of the fundamental aspects of mineral ion metabolism under physiological and pathological conditions, including calcification.
Acknowledgments
Part of the original research works are supported by the grants (R01-073944 to BL) and (R01-077276 to MSR) provided by NIH (NIDDK). Fahad Memon is a sophomore at Boston University, Boston, MA, and majoring Biomedical Engineering.
References
- 1.Sitara D, Razzaque MS, St-Arnaud R, Huang W, Taguchi T, Erben RG, Lanske B. Genetic ablation of vitamin D activation pathway reverses biochemical and skeletal anomalies in Fgf-23-null animals. Am J Pathol. 2006;169:2161–70. doi: 10.2353/ajpath.2006.060329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291:E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 3.Quarles LD. FGF23, PHEX, and MEPE regulation of phosphate homeostasis and skeletal mineralization. Am J Physiol Endocrinol Metab. 2003;285:E1–9. doi: 10.1152/ajpendo.00016.2003. [DOI] [PubMed] [Google Scholar]
- 4.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194:1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ADHR_Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. The ADHR Consortium. Nat Genet. 2000;26:345–8. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 6.Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–5. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–63. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 8.Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–5. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38:1248–50. doi: 10.1038/ng1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert TA, Zhang F, Linhardt RJ, Yu X, White KE, Inagaki T, Kliewer SA, Yamamoto M, Kurosu H, Ogawa Y, Kuro-o M, Lanske B, Razzaque MS, Mohammadi M. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417–28. doi: 10.1128/MCB.02249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–3. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–4. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 13.Torres PU, Prie D, Molina-Bletry V, Beck L, Silve C, Friedlander G. Klotho: an antiaging protein involved in mineral and vitamin D metabolism. Kidney Int. 2007;71:730–7. doi: 10.1038/sj.ki.5002163. [DOI] [PubMed] [Google Scholar]
- 14.Nabeshima Y. Toward a better understanding of Klotho. Sci Aging Knowledge Environ. 2006:pe11. doi: 10.1126/sageke.2006.8.pe11. [DOI] [PubMed] [Google Scholar]
- 15.Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. Premature ageing-like phenotype in fibroblast growth factor 23 null mice is a vitamin-D mediated process. FASEB J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Razzaque MS, Lanske B. Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med. 2006;12:298–305. doi: 10.1016/j.molmed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 18.El-Abbadi M, Giachelli CM. Mechanisms of vascular calcification. Adv Chronic Kidney Dis. 2007;14:54–66. doi: 10.1053/j.ackd.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Towler DA. Inorganic pyrophosphate: a paracrine regulator of vascular calcification and smooth muscle phenotype. Arterioscler Thromb Vasc Biol. 2005;25:651–4. doi: 10.1161/01.ATV.0000158943.79580.9d. [DOI] [PubMed] [Google Scholar]
- 20.El-Abbadi M, Giachelli CM. Arteriosclerosis, calcium phosphate deposition and cardiovascular disease in uremia: current concepts at the bench. Curr Opin Nephrol Hypertens. 2005;14:519–24. doi: 10.1097/01.mnh.0000168335.29381.23. [DOI] [PubMed] [Google Scholar]
- 21.Raggi P, Ali O. Phosphorus restriction and control of coronary calcification as assessed by electron beam tomography. Curr Opin Nephrol Hypertens. 2002;11:391–5. doi: 10.1097/00041552-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–54. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 23.Braun J, Asmus HG, Holzer H, Brunkhorst R, Krause R, Schulz W, Neumayer HH, Raggi P, Bommer J. Long-term comparison of a calcium-free phosphate binder and calcium carbonate--phosphorus metabolism and cardiovascular calcification. Clin Nephrol. 2004;62:104–15. doi: 10.5414/cnp62104. [DOI] [PubMed] [Google Scholar]
- 24.Yang H, Curinga G, Giachelli CM. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int. 2004;66:2293–9. doi: 10.1111/j.1523-1755.2004.66015.x. [DOI] [PubMed] [Google Scholar]
- 25.Lomashvili KA, Khawandi W, O’Neill WC. Reduced plasma pyrophosphate levels in hemodialysis patients. J Am Soc Nephrol. 2005;16:2495–500. doi: 10.1681/ASN.2004080694. [DOI] [PubMed] [Google Scholar]
- 26.Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99:9445–9. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poole KE, Reeve J. Parathyroid hormone - a bone anabolic and catabolic agent. Curr Opin Pharmacol. 2005;5:612–7. doi: 10.1016/j.coph.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 28.Towler DA, Shao JS, Cheng SL, Pingsterhaus JM, Loewy AP. Osteogenic regulation of vascular calcification. Ann N Y Acad Sci. 2006;1068:327–33. doi: 10.1196/annals.1346.036. [DOI] [PubMed] [Google Scholar]
- 29.Hur DJ, Raymond GV, Kahler SG, Riegert-Johnson DL, Cohen BA, Boyadjiev SA. A novel MGP mutation in a consanguineous family: review of the clinical and molecular characteristics of Keutel syndrome. Am J Med Genet A. 2005;135:36–40. doi: 10.1002/ajmg.a.30680. [DOI] [PubMed] [Google Scholar]
- 30.Reddy J, Bilezikian JP, Smith SJ, Mosca L. Reduced bone mineral density is associated with breast arterial calcification. J Clin Endocrinol Metab. 2008;93:208–11. doi: 10.1210/jc.2007-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcovitz PA, Tran HH, Franklin BA, O’Neill WW, Yerkey M, Boura J, Kleerekoper M, Dickinson CZ. Usefulness of bone mineral density to predict significant coronary artery disease. Am J Cardiol. 2005;96:1059–63. doi: 10.1016/j.amjcard.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 32.Lanske B, Razzaque MS. Premature aging in klotho mutant mice: cause or consequence? Ageing Res Rev. 2007;6:73–9. doi: 10.1016/j.arr.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Razzaque MS, St-Arnaud R, Taguchi T, Lanske B. FGF-23, vitamin D and calcification: the unholy triad. Nephrol Dial Transplant. 2005;20:2032–5. doi: 10.1093/ndt/gfh991. [DOI] [PubMed] [Google Scholar]
- 34.Lanske B, Razzaque MS. Mineral metabolism and aging: the fibroblast growth factor 23 enigma. Curr Opin Nephrol Hypertens. 2007;16:311–8. doi: 10.1097/MNH.0b013e3281c55eca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36:579–81. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- 36.Kato K, Jeanneau C, Tarp MA, Benet-Pages A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281:18370–7. doi: 10.1074/jbc.M602469200. [DOI] [PubMed] [Google Scholar]
- 37.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–91. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–35. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 39.Inaba M, Okuno S, Imanishi Y, Yamada S, Shioi A, Yamakawa T, Ishimura E, Nishizawa Y. Role of fibroblast growth factor-23 in peripheral vascular calcification in non-diabetic and diabetic hemodialysis patients. Osteoporos Int. 2006;17:1506–13. doi: 10.1007/s00198-006-0154-6. [DOI] [PubMed] [Google Scholar]
- 40.Stompor T. An overview of the pathophysiology of vascular calcification in chronic kidney disease. Perit Dial Int. 2007;27 (Suppl 2):S215–22. [PubMed] [Google Scholar]
- 41.Cozzolino M, Mazzaferro S, Pugliese F, Brancaccio D. Vascular Calcification and Uremia: What Do We Know? Am J Nephrol. 2007;28:339–346. doi: 10.1159/000111827. [DOI] [PubMed] [Google Scholar]
- 42.DeLoach SS, Berns JS. Arterial stiffness and vascular calcification in dialysis patients: new measures of cardiovascular risk. Semin Dial. 2007;20:477–9. doi: 10.1111/j.1525-139X.2007.00332.x. [DOI] [PubMed] [Google Scholar]
- 43.Roos M, Lutz J, Salmhofer H, Luppa P, Knauss A, Braun S, Martinof S, Schomig A, Heemann U, Kastrati A, Hausleiter J. Relation between plasma fibroblast growth factor-23, serum fetuin-A levels and coronary artery calcification evaluated by multislice computed tomography in patients with normal kidney function. Clin Endocrinol (Oxf) 2008;68:660–5. doi: 10.1111/j.1365-2265.2007.03074.x. [DOI] [PubMed] [Google Scholar]
- 44.Razzaque MS. Klotho and Na+, K+-ATPase activity: solving the calcium metabolism dilemma? Nephrol Dial Transplant. 2008;23:459–461. doi: 10.1093/ndt/gfm702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshida T, Fujimori T, Nabeshima Y. Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1alpha-hydroxylase gene. Endocrinology. 2002;143:683–9. doi: 10.1210/endo.143.2.8657. [DOI] [PubMed] [Google Scholar]
- 46.Miyamoto K, Ito M, Tatsumi S, Kuwahata M, Segawa H. New aspect of renal phosphate reabsorption: the type IIc sodium-dependent phosphate transporter. Am J Nephrol. 2007;27:503–15. doi: 10.1159/000107069. [DOI] [PubMed] [Google Scholar]
- 47.Lanske B, Razzaque MS. Vitamin D and aging: old concepts and new insights. J Nutr Biochem. 2007;18:771–7. doi: 10.1016/j.jnutbio.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Razzaque MS. Can fibroblast growth factor 23 fine-tune therapies for diseases of abnormal mineral ion metabolism? Nat Clin Pract Endocrinol Metab. 2007;3:788–9. doi: 10.1038/ncpendmet0667. [DOI] [PubMed] [Google Scholar]