

Abstract
Sleep is an essential process conserved from flies to humans. The importance of sleep is underscored by its tight homeostatic control. Here, through a forward-genetic screen, we identify a novel gene, sleepless, required for sleep in Drosophila. sleepless encodes a brain-enriched, glycosyl-phosphatidylinositol-anchored protein. Loss of SLEEPLESS protein causes an extreme (>80%) reduction in sleep. Furthermore, a moderate reduction in SLEEPLESS protein has minimal effects on baseline sleep, but markedly reduces recovery sleep following sleep deprivation. Genetic and molecular analyses reveal that quiver, a mutation that impairs Shaker-dependent K+ current, is an allele of sleepless. Consistent with this finding, Shaker protein level is reduced in sleepless mutants. We propose that SLEEPLESS is a signaling molecule that connects sleep drive to lowered membrane excitability.
Insufficient and poor quality sleep is an increasing problem in industrialized nations. Chronic sleep problems diminish quality of life, reduce workplace productivity, and contribute to fatal accidents (1). Although the biological needs fulfilled by sleep are unclear (2), they are likely to be important because sleep is conserved from flies to humans (3-7), and prolonged sleep deprivation can lead to lethality (8-10). Identifying mechanisms that control sleep may lead to novel approaches for improving sleep quality.
Sleep is regulated by two main processes: circadian and homeostatic (11, 12). The circadian clock regulates the timing of sleep, whereas the homeostatic mechanism controls sleep need. Homeostatic pressure to sleep increases with time spent awake and decreases with time spent asleep. Homeostatic control is thought to influence sleep under normal (baseline) conditions as well as recovery (rebound) sleep following deprivation. However, the molecular mechanisms underlying homeostatic regulation of sleep remain unclear.
A powerful approach to unraveling a poorly understood biological process is to conduct unbiased genetic screens to identify novel molecules required for that process. The Drosophila model for sleep is well-suited for such an approach, which proved invaluable for elucidation of the molecular basis of the circadian clock. Although several Drosophila genes have been implicated in sleep regulation (for example, 13-15), only one of these, the gene encoding the Shaker (Sh) K+ channel, was isolated as a result of a genetic screen (16). A mutation in this gene causes one of the shortest-sleeping phenotypes known, validating the use of screens and suggesting that control of membrane excitability is a critical requirement for sleep. However, the mechanisms by which sleep homeostatic inputs regulate neuronal excitability remain unknown.
Here, using a large-scale, unbiased genetic screen, we identify a novel gene, sleepless (sss), which is required in Drosophila for both normal baseline sleep and rebound sleep following deprivation. We find that sss encodes a brain-enriched, glycosyl-phosphatidylinositol (GPI)-anchored membrane protein. We also show that quiver (qvr), a mutation causing impaired Sh-dependent K+ current (17, 18), is an allele of sss, and that Sh protein level is reduced in sss mutant flies. We propose that SSS protein signals homeostatic sleep drive by enhancing K+ channel activity and thus reducing neuronal excitability.
Identification of sss
To identify novel genes involved in sleep regulation, we carried out a forward-genetic screen for Drosophila mutants with reduced daily sleep. We screened approximately 3,500 mutant lines bearing transposon insertions. A histogram summarizing the daily sleep of these lines is shown in Fig. 1A. We selected for further study the mutant line with the lowest amount of daily sleep, which we named sleepless (sss). In order to homogenize the genetic background, we outcrossed this strain five times into an isogenic wild-type strain, iso31, a line generated specifically for use in behavioral experiments (19). As shown in Fig. 1B, both daytime and nighttime sleep are severely reduced in both male and female sss mutants compared to background controls. Indeed, a small percentage of sss flies (∼9% for both males and females) exhibits no sleep at all in our assay, a phenotype never seen in control flies. To our knowledge, sss mutants have the most extreme reduction in daily sleep (>85% for males and >80% for females; Fig. 1C) attributable to a single gene mutation.
Fig. 1.

Sleep phenotype of sss mutants. (A) Histogram showing the distribution of daily sleep for ∼3,500 mutant lines (∼8 female flies per line). For each line, daily sleep is shown as the difference from the mean of a group of about 100−250 lines tested simultaneously. The arrow indicates the sss mutant line. (B) Sleep profile in 30-min intervals for sss flies (open diamonds) versus background controls (ctrl, closed diamonds). Data for male (M) and female (F) flies are shown. The bar below the x-axis indicates light (white) and dark (black) periods. (C) Daily sleep amount for ctrl (162 males and 148 females), ctrl/sss (111 males and 113 females), and sss flies (146 males and 148 females). Data from the same animals are shown in (C-F). (D-F) Activity counts/minute awake (D), sleep bout duration (E), and daily number of sleep bouts (F) for male and female ctrl, ctrl/sss, and sss flies. In this and subsequent figures, error bars represent SEM. *P < 0.05; **P < 0.0001. For (C), (E), and (F), significance level is shown for sss mutants compared to both ctrl and ctrl/sss flies. For (D), significance level is shown for pairwise comparisons as indicated by lines. In (E), sleep bout duration, which is not normally distributed, is presented as simplified box plots. The line inside each box indicates the median, and the top and bottom represent 75th and 25th percentiles, respectively. Approximately 9% of animals exhibiting zero sleep were excluded from calculation of sleep bout duration.
Despite this extreme reduction in daily sleep, waking activity (defined as activity counts/minute awake) is not significantly elevated in this mutant (Fig. 1D), suggesting that the mutant is not hyperactive when awake (20). The marked decrease in sleep amount is largely due to a sharp reduction in the duration of sleep bouts (Fig. 1E). However, decreased sleep in the sss mutant is also attributable in part to a significant reduction in the number of daily sleep bouts (Fig. 1F). These phenotypes are recessive in mutant animals since flies bearing one copy of the sss mutation behave similarly to background controls (Fig. 1, C through F).
SSS is a novel brain-enriched, GPI-anchored protein
sss mutants bear a P-element insertion (EY04063, which we refer to as P1) in the open reading frame of a novel gene designated CG33472 by the Drosophila Genome Project. The genomic structure of this gene consists of 2 non-coding exons and 5 coding exons, the last of which also contains a ∼3.9 kb predicted 3’ untranslated region (3’UTR; Fig. 2A). In addition to the original P1 insertion line, there is a second line, which we call P2, bearing a transposon insertion (f01257) in the 3’UTR. The SSS protein is predicted to contain a signal peptide, an N-type glycosylation site, and a potential GPI attachment site (Fig. 2, B and C). SSS is well conserved in other insect species, and there is a potential C. elegans homolog (F31F6.8 in Wormbase, 46% similarity for amino acids 51−133), but no obvious vertebrate homologs. Nonetheless, there may be functional vertebrate homologs with conserved downstream pathways.
Fig. 2.

sss encodes a novel brain-enriched, GPI-anchored protein. (A) Schematic of the genomic structure of the sss locus. Non-coding regions of the cDNA are shaded, while coding regions are shown in white. (B) Schematic of structural features of the SSS protein. The primary sequence contains a predicted signal peptide, a N-type glycosylation site (ψ), and a potential GPI attachment site (*). (C) Amino acid sequence of SSS. Amino acids 1−32 comprise the predicted signal peptide (shown boxed), and the predicted N-type glycosylation site is underlined. * denotes the predicted GPI attachment site. (D) Glycosylation of the SSS protein. Western blot analysis with anti-SSS antibody reveals two bands detected in head extracts from wild-type (ctrl) but not sss flies. Deglycosylation of head extracts by treatment with PNGase F results in detection of a single band. Because our antibody to SSS does not recognize glycosylated SSS well, Western blots were treated with PNGase F before being probed with antibody to SSS. In this and subsequent Western blots, antibody to MAP kinase (MAPK) is used to control for loading. (E) Surface expression of SSS in cultured Drosophila cells. S2R+ cells were transfected with a pIZ-sss construct, and stained with or without permeabilization to assay for total or surface expression, respectively. Transfection with the pIZ vector alone shows specificity of our SSS antibody. (F) Reduced surface expression of SSS after PI-PLC treatment. S2R+ cells transfected with a pIZ-sss construct were stained without permeabilization after PI-PLC(+) or mock(−) treatment. (G) Release of SSS into the culture medium by PI-PLC. Western blot analysis of S2R+ cells transfected with pIZ-sss was performed after PI-PLC(+) or mock(−) treatment. (H) Enrichment of SSS expression in brain and head compared to body. An equal amount of total protein (∼40 ug) was loaded per lane. The experiments in (D) through (H) were performed 3−4 times with similar results.
To characterize the SSS protein, we generated an antibody using a peptide antigen (see Materials and Methods). This antibody recognizes two bands on Western blots of wild-type head extracts that are not detectable in sssP1 mutant extracts (Fig. 2D), suggesting that sssP1 is a severe hypomorph or null allele. Since SSS contains a consensus site for N-type glycosylation, we deglycosylated proteins from head extracts and examined SSS mobility by Western blotting. Under these conditions, only a single band of a lower apparent molecular weight than the two untreated bands is detectable (Fig. 2D), indicating that SSS is glycosylated in vivo. Since sss also contains a potential GPI attachment site, we next examined subcellular localization of SSS. Transfection of Drosophila S2R+ cells with a wild-type sss construct and staining with the SSS antibody under non-permeabilizing conditions reveal a subset of the SSS protein expressed on the cell surface (Fig. 2E). Treatment of the cells with phosphatidylinositol-specific phospholipase C (PI-PLC) results in severe reduction of surface expression (Fig. 2F) and release of the SSS protein into the culture medium (Fig. 2G). These results show that the SSS protein is attached to the extracellular surface of the plasma membrane with a GPI anchor and can be released by cleavage with PLC.
Using our anti-SSS antibody, we find that SSS protein levels are enriched in fly brain and head compared to body (Fig. 2H). Consistent with these findings, sss mRNA expression is enriched 23−42 fold in brain compared to whole fly (Adult Drosophila Gene Expression Atlas (21)). SSS protein levels do not cycle in a circadian fashion, nor do they change following sleep deprivation (fig. S1, A and B, see Discussion).
Genetic analysis of sleepless
To determine if the sleep phenotype maps to the sss locus, we crossed sssP1 to two deficiencies that remove the locus. As predicted, both deficiencies fail to complement the short-sleeping phenotype of sssP1 (fig. S2A and B). In order to confirm that the sleep phenotype in sssP1 mutants is caused by disruption of the sss gene, we mobilized the P-element to generate precise and imprecise excision lines. Precise excision of the P-element restores daily sleep amount in sss mutants to wild-type levels (Fig. 3A and fig. S2C). We also obtained an imprecise excision allele (Δ40) that removes part of the sss coding region and is likely to be a null allele (fig. S2D). Consistent with this interpretation, sssΔ40 mutants produce an undetectable level of the SSS protein (Fig. 3B). Sleep in this mutant is reduced as severely as in the P1 mutant, and the phenotype maps to the sss gene since the Δ40 allele fails to complement the P1 allele (Fig. 3A and fig. S2C).
Fig. 3.
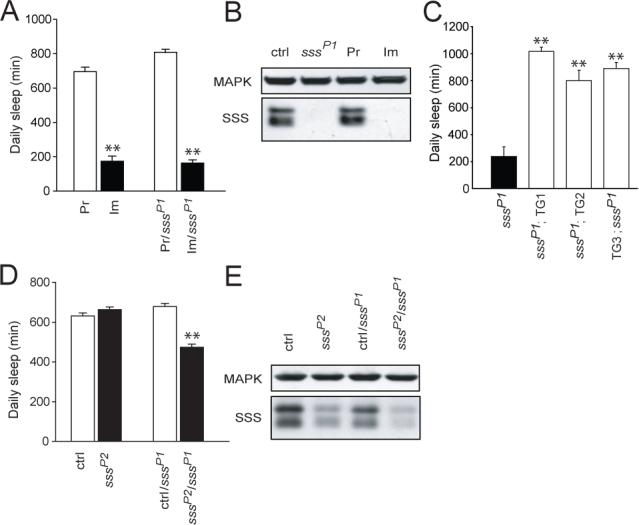
Genetic analysis of sss. (A) Daily sleep amount for precise excision (Pr, n=26), sssΔ40 imprecise excision (Im, n=15), precise/sssP1 (Pr/sssP1, n=24), and imprecise/sssP1 (Im/sssP1, n=35) female flies. (B) Western blot analysis of SSS protein levels. Similar levels of SSS protein are seen in head extracts from background control (ctrl) and precise excision (Pr) flies. SSS protein is undetectable in sssP1 and sssΔ40 imprecise excision (Im) flies. Similar results were obtained in 2 additional experiments. (C) Daily sleep amount for female sssP1 mutant flies with (sssP1;TG1−3, n=15,8,16, respectively) or without (sssP1, n=16) a genomic sss transgene. TG1−3 refer to three independent transgene insertions, and either 1 or 2 copies of the transgene were present in the flies tested. (D) Daily sleep amount for sssP2 (n=110) versus background control (ctrl, n=80), as well as ctrl/sssP1 (n=80) versus sssP2/sssP1 (n=112) female flies. (E) Reduced levels of SSS protein in sssP2 and transheterozygous sssP2/sssP1 flies. Similar results were obtained in 3 additional experiments. Data from male flies of the genotypes shown in (A), (C), and (D) are available in fig S2. *P < 0.05; **P < 0.0001.
We next tested whether expression of wild-type SSS from a transgene can rescue the sleep phenotype of sssP1 mutants. Daily sleep amount is fully rescued to wild-type levels in sssP1 mutants carrying a genomic sss transgene (Fig. 3C and fig. S2E). Together with deficiency and excision experiments, the rescue data provide strong evidence that disruption of the sss gene is responsible for the marked reduction in sleep in sssP1 mutants.
As described above, sssP2 mutants harbor an independent transposon insertion in the 3'UTR of the sss gene. Homozygous sssP2 mutant females have similar amounts of daily sleep as controls, while mutant males have slightly lower amounts of sleep than controls (Fig. 3D and fig. S2F). In contrast, sssP2/sssP1 transheterozygous mutants have a ∼30% reduction in daily sleep compared to control/sssP1 flies. These data suggest that the P2 insertion is a weaker allele than the original P1 insertion. To examine the biochemical basis of this possibility, we performed Western analysis on head lysates from mutant and control animals. As noted above, the P1 insertion severely reduces baseline sleep and renders SSS undetectable (Figs. 2D and 3B). In contrast, the P2 insertion, which has minimal effect on baseline sleep, causes a moderate reduction in the level of SSS protein compared to control (Fig. 3E). Finally, transheterozygous sssP1/sssP2 animals, which exhibit a ∼30% reduction in sleep, have a greatly reduced, but still detectable level of SSS protein. These data suggest that the amount of daily sleep is correlated with the level of SSS protein and that large reductions of SSS protein are necessary to cause a significant change in daily sleep.
Reduced homeostatic response in sss mutants
We next sought to determine whether sss mutants have defects in their homeostatic response to sleep deprivation. We did not observe rebound sleep in sssP1 flies, but sssP1 flies do not have much sleep to deprive. Thus, we tested sssP1/sssP2 transheterozygous animals, which still have moderate amounts of sleep, as well as sssP2 homozygotes, which have essentially normal amounts of sleep. Mechanical stimulation results in equivalent sleep loss in sssP2 homozygous flies and controls and in moderately reduced sleep loss in sssP1/sssP2 flies compared with controls (Fig. 4A and fig. S3A). Whereas control flies show substantial rebound sleep following deprivation, sssP1/sssP2 animals have little or none (Fig. 4B and fig. S3B). Unexpectedly, a similar lack of rebound sleep is observed in sssP2 homozygous flies. In addition, upon lights on, control animals go to sleep faster following deprivation, but this effect is significantly less pronounced or non-existent in sssP2 and sssP1/sssP2 mutants (Fig. 4C and fig. S3C).
Fig. 4.

Reduced homeostatic response to sleep deprivation in female sss mutants. (A) Amount of sleep lost during 6 or 12 hours of deprivation by the end of the dark period for background control (ctrl), sssP2, ctrl/sssP1, and sssP2/sssP1 flies. Data from 13−56 female flies are presented. (B) Amount of sleep gained during 6 hours of recovery following deprivation as in (A). (C) Change in sleep latency following deprivation, compared to undisturbed controls as in (A). Data from male files are shown in fig. S3. *P < 0.05; **P < 0.001.
Although other genes have been suggested to play a role in homeostatic regulation of sleep, assessment of rebound sleep in animals bearing mutations in these genes is often confounded by concomitant reductions in baseline sleep (13, 16, 22, 23). The amount of rebound sleep generally increases with sleep lost (24, 25). Thus, when comparing the effects of sleep deprivation in animals with different amounts of baseline sleep (which leads to loss of different amounts of sleep), it is unclear whether rebound sleep should be compared in absolute terms or relative to amount of sleep lost. We have circumvented this problem by studying the contribution of SSS to sleep homeostasis using the sssP2 mutant. The finding that sssP2 animals exhibit markedly reduced rebound sleep, but minimally affected baseline sleep, provides strong evidence that sleep homeostasis is impaired in these mutants.
Effect of sss on other behaviors and longevity
To further characterize sss mutants, we examined several other behavioral phenotypes. Since mutations in certain central clock genes cause baseline and rebound sleep phenotypes (9, 26-29), we analyzed the circadian rhythm phenotypes of sss mutants. Whereas sssP1 mutants exhibit weak rhythms, almost all sssP1/sssP2 transheterozygous mutants, which display a roughly 30% reduction in daily sleep time, are rhythmic (Fig. 5A and B, Supplementary Table 1). Furthermore, daily oscillations in the level of PERIOD (PER) protein in the ventral lateral neurons (clock cells) remain intact in sssP1 mutants (Fig. 5C), suggesting that the reduced behavioral rhythmicity seen in these mutants is not due to a defect in the central clock.
Fig. 5.
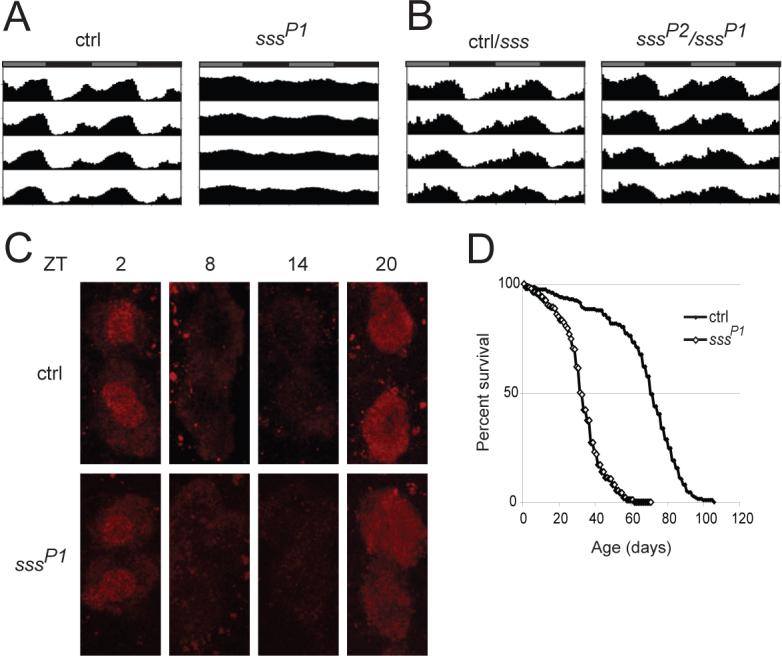
Circadian rhythm and longevity phenotypes of sss mutants. (A) Average activity records for background control (ctrl, n=64) and sssP1 male flies (n=81) assayed in DD. The activity records are double plotted so that each horizontal line represents data for 2 days. The gray and black bars above each activity record indicate subjective day and night, respectively. (B) Activity records showing average activity in DD for ctrl/sssP1 and ctrl/sssP2 (n=76) versus sssP2/sssP1 (n=65) male flies. Circadian data for ctrl/sssP1 and ctrl/sssP2 were statistically similar and thus pooled. (C) Cycling of PER protein in large ventral lateral neurons in control (ctrl) and sssP1 mutants. Ventral lateral neurons for ctrl and sssP1 animals are stained for PER at indicated Zeitgeber times (ZT). PER protein level is elevated at ZT2 and ZT20, and low at ZT8 and ZT14. (D) Survivorship curves of background control (ctrl, closed diamonds) and sssP1 (open diamonds) flies. Female sss flies (n=187) show a significantly shorter lifespan (P < 0.0001) than controls (n=198). Data from male flies are shown in fig. S5.
Several other behaviors that we tested also appear normal. We find that the phototactic responses of sssP1 mutants are similar to those of controls (fig. S4A), and sssP1 mutants perform as well as controls in a taste discrimination assay (fig. S4B). sssP1 flies (n=43) do not exhibit a bang-sensitive paralytic phenotype, while 89% (n=56) of easily shocked (eas1) flies used as a positive control do. On the other hand, the sssP1 mutants appear somewhat uncoordinated, and fewer mutants are able to climb a specific distance in given times compared to controls (fig. S4C). It is noteworthy, however, that despite their apparent difficulties with coordination, sssP1 mutants spend more time walking than controls and are capable of flying and mating. Consistent with the widely-held view that sleep serves essential biological functions, sssP1 mutants also exhibit a shortened lifespan compared with background controls (Fig. 5D and fig. S5).
sss is allelic to qvr and affects Sh expression
Because two short-sleeping mutants, Sh and Hyperkinetic (Hk), exhibit ether-induced leg shaking we assayed sss mutants for this phenotype (16, 22). We find that both sssP1 and sssP2 mutants show ether-induced leg shaking. Notably, qvr, a mutant for which the underlying molecular defect is unknown, also has a leg-shaking phenotype, and this phenotype has been mapped close to sss (17). Since qvr mutants exhibit impaired Sh-dependent K+ current (18), identification of qvr as an allele of sss would implicate Sh as an effector of SSS function. Genetic and molecular analyses confirm that qvr is indeed an allele of sss. The qvr mutation fails to complement sssP1 for the leg-shaking phenotype. Similarly, after being outcrossed five times, qvr mutants show a significant decrease in sleep compared with wild-type controls, and sssP1/qvr transheterozygotes show a further reduction in sleep (Fig. 6A and fig. S6).
Fig. 6.

sss is allelic to qvr and affects Sh expression. (A) Daily sleep amount for qvr (qvr, n=31), versus background control (ctrl, n=32), as well as ctrl/sssP1 (n=30) versus qvr/sssP1 (n=32) female flies. **P < 0.0001. (B) Altered sss transcripts in qvr mutants. RT-PCR products obtained with qvr and background control (ctrl) RNA and with water used as a negative control (neg). (C) Schematic representation of sss transcripts in qvr mutants. qvr 1, 2, and 3 correspond to the top, middle, and bottom bands, respectively. In background control (ctrl) transcripts, 163 nucleotides of Intron 6 are spliced out. In contrast, the entire intron is present in qvr 1 transcripts. In qvr 2 and 3 transcripts, splice donor sites differ from the one used in wild-type control transcripts, as indicated by the nucleotide numbers for splice sites. (D) Sequence change in qvr genomic DNA in Intron 6 of sss. The fifth nucleotide in Intron 6 has a G to A transition. (E) Altered expression of SSS in qvr mutants. Fly head extracts from background control (ctrl), qvr, and sssP1 flies were analyzed by Western blotting with anti-SSS antibody. (F) Reduced expression of Sh in sss mutants. Western blot analysis of head extracts with anti-Sh antibody reveals a Sh-specific band that is substantially reduced in sssP1 mutants compared with background control (ctrl) flies. Sh14 flies were used to identify a Sh-specific band. Non-specific bands (*) may have obscured additional Sh bands. The experiments in (E) and (F) were performed 3 times with similar results.
We next investigated the molecular basis of the qvr mutation. Reverse-transcriptase polymerase chain reaction (RT-PCR) of sss transcripts in qvr mutants produces three bands, whereas that of wild-type sss transcripts produces a single band (Fig. 6B), indicating splicing defects in qvr mutants. None of the three qvr bands show the same electrophoretic mobility as the wild-type control band. Sequencing of the RT-PCR products reveals altered splicing of the last intron (Intron 6) of sss in the qvr mutant (Fig. 6C). A single base change found in the intron is likely to be responsible for the defective splicing (Fig. 6D). Only one of the three qvr transcripts (qvr 2) is predicted to be in frame (resulting in an insertion of 21 amino acids), and thus has the potential to produce functional SSS protein. Western analysis of qvr mutants reveals a small amount of SSS with a slightly higher apparent molecular weight than wild-type SSS protein, which may correspond to the product of the in-frame qvr 2 transcript (Fig. 6E).
Since qvr mutants were shown to have severely reduced Sh-dependent K+ current (18), we examined whether the Sh protein level is affected in sss mutants. We find that one form of Sh protein is expressed at a substantially reduced level in sssP1 mutants compared with wild-type flies (Fig. 6F), suggesting that the effect of SSS on Sh is at least in part through its protein expression. These results establish SSS as an important regulator of the Sh K+ channel.
Discussion
In summary, we have identified a novel Drosophila gene required for homeostatic regulation of sleep under normal conditions and following sleep deprivation. Although genes have been identified that regulate sleep-wake stability and baseline sleep amount, few have been demonstrated to be important for sleep rebound (13, 15, 30-34). Thus, further analysis of SSS function may provide a rare opportunity to gain mechanistic insight into the homeostatic regulation of sleep.
It is worth noting that sssP2 animals show a moderate reduction in SSS protein and a minimal reduction in baseline sleep, but have severely reduced sleep rebound. The differential requirement for SSS protein in normal versus rebound sleep may be explained in the context of the two-process model of sleep regulation, where sleep is postulated to be controlled by the opposing influences of circadian waking drive and homeostatic sleep drive (11, 35). In this context, for early-morning rebound sleep to occur, a strong homeostatic signal promoting sleep would be required to counteract a strong circadian input keeping the flies awake. At night when circadian waking drive is weaker or absent, a relatively low level of homeostatic input may suffice to allow flies to sleep. The moderate level of SSS protein in sssP2 mutants may be within the range where sleep is possible when a wake-promoting circadian signal is low (at night), but not when it is high (in the early morning). In contrast, sssP1 and sssΔ40 mutants, which have undetectable levels of SSS expression, display severe reductions in both baseline and rebound sleep. In these mutants, the sleep-promoting signal may be too low to allow flies to sleep even when the circadian waking drive is weak at night.
Clues to the role of SSS at the cellular level come from our biochemical characterization of this molecule. The SSS protein is a GPI-anchored membrane protein enriched in the brain. GPI-anchored proteins can function as ligands or co-receptors and can also act as diffusible signals following cleavage of the GPI anchor (36, 37). Although we are unable to detect circadian or homeostatic regulation of the total levels of SSS protein, such regulation may occur at the level of cleavage of the GPI anchor. Regulation of release is known to be controlled by time of day for other proteins that do not cycle in overall levels, such as pigment-dispersing factor, a molecular output of clock neurons (38). Alternatively, SSS may be regulated in a subset of cells that express it, which would be undetectable on our western blots.
A potential mechanism by which SSS regulates sleep is suggested by our finding that qvr is an allele of sss and that the Sh protein level is reduced in sss mutants. Furthermore, qvr mutants exhibit markedly impaired Sh-dependent K+ current at the larval neuromuscular junction (18). Thus we propose that SSS lowers membrane excitability by modulating K+ channel expression and activity. It is striking that among thousands of mutants screened in Drosophila, two with the strongest sleep phenotypes affect the Sh K+ channel (16) and its putative regulator, sss. Reduced membrane excitability may thus be a central feature of sleep. Collectively, our data suggest that SSS is a signaling molecule that links homeostatic sleep drive to neuronal excitability.
Supplementary Material
Supporting Online Material
Materials and Methods
Fly stocks
Flies were maintained on standard food containing molasses, cornmeal, and yeast at room temperature. In our screen for mutants with reduced sleep, a total of 3473 transposon lines were used. The strains used in the original screen and subsequent experiments (except for the P2 insertion) were obtained from the Bloomington Stock Center (Bloomington, Indiana) and the Drosophila Gene Disruption Project (1) (http://flypush.imgen.bcm.tmc.edu/pscreen/). The P2 insertion (f01257) was obtained from the Exelixis collection at the Artavanis-Tsakonas laboratory (Harvard University). eas1 and qvr mutants were obtained from Dr. C.-F. Wu. Sh14 and Hk1 flies were obtained from the Bloomington Stock Center. Shmns was a gift from Dr. C. Cirelli. For the screen, each line was outcrossed to the iso31 background twice and balanced before testing homozygotes. For subsequent examination of sss mutants, sssP1, sssP2, and sssqvr were outcrossed to the iso31 background 5 times, and balanced mutant and sibling control lines were established for each allele. Transgenic fly lines bearing the genomic sss rescue construct (TG1−3) were generated by standard techniques (2) in an iso31 background (Rainbow Transgenics).
Generation of excision lines
Precise and imprecise excision lines were derived from the sssP1 line by mobilizing the P element using the Δ2−3 recombinase. By screening 49 excision lines by PCR amplification and sequencing, we obtained several precise excision lines and one imprecise excision line. The imprecise excision line (Δ40) removes 1069 base pairs (from +1756 to +2824 of the sss genomic region relative to the translational start site). The cDNA from the Δ40 line was sequenced between the translational start site and what corresponds to the stop codon in a wild-type strain; the Δ40 protein is predicted to include the first 35 amino acids of SSS and 24 amino acids unrelated to SSS before encountering a stop codon. As the first 32 amino acids of SSS constitute the signal peptide, only 3 out of 126 amino acids of the mature protein are expected to be intact in the Δ40 line, and thus the allele is likely to be null. In an initial experiment, three precise excision lines were assayed for sleep, and since they all had sleep amounts similar to wild-type control lines, one of them was selected for further characterization.
Sleep and circadian assays
Flies were entrained to a 12 hr:12 hr light:dark (LD) cycle for at least two days before being assayed for sleep in glass tubes containing 5% sucrose and 2% agarose using the Drosophila Activity Monitoring System (Trikinetics) in an incubator at 25°C. For the screen, up to 8 female flies of 5 to 10 days of age were tested per line. In subsequent experiments on sss mutants, 4- to 7-day old male and female flies were monitored for sleep behavior. For sleep measurements, activity counts were collected in 30-sec or 1-min bins in LD for 2 days, and a moving window was used to identify sleep as periods of inactivity lasting at least 5 minutes (3, 4). Sleep parameters were computed using MATLAB-based (MathWorks) custom software. For analysis of circadian behavior, activity counts were collected in 30-min bins in DD over a 6-day period and analyzed using ClockLab (Actimetrics) as previously described (5). One-way analyses of variance (ANOVAs) with genotype as a between-subject factor (and if there was a significant effect) followed by post-hoc comparisons with the Bonferroni correction were used to compare sleep and circadian parameters of more than two genotypes. For comparisons of two genotypes, unpaired t-tests with unequal variances were used. For analysis of sleep bout duration, which is not normally distributed, Mann-Whitney U test was used.
Sleep deprivation
Mechanical stimulation was applied for 2 seconds at random intervals averaging 20 seconds by a custom-built device to deprive flies of sleep for six hours (ZT 18−24) or 12 hours (ZT 12−24) in the second half of the night. Locomotor activity was monitored during mechanical stimulation, and only data from flies that were deprived of sleep by at least 75% compared with baseline conditions were included. Rebound sleep was calculated as the difference in the amount of sleep between the deprived and undisturbed control animals during the first 6 hours following deprivation. To account for individual differences in baseline sleep, pre-deprivation sleep was subtracted from post-deprivation sleep at ZT0−6 for each fly. Similarly, change in sleep latency due to deprivation was computed as the difference in latency to sleep between the deprived and undisturbed animals. Pre-deprivation sleep latency was subtracted from post-deprivation latency at ZT0 to account for individual differences. Two-way ANOVAs with genotype and deprivation as between-subject variables were performed to assess statistical significance of differences in rebound sleep and latency change between control and mutant strains.
Other behavioral assays
For assessment of general behaviors, 5−10 day old female flies were used (unless noted otherwise). Experimental flies were allowed to recover from CO2 anesthesia for at least 1 day prior to testing. To measure phototaxis, a modified version of the fast phototaxis assay was used (6). In the dark, flies were quickly tapped down into a 17×100 mm tube connected to another similar tube, both of which were then laid in a horizontal position. Flies were exposed to light (15W fluorescent bulb) either proximal or distal to the original tube to assess propensity to run away from or towards the light, respectively. After 30 seconds, the number of flies in the original tube was counted. To assess the ability of flies to distinguish between attractive and aversive tastes, animals were given a modified two-choice preference test (7). 2% agarose plus 1 mM or 5 mM sucrose was evenly split across the bottoms of 17×100 mm vials. Each of the two food sources was supplemented with either red or blue food coloring, and in one set of experiments 1 mM quinine (Sigma) was added to the higher concentration of sugar. Flies were starved for 12−16 hrs, then added to vials and allowed to feed for 1 hr in the dark (to avoid influence of food color). After feeding, animals were frozen and examined visually for feeding preference by assessing the color of their abdomens. To assess bang-sensitivity, male flies were vortexed in vials at maximum speed for 10 seconds and examined for paralysis; eas1 flies were used as a positive control. For climbing assays, flies were gently tapped down into a vertical 17×100 mm tube, and the number of flies able to climb 9 cm in 5 and 10 seconds was counted. To elicit ether-induced leg shaking, we anesthetized flies using diethyl ether (Sigma) and observed for the characteristic high-frequency leg-shaking phenotype. Shmns flies were used as a positive control for ether-induced leg-shaking. Differences in all general behavioral assays were assessed statistically using Chi-square tests.
Longevity assay
Background control and sssP1 mutant flies were maintained in a 12 hr:12 hr LD cycle at 25°C. Groups of about 30 flies (males and females mixed) were collected into vials within 24 hr of eclosion. Flies were transferred to fresh vials and the number of dead flies counted every 2 days. Log-rank tests were performed to compare longevity of sss flies to that of control flies.
Molecular Biology
mRNA from adult fly heads was isolated using the Ultraspec RNA Isolation System (Biotecx) and reverse transcribed using Superscript III (Invitrogen). sss cDNA was amplified by RT-PCR using primers encoded by 5’-GGT TGG CCA GTA GTA ACT GGG AC-3’ and 5’-GTC GAC GAG CCT AAC ACT TTC TAT CTG CTG AGC-3’. Three independent clones derived from multiple PCRs were subcloned using the TOPO TA-cloning system (Invitrogen) and sequenced in both directions. The cloned sss open reading frame is the same as the predicted sequence CG33472-RB in Flybase except for a few base changes. We did not observe RT-PCR products corresponding to the other predicted sequence CG33472-RA, suggesting that it is either rare or artifactual.
To construct pIZ-sss, the cloned sss cDNA was PCR amplified using the following primers: 5’-CGG AAT TCC GGC AAG ATG TGG ACG C-3’ and 5’-AAC TCG AGC TAT CTG CTG AGC AAT TGA CC-3’. The PCR fragment was then inserted into the pIZ/V5-His vector (Invitrogen), and the construct was verified by sequencing.
To generate the genomic sss rescue construct, ∼9.8 kb of genomic sequence containing the entire 5’UTR and 3’UTR was recombined into the P[acman] vector by gap repair as described (8). Primers to amplify the left homology arm (LA) were designed ∼400 bp upstream of the start of the 5’UTR and were as follows: 5’-CTT GTA CTC TCA TGC GCT C-3’ and 5’-CCA CAA CAC TTT AGT GCA TCG C-3’. Primers to amplify the right homology arm (RA) were designed ∼300 bp downstream of the end of the 3’UTR and were as follows: 5’-GGT GCT TCC AAC TCG CTT TGC-3’ and 5’-CGT GCG AGC TAT CGG AAA CAC TC-3’. LA and RA were cloned into the P[acman] vector and confirmed by sequencing. Recombination was induced between linearized P[acman]-LA/RA and BACR09A11 (Children's Hospital Oakland Research Institute), and the desired recombinant was detected by PCR and then partially sequenced to confirm recombination.
To determine the molecular basis of the qvr mutation, we sequenced the coding region and intron-exon boundaries of the genomic DNA of qvr mutants and wild-type control flies, and did not find any sequence difference that would cause an amino acid substitution. We observed a few base changes in introns, however, and to determine if splicing is altered, we amplified sss cDNA in qvr mutants by RT-PCR using primers encoded by 5’-CGG AAT TCC GGC AAG ATG TGG ACG C-3’ and 5’-AAC TCG AGC TAT CTG CTG AGC AAT TGA CC-3’. Three distinct bands were observed in qvr mutants compared with a single band in wild-type flies. All three qvr bands were sequenced, revealing altered splicing of Intron 6. Two of the three transcripts are predicted to introduce a frame shift, but one of them is predicted to be in frame resulting in a 21 amino acid insertion.
Transient transfection and PI-PLC treatment
Drosophila S2R+ (9) cells were transfected with pIZ-sss (150 ng) in 24-well plates using Effectene (Qiagen). Cells were maintained at room temperature for two or three days before being processed for Western analysis or immunostaining. For PI-PLC treatment, cells were washed in PBS (10 mM phosphate buffer, pH 7.2, 0.15 M NaCl) once, and were incubated with or without PI-PLC (1 U/mL, Sigma) in PBS for 1 hr at 28°C.
Western analysis and antibody production
Western blot analysis of S2R+ cell lysates and fly head extracts was performed as described (5, 10). For comparing SSS expression levels of different genotypes, head extracts from 8 females were loaded per lane. For comparison of different tissues, an equal amount of total protein (∼40 ug) was loaded per lane. To assay release of SSS by PI-PLC treatment, protein in the medium was concentrated about 20-fold using a Microcon YM-10 filter (Millipore), and 100% of the concentrated medium or 8% of the cell extract was loaded per lane. The PA0681 rabbit antibody to SSS was raised against a peptide: DSWTDARCKDPFNYTALPR (Open Biosystems). We did not detect specific staining by the antibody in whole-mount brain samples, probably because the antibody poorly recognizes glycosylated SSS. We were able to circumvent this problem in Western analysis by first deglycosylating blots using Peptide N-Glycosidase F (PNGase F, New England Biolabs) before incubating them with the antibody to SSS. Antibodies to SSS, Sh (DN16, Santa Cruz biotechnology), and MAPK (Sigma) were used at 1:500, 1:1000, and 1:2500, respectively.
Immunostaining
Flies entrained to a 12 hr:12 hr LD cycle were collected at ZT2, 8, 14, and 20, and immunostaining of whole-mount brain samples was performed as described (10). Samples were incubated with antibodies to PER (UPR34) at 1:1500 and Pigment Dispensing Factor (PDF, HH74) at 1:1000. PDF staining was used to identify ventral lateral neurons. Four to six fly brains were examined per condition. For immunostaining of S2R+ cells, transfected cells were fixed with 4% formaldehyde in PBS for 30 min at room temperature. After three quick washes with PBS, non-specific binding was blocked with culture medium (10% fetal bovine serum in Schneider's medium, Gibco). Cells were then incubated with antibody to SSS in culture medium at 1:250 for 1 hr, washed with culture medium for 15 min three times, and incubated with Cy3-conjugated anti-rabbit antibody at 1:500 for 1hr, followed by three washes with culture medium. To permeabilize cells, 0.1% Triton X-100 was added to the culture medium during fixing, antibody incubation and washing. Immunostained samples were imaged with a Leica confocal microscope.
Supplementary Table 1. Circadian rhythm parameters of sss mutants and controls.
References for Supporting Online Material
1. H. J. Bellen et al., Genetics 167, 761 (2004).
2. G. M. Rubin, A. C. Spradling, Science 218, 348 (1982).
3. P. J. Shaw, C. Cirelli, R. J. Greenspan, G. Tononi, Science 287, 1834 (2000).
4. R. Andretic, P. J. Shaw, Methods Enzymol 393, 759 (2005).
5. J. A. Williams, H. S. Su, A. Bernards, J. Field, A. Sehgal, Science 293, 2251 (2001).
6. S. Benzer, Proc Natl Acad Sci U S A 58, 1112 (1967).
7. S. J. Moon, M. Kottgen, Y. Jiao, H. Xu, C. Montell, Curr Biol 16, 1812 (2006).
8. K. J. Venken, Y. He, R. A. Hoskins, H. J. Bellen, Science 314, 1747 (2006).
9. S. Yanagawa, J. S. Lee, A. Ishimoto, J Biol Chem 273, 32353 (1998).
10. S. Sathyanarayanan, X. Zheng, R. Xiao, A. Sehgal, Cell 116, 603 (2004).
fig. S1. Regulation of SSS protein. (A) Circadian profile of SSS protein in head extracts. Wild-type fly heads were collected at indicated Zeitgeber times (ZT), and SSS levels were determined by Western blot analysis. (B) SSS protein levels in head extracts do not change in response to sleep deprivation. The SSS protein level of wild-type (iso31) flies that were deprived of sleep for 8 hours during ZT 12−20 (Dep) is comparable to that of wild-type flies that were not deprived (Non-dep). Each of these experiments was performed 3 times with similar results.
fig. S2. Genetic analysis of sss flies. (A-B) Deficiency mapping of the sss mutation. Daily sleep in minutes is shown for Df(2R)ED2219 (Df1) and Df(2R)en-B (Df2) flies crossed to either background control (ctrl) or sssP1 flies. 21−65 female (A) and 18−42 male (B) flies were tested in each condition. (C) Daily sleep amount for precise excision (Pr; n=25), sssΔ40 imprecise excision (Im, n=18) , precise/sssP1 (Pr/sssP1, n=29), and imprecise/sssP1 (Im/sssP1, n=33) male flies. (D) Schematic of the sss genomic region and the imprecise excision allele, Δ40. The bracket indicates bases deleted in the Δ40 allele. (E) Daily sleep amount for male sssP1 mutant flies with (sssP1;TG1−3, n=16, 8, 15, respectively) or without (sssP1, n=13) a genomic sss transgene. TG1−3 refer to three independent transgenic insertions and either 1 or 2 copies of the transgene were present in the flies tested. (F) Daily sleep amount for sssP2 (n=106) versus background control (ctrl, n=80), as well as ctrl/sssP1 (n=79) versus sssP2/sssP1 (n=112) male flies. *P < 0.05; **P < 0.0001.
fig. S3. Reduced homeostatic response to sleep deprivation in male sss mutants. (A) Amount of sleep lost during 6 or 12 hours of deprivation at the end of the dark period for background control (ctrl), sssP2, ctrl/sssP1, and sssP2/sssP1 flies. Data from 18−60 male flies are presented. (B) Amount of sleep gained during 6 hours of recovery following deprivation as in (A). (C) Change in sleep latency following deprivation, compared to undisturbed controls as in (A). *P < 0.05; **P < 0.001.
fig. S4. General behavioral assays for sss mutants. (A) Percent of control and sssP1 flies that run toward a distal light source or away from a proximal light source is shown. 79−82 flies were tested in each condition. (B) Percent of control and sssP1 flies that choose 5 mM over 1 mM sucrose is shown. Addition of food coloring (red or blue) or quinine to the 5 mM sucrose condition is denoted below the results of each of three experiments. No preference is observed for color of food. Control and mutant flies have an equivalent preference for 5 mM over 1 mM sucrose and an equivalent avoidance of 1 mM quinine in the presence of the higher concentration of sugar. 26−95 animals were used in each condition. (C) Percent of control and sssP1 flies that climb 9 cm in either 5 or 10 seconds is shown. 74−83 animals were tested in each condition. In all three panels, white depicts control and black depicts sssP1 animals. *P < 0.05; **P < 0.0001.
Fig. S5. Survivorship curves of background control (ctrl, closed diamonds) and sssP1 (open diamonds) flies. Male sssP1 flies (n=154) show a significantly shorter lifespan (P < 0.0001) than male control flies (n=161).
fig. S6. Daily sleep amount for qvr (n=32) versus background control (ctrl, n=29), as well as ctrl/sssP1 (n=30) versus qvr/sssP1 (n=31) male flies. **P < 0.0001.
Footnotes
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. Their manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References and notes
- 1.Rosekind MR. Sleep Med. 2005;6(Suppl 1):S21. doi: 10.1016/s1389-9457(05)80005-x. [DOI] [PubMed] [Google Scholar]
- 2.Siegel JM. Nature. 2005;437:1264. doi: 10.1038/nature04285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell SS, Tobler I. Neurosci Biobehav Rev. 1984;8:269. doi: 10.1016/0149-7634(84)90054-x. [DOI] [PubMed] [Google Scholar]
- 4.Hendricks JC, et al. Neuron. 2000;25:129. doi: 10.1016/s0896-6273(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 5.Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. Science. 2000;287:1834. doi: 10.1126/science.287.5459.1834. [DOI] [PubMed] [Google Scholar]
- 6.Greenspan RJ, Tononi G, Cirelli C, Shaw PJ. Trends Neurosci. 2001;24:142. doi: 10.1016/s0166-2236(00)01719-7. [DOI] [PubMed] [Google Scholar]
- 7.Yokogawa T, et al. PLoS Biol. 2007;5:2379. doi: 10.1371/journal.pbio.0050277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rechtschaffen A, Gilliland MA, Bergmann BM, Winter JB. Science. 1983;221:182. doi: 10.1126/science.6857280. [DOI] [PubMed] [Google Scholar]
- 9.Shaw PJ, Tononi G, Greenspan RJ, Robinson DF. Nature. 2002;417:287. doi: 10.1038/417287a. [DOI] [PubMed] [Google Scholar]
- 10.Pitman JL, McGill JJ, Keegan KP, Allada R. Nature. 2006;441:753. doi: 10.1038/nature04739. [DOI] [PubMed] [Google Scholar]
- 11.Borbely AA, Achermann P. J Biol Rhythms. 1999;14:557. doi: 10.1177/074873099129000894. [DOI] [PubMed] [Google Scholar]
- 12.Saper CB, Cano G, Scammell TE. J Comp Neurol. 2005;493:92. doi: 10.1002/cne.20770. [DOI] [PubMed] [Google Scholar]
- 13.Kume K, Kume S, Park SK, Hirsh J, Jackson FR. J Neurosci. 2005;25:7377. doi: 10.1523/JNEUROSCI.2048-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendricks JC, et al. Nat Neurosci. 2001;4:1108. doi: 10.1038/nn743. [DOI] [PubMed] [Google Scholar]
- 15.Foltenyi K, Greenspan RJ, Newport JW. Nat Neurosci. 2007;10:1160. doi: 10.1038/nn1957. [DOI] [PubMed] [Google Scholar]
- 16.Cirelli C, et al. Nature. 2005;434:1087. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]
- 17.Humphreys JM, Duyf B, Joiner ML, Phillips JP, Hilliker AJ. Genome. 1996;39:749. doi: 10.1139/g96-094. [DOI] [PubMed] [Google Scholar]
- 18.Wang JW, Humphreys JM, Phillips JP, Hilliker AJ, Wu CF. J Neurosci. 2000;20:5958. doi: 10.1523/JNEUROSCI.20-16-05958.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryder E, et al. Genetics. 2004;167:797. doi: 10.1534/genetics.104.026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andretic R, Shaw PJ. Methods Enzymol. 2005;393:759. doi: 10.1016/S0076-6879(05)93040-1. [DOI] [PubMed] [Google Scholar]
- 21.Chintapalli VR, Wang J, Dow JA. Nat Genet. 2007;39:715. doi: 10.1038/ng2049. [DOI] [PubMed] [Google Scholar]
- 22.Bushey D, Huber R, Tononi G, Cirelli C. J Neurosci. 2007;27:5384. doi: 10.1523/JNEUROSCI.0108-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu WP, et al. Sleep. 2007;30:247. [PMC free article] [PubMed] [Google Scholar]
- 24.Huber R, et al. Sleep. 2004;27:628. doi: 10.1093/sleep/27.4.628. [DOI] [PubMed] [Google Scholar]
- 25.Huber R, Deboer T, Tobler I. Brain Res. 2000;857:8. doi: 10.1016/s0006-8993(99)02248-9. [DOI] [PubMed] [Google Scholar]
- 26.Naylor E, et al. J Neurosci. 2000;20:8138. doi: 10.1523/JNEUROSCI.20-21-08138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wisor JP, et al. BMC Neurosci. 2002;3:20. doi: 10.1186/1471-2202-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laposky A, et al. Sleep. 2005;28:395. doi: 10.1093/sleep/28.4.395. [DOI] [PubMed] [Google Scholar]
- 29.Hendricks JC, et al. J Biol Rhythms. 2003;18:12. doi: 10.1177/0748730402239673. [DOI] [PubMed] [Google Scholar]
- 30.Lin L, et al. Cell. 1999;98:365. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 31.Chemelli RM, et al. Cell. 1999;98:437. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- 32.Wisor JP, et al. J Neurosci. 2001;21:1787. doi: 10.1523/JNEUROSCI.21-05-01787.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kramer A, et al. Science. 2001;294:2511. doi: 10.1126/science.1067716. [DOI] [PubMed] [Google Scholar]
- 34.Kapfhamer D, et al. Nat Genet. 2002;32:290. doi: 10.1038/ng991. [DOI] [PubMed] [Google Scholar]
- 35.Edgar DM, Dement WC, Fuller CA. J Neurosci. 1993;13:1065. doi: 10.1523/JNEUROSCI.13-03-01065.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hattori M, Osterfield M, Flanagan JG. Science. 2000;289:1360. doi: 10.1126/science.289.5483.1360. [DOI] [PubMed] [Google Scholar]
- 37.Paratcha G, et al. Neuron. 2001;29:171. doi: 10.1016/s0896-6273(01)00188-x. [DOI] [PubMed] [Google Scholar]
- 38.Park JH, et al. Proc Natl Acad Sci U S A. 2000;97:3608. doi: 10.1073/pnas.070036197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.We thank S. Artavanis-Tsakonas, C.-F. Wu, and C. Cirelli for fly strains. We are grateful to Y. He, H. Bellen, and the Bloomington Stock Center for sending stocks for the screen. This work was supported in part by a grant from the NIH (AG017628) to A.S. and K.K., a University Research Foundation award from the University of Pennsylvania (K.K.), and a Career Award for Medical Scientists from the Burroughs-Wellcome Foundation (M.W.). A.S. is an Investigator of the Howard Hughes Medical Institute.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Online Material
Materials and Methods
Fly stocks
Flies were maintained on standard food containing molasses, cornmeal, and yeast at room temperature. In our screen for mutants with reduced sleep, a total of 3473 transposon lines were used. The strains used in the original screen and subsequent experiments (except for the P2 insertion) were obtained from the Bloomington Stock Center (Bloomington, Indiana) and the Drosophila Gene Disruption Project (1) (http://flypush.imgen.bcm.tmc.edu/pscreen/). The P2 insertion (f01257) was obtained from the Exelixis collection at the Artavanis-Tsakonas laboratory (Harvard University). eas1 and qvr mutants were obtained from Dr. C.-F. Wu. Sh14 and Hk1 flies were obtained from the Bloomington Stock Center. Shmns was a gift from Dr. C. Cirelli. For the screen, each line was outcrossed to the iso31 background twice and balanced before testing homozygotes. For subsequent examination of sss mutants, sssP1, sssP2, and sssqvr were outcrossed to the iso31 background 5 times, and balanced mutant and sibling control lines were established for each allele. Transgenic fly lines bearing the genomic sss rescue construct (TG1−3) were generated by standard techniques (2) in an iso31 background (Rainbow Transgenics).
Generation of excision lines
Precise and imprecise excision lines were derived from the sssP1 line by mobilizing the P element using the Δ2−3 recombinase. By screening 49 excision lines by PCR amplification and sequencing, we obtained several precise excision lines and one imprecise excision line. The imprecise excision line (Δ40) removes 1069 base pairs (from +1756 to +2824 of the sss genomic region relative to the translational start site). The cDNA from the Δ40 line was sequenced between the translational start site and what corresponds to the stop codon in a wild-type strain; the Δ40 protein is predicted to include the first 35 amino acids of SSS and 24 amino acids unrelated to SSS before encountering a stop codon. As the first 32 amino acids of SSS constitute the signal peptide, only 3 out of 126 amino acids of the mature protein are expected to be intact in the Δ40 line, and thus the allele is likely to be null. In an initial experiment, three precise excision lines were assayed for sleep, and since they all had sleep amounts similar to wild-type control lines, one of them was selected for further characterization.
Sleep and circadian assays
Flies were entrained to a 12 hr:12 hr light:dark (LD) cycle for at least two days before being assayed for sleep in glass tubes containing 5% sucrose and 2% agarose using the Drosophila Activity Monitoring System (Trikinetics) in an incubator at 25°C. For the screen, up to 8 female flies of 5 to 10 days of age were tested per line. In subsequent experiments on sss mutants, 4- to 7-day old male and female flies were monitored for sleep behavior. For sleep measurements, activity counts were collected in 30-sec or 1-min bins in LD for 2 days, and a moving window was used to identify sleep as periods of inactivity lasting at least 5 minutes (3, 4). Sleep parameters were computed using MATLAB-based (MathWorks) custom software. For analysis of circadian behavior, activity counts were collected in 30-min bins in DD over a 6-day period and analyzed using ClockLab (Actimetrics) as previously described (5). One-way analyses of variance (ANOVAs) with genotype as a between-subject factor (and if there was a significant effect) followed by post-hoc comparisons with the Bonferroni correction were used to compare sleep and circadian parameters of more than two genotypes. For comparisons of two genotypes, unpaired t-tests with unequal variances were used. For analysis of sleep bout duration, which is not normally distributed, Mann-Whitney U test was used.
Sleep deprivation
Mechanical stimulation was applied for 2 seconds at random intervals averaging 20 seconds by a custom-built device to deprive flies of sleep for six hours (ZT 18−24) or 12 hours (ZT 12−24) in the second half of the night. Locomotor activity was monitored during mechanical stimulation, and only data from flies that were deprived of sleep by at least 75% compared with baseline conditions were included. Rebound sleep was calculated as the difference in the amount of sleep between the deprived and undisturbed control animals during the first 6 hours following deprivation. To account for individual differences in baseline sleep, pre-deprivation sleep was subtracted from post-deprivation sleep at ZT0−6 for each fly. Similarly, change in sleep latency due to deprivation was computed as the difference in latency to sleep between the deprived and undisturbed animals. Pre-deprivation sleep latency was subtracted from post-deprivation latency at ZT0 to account for individual differences. Two-way ANOVAs with genotype and deprivation as between-subject variables were performed to assess statistical significance of differences in rebound sleep and latency change between control and mutant strains.
Other behavioral assays
For assessment of general behaviors, 5−10 day old female flies were used (unless noted otherwise). Experimental flies were allowed to recover from CO2 anesthesia for at least 1 day prior to testing. To measure phototaxis, a modified version of the fast phototaxis assay was used (6). In the dark, flies were quickly tapped down into a 17×100 mm tube connected to another similar tube, both of which were then laid in a horizontal position. Flies were exposed to light (15W fluorescent bulb) either proximal or distal to the original tube to assess propensity to run away from or towards the light, respectively. After 30 seconds, the number of flies in the original tube was counted. To assess the ability of flies to distinguish between attractive and aversive tastes, animals were given a modified two-choice preference test (7). 2% agarose plus 1 mM or 5 mM sucrose was evenly split across the bottoms of 17×100 mm vials. Each of the two food sources was supplemented with either red or blue food coloring, and in one set of experiments 1 mM quinine (Sigma) was added to the higher concentration of sugar. Flies were starved for 12−16 hrs, then added to vials and allowed to feed for 1 hr in the dark (to avoid influence of food color). After feeding, animals were frozen and examined visually for feeding preference by assessing the color of their abdomens. To assess bang-sensitivity, male flies were vortexed in vials at maximum speed for 10 seconds and examined for paralysis; eas1 flies were used as a positive control. For climbing assays, flies were gently tapped down into a vertical 17×100 mm tube, and the number of flies able to climb 9 cm in 5 and 10 seconds was counted. To elicit ether-induced leg shaking, we anesthetized flies using diethyl ether (Sigma) and observed for the characteristic high-frequency leg-shaking phenotype. Shmns flies were used as a positive control for ether-induced leg-shaking. Differences in all general behavioral assays were assessed statistically using Chi-square tests.
Longevity assay
Background control and sssP1 mutant flies were maintained in a 12 hr:12 hr LD cycle at 25°C. Groups of about 30 flies (males and females mixed) were collected into vials within 24 hr of eclosion. Flies were transferred to fresh vials and the number of dead flies counted every 2 days. Log-rank tests were performed to compare longevity of sss flies to that of control flies.
Molecular Biology
mRNA from adult fly heads was isolated using the Ultraspec RNA Isolation System (Biotecx) and reverse transcribed using Superscript III (Invitrogen). sss cDNA was amplified by RT-PCR using primers encoded by 5’-GGT TGG CCA GTA GTA ACT GGG AC-3’ and 5’-GTC GAC GAG CCT AAC ACT TTC TAT CTG CTG AGC-3’. Three independent clones derived from multiple PCRs were subcloned using the TOPO TA-cloning system (Invitrogen) and sequenced in both directions. The cloned sss open reading frame is the same as the predicted sequence CG33472-RB in Flybase except for a few base changes. We did not observe RT-PCR products corresponding to the other predicted sequence CG33472-RA, suggesting that it is either rare or artifactual.
To construct pIZ-sss, the cloned sss cDNA was PCR amplified using the following primers: 5’-CGG AAT TCC GGC AAG ATG TGG ACG C-3’ and 5’-AAC TCG AGC TAT CTG CTG AGC AAT TGA CC-3’. The PCR fragment was then inserted into the pIZ/V5-His vector (Invitrogen), and the construct was verified by sequencing.
To generate the genomic sss rescue construct, ∼9.8 kb of genomic sequence containing the entire 5’UTR and 3’UTR was recombined into the P[acman] vector by gap repair as described (8). Primers to amplify the left homology arm (LA) were designed ∼400 bp upstream of the start of the 5’UTR and were as follows: 5’-CTT GTA CTC TCA TGC GCT C-3’ and 5’-CCA CAA CAC TTT AGT GCA TCG C-3’. Primers to amplify the right homology arm (RA) were designed ∼300 bp downstream of the end of the 3’UTR and were as follows: 5’-GGT GCT TCC AAC TCG CTT TGC-3’ and 5’-CGT GCG AGC TAT CGG AAA CAC TC-3’. LA and RA were cloned into the P[acman] vector and confirmed by sequencing. Recombination was induced between linearized P[acman]-LA/RA and BACR09A11 (Children's Hospital Oakland Research Institute), and the desired recombinant was detected by PCR and then partially sequenced to confirm recombination.
To determine the molecular basis of the qvr mutation, we sequenced the coding region and intron-exon boundaries of the genomic DNA of qvr mutants and wild-type control flies, and did not find any sequence difference that would cause an amino acid substitution. We observed a few base changes in introns, however, and to determine if splicing is altered, we amplified sss cDNA in qvr mutants by RT-PCR using primers encoded by 5’-CGG AAT TCC GGC AAG ATG TGG ACG C-3’ and 5’-AAC TCG AGC TAT CTG CTG AGC AAT TGA CC-3’. Three distinct bands were observed in qvr mutants compared with a single band in wild-type flies. All three qvr bands were sequenced, revealing altered splicing of Intron 6. Two of the three transcripts are predicted to introduce a frame shift, but one of them is predicted to be in frame resulting in a 21 amino acid insertion.
Transient transfection and PI-PLC treatment
Drosophila S2R+ (9) cells were transfected with pIZ-sss (150 ng) in 24-well plates using Effectene (Qiagen). Cells were maintained at room temperature for two or three days before being processed for Western analysis or immunostaining. For PI-PLC treatment, cells were washed in PBS (10 mM phosphate buffer, pH 7.2, 0.15 M NaCl) once, and were incubated with or without PI-PLC (1 U/mL, Sigma) in PBS for 1 hr at 28°C.
Western analysis and antibody production
Western blot analysis of S2R+ cell lysates and fly head extracts was performed as described (5, 10). For comparing SSS expression levels of different genotypes, head extracts from 8 females were loaded per lane. For comparison of different tissues, an equal amount of total protein (∼40 ug) was loaded per lane. To assay release of SSS by PI-PLC treatment, protein in the medium was concentrated about 20-fold using a Microcon YM-10 filter (Millipore), and 100% of the concentrated medium or 8% of the cell extract was loaded per lane. The PA0681 rabbit antibody to SSS was raised against a peptide: DSWTDARCKDPFNYTALPR (Open Biosystems). We did not detect specific staining by the antibody in whole-mount brain samples, probably because the antibody poorly recognizes glycosylated SSS. We were able to circumvent this problem in Western analysis by first deglycosylating blots using Peptide N-Glycosidase F (PNGase F, New England Biolabs) before incubating them with the antibody to SSS. Antibodies to SSS, Sh (DN16, Santa Cruz biotechnology), and MAPK (Sigma) were used at 1:500, 1:1000, and 1:2500, respectively.
Immunostaining
Flies entrained to a 12 hr:12 hr LD cycle were collected at ZT2, 8, 14, and 20, and immunostaining of whole-mount brain samples was performed as described (10). Samples were incubated with antibodies to PER (UPR34) at 1:1500 and Pigment Dispensing Factor (PDF, HH74) at 1:1000. PDF staining was used to identify ventral lateral neurons. Four to six fly brains were examined per condition. For immunostaining of S2R+ cells, transfected cells were fixed with 4% formaldehyde in PBS for 30 min at room temperature. After three quick washes with PBS, non-specific binding was blocked with culture medium (10% fetal bovine serum in Schneider's medium, Gibco). Cells were then incubated with antibody to SSS in culture medium at 1:250 for 1 hr, washed with culture medium for 15 min three times, and incubated with Cy3-conjugated anti-rabbit antibody at 1:500 for 1hr, followed by three washes with culture medium. To permeabilize cells, 0.1% Triton X-100 was added to the culture medium during fixing, antibody incubation and washing. Immunostained samples were imaged with a Leica confocal microscope.
Supplementary Table 1. Circadian rhythm parameters of sss mutants and controls.
References for Supporting Online Material
1. H. J. Bellen et al., Genetics 167, 761 (2004).
2. G. M. Rubin, A. C. Spradling, Science 218, 348 (1982).
3. P. J. Shaw, C. Cirelli, R. J. Greenspan, G. Tononi, Science 287, 1834 (2000).
4. R. Andretic, P. J. Shaw, Methods Enzymol 393, 759 (2005).
5. J. A. Williams, H. S. Su, A. Bernards, J. Field, A. Sehgal, Science 293, 2251 (2001).
6. S. Benzer, Proc Natl Acad Sci U S A 58, 1112 (1967).
7. S. J. Moon, M. Kottgen, Y. Jiao, H. Xu, C. Montell, Curr Biol 16, 1812 (2006).
8. K. J. Venken, Y. He, R. A. Hoskins, H. J. Bellen, Science 314, 1747 (2006).
9. S. Yanagawa, J. S. Lee, A. Ishimoto, J Biol Chem 273, 32353 (1998).
10. S. Sathyanarayanan, X. Zheng, R. Xiao, A. Sehgal, Cell 116, 603 (2004).
fig. S1. Regulation of SSS protein. (A) Circadian profile of SSS protein in head extracts. Wild-type fly heads were collected at indicated Zeitgeber times (ZT), and SSS levels were determined by Western blot analysis. (B) SSS protein levels in head extracts do not change in response to sleep deprivation. The SSS protein level of wild-type (iso31) flies that were deprived of sleep for 8 hours during ZT 12−20 (Dep) is comparable to that of wild-type flies that were not deprived (Non-dep). Each of these experiments was performed 3 times with similar results.
fig. S2. Genetic analysis of sss flies. (A-B) Deficiency mapping of the sss mutation. Daily sleep in minutes is shown for Df(2R)ED2219 (Df1) and Df(2R)en-B (Df2) flies crossed to either background control (ctrl) or sssP1 flies. 21−65 female (A) and 18−42 male (B) flies were tested in each condition. (C) Daily sleep amount for precise excision (Pr; n=25), sssΔ40 imprecise excision (Im, n=18) , precise/sssP1 (Pr/sssP1, n=29), and imprecise/sssP1 (Im/sssP1, n=33) male flies. (D) Schematic of the sss genomic region and the imprecise excision allele, Δ40. The bracket indicates bases deleted in the Δ40 allele. (E) Daily sleep amount for male sssP1 mutant flies with (sssP1;TG1−3, n=16, 8, 15, respectively) or without (sssP1, n=13) a genomic sss transgene. TG1−3 refer to three independent transgenic insertions and either 1 or 2 copies of the transgene were present in the flies tested. (F) Daily sleep amount for sssP2 (n=106) versus background control (ctrl, n=80), as well as ctrl/sssP1 (n=79) versus sssP2/sssP1 (n=112) male flies. *P < 0.05; **P < 0.0001.
fig. S3. Reduced homeostatic response to sleep deprivation in male sss mutants. (A) Amount of sleep lost during 6 or 12 hours of deprivation at the end of the dark period for background control (ctrl), sssP2, ctrl/sssP1, and sssP2/sssP1 flies. Data from 18−60 male flies are presented. (B) Amount of sleep gained during 6 hours of recovery following deprivation as in (A). (C) Change in sleep latency following deprivation, compared to undisturbed controls as in (A). *P < 0.05; **P < 0.001.
fig. S4. General behavioral assays for sss mutants. (A) Percent of control and sssP1 flies that run toward a distal light source or away from a proximal light source is shown. 79−82 flies were tested in each condition. (B) Percent of control and sssP1 flies that choose 5 mM over 1 mM sucrose is shown. Addition of food coloring (red or blue) or quinine to the 5 mM sucrose condition is denoted below the results of each of three experiments. No preference is observed for color of food. Control and mutant flies have an equivalent preference for 5 mM over 1 mM sucrose and an equivalent avoidance of 1 mM quinine in the presence of the higher concentration of sugar. 26−95 animals were used in each condition. (C) Percent of control and sssP1 flies that climb 9 cm in either 5 or 10 seconds is shown. 74−83 animals were tested in each condition. In all three panels, white depicts control and black depicts sssP1 animals. *P < 0.05; **P < 0.0001.
Fig. S5. Survivorship curves of background control (ctrl, closed diamonds) and sssP1 (open diamonds) flies. Male sssP1 flies (n=154) show a significantly shorter lifespan (P < 0.0001) than male control flies (n=161).
fig. S6. Daily sleep amount for qvr (n=32) versus background control (ctrl, n=29), as well as ctrl/sssP1 (n=30) versus qvr/sssP1 (n=31) male flies. **P < 0.0001.