

Abstract
Objectives
Increased formation of reactive oxygen species (ROS) has been identified as a causative factor in endothelial dysfunction by reducing NO bioavailability and uncoupling endothelial nitric oxide synthase (eNOS). However, the specific contribution of ROS to endothelial function is not well understood.
Methods and Results
A major source of intracellular ROS is the NADPH oxidase (Nox) family of enzymes. The goal of the current study was to directly assess the contribution of NADPH oxidase derived superoxide to eNOS function by expressing Nox5, a single gene product that constitutively produces superoxide within cells. Paradoxically, we found that instead of inhibiting eNOS, co-expression of Nox5 increased NO release from both bovine and human endothelial cells. To establish the functional significance of this observation in intact blood vessels, the endothelium of mouse aorta was transduced with Nox5 or control adenoviruses. Nox5 potently inhibited Ach-induced relaxation and potentiated contractile responses to phenylephrine. In precontracted aortae, acute exposure to superoxide dismutase induced significant vascular relaxation in vessels exposed to Nox5 versus control and unmasked the ability of Nox5 to activate eNOS in blood vessel endothelium.
Conclusions
These findings suggest that ROS inhibit eNOS function via consumption of NO rather than direct inhibition of enzymatic activity.
Keywords: nitric oxide, endothelial nitric oxide synthase, NADPH oxidase, endothelial cells, superoxide dismutase
Reactive oxygen species (ROS) are leading candidates in the etiology of endothelial dysfunction and ensuing cardiovascular disease1. Increased superoxide formation in endothelial cells (EC) has been identified as a causative factor in this process by reducing NO bioavailability, uncoupling eNOS via BH4 depletion or homodimer disruption and also by altering redox-sensitive signaling cascades. However, elevated superoxide production is frequently accompanied by changes in blood pressure, cellular signaling, hormones and the composition of the extracellular milieu making it difficult to ascertain the independent effects of intracellular ROS. For example, endothelial function is reduced in animal models of type II diabetes, angiotensin-dependent hypertension and atherosclerosis and this deficit is accompanied by significant increases in superoxide formation2-4. However, whether increased superoxide is the causative factor, a participant or requires the cooperation of other factors present in the extracellular milieu is not yet known.
Within vascular cells, there are several sources of ROS including the mitochondrial electron transport chain, xanthine oxidase, arachidonic acid oxygenases (lipogenase, cyclooxygenase, cytochrome P450), uncoupled eNOS and NADPH oxidases (Noxs)5. However, superoxide production is an unintended secondary product in all of these enzymes except for the Nox isoforms which have acquired the unique capacity to exclusively synthesize superoxide. Endothelial cells primarily express Nox2 and Nox46, 7 and changes in the expression and/or activity of these enzymes have been reported in diabetes8, 9 hypertension and in response to angiotensin ∥10-12 }. The functional consequences of superoxide production from NADPH oxidases versus other cellular sources such as the mitochondria are not well defined. Increased superoxide anion and other reactive oxygen species have been proposed to contribute to endothelial dysfunction3, elevated blood pressure13, 14 and increased cellular proliferation and hypertrophy 15, 16. The restricted intracellular distribution of Noxs to specific locations such as the endoplasmic reticulum (ER) suggests that they may have important roles in modulating discrete aspects of intracellular signaling. However, the individual role of NADPH oxidases in these processes remains uncertain due to the presence of multiple Nox isoforms and their binding partners and the lack of specific inhibitors17.
Thus, the goal of our current study is to identify the functional consequences of increased Nox activity in the vascular endothelium as a functional correlate to the elevated Nox activity seen in cardiovascular disease. To achieve this we have adopted a novel genetic approach of using the Nox5 gene to produce superoxide at a time and place of our choosing. This strategy has several advantages. Firstly, the ability of Nox5 to produce superoxide is contained within a single gene. This is important, as the protein-binding partners required to activate the other Nox isoforms are not necessary and thus their absence or inactivity cannot impede superoxide formation14. For example, Nox2 requires the co-expression of at least 4 different gene products just to have the capacity to produce superoxide and then requires a stimulus, such as PMA to induce activity. Secondly, Nox5 is a calcium-activated enzyme and produces low levels of superoxide constitutively and therefore is active in the vast majority of vascular cells18. Thirdly, the intracellular distribution of Nox5 is consistent with that described for Nox4 and Nox2 in endothelium19-22 and consequently the location of superoxide production from Nox5 should reliably replicate that derived from other Nox(s).
Materials and Methods
Materials
All chemicals and reagents were purchased from Sigma. Antibodies: GAPDH (Ambion), HA (Roche), hsp90, phosphorylated eNOS, (Cell Signaling, Millipore).
Animals
C57bl6 mice (Jackson laboratories) were used in accordance with the guidelines for animal use of the Institutional Animal Care and Use Committee of the Medical College of Georgia.
Cell Culture and Transfection
COS-7 cells, bovine aortic endothelial cells (BAECs), human aortic endothelial cells (HAEC) were grown as previously described 23. Replication-deficient adenoviruses encoding the control viruses GFP, β-gal or HA-Nox5 were generated as described previously19, 23. Adenoviral gene delivery into blood vessel endothelium has been described in detail elsewhere24
NO Release
Thirty-six hours after transfection or adenoviral transduction, net NO release was calculated by NO specific chemiluminescence after subtracting background levels from cells treated with L-NMMA, cells without eNOS and unstimulated basal release as described previously25.
cGMP Reporter Assay
HAECs were grown on glass coverslips and acutely placed over the top of HAVSMCs, a source of soluble guanylyl cyclase (sGC). The HAECs were stimulated with 1μM ionomycin in the presence of 100U/ml SOD and 300μM IBMX for 10 minutes. cGMP content was measured in HVSMC using a cGMP specific EIA (Cayman).
Isometric Measurements of Aortic Tone
Aortic rings (1mm in length) were mounted on 2 wires in a 6-mL chamber vessel myograph (Danish Myo Technology) with 1g basal tension. After equilibration, rings were tested for reproducible contraction using 32mM KCl. Concentration-response curves were then constructed to phenylephrine (PE) and subsequently to ACh in vessels precontracted with a submaximal concentration of PE 26.
Statistical Analysis
Data are expressed as means ± S.E.M. Comparisons were made using two-tailed Student's t test or analysis of variance with a post-hoc test where appropriate. Differences were considered significant at p < 0.05.
Results
Nox5 increases eNOS activity in both transfected COS-7 cells and endothelial cells
To evaluate the direct interaction between eNOS and NADPH oxidase, we transfected COS-7 cells with cDNAs encoding eNOS, with or without the epitope tagged, HA-Nox5. In contrast to our expectations, we found that Nox5 increased eNOS activity as determined by the detection of nitrite (NO2−) in the extracellular media. This was apparent under both basal and 100nM thapsigargin stimulated conditions (data not shown). We next extended these studies to bovine aortic endothelial cells (BAEC), which were transduced with control (β-gal) or Nox5 expressing adenoviruses. Increased expression of Nox5 (MOI ranging from 4 to 200) resulted in the dose-dependent increases in superoxide production (Fig. 1A) and corresponding dose-dependent increases in NO release (Fig. 1B). The Nox5-dependent increase in superoxide production in BAEC was sensitive to DPI and SOD inhibition, but was not inhibited by L-NAME, catalase or allopurinol (Supplemental Figure 1). The increase in eNOS activity occurred without variation in total eNOS expression (Fig. 1B lower panel) suggesting that the mechanisms responsible for the increase in NO release are post-translational. Furthermore, co-expression of active Nox1 was also capable of increasing eNOS activity (Supplemental Figure 2). Our next goal was to assess the effect of increased ROS on the release of biologically active NO. To achieve this we employed a co-culture cGMP bioassay in which glass coverslips coated with human aortic endothelial cells (HAECs) are placed over the top of human aortic vascular smooth muscle cells (HAVSMCs, a source of soluble guanylyl cyclase (sGC) that functions as a bio-detector). cGMP content in HAVSMCs was then measured using a cGMP specific EIA. In the presence of extracellular SOD, selective expression of Nox5 in HAEC increased cGMP production under both basal conditions and following ionomycin stimulation, data consistent with that obtained in BAEC and COS cells. eNOS expression in HAEC was unchanged (data not shown). These findings demonstrate that the increase in cGMP levels in VSMC derive exclusively from Nox5 modulation of endothelial eNOS activity. In the absence of extracellular SOD, basal cGMP production dropped significantly to levels equivalent to L-NAME treatment (11.4746+/−1.1357 versus 5.4924+/−0.8834pmol/ml). Collectively, these data show that increased superoxide via expression of Nox5 activates, rather than inhibits, eNOS activity and that the major inhibitory action of superoxide anion is to consume biologically active NO.
Fig. 1. Nox5 increases eNOS activity in endothelial cells.

In A, BAEC were transduced with control or Nox5 adenovirus at different MOIs (4 to 200). 36hrs post transduction, ROS production was monitored by chemiluminescence in the presence and absence of DPI (10μM). In B, NO release was measured from BAECs as described. The relative expression of eNOS and Nox5 (HA) was determined via Western blotting (lower panels). GAPDH was used as a loading control. The data are presented as mean±SE (n=5-6). *P<0.05 vs control or DPI. In C, HAECs were transduced with control or Nox5 adenovirus and 36hrs post-transduction, NO release was determined via cGMP accumulation in HASMC reporter cells. Cells were stimulated with ionomycin (1μM) and cGMP accumulation in SMC reporter cells was measured in the presence of SOD (100U/ml). The data are presented as mean±SE (n=4-6). *P<0.05 vs the untreated control, # in the presence of L-NAME and + P<0.05 vs β-gal.
Nox5 induces endothelial dysfunction in isolated blood vessels
To establish the vascular significance of this relationship in the intact blood vessel, the endothelium of mouse aorta was transduced with control or Nox5 adenovirus. Adenovirus was delivered into the lumen of the aorta as previously described24. This procedure facilitates the selective uptake of virus into the endothelium as shown in Fig. 2A. To further confirm the validity of this approach, we first determined whether we could detect the expression and activity of Nox5 in the aorta and also determine whether it affected eNOS expression. As shown in Fig. 2C, expression of Nox5 can be detected in blood vessels transduced with the Nox5 adenovirus. This correlated with an increase in ROS production (Fig. 2B) but did not significantly modify the level of eNOS expression or phosphorylation compared to control virus (Fig. 2C). We next examined the functional responses of the transduced blood vessels using a myograph to quantify changes in isometric tension. Nox5 potentiated contractile responses to phenylephrine (Fig. 2D) and potently inhibited endothelium-dependent relaxation in response to acetylcholine (Fig. 2E). In contrast, endothelial expression of Nox5 did not modify responses to the NO donor, sodium nitroprusside (SNP) (Fig. 2F). These are well established characteristics of a dysfunctional endothelium and importantly, they occur without changes in the expression level of eNOS (Fig. 2C).
Fig. 2. Endothelial dysfunction in blood vessels expressing Nox5.

The endothelium of mouse aorta was transduced via luminal delivery of control (β-gal/GFP) or Nox5 adenovirus (6×1011 particles/ml). In A, en face presentation of a β-gal transduced aorta showing extent of endothelial cell uptake. In B, transduction of mouse aorta with Nox5 adenovirus increases superoxide production. In C, the relative expression of eNOS, phosphorylated eNOS and Nox5 (HA) in transduced vessels. In D, contractile responses to phenylephrine (PE, 10−9−10−5M) in blood vessels expressing Nox5 versus GFP. In E, vasorelaxant responses to acetylcholine (ACh, 10−9−10−5M). In F, vasorelaxation to sodium nitroprusside (SNP, 10−9−10−5M). Data is presented as mean force generated (2D) and as the means ± SE of the percentage of PE-induced tone (2E–F). *P<0.05 vs control vessels (n=5).
Mechanism of Endothelial Dysfunction induced by Nox5.
To determine whether Nox5-derived ROS competes with NO in a stoichiometric manner or simply “stuns” the endothelium to produce an all or none effect, we next performed a titration experiment. In phenylephrine pre-constricted aortic rings, progressively lower (logarithmic) titers of Nox5 adenovirus impaired Ach-induced relaxation in a dose-dependent manner (Fig. 3A-C). The highest concentration of Nox5 (3 fold lower versus that used in Fig. 2) completely inhibited Ach-dependent vasorelaxation versus control (Fig. 3A), whereas a further 3 fold lower concentration of Nox5 resulted in approximately 50% impairment of relaxation (Fig 3B). Eventually a concentration was reached that was without effect (Fig. 3C). The extent of Nox5-inhibition is summarized in Fig. 3D. In the next experiment we determined whether inhibition could be reversed with SOD. Blood vessels were transduced with Nox5 or β-gal control virus (1.8×1011 particles/ml) and then pretreated with either SOD (100U/ml) or vehicle. Consistent with that shown previously, Nox5 potently inhibited endothelium-dependent relaxation to Ach (Fig. 3E) and this deficit was reversed with SOD pretreatment (Fig 3E-F).
Fig. 3. Endothelial dysfunction in blood vessels expressing Nox5 is dose-dependent and SOD sensitive.

Murine aortae were transduced with 3 fold progressively lower (1.8×1011−2×1010) concentrations of Nox5 or control (GFP) adenovirus (Fig. 3A-C). Blood vessels were precontracted with a submaximal concentration of PE (10−6M) and endothelium-dependent relaxation initiated with acetylcholine (Ach, 10−9 to10−5M). Data is presented as the means ± SE of the percentage of PE-induced tone. *P<0.05 vs the control (n=4). A summary of the degree of inhibition is shown in D. In E-F, mouse aortae were transduced with control or Nox5 adenovirus (1.8×1011 particles/ml). Aortic rings were exposed to SOD 100U/ml prior to preconstriction with PE(10−6M). Endothelium-dependent relaxation was recorded in response to acetylcholine (Ach, 10−9−10−5M) as described. Data is presented as the means ± SE of the percentage of PE-induced tone. *P<0.05 vs the control (n=4).
Extracellular SOD reveals eNOS activation in blood vessels expressing Nox5
To evaluate whether Nox5 has a direct effect on eNOS function in blood vessel endothelium, we exposed aortae to β-gal (control) or Nox5 adenovirus (6×1011particles/ml). Blood vessels were preconstricted with PE as described previously and at the plateau of contraction SOD was administered acutely. This is shown graphically by representative traces of isometric tension in Fig. 4. In blood vessels expressing Nox5, SOD induced an immediate and striking relaxation (45%, Fig. 4B and C) that was much greater than that observed in β-gal treated vessels (5%, Fig. 4A and C).
Fig. 4. Acute exposure of extracellular SOD reveals eNOS activation in blood vessels expressing Nox5.

Aortic endothelium was transduced with control or Nox5 adenovirus (6×1011particles/ml). 36hrs later, aortic rings were precontracted with PE and acutely exposed to 100U/ml SOD and the degree of relaxation recorded. Representative traces of the changes in isometric tension are shown in A-B. In C, data is summarized as the percentage of PE-induced tone in response to SOD in control and Nox5 transduced blood vessels. *P<0.05 vs the control (n=5).
Nox5 does modify not modify the phosphorylation of eNOS or induce uncoupling
The multi-site phosphorylation of eNOS is an important post-translational mechanism regulating its activity and calcium-sensitivity. To further our understanding of the mechanisms by which Nox5 activates eNOS, we first examined whether the level of eNOS phosphorylation is modified by co-expression of Nox5. There was no significant difference in the level of phosphorylated bovine eNOS (S116, T-497, S-617, S-635 and S-1179) between Nox5 and control cDNA (GFP) expressing COS-7 cells (data not shown). Similarly, as shown in Fig. 5A, Nox5 did not modify eNOS expression or phosphorylation versus control (β-gal) virus in BAEC. Increased superoxide levels have been proposed to reduce tetrahydrobiopterin (BH4) levels and uncouple eNOS. To assess whether depletion of BH4 further restrains the ability of Nox5 to activate eNOS, we administered the BH4 donor sepiapterin to cells expressing Nox5 and measured NO release. There was no significant difference in the ability of sepiapterin to enhance NO release from cells regardless of ROS production (Fig. 5B). We next investigated whether the ability of Nox5 to activate eNOS derives from extra or intracellular superoxide production. NO release was measured from populations of COS cells expressing either eNOS or Nox5 in separate cells or both proteins co-expressed within the same cell. Cells expressing both Nox5 and eNOS produced more NO compared to cells expressing equivalent amounts of eNOS only or those in which Nox5 and eNOS are expressed in separate cells. These findings suggest that the extracellular release of superoxide is insufficient to activate eNOS (Supplemental figure 3). To determine whether increased ROS uncouples eNOS we next measured the eNOS monomer/dimer ratio and the relative association of hsp90 in endothelial cells expressing Nox5. Increased production of ROS did not influence the ratio of eNOS monomer/ dimer suggesting that the low level ROS production from NADPH oxidase is not sufficient in itself to disrupt the eNOS dimer (Fig. 5C). We next immunoprecipitated eNOS from cells expressing Nox5 or a control gene and the relative amount of hsp90 bound to eNOS was determined by immunoblotting. As shown in Fig. 5D, increased expression of Nox5 and attendant ROS production resulted in an increased association of eNOS with hsp90. These data argue in favor of increased coupling of eNOS and accordingly, increased NO production.
Fig. 5. Nox5-derived ROS induce post-translational changes in eNOS.
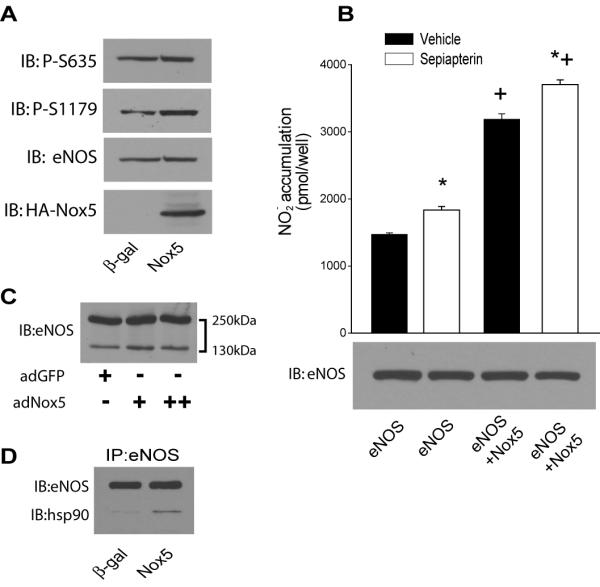
In A, BAEC were transduced with Nox5 or control (β-gal) adenovirus and changes in eNOS phosphorylation determined at S1179 and S635 relative to total eNOS. In B, cells expressing a control gene (RFP) or Nox5 were treated with sepiapterin (10μM) and NO release recorded over 24hrs. In C, BAEC were transduced with Nox5 adenovirus (37.5 – 200MOI) and the relative ratio of eNOS dimer/monomer determined by low temperature SDS-PAGE. In D, eNOS was immunoprecipitated from cells in the presence and absence of Nox5 and immunoblotted for eNOS (upper band) and bound hsp90 (lower band). Results are representative of >3 individual experiments.
Discussion
The goal of the present study was to directly assess the contribution of elevated superoxide anion to endothelial cell function in vitro and in isolated blood vessels. Elevated ROS resulting from increased expression and/or activity of Nox enzymes has been identified as a key mechanism contributing to endothelial and vascular dysfunction2, 27-30. However, because disease states result in many accompanying changes to the extracellular milieu, the direct causative effects of Nox-derived superoxide on vascular cells are poorly understood. A common approach in the past has been to acutely apply supraphysiological concentrations of ROS outside the cell and assess the cellular response31-33. Other approaches using redox cycling agents that elevate intracellular superoxide may not faithfully mimic the amount or location of Nox derived superoxide34. Therefore, we took the approach of expressing a novel Nox isoform, Nox5, in the vascular endothelium. This approach allows for the production of more biologically relevant concentrations of superoxide from a defined intracellular area. We found that in contrast to what we had expected, elevated Nox expression and ROS production dose-dependently increased eNOS activity. However, the increased production of superoxide from Nox5 consumed NO and abrogated the ability of both cultured endothelial cells and blood vessel to deliver biologically active NO. These findings suggest that increased intracellular production of superoxide-derived from NADPH oxidase does not inhibit eNOS activity directly, but instead prevents the extracellular actions of NO.
We originally hypothesized that Nox5 would decrease eNOS activity because numerous studies have shown that ROS decrease eNOS activity by reducing BH4 bioavailability30, 35, uncoupling eNOS36 and disrupting the eNOS homodimer33. However, we found increased activation of eNOS in a range of cells in vitro and also in intact blood vessels. Many of these previous studies may not have detected the increased activity of eNOS as they directly measured intact NO (which is consumed by superoxide) or vascular function. However, in the current study, we measured the level of nitrite (NO2−) which results from the spontaneous breakdown of NO and peroxynitrite in aqueous media37. Therefore, even if the majority of the NO produced is scavenged by O2−, the elevated production of NO can be efficiently recorded via detection of nitrite. Indeed, increased formation of nitrite in blood vessels from atherosclerotic animals has been shown and this occurs despite impaired endothelium-dependent relaxation38. Increased ROS have also been shown to increase NO release in blood vessels and cultured endothelial cells39, 40 . The ability of increased Nox expression and accompanying superoxide production to activate eNOS provides a mechanism to account for this phenomenon.
eNOS is acutely regulated by a number of post-translational mechanisms including phosphorylation, protein-protein interactions and subcellular location41, 42. To address the mechanisms by which Nox5 stimulates eNOS activity, we first examined whether Nox5 affects eNOS phosphorylation at different sites and no changes were observed. The elevated intracellular superoxide and NO that accompanies Nox5 expression in endothelial cells also did not disrupt the eNOS homodimer suggesting that factors beyond NADPH oxidase derived superoxide must contribute to monomer formation seen in disease states. ROS have also been shown to reduce cellular BH4 levels, but cells expressing Nox5 had sufficient BH4 levels to support increased eNOS activity and supplementation with a BH4 donor did not reveal a significant deficit of BH4. Decreased association of hsp90 with eNOS is known to induce what has been termed “uncoupling” and results in increased superoxide formation from eNOS at the expense of NO 43. However, the converse relationship is poorly understood. In this study we found that elevated superoxide production increases the association of hsp90 with eNOS and thereby facilitates rather than inhibits NO synthesis.
In summary, we found that increased superoxide production via expression of Nox5 paradoxically enhanced overall eNOS activity in a variety of different cell types and in intact blood vessels. The increase in activity occurred despite a decrease in the amount of biologically available NO reaching adjacent cells. The functional significance of this is not yet clear, but may represent a feedback system that enhances NOS enzymatic activity to counterbalance a decrease in the amount biologically available NO. Importantly, the ability of Nox-derived superoxide to increase eNOS activity may accelerate the formation of peroxynitrite and have deleterious effects on vascular cells.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health and the American Heart Association.
References
- 1.Harrison DG. Endothelial function and oxidant stress. Clin Cardiol. 1997 Nov;20(11 Suppl 2):II-11–17. [PubMed] [Google Scholar]
- 2.Frisbee JC, Stepp DW. Impaired NO-dependent dilation of skeletal muscle arterioles in hypertensive diabetic obese Zucker rats. Am J Physiol Heart Circ Physiol. 2001 Sep;281(3):H1304–1311. doi: 10.1152/ajpheart.2001.281.3.H1304. [DOI] [PubMed] [Google Scholar]
- 3.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004 Nov;287(5):R1014–1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 4.Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008 Feb;31(Suppl 2):S170–180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 5.Wolin MS, Gupte SA, Oeckler RA. Superoxide in the vascular system. J Vasc Res. 2002 May-Jun;39(3):191–207. doi: 10.1159/000063685. [DOI] [PubMed] [Google Scholar]
- 6.Van Buul JD, Fernandez-Borja M, Anthony EC, Hordijk PL. Expression and localization of NOX2 and NOX4 in primary human endothelial cells. Antioxid Redox Signal. 2005 Mar-Apr;7(34):308–317. doi: 10.1089/ars.2005.7.308. [DOI] [PubMed] [Google Scholar]
- 7.Ago T, Kitazono T, Ooboshi H, Iyama T, Han YH, Takada J, Wakisaka M, Ibayashi S, Utsumi H, Iida M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation. 2004 Jan 20;109(2):227–233. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]
- 8.Schnackenberg CG. Oxygen radicals in cardiovascular-renal disease. Curr Opin Pharmacol. 2002 Apr;2(2):121–125. doi: 10.1016/s1471-4892(02)00133-9. [DOI] [PubMed] [Google Scholar]
- 9.Cai H, Li Z, Dikalov S, Holland SM, Hwang J, Jo H, Dudley SC, Jr., Harrison DG. NAD(P)H oxidase-derived hydrogen peroxide mediates endothelial nitric oxide production in response to angiotensin II. J Biol Chem. 2002 Dec 13;277(50):48311–48317. doi: 10.1074/jbc.M208884200. [DOI] [PubMed] [Google Scholar]
- 10.Berry C, Hamilton CA, Brosnan MJ, Magill FG, Berg GA, McMurray JJ, Dominiczak AF. Investigation into the sources of superoxide in human blood vessels: angiotensin II increases superoxide production in human internal mammary arteries. Circulation. 2000 May 9;101(18):2206–2212. doi: 10.1161/01.cir.101.18.2206. [DOI] [PubMed] [Google Scholar]
- 11.Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci U S A. 1997 Dec 23;94(26):14483–14488. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanna IR, Taniyama Y, Szocs K, Rocic P, Griendling KK. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid Redox Signal. 2002 Dec;4(6):899–914. doi: 10.1089/152308602762197443. [DOI] [PubMed] [Google Scholar]
- 13.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005 Oct 25;112(17):2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 14.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007 Jan;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 15.Ushio-Fukai M, Zafari AM, Fukui T, Ishizaka N, Griendling KK. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J Biol Chem. 1996 Sep 20;271(38):23317–23321. doi: 10.1074/jbc.271.38.23317. [DOI] [PubMed] [Google Scholar]
- 16.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998 Sep;32(3):488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 17.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008 Feb;51(2):211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 18.Banfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004 Oct 29;279(44):46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- 19.Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ. Novel mechanism of activation of NADPH oxidase 5(NOX5): Calcium-sensitization via phosphorylation. J Biol Chem. 2006 Dec 12; doi: 10.1074/jbc.M608966200. [DOI] [PubMed] [Google Scholar]
- 20.Petry A, Djordjevic T, Weitnauer M, Kietzmann T, Hess J, Gorlach A. NOX2 and NOX4 mediate proliferative response in endothelial cells. Antioxid Redox Signal. 2006 Sep-Oct;8(910):1473–1484. doi: 10.1089/ars.2006.8.1473. [DOI] [PubMed] [Google Scholar]
- 21.Belaiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, Hess J, Pogrebniak A, Bickel C, Gorlach A. NOX5 variants are functionally active in endothelial cells. Free Radic Biol Med. 2007 Feb 15;42(4):446–459. doi: 10.1016/j.freeradbiomed.2006.10.054. [DOI] [PubMed] [Google Scholar]
- 22.Bayraktutan U, Blayney L, Shah AM. Molecular characterization and localization of the NAD(P)H oxidase components gp91-phox and p22-phox in endothelial cells. Arterioscler Thromb Vasc Biol. 2000 Aug;20(8):1903–1911. doi: 10.1161/01.atv.20.8.1903. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D. Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arterioscler Thromb Vasc Biol. 2006 May;26(5):1015–1021. doi: 10.1161/01.ATV.0000216044.49494.c4. [DOI] [PubMed] [Google Scholar]
- 24.Scotland RS, Morales-Ruiz M, Chen Y, Yu J, Rudic RD, Fulton D, Gratton JP, Sessa WC. Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ Res. 2002 May 3;90(8):904–910. doi: 10.1161/01.res.0000016506.04193.96. [DOI] [PubMed] [Google Scholar]
- 25.Fulton D, Babbitt R, Zoellner S, Fontana J, Acevedo L, McCabe TJ, Iwakiri Y, Sessa WC. Targeting of endothelial nitric-oxide synthase to the cytoplasmic face of the Golgi complex or plasma membrane regulates Akt-versus calcium-dependent mechanisms for nitric oxide release. J Biol Chem. 2004 Jul 16;279(29):30349–30357. doi: 10.1074/jbc.M402155200. [DOI] [PubMed] [Google Scholar]
- 26.Romanko OP, Stepp DW. Reduced constrictor reactivity balances impaired vasodilation in the mesenteric circulation of the obese Zucker rat. Am J Physiol Heart Circ Physiol. 2005 Nov;289(5):H2097–2102. doi: 10.1152/ajpheart.00213.2005. [DOI] [PubMed] [Google Scholar]
- 27.Papaharalambus CA, Griendling KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc Med. 2007 Feb;17(2):48–54. doi: 10.1016/j.tcm.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tesfamariam B, Cohen RA. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol. 1992 Aug;263(2 Pt 2):H321–326. doi: 10.1152/ajpheart.1992.263.2.H321. [DOI] [PubMed] [Google Scholar]
- 29.Jung O, Schreiber JG, Geiger H, Pedrazzini T, Busse R, Brandes RP. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004 Apr 13;109(14):1795–1801. doi: 10.1161/01.CIR.0000124223.00113.A4. [DOI] [PubMed] [Google Scholar]
- 30.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003 Apr;111(8):1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubanyi GM, Vanhoutte PM. Oxygen-derived free radicals, endothelium, and responsiveness of vascular smooth muscle. Am J Physiol. 1986 May;250(5 Pt 2):H815–821. doi: 10.1152/ajpheart.1986.250.5.H815. [DOI] [PubMed] [Google Scholar]
- 32.Shah KA, Samson SE, Grover AK. Effects of peroxide on endothelial nitric oxide synthase in coronary arteries. Mol Cell Biochem. 1998 Jun;183(12):147–152. doi: 10.1023/a:1006828205261. [DOI] [PubMed] [Google Scholar]
- 33.Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002 Mar;109(6):817–826. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas DD, Ridnour LA, Espey MG, Donzelli S, Ambs S, Hussain SP, Harris CC, DeGraff W, Roberts DD, Mitchell JB, Wink DA. Superoxide fluxes limit nitric oxide-induced signaling. J Biol Chem. 2006 Sep 8;281(36):25984–25993. doi: 10.1074/jbc.M602242200. [DOI] [PubMed] [Google Scholar]
- 35.Channon KM. Tetrahydrobiopterin: regulator of endothelial nitric oxide synthase in vascular disease. Trends Cardiovasc Med. 2004 Nov;14(8):323–327. doi: 10.1016/j.tcm.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 36.Stepp DW, Ou J, Ackerman AW, Welak S, Klick D, Pritchard KA., Jr Native LDL and minimally oxidized LDL differentially regulate superoxide anion in vascular endothelium in situ. Am J Physiol Heart Circ Physiol. 2002 Aug;283(2):H750–759. doi: 10.1152/ajpheart.00029.2002. [DOI] [PubMed] [Google Scholar]
- 37.Lewis RS, Deen WM. Kinetics of the reaction of nitric oxide with oxygen in aqueous solutions. Chem Res Toxicol. 1994 Jul-Aug;7(4):568–574. doi: 10.1021/tx00040a013. [DOI] [PubMed] [Google Scholar]
- 38.Minor RL, Jr., Myers PR, Guerra R, Jr., Bates JN, Harrison DG. Diet-induced atherosclerosis increases the release of nitrogen oxides from rabbit aorta. J Clin Invest. 1990 Dec;86(6):2109–2116. doi: 10.1172/JCI114949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pagano PJ, Griswold MC, Najibi S, Marklund SL, Cohen RA. Resistance of endothelium-dependent relaxation to elevation of O(-)(2) levels in rabbit carotid artery. Am J Physiol. 1999 Nov;277(5 Pt 2):H2109–2114. doi: 10.1152/ajpheart.1999.277.5.H2109. [DOI] [PubMed] [Google Scholar]
- 40.Cosentino F, Hishikawa K, Katusic ZS, Luscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation. 1997 Jul 1;96(1):25–28. doi: 10.1161/01.cir.96.1.25. [DOI] [PubMed] [Google Scholar]
- 41.Sessa WC. eNOS at a glance. J Cell Sci. 2004 May 15;117(Pt 12):2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 42.Oess S, Icking A, Fulton D, Govers R, Muller-Esterl W. Subcellular targeting and trafficking of nitric oxide synthases. Biochem J. 2006 Jun 15;396(3):401–409. doi: 10.1042/BJ20060321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu H, Shi Y, Wang J, Jones D, Weilrauch D, Ying R, Wakim B, Pritchard KA., Jr A heat shock protein 90 binding domain in endothelial nitric-oxide synthase influences enzyme function. J Biol Chem. 2007 Dec 28;282(52):37567–37574. doi: 10.1074/jbc.M706464200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.