

Abstract
The natural proteasome inhibitor salinosporamide A from the marine bacterium Salinispora tropica is a promising drug candidate for the treatment of multiple myeloma and mantle cell lymphoma. Using a comprehensive approach that combined chemical synthesis with metabolic engineering, we generated a series of salinosporamide analogues with altered proteasome binding affinity. One of the engineered compounds is equipotent to salinosporamide A in inhibition of the chymotrypsin-like activity of the proteasome, yet, exhibits superior activity in the cell-based HCT-116 assay.
Introduction
Ubiquitin-mediated proteolysis has evolved as the primary mechanism for protein degradation and maintenance of protein homeostasis in eukaryotic cells.1,2 Ligation with ubiquitin initiates the ATP-driven process which is consummated within the cylindrical core of the 20S proteasome,3 where chymotrypsin-like (CT-L), trypsin-like (T-L), and caspase-like (CA-L) activities allow the hydrolysis of structurally diverse substrates. The catalytic functions of the 20S proteasome are physically separated on three distinct β-subunits (β5, β2, β1), all of which possess an N-terminal threonine residue. The γ-hydroxyl groups of the latter induce peptide cleavage by nucleophilic attack onto the carbonyl group of an amide bond. Subsequent hydrolysis of the resulting acyl-ester mediated by a water molecule regenerates Thr1-Oγ for another reaction.4 In this way, a protein is dissected to a set of peptides for further degradation by peptidases in the cytosol.
In recent years, the 20S proteasome has emerged as a therapeutic target due to its involvement in NF-κB activation (by cleaving the physiological NF-κB antagonist IκB).5 Proteasome inhibition has thus important implications in numerous inflammatory diseases as well as malignancies.6,7 Actinomycete bacteria produce a number of metabolites that target the proteasome.8 These natural inhibitors can be roughly divided into two structural groups, featuring either a peptidic scaffold or a β-lactone ring system. The binding mode and the molecular mechanism of the latter have been elucidated in structural studies of the yeast 20S proteasome in complex with the two drug leads salinosporamide A (1) and omuralide (2) revealing that the β-lactone group forms a covalent adduct with the N-terminal threonine residue via an ester linkage in the active site.9,10 Albeit sharing the same pharmacophore, 1 exhibits superior potency as compared to 2. SAR studies clearly highlighted the importance of leaving groups at the ethyl side chain of 1 to preserve the improved bioactivity.11–14 Such substituents allow the intramolecular formation of a tetrahydrofuran ring by nucleophilic attack of the unmasked C-3 hydroxy group to C-13 after β-lactone hydrolysis and acylation by the catalytically active threonine residue.10 The enzyme-ligand complex formed by this reaction cascade prevents hydrolytic water from entering the active site and/or reformation of the β-lactone ring, and thus renders 1 irreversibly bound to the proteasome. Another factor that may add to the enhanced potency of 1 is the C-5 substituent or P1 residue, which causes additional hydrophobic interactions with the proteasome S1 specificity pocket,10 potentially increasing the residence time of the ligand. Few derivatives of 1 bearing a C-5 substituent different from cyclohexene have been reported to date,11 impeding a confirmation of this hypothesis.
We recently reported the discovery of the salinosporamide biosynthetic gene cluster (sal) from the genome sequence of the producing bacterium Salinispora tropica CNB-440.15,16 Bioinformatic analysis supported the originally proposed pathway to the cyclohexenyl unit of 1,17 in which a new branching point in the phenylalanine pathway leads to the formation of dihydroprephenate (Scheme 1). Following decarboxylative dehydration to 2,3-dihydrophenylpyruvate, the latter would undergo a 1,4-reduction and transamination to yield 3-R-cyclohex-2-enyl-l-alanine. The sal locus harbors many of the desired genes for such a putative pathway, including a copy of the shikimate biosynthesis initiating enzyme 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase (salU), a prephenate dehydratase homolog (salX), and an aliphatic L-amino acid aminotransferase (salW). Inactivation of the pathway-specific gene salX led to the elimination of all natural salinosporamides and the concomitant production of antiprotealide (3) in which leucine substitutes for the nonproteinogenic cyclohexenylalanine residue.18,19 This result suggested that the gate-keeping biosynthetic enzymes responsible for the activation and incorporation of the native sal-specific amino acid residue may exhibit relaxed substrate specificity toward other aliphatic amino acids.20 In this model study, two salinosporamide derivatives featuring a saturated cyclohexane (4) or cyclopentane (5) ring were prepared by mutasynthesis. Loss of the double bond in the cyclohexenyl moiety led to a 10-fold decrease in proteasome inhibition,11,18 whereas the ring contraction from 4 to 5 was surprisingly well tolerated. When evaluated against the cancer cell line HCT-116, 5 was ~2 times more active than 4, suggesting that an olefinic, pentacyclic derivative of 1 may have enhanced potency. We surmised that this structural feature, as well as other functional groups, could be introduced onto the C-5 position of the salinosporamide scaffold exploiting the metabolic plasticity of the biosynthetic machinery. We hence further explored the generality of this biosynthetic system to accommodate other amino acid substrates and report here the structures and activities of five new salinosporamide derivatives (Table 1).
Scheme 1.

Proposed Biosynthesis of the Amino Acid Building Block of 1 via a Shunt in the Phenylalanine Pathway.
Table 1.
Salinosporamides Engineered in S. tropica and their Proteasome Inhibitory Activities Compared to 1 and 3a,b
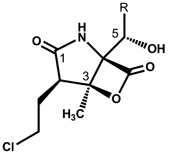 | |||
|---|---|---|---|
| compd | R | CT-L YP IC50 [nM] |
HCT-116 IC50 [nM] |
| 1 | 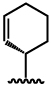 |
1.9 ± 0.2 | 16 ± 5.0 |
| 10 | 2.2 ± 0.1 | 5.9 ± 1.6 | |
| 5 | 9.3 ± 1.6 | 54 ± 22 | |
| 4 | 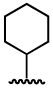 |
27.5 ± 3.7 | 176 ± 59 |
| 6 | 93.4 ± 4.3 | 188 ± 66 | |
| 3 | 101 ± 15 | 777 ± 202 | |
| 9 | 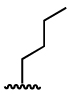 |
132 ± 19 | 1108 ± 187 |
| 8 | 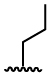 |
245 ± 38 | 1094 ± 137 |
| 7 | 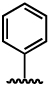 |
1029 ± 419 | 6877 ± 156 |
IC50 values represent the mean ± standard deviation of three or more experiments.
CT-L YP: 20S yeast proteasome fluorimetric kinetic assay of chymotrypsin-like activity; HCT-116: cytotoxicity assay against human colon tumor cell line 116.
Results and Discussion
The primary objective of this study was to evaluate the effects of ring contraction, oxidation, aromatization, or linearization of the P1 residue of salinosporamide analogs in terms of proteasome inhibition and cytotoxicity. Additionally, we were anxious to probe the structural boundaries for the in vivo production of salinosporamide analogues. For these purposes, selected 3-substituted alanine derivatives and branched-chain amino acids were administered to cultures of the S. tropica salX mutant strain. To complement the commercially available pool of proteinogenic and non-proteinogenic amino acids, three additional substrates were synthesized. Racemic 3-cyclopropylalanine and 3-cyclobutylalanine were prepared by alkylation of diethyl formamidomalonate with (bromomethyl)cyclopropane and (bromomethyl)cyclobutane, respectively.21 3-R,S-Cyclopent-2-enyl-d,l-alanine was synthesized in a Strecker-type reaction from 2-cyclopent-2-enyl-1-acetaldehyde,22 readily available by reduction/oxidation of 2-cyclopent-2-enyl-1-acetic acid.23
In total, 16 amino acid precursors (see Supporting Information) were independently fed to cultures of the salX mutant of S. tropica, five of which were unequivocally incorporated into novel salinosporamides (6–10) as evidenced by LC/MS analyses of the fermentation broths. In addition, three substrates led to the known metabolites 3–5 as previously reported.18 All compounds were isolated and fully characterized by NMR and high resolution MS. In general, increased hydrophobicity favored the recruitment of an amino acid, exemplarily shown by the incorporation of (S)-2-aminohexanoic acid, (S)-2-aminoheptanoic acid and 3-cyclobutyl-l-alanine as opposed to (S)-2-aminobutyric acid, (S)-2-aminopentanoic acid and 3-cyclopropyl-l-alanine, respectively. Moreover, it became evident that the salinosporamide biosynthetic enzymes tolerate both secondary and tertiary carbons in the γ position of the amino acid precursor, whereas a substitution in the β position is not accepted as it may prevent the installation of the β-hydroxy group at this site by the cytochrome P450 enzyme SalD.18 Thus l-valine and l-isoleucine are not substrates of the biosynthetic machinery. Contrary to previous reports,18 we encountered a salinosporamide analogue (7) that was derived from l-phenylalanine, even though the yield was extremely low (see Supporting Information). As already observed in our previous study,18 only l-configured amino acids were incorporated into the respective final products. Most interestingly, in case of the feeding study of 3-R,S-cyclopent-2-enyl-d,l-alanine, only a single, optically pure salinosporamide product (10) with the same absolute configuration as 1 was detected.24 This clearly demonstrates the high selectivity of the salinosporamide biosynthesis enzymes concerning stereochemical differences of the precursor amino acids, in contrast to the quite relaxed substrate specificity in terms of the alkyl/alkenyl substitution pattern of the precursors employed.
Based on their IC50 values in a 20S yeast proteasome fluorimetric kinetic assay of CT-L activity, the C-5 modified salinosporamides fall into three distinct groups (Table 1). Those featuring an alicyclic side chain were found to be the most active proteasome inhibitors with 1 ≥ 10 > 5 > 4 > 6, followed by the analogues with an aliphatic side chain. The rank order of the latter was established as 3 > 9 > 8. Noteworthy, the isopropyl-bearing antiprotealide (3) exhibited greater in vitro potency than such salinosporamides that were derived from linear amino acids (8, 9). While the ~2 fold increase in activity from 8 to 9 is likely ascribed to growing van der Waals attraction within the S1 binding site, the enhanced activity of 3 requires a different explanation. As opposed to the linear hydrocarbon chain of 9, the isopropyl group in 3 is less lipophilic, yet, may serve as an anchor prolonging the residence time in the binding pocket. The importance of a tertiary carbon at C-6 for biological activity could be confirmed within the entire series of tested compounds. The slight increase of CT-L inhibition from 3 to 6 in comparison with the abrupt rise in activity from 6 to 4 or 5 indicates that both isopropyl and cyclobutyl residues are too small to fill the binding pocket and establish persistent interactions. Among the saturated alicycles, the cyclopentyl ring exhibited the highest affinity to the β5 subunit of the proteasome and the corresponding derivative 5 was about three times more active than the cyclohexyl analogue 4.25 The difference in bioactivity vanished after insertion of a double bond into both ring systems adjacent to C-6, introducing a new stereocenter and altering bond lengths as well as bond angles as compared to the saturated 5- and 6-membered rings.26 By confining the ring flexibility, both 1 and 10 are oriented in a position, which perfectly meets the structural requirements for binding to the CT-L site. On the contrary, substitution of the natural cyclohexenyl moiety by phenyl as in 7 significantly attenuated proteasome inhibition, which is consistent with data previously reported from a synthetic omuralide derivative.27 We argue that the β5 S1 binding pocket can only temporarily accommodate substrates with aromatic side chains, as the lack of flexibility impairs two-way hydrophobic interactions between the C-5 substituent and the S1 site.
To further evaluate the effects of proteasome inhibition on cell viability and proliferation, all compounds were tested against the human cancer cell line HCT-116. The rank order of enzyme and cellular potency was almost identical in all salinosporamides, except that 10 was nearly three times more potent than 1 in the cell-based assay. As cell-based assays often allow a first approximation on the cell permeability of an inhibitor,28,29 it is possible that increased uptake of 10 has conferred the modest gain in activity. An alternative explanation would involve differences in intracellular metabolism of the compounds. The assumption that reduction of the ring size affects cellular uptake is further corroborated by a comparison of the activities of 4, 5, and 6. In the HCT-116 assay, 6 is nearly as active as 4, suggesting that the biopharmaceutical properties of the former are more favorable. It is interesting to note, however, that the series of derivatives containing an aliphatic side chain were only weakly cytotoxic, albeit being nanomolar inhibitors of CT-L activity. Additional profiling in proteasome inhibition assays of T-L and CA-L activity as well as in the parallel artificial membrane-permeability assay will be required to determine to which extent enhanced inhibition across other proteolytic subunits of the proteasome, lipophilicity or off-target effects contribute to the cytotoxicity of 10.
Conclusions
Precursor-directed biosynthesis using a cyclohex-2-enylalanine-deficient mutant of S. tropica CNB-440 has expanded the structural diversity within the salinosporamide series of natural products. In terms of mutasynthesis, substitution is highly permissive at C-5 (with 8 out of 16 substrates tested being incorporated) providing a handle to modulate affinity for the S1 recognition domain and allow further insight into the binding of β-lactone proteasome inhibitors. A synthetic strategy to access the novel metabolites described in this paper would require individual optimization of reaction conditions from the synthetic branching point involving the introduction of respective C-5 residues through to the final product and lead to the repeated use of identical linear transformations.30 The work presented here, by contrast, allows for the fast generation of a focused library of side-chain modified salinosporamide derivatives by simply feeding readily available amino acid precursors. Despite previous intensive efforts to improve the bioactivity of 1, as exemplified by several SAR studies,11–14 this mutasynthetic approach for the generation of molecular diversity at C-5 led to the production of the first salinosporamide-based proteasome inhibitor, the cyclopentenyl substituted derivative salinosporamide X7 (10), with equal to slightly improved cytotoxic potency as compared to the natural product 1.
Experimental Section
Engineering of salinosporamide derivatives
The salX mutant strain of S. tropica CNB-440 was cultured at 27° C in shaked Fernbach flasks containing a seawater-based medium [10 g starch, 4 g yeast extract, 2 g peptone, 5 mL Fe2(SO4)3×4H2O (8 g/L in deionized water), 5 mL KBr (20 g/L in deionized water)] supplemented with 20 g/L Amberlite XAD-7 resin. Amino acid substrates were added aseptically as filter-sterilized aqueous solutions (50–100 mg/L) after 24 h of growth. Salinosporamide derivatives were obtained by acetone elution from the polymer resin and subsequent purification via RP-HPLC (Luna C-18, 5 µm, 250 × 10 mm, flow rate 2.5 mL/min, detection at 210 nm; Suppl. Table 1).
Assessment of compound purity
The isolated compound was dissolved at 0.1 mg/mL in acetonitrile. 5 µL of the ensuing solution were examined by LC/MS, and compared to an injection of 10 µL of the acetonitrile dissolution solvent run immediately prior to the compounds. Chromatography was achieved on a Phenomenex C18 column (150 × 4.6 mm; 5 µm particle size) using an MeCN/water gradient: 0% MeCN for 1 min, 0–35% MeCN over 7 min, isocratic 35% MeCN over 11 min, 35–100% MeCN over 8 min. Elution was at 0.7 mL/min. UV spectra were recorded between 190 and 400 nm, with extracted chromatograms taken at 210, 254 and 280 nm. Mass spectra were collected scanning 100 to 2,000 atomic mass units in the positive mode. The purity of all isolated compounds determined by HPLC was >95% at all wavelengths (compared to sum of total peak areas).
Salinosporamide X3 (6)
white solid; 1H NMR (600 MHz, CDCl3): δ (multiplicity assignment, coupling constants, position) 6.67 (br s, NH), 4.00 (d, J = 6.0 Hz, H-5), 3.96 (ddd, J = 11.1, 7.7, 5.1 Hz, H-11a), 3.76 (ddd, J = 11.1, 7.3, 5.1 Hz, H-11b), 2.82 (t, J = 7.2 Hz, H-2), 2.68 (m, H-6), 2.26 (dddd, J = 14.9, 7.3, 7.2, 5.1 Hz, H-10a), 2.11 (dddd, J = 14.9, 7.7, 7.2, 5.1 Hz, H-10b), 2.03 (m, H-7), 2.03 (m, H-9), 2.01 (m, H-8a), 1.84 (m, H-8b), 1.82 (s, H3-12); 13C NMR (150 MHz, CDCl3): δ (position) 176.5 (C-1), 167.3 (C-13), 85.4 (C-3), 77.9 (C-4), 68.7 (C-5), 44.6 (C-2), 42.3 (C-11), 37.0 (C-6), 28.0 (C-10), 24.6 (C-7), 24.6 (C-9), 19.4 (C-12), 18.5 (C-8); HR ESITOFMS calcd for C13H19ClNO4 [M+H]+ 288.0997, found 288.1005.
Salinosporamide X4 (7)
white solid; 1H NMR (600 MHz, CDCl3): δ (multiplicity assignment, coupling constants, position) 7.48 (d, J = 7.7 Hz, H-7), 7.45 (t, J = 7.7, 7.1 Hz, H-8), 7.40 (t, J = 7.1 Hz, H-9), 5.91 (br s, NH), 5.30 (s, H-5), 3.96 (ddd, J = 11.2, 7.6, 5.2 Hz, H-11a), 3.77 (ddd, J = 11.2, 7.2, 5.2 Hz, H-11b), 2.90 (dd, J = 7.6, 6.8 Hz, H-2), 2.27 (dddd, J = 15.0, 7.6, 7.2, 5.2 Hz, H-10a), 2.11 (dddd, J = 15.0, 7.6, 6.8, 5.2 Hz, H-10b), 1.91 (s, H3-12); 13C NMR (150 MHz, CDCl3): δ (position) 175.1 (C-1), 166.7 (C-13), 136.4 (C-6), 129.0 (C-9), 128.9 (C-8), 126.1 (C-7), 85.5 (C-3), 77.4 (C-4), 68.4 (C-5), 44.9 (C-2), 42.1 (C-11), 27.9 (C-10), 19.5 (C-12); HR ESITOFMS calcd for C15H15ClNO4 [M-H]− 308.0695, found 308.0691.
Salinosporamide X5 (8)
; white solid; 1H NMR (600 MHz, CDCl3): δ (multiplicity assignment, coupling constants, position) 7.67 (br s, NH), 4.06 (dd, J = 10.5, 2.3 Hz, H-5), 3.95 (ddd, J = 11.3, 7.3, 4.9 Hz, H-10a), 3.74 (ddd, J = 11.3, 7.4, 4.9 Hz, H-10b), 2.86 (t, J = 7.2 Hz, H-2), 2.26 (dddd, J = 14.8, 7.4, 7.2, 4.9 Hz, H-9a), 2.12 (dddd, J = 14.8, 7.3, 7.2, 4.9 Hz, H-9b), 1.81 (s, H3-11), 1.77 (m, H-6a), 1.66 (m, H-7a), 1.49 (m, H-6b), 1.47 (m, H-7b), 0.98 (t, J = 7.2 Hz, H3-8); 13C NMR (150 MHz, CDCl3): δ (position) 177.6 (C-1), 168.1 (C-12), 85.8 (C-3), 78.7 (C-4), 66.4 (C-5), 45.1 (C-2), 42.2 (C-10), 34.0 (C-6), 28.1 (C-9), 19.3 (C-11), 19.0 (C-7), 13.7 (C-8); HR ESITOFMS calcd for C12H19ClNO4 [M+H]+ 276.0997, found 276.1000.
Salinosporamide X6 (9)
; white solid; 1H NMR (600 MHz, CDCl3): δ (multiplicity assignment, coupling constants, position) 7.89 (br s, NH), 4.03 (dd, J = 10.8, 2.1 Hz, H-5), 3.93 (ddd, J = 11.6, 7.4, 5.2 Hz, H-11a), 3.73 (ddd, J = 11.6, 7.4, 5.0 Hz, H-11b), 2.85 (t, J = 7.3 Hz, H-2), 2.25 (dddd, J = 14.7, 7.4, 7.3, 5.2 Hz, H-10a), 2.11 (dddd, J = 14.7, 7.4, 7.3, 5.0 Hz, H-10b), 1.80 (s, H3-12), 1.80 (m, H-6a), 1.58 (m, H-7a), 1.49 (m, H-6b), 1.40 (m, H-8a), 1.39 (m, H-7b), 1.35 (m, H-8b), 0.93 (t, J = 7.1 Hz, H3-9); 13C NMR (150 MHz, CDCl3): δ (position) 177.9 (C-1), 168.1 (C-13), 85.7 (C-3), 78.9 (C-4), 66.6 (C-5), 45.2 (C-2), 42.2 (C-11), 31.6 (C-6), 28.1 (C-10), 27.8 (C-7), 22.3 (C-8), 19.3 (C-12), 13.9 (C-9); HR ESITOFMS calcd for C13H20ClNO4 [M+H]+ 290.1154, found 290.1150.
Salinosporamide X7 (10)
white solid; ; 1H NMR (600 MHz, CDCl3): δ (multiplicity assignment, coupling constants, position, HMBCs, NOESYs) 7.39 (br s, NH, C-2, C-3, C-4, H-6, H-7), 6.07 (dddd, J = 5.8, 2.2, 2.2, 2.2 Hz, H-8, C-6, C-7, C-9, C-10, H-9a), 5.76 (dddd, J = 5.8, 2.2, 2.2, 2.2 Hz, H-7, C-6, C-8, C-9, C-10, H-6, N-H), 3.97 (d, J = 5.7 Hz, H-5, C-3, C-6, C-7, C-10, C-14, H-6, H-13), 3.96 (ddd, J = 11.2, 7.7, 5.1 Hz, H-12a, C-2, C-11, H-11a, H-11b, H-12b), 3.76 (ddd, J = 11.2, 7.2, 5.1 Hz, H-12b, C-2, C-11, H-11a, H-11b, H-12a), 3.11 (m, H-6, C-4, C-5, C-7, C-8, C-9, C-10, H-5, H-7, H-10a, H-10b, N-H), 2.83 (dd, J = 7.7, 6.7 Hz, H-2, C-1, C-3, C-11, C-12, C-13, H-13), 2.43 (m, H-9a, C-5, C-6, C-7, C-8, C-10, H-8, H-9b, H-10b), 2.33 (m, H-9b, C-5, C-6, C-7, C-8, C-10, H-9a), 2.26 (dddd, J = 14.8, 7.7, 7.2, 5.1 Hz, H-11a, C-1, C-2, C-3, C-12, H-11b, H-12a, H-12b), 2.18 (dddd, J = 13.1, 9.1, 9.1, 5.2 Hz, H-10a, C-5, C-6, C-7, C-8, C-9, H-6, H-10b), 2.10 (dddd, J = 14.8, 7.7, 6.7, 5.1 Hz, H-11b, C-1, C-2, C-3, C-12, H-11a, H-12a, H-12b), 1.81 (s, H3-13, C-2, C-3, C-4, C-14, H-2, H-5), H 1.80 (m, H-10b, C-5, C-6, C-7, C-8, C-9, H-6, H-9a, H-10a); 13C NMR (150 MHz, CDCl3): δ (position) 177.8 (C-1), 167.8 (C-14), 137.3 (C-8), 127.9 (C-7), 85.9 (C-3), 78.7 (C-4), 69.9 (C-5), 47.7 (C-6), 44.6 (C-2), 42.5 (C-12), 32.1 (C-9), 28.1 (C-11), 27.9 (C-10), 19.6 (C-13); HR ESITOFMS calcd for C14H19ClNO4 [M+H]+ 300.0997, found 300.0991.
Proteasome inhibition assays
Proteasome inhibition assays were carried out using yeast 20S proteasome and the fluorogenic substrate Suc-LLVY-AMC for chymotrypsin-like activity, both from Enzo Life Sciences. Serial dilutions of each inhibitor were added to 0.5 nM proteasome in assay buffer (25 mM Tris-HCl pH 7.5, 0.5 mM EDTA, and 0.03% SDS) and incubated at 37 °C for 15 min. The 96-well plate was placed on ice and substrate was added to a final concentration of 40 µM. Plates were incubated in the dark at 37 °C for 30 min and then placed on ice. Proteasome activities were measured by reading the fluorescence of the cleaved substrate at 355 nm (excitation) and 460 nm (emission) at 37 °C using SpectraMax M2 (Molecular Devices). IC50 values (compound concentration at which 50% maximal relative activity is inhibited) were calculated using SigmaPlot software and a standard four parameter sigmoidal fit curve, i.e. “Hill, 4 Parameter” (Figure S1 and Figure S2). Assays were performed in triplicate and IC50 values were obtained using the mean of all collected data sets ± standard deviation.
Human colon tumor (HCT-116) cytotoxicity assay
Phenazine methosulfate (PMS, Promega) was dissolved at 0.92 mg/mL in Dulbecco’s phosphate-buffered saline (DPBS) with Ca2+ and Mg2+ (Invitrogen). 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS, Promega) was dissolved at 2 mg/mL in DPBS. 1 mL of PMS solution was then added to 20 mL of MTS solution. HCT-116 cells were diluted in McCoy’s 5A media to a concentration of 2.5 * 104 cells/mL. Aliquot samples were transferred to 96-well plates (Costar flat-bottom culture plates) and incubated overnight at 37 °C in 5% CO2/air. Test compounds were added to the plates in DMSO at a concentration of 1 mM and serially diluted. Etoposide VP-16 (Sigma) and DMSO (solvent) were used as positive and negative controls. The plates were then further incubated for another 72 h, and at the end of this period, a CellTiter 96 Aqueous Nonradioactive Cell Proliferation Assay (Promega) was used to assess cell viability. Inhibition concentration (IC50) values were deduced from the bioreduction of MTS/PMS by living cells into a formazan product. MTS/PMS was first applied to the sample wells, followed by incubation for 3 h. The quantity of the formazan product (in proportion to the number of living cells) in each well was determined in an EMax microplate reader (Molecular Devices) set to 490 nm. IC50 values were calculated using the analysis program SOFTMax.
Supplementary Material
Chart 1.

Acknowledgment
We thank UCSD colleagues William Fenical for valuable discussions, Alexandra Besser for cytotoxicity data, Yuan Liu for technical assistance and Ryan P. McGlinchey for providing the S. tropica salX mutant strain. M.N. and T.A.M.G. are postdoctoral fellows of the German Academic Exchange Service (DAAD). This work was generously supported by a grant from the NIH (CA127622 to B.S.M.).
Abbreviations
- CA-L
caspase-like
- CT-L
chymotrypsin-like
- sal
salinosporamide biosynthetic gene cluster
- T-L
trypsin-like
Footnotes
Supporting Information Available: Isolation table, proteasome inhibitory graphs, and NMR spectra for compounds 6–10. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Goldberg AL. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem. Soc. Trans. 2007;35:12–17. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 2.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell. Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 3.Smith DM, Chang S-C, Park S, Finley D, Cheng Y, Goldberg AL. Docking of the proteasomal ATPases’ carboxyl termini in the 20S proteasome’s α ring opens the gate for substrate entry. Mol. Cell. 2007;27:731–744. doi: 10.1016/j.molcel.2007.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ. Catalytic mechanism and assembly of the proteasome. Chem. Rev. 2009;109:1509–1536. doi: 10.1021/cr8004857. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009;49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- 6.Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin. Cancer Res. 2008;14:1649–1657. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 7.Meiners S, Ludwig A, Stangl V, Stangl K. Proteasome inhibitors: poisons and remedies. Med. Res. Rev. 2008;28:309–327. doi: 10.1002/med.20111. [DOI] [PubMed] [Google Scholar]
- 8.Moore BS, Eustáquio AS, McGlinchey RP. Advances in and applications of proteasome inhibitors. Curr. Opin. Chem. Biol. 2008;12:434–440. doi: 10.1016/j.cbpa.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borissenko L, Groll M. 20S proteasome and ist inhibitors: crystallographic knowledge for drug development. Chem. Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]
- 10.Groll M, Huber R, Potts BCM. Crystal structures of salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important consequences of β-lactone ring opening and a mechanism for irreversible binding. J. Am. Chem. Soc. 2006;128:5136–5141. doi: 10.1021/ja058320b. [DOI] [PubMed] [Google Scholar]
- 11.Macherla VR, Mitchell SS, Manam RR, Reed KA, Chao T-H, Nicholson B, Deyanat-Yazdi G, Mai B, Jensen PR, Fenical WF, Neuteboom STC, Lam KS, Palladino MA, Potts BCM. Structure-activity relationship studies of salinosporamide A (NPI-0052), a novel marine derived proteasome inhibitor. J. Med. Chem. 2005;48:3684–3687. doi: 10.1021/jm048995+. [DOI] [PubMed] [Google Scholar]
- 12.Manam RR, McArthur KA, Chao T-H, Weiss J, Ali JA, Palombella VJ, Groll M, Lloyd GK, Palladino MA, Neuteboom STC, Macherla VR, Potts BCM. Leaving groups prolong the duration of 20S proteasome inhibition and enhance the potency of salinosporamides. J. Med. Chem. 2008;51:6711–6724. doi: 10.1021/jm800548b. [DOI] [PubMed] [Google Scholar]
- 13.Eustáquio AS, Moore BS. Mutasynthesis of fluorosalinosporamide, a potent and reversible inhibitor of the proteasome. Angew. Chem. Int. Ed. 2008;47:3936–3938. doi: 10.1002/anie.200800177. [DOI] [PubMed] [Google Scholar]
- 14.Reddy LR, Fournier JF, Reddy BVS, Corey EJ. An efficient, stereocontrolled synthesis of a potent omuralide-salinosporin hybrid for selective proteasome inhibition. J. Am. Chem. Soc. 2005;127:8974–8976. doi: 10.1021/ja052376o. [DOI] [PubMed] [Google Scholar]
- 15.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc. Natl. Acad. Sci. USA. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eustáquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA, Moore BS. Biosynthesis of the salinosporamide A polyketide synthase substrate chloroethylmalonyl-CoA from S-adenosyl-L-methionine. Proc. Natl. Acad. Sci. USA. 2009;106:12295–12300. doi: 10.1073/pnas.0901237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beer LL, Moore BS. Biosynthetic convergence of salinosporamides A and B in the marine actinomycete Salinispora tropica. Org. Lett. 2007;9:845–848. doi: 10.1021/ol063102o. [DOI] [PubMed] [Google Scholar]
- 18.McGlinchey RP, Nett M, Eustáquio AS, Asolkar RN, Fenical W, Moore BS. Engineered biosynthesis of antiprotealide and other unnatural salinosporamide proteasome inhibitors. J. Am. Chem. Soc. 2008;130:7822–7823. doi: 10.1021/ja8029398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antiprotealide was subsequently also reported as a minor congener of salinosporamide A in fermentations of wild-type S. tropica (see Manam RR, Macherla VR, Tsueng G, Dring CW, Weiss J, Neuteboom STC, Lam KS, Potts BCM. Antiprotealide is a natural product. J. Nat. Prod. 2009;72:295–297. doi: 10.1021/np800578e.
- 20.Nett M, Moore BS. Exploration and engineering of biosynthetic pathways in the marine actinomycete Salinispora tropica. Pure Appl. Chem. 2009;81:1075–1084. [Google Scholar]
- 21.Brackmann F, de Meijere A. Natural occurrence, syntheses, and applications of cyclopropyl-group-containing α-amino acids. 2. 3,4- and 4,5-methanoamino acids. Chem. Rev. 2007;107:4538–4583. doi: 10.1021/cr0784083. [DOI] [PubMed] [Google Scholar]
- 22.Porter TH, Gipson RM, Shive W. Syntheses and biological activities of some cycloalkenealanines. J. Med. Chem. 1968;11:263–266. doi: 10.1021/jm00308a016. [DOI] [PubMed] [Google Scholar]
- 23.Oppolzer W, Siles E, Snowden RL, Bakker BH, Petrzilka M. Intramolecular N-alkenylnitrone-additions regio and stereochemistry. Tetrahedron. 1985;41:3497–3509. [Google Scholar]
- 24.The full stereostructure of 10 was elucidated by 2D NOESY NMR spectroscopy (relative configuration) and measurement of its optical rotation (absolute configuration) in comparison with the data of salinosporamide A (see Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR, Fenical W, Salinosporamide A. A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the genus Salinospora. Angew. Chem. Int. Ed. 2003;42:355–357. doi: 10.1002/anie.200390115.
- 25.The differences in the reported absolute values of the activity data in relation to previously published data (see ref. 18) are likely to be ascribed to the usage of a new yeast 20S proteasome batch. The availability of larger amounts of 3 and 5 in this study (≥10 mg for every compound) as compared to ref. 18 allowed for the generation of more reliable dilution series and hence to more accurate test results which may explain the discrepancies between the two reports.
- 26.Agapito F, Nunes PM, Cabral BJC, dos Santos RMB, Simões JAM. Energetic differences between the five- and six-membered ring hydrocarbons: strain energies in the parent and radical molecules. J. Org. Chem. 2008;73:6213–6223. doi: 10.1021/jo800690m. [DOI] [PubMed] [Google Scholar]
- 27.Corey EJ, Li W-DZ. Total synthesis and biological activity of lactacystin, omuralide and analogs. Chem. Pharm. Bull. 1999;47:1–10. doi: 10.1248/cpb.47.1. [DOI] [PubMed] [Google Scholar]
- 28.Remiszewski SW, Sambucetti LC, Atadja P, Bair KW, Cornell WD, Green MA, Lulu Howell K, Jung M, Kwon P, Trogani N, Walker H. Inhibitors of human histone deacetylase: synthesis and enzyme and cellular activity of straight chain hydroxamates. J. Med. Chem. 2002;45:753–757. doi: 10.1021/jm015568c. [DOI] [PubMed] [Google Scholar]
- 29.Rydzewski RM, Burrill L, Mendonca R, Palmer JT, Rice M, Tahilramani R, Bass KE, Leung L, Gjerstad E, Janc JW, Pan L. Optimization of subsite binding to the β5 subunit of the human 20S proteasome using vinyl sulfones and 2-keto-1,3,4-oxadiazoles: syntheses and cellular properties of potent, selective proteasome inhibitors. J. Med. Chem. 2006;49:2953–2968. doi: 10.1021/jm058289o. [DOI] [PubMed] [Google Scholar]
- 30.Shibasaki M, Kanai M, Fukuda N. Total synthesis of lactacystin and salinosporamide A. Chem. Asian J. 2007;2:20–38. doi: 10.1002/asia.200600310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.