

Abstract
Acute liver failure in infancy accompanied by lactic acidemia was previously shown to result from mtDNA depletion. We report on 13 unrelated infants who presented with acute liver failure and lactic acidemia with normal mtDNA content. Four died during the acute episodes, and the survivors never had a recurrence. The longest follow-up period was 14 years. Using homozygosity mapping, we identified mutations in the TRMU gene, which encodes a mitochondria-specific tRNA-modifying enzyme, tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase. Accordingly, the 2-thiouridylation levels of the mitochondrial tRNAs were markedly reduced. Given that sulfur is a TRMU substrate and its availability is limited during the neonatal period, we propose that there is a window of time whereby patients with TRMU mutations are at increased risk of developing liver failure.
Main Text
Acute liver failure in infancy is a life-threatening condition manifested by poor feeding, vomiting, jaundice, distended abdomen, hemorrhagic diathesis, irritability, and hypoactivity. Routine laboratory investigations reveal elevated liver transaminases, hypoglycemia, coagulopathy, hyperammonemia, and direct hyperbilirubinemia. The differential diagnosis includes viral infections, intoxications, and inborn errors of metabolism. The finding of hyperlactatemia directs the diagnosis toward mitochondrial respiratory chain disorders, and in about half of the patients there is a defect in the mtDNA synthesis machinery, resulting in mtDNA depletion (MIM 251880). This was heretofore attributed to mutations in three genes: DGUOK (MIM 601465), POLG (MIM 174763), and MPV17 (MIM 137960).1–3
In the past 14 years, we have encountered eight patients in seven unrelated families of Yemenite Jewish origin, who presented in infancy with acute liver failure. All were born at term, had birth weights appropriate for gestational age, and had physiologic hyperbilirubinemia that resolved in a normal manner. All were reportedly healthy during the early neonatal period but were admitted at 2–4 months because of irritability, poor feeding, and vomiting. On physical examination, all were found to be well-nourished but lethargic, with pale-gray skin color, jaundiced sclerae, distended abdomen, and hepatomegaly. All of the patients required intensive care for several weeks, with supportive nutrition and blood products given as compensation for coagulopathy and active GIT bleeding. Liver transplantation was considered but was not performed in any of the patients.
Laboratory investigation disclosed acute liver failure (clinical and biochemical data presented in Table 1) with severe coagulopathy that included low factor 5 and 11 and was not corrected by vitamin K supplementation, low albumin, direct hyperbilirubinemia, metabolic acidosis, hyperlactatemia, and high alpha-fetoprotein. Blood ammonia level was normal or slightly elevated, and plasma amino acid profile was noted for high phenylalanine, tyrosine, methionine, glutamine, and alanine. Urinary organic acid analysis revealed massive excretion of lactate, phenylalanine and tyrosine metabolites, and ketotic dicarboxylic and 3-hydroxydicarboxylic aciduria. Serology for hepatitis viruses and body fluid cultures failed to detect an infectious etiology. Abdominal ultrasound disclosed enlarged homogenous liver with normal diameter of the bile ducts and the portal vein.
Table 1.
Clinical and Biochemical Data of the Patients
| Patient | Origin | Age at Presentation | Outcome | Peak Values |
||||
|---|---|---|---|---|---|---|---|---|
| ALT (IU/L) | GGT (IU/L) | INR | T-Bil (mg%) | Lactate (mM) | ||||
| 2624 | Y-J | 6 mo | A&W at 2 yrs | 367 | 356 | 2.6 | 3.3 | 5.5 |
| 3032 | Y-J | 4 mo | A&W at 9 mo | 169 | 621 | 5.7 | 4.5 | |
| 1432 | Y-J | 2 mo | A&W at 10 yrs | 1150 | 3.4 | 10 | 20 | |
| 1116 | Y-J | 3 mo | A&W at 10 yrs | 293 | 139 | 9.7 | 6.6 | |
| 111 | Y-J | 4 mo | A&W at 8 yrs | 417 | 3.0 | 7.0 | ||
| 421 | Y-J | 4 mo | A&W at 14 yrs | 430 | 3.0 | 4.3 | 20 | |
| 2859 | Y-J | 3 mo | death at 4 mo | 400 | 157 | 7.0 | 24.0 | 30 |
| 2375 | Y-J | 6 mo | A&W at 2 yrs | 532 | 305 | 3.6 | 7.5 | 3.2 |
| 2006 | Arab | 1 mo | death at 2 mo | 1193 | 77 | 3.4 | 14.4 | 19 |
| 3015 | Arab | 6 mo | A&W at 2 yrs | |||||
| 1910 | Ashk. | 1 day | A&W at 5 yrs | 1146 | 270 | 2.3 | 0.1 | 20 |
| Akh | Alger | 1 day | death at 3 mo | 93 | 13.2 | 7.0 | ||
| Aza | Alger | 2 days | death at 4 mo | 229 | 6.3 | 10.0 | ||
| control | <52 | <142 | <1.0 | <0.4 | <2 | |||
Abbreviations are as follows: ALT, alanine aminotransferase; GGT, gamma glutamyl transpeptidase; T-Bil, total bilirubin; INR, international normalized ratio; Y-J, Yemenite Jewish; Ashk, Ashkenazi-Jewish; Alger, Algerian; A&W, alive and well.
Clinical and biochemical improvement started after 2–3 weeks, and liver functions returned to normal within 3-4 months. Nonetheless, liver size had normalized only after 3 months to 3 years. Seven patients survived the acute episode, were observed on a long term follow-up (the oldest currently 14 years of age) to be developing normally, and never experienced a similar episode. One patient (2859) died of intractable lactic acidosis and multiple organ failure. During the acute phase, there was usually no indication of extrahepatic involvement, as evidenced by normal electrolytes, creatinine and renal function, blood count, bone marrow aspiration, creatine phosphokinase (CPK), electromyography (EMG), echocardiogram, ophthalmologic examination, brain magnetic resonance imaging (MRI), electroencephalogram (EEG), and nerve conduction velocity (NCV). An exception was patient 1116, who suffered from dilated cardiomyopathy with impaired myocardial contractility and from nephromegaly with massive proteinuria that resolved only after several months.
During the acute phase, liver biopsy, performed in two patients, revealed minimal chronic inflammation and mild focal proliferation of bile ductules with variable portal and sinusoidal fibrosis. In the parenchyma, extensive oncocytic change in the hepatocytes was noted, as well as focal macrovesicular steatosis and focal ballooning of their cytoplasm (Figure 1A). Iron stain revealed slight accumulation of pigment, primarily within the hepatocytes. In the liver sample of patient 3015, obtained when the patient was 9 months of age, during which time the patient was still symptomatic, the liver architecture was markedly disrupted by micronodule formation separated by delicate fibrous septae. The nodules were composed of enlarged hepatocytes, with thickening of the liver plates and hepatocanalicular cholestasis (Figure 1B). The pathological and histochemical examinations of muscle tissue obtained from three patients were invariably normal.
Figure 1.
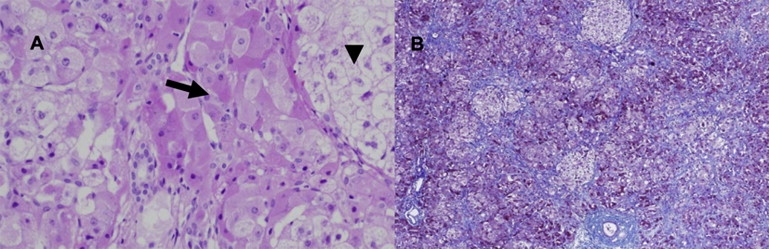
Histopathological Findings in Liver Tissue
(A) Liver tissue showing marked oncocytic change in the hepatocytes (arrow) and focal ballooning degeneration of hepatocytes (arrowhead) (H&E).
(B) Hepatic tissue with markedly disrupted architecture characterized by nodule formation with prominent sinusoidal fibrosis (Masson Trichrome stain).
The enzymatic activities of the mitochondrial respiratory chain complexes I–IV in liver homogenate and in mitochondria isolated from the patients' muscles were determined by standard spectrophotometric methods.4 In liver obtained during the acute phase, the activities of complexes I, III, and IV normalized to citrate synthase activity were markedly reduced; only complex II activity was relatively preserved (Table 2). The mitochondrial respiratory chain activities were normal in homogenate of the liver tissue obtained six months after the onset of the acute episode in patient 1116. In mitochondria isolated from the acute phase muscle tissue, only complex IV activity was slightly reduced.
Table 2.
Mitochondrial Enzymatic Activities, mtDNA Content, and TRMU Genotype of the Patients
| Patient | Tissue | Citrate Synthase | Complex I | Complex II | Complex II+III | Complex IV | mtDNA Content | TRMU Genotype |
|---|---|---|---|---|---|---|---|---|
| 2624 | L | 270% | 29% | 66% | 43% | 15% | Y77H/Y77H | |
| M | 21% | 71% | 95% | 76% | 47% | |||
| 3032 | L | 238% | 7% | 51% | 8% | 22% | 143% | Y77H/Y77H |
| 1432 | L | 211% | 75% | 34% | 10% | 78% | Y77H/a | |
| M | 38% | 132% | 108% | 86% | ||||
| 1116 | L∗ | 65% | 260% | 141% | 103% | Y77H/Y77H | ||
| M | 64% | 75% | 63% | 60% | ||||
| 111 | N.A. | Y77H/Y77H | ||||||
| 421 | N.A. | Y77H/Y77H | ||||||
| 2859 | L | 208% | 11% | 65% | 12% | 16% | 380% | Y77H/c.706-1G>Ab |
| 2375 | N.A. | L233F/A10S | ||||||
| 2006 | L | 148% | 25% | 70% | 17% | 250% | V279M/c.500-510del | |
| 3015 | L | 302% | 8% | 80% | 39% | 14% | 104% | G272D/G272D |
| 1910 | M | 75% | 42% | 97% | 89% | 29% | 107% | G14S/c |
| Akh | M | 12% | 44% | 17% | M1K/M1K | |||
| Aza | M | 68% | 14% | 47% | 22% | 38% | M1K/M1K |
Tissue samples (L, liver; M, muscle) were obtained during the acute phase, with the exception of patient 1116, whose liver (L∗) was obtained 6 mo after the acute episode. N.A. denotes not available. All enzymatic activities are given as a percentage of the control mean and are normalized for citrate synthase activity. The citrate synthase activity and mtDNA content are given as a percentage of the control mean.
A second mutation was not identified in the 11 exons of the TRMU gene, and cDNA of this patient was not available.
This mutation resulted in exon 3 skipping (107 bp).
The patient was heterozygous for the G14S mutation, but the patient's cDNA consisted of only the paternal allele carrying this mutation.
The markedly reduced activities of complexes I, III, and IV in liver homogenate and the relatively normal activity of complex II—the only complex that is encoded solely by the nuclear genome—suggested a defect in the synthesis of the mtDNA-encoded proteins. The normal ratio of mtDNA to nuclear DNA in the patients' liver, as determined by real-time PCR (Table 2), ruled out mtDNA depletion. The mtDNA transcription was investigated in patient 2859 fibroblasts by determination of the abundance of the 12S and 16S rRNA transcripts and of the COX2 mRNA. The normal results of these analyses (data not shown) not only indicated intact transcription but have also excluded a defect in the mitochondrial ribosomal assembly, which would lead to a severe reduction of the rRNA transcripts.5 Assuming a defect in mitochondrial translation, we determined the sequence of the 22 mitochondrial tRNA genes and the two rRNA genes in patient 2859 liver but did not identify any mutation, suggesting a defect in a nuclear-encoded mitochondrial translation factor. We next quantified mitochondrial translation by pulse-chase incorporation of 35S-methionine into mitochondrially synthesized polypeptides in fibroblasts of three patients, in the presence of 0.5 mg/ml of emetine for inhibition of cytoplasmic translation, as previously described.6 To assure correct quantification, we performed immunoblotting of tubulin in the same samples. In all three patients' fibroblasts, the overall mitochondrial translation level was reproducibly twice lower than that in control cells (Figure 2).
Figure 2.
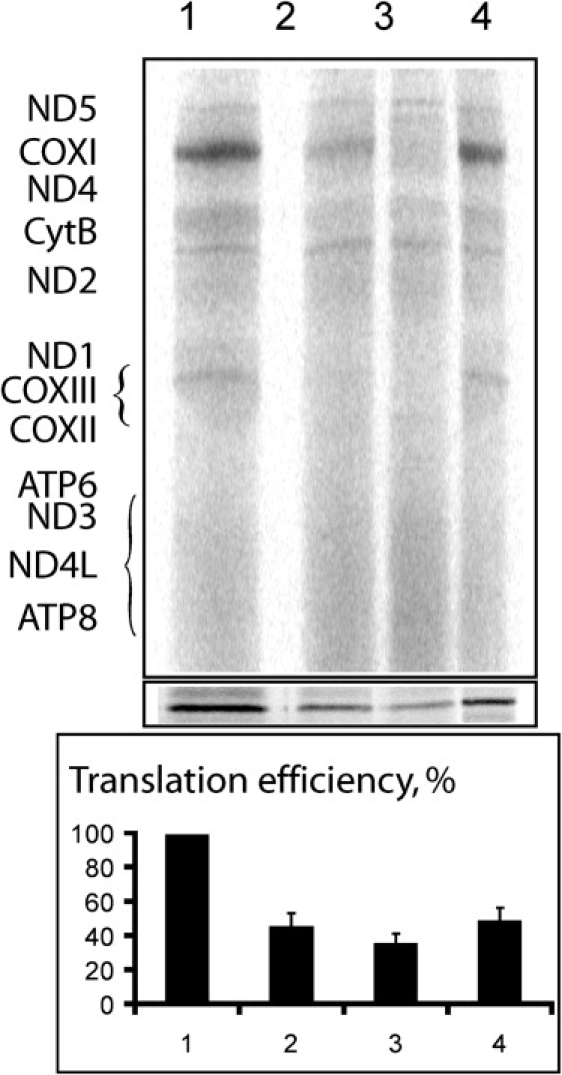
Analysis of Mitochondrial Translation in the Patients' Fibroblasts
The mitochondrial translation products on SDS-PAAG are indicated according to a standard pattern.7 Assays were performed in the fibroblasts of a control (lane 1) and three patients (lanes 2–4 for patients 2624, 2859, and 1910, respectively). The relative values were normalized to tubulin (panel below the autoradiographs) and are presented as a diagram. Error bars represent the results of two independent experiments.
In order to localize the mutated gene, we performed homozygosity mapping with the DNA of patients 3032 and 2624, using the GeneChip Human Mapping 250K Nsp Array of Affymetrix, as previously described.8 All experiments involving DNA of the patients, their relatives, healthy controls, and patients' cells were approved by the Hadassah Ethical Review Committee. This analysis disclosed two nonoverlapping homozygous regions > 5 Mb in each sample. The only genomic region of identical homozygous markers was a 3.06 Mb region on chromosome 22, from 43.49 Mb to 46.55 Mb, which included 223 SNP markers (from rs5765930 to rs7292036). Within this region, there were 27 open reading frames, including TRMU (MIM 610230), which encodes the mitochondria-specific tRNA-modifying enzyme, tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase. Sequence determination of the 11 exons of TRMU and their flanking intronic regions identified a homozygous mutation, c.232T>C, which changes the highly conserved Tyr77 to His (Y77H). Five patients were homozygous for the mutation and two were heterozygous. Because the mutation created an MslI restriction site, we used this enzyme for the screening of 120 anonymous individuals of Yemenite Jewish origin and identified three carriers. Patient 2859, who was heterozygous for the Y77H mutation on the maternal allele, carried a second mutation, c.706-1G>A, on her paternal allele, which resulted in skipping of exon 3. The only Yemenite Jewish patient who did not carry the Y77H mutation, patient 2375, was compound heterozygous for c.697C>T (L233F) and c.28G>T (A10S), both changing highly conserved residues. Because the patient cDNA and parental DNA were not available, we could not assign the phase of the mutations. We then screened the TRMU gene for mutations in DNA of patients of non-Yemenite-Jewish origin who presented with infantile liver failure and a similar pattern of respiratory chain defects and identified five additional mutations in five unrelated patients (Table 1 and Table 2). Four mutations, c.2T>A (M1K), c.40G>A (G14S), c.835G>A (V279M), and c.815G>A (G272D), changed highly conserved residues, and the fifth, c.500-510del, was a frame-shift mutation. Patient 1910 carried the G14S mutation on his paternal allele, but homozygosity for this mutation was present in cDNA produced from his fibroblasts, suggesting a nonexpressing maternal allele. No mutation was detected in the promoter region and at the ∼1100 nucleotide, which separates TRMU from the neighboring 5′ gene. The M1K mutation was identified in two Algerian patients, the G14S mutation was found in an Ashkenazi Jewish patient, and the rest of the mutations were detected in patients of Arabic ethnicity. We did not detect any carrier for the M1K mutation among 106 individuals of North African origin. Altogether, we identified nine mutations in 13 patients who presented with acute liver failure during infancy (Figure 3). Of note, no mutations were detected in the TRMU gene of 17 unrelated patients of North African, Jewish, and Arabic origin having a similar pattern of enzymatic defects and presenting with isolated mitochondrial liver disease immediately after birth, nor in three patients with chronic extrahepatic involvement, indicating that mutations in the TRMU gene primarily affect the liver at a specific window of time.
Figure 3.
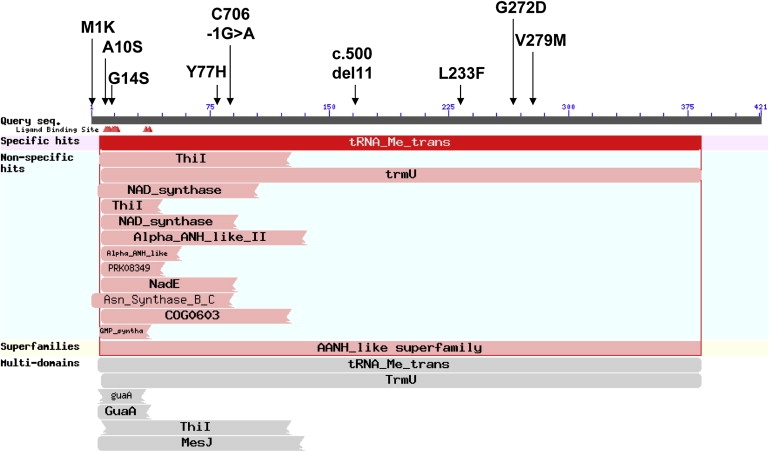
The Mutations Identified in the TRMU Gene
The mutations identified in the TRMU gene of patients with acute liver failure, depicted on a schematic representation of the conserved domains (NCBI conserved domains website). G14 is one of six residues (red arrowheads) that form the P loop motif (SGGXDS), which is an ATP-binding motif commonly found in enzymes responsible for RNA modifications.14
The human TRMU gene encodes 421-aa-long protein that participates in the modification of mitochondrial tRNAs and is therefore important for mitochondrial translation. Specifically, it is responsible for the 2-thiolation of the wobble position of the mitochondrial tRNA-Lys, tRNA-Gln, and tRNA-Glu. We therefore studied the 2-thiouridylation at the wobble nucleotide of these three tRNAs in patients 2624, 2859, and 1910. This was tested by retardation in an electrophoretic system consisting of a 10% PAAG with 7 M urea, tris-borate buffer polymerized in the presence of 50 μg/ml of (N-)Acroylamino-phenyl-mercuric chloride) (APM), which was synthesized by the procedure described by Igloi.9 Total cellular RNA was isolated with Trizol-reagent (Invitrogen). RNA hybridization was performed as described by Shigi et al.,10 with the following [32P]-5′-end-labeled oligonucleotide probes: mt-tRNA-Lys, GGTTCTCTTAATCTTTAAC; mt-tRNA-Glu, CCACGACCAATGATATG; mt-tRNA-Gln, CGAACCCATCCCTGAG, and cy-tRNA-Lys, ACTTGAACCCTGGACC. In this system, the thiolated tRNAs are covalently retained by Hg-groups incorporated in the polyacrylamide gel and have lower mobility than nonthiolated ones. For the purpose of quantification, hybridizations were performed in parallel after separation of the same samples on gels without APM. The results of this analysis clearly disclosed that the amount of the thio-modified mitochondrial tRNAs is severely reduced in all three patients, whereas the pattern of hybridization obtained for the cytosolic tRNA (cy-tRNA-Lys) modified by another enzyme was similar in control and patient cells (Figure 4). Finally, the pattern of hybridization obtained for the mitochondrial tRNA-Leu, which is not subjected to thio-modification, was similar in control and patient cells (data not shown).
Figure 4.
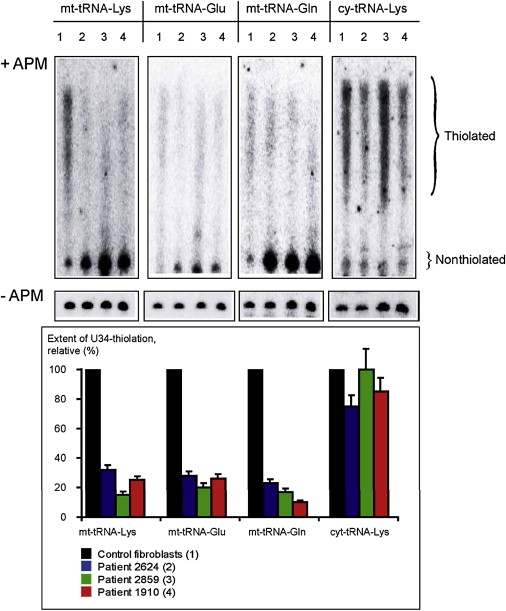
Thio-Modification in Mitochondrial tRNAs
Analysis of thio-modification at position 2 of the wobble uridine via RNA hybridization of mitochondrial (mt-tRNA-Lys, mt-tRNA-Glu, and mt-tRNA-Gln) and cytoplasmic (cy-tRNA-Lys) tRNAs separated in APM-containing gels (+APM, upper panel). For quantification, the same amount of RNA obtained from patient and control fibroblasts was separated in gels without APM (-APM, middle panel). The retarded diffused zones correspond to the thiolated and nonthiolated versions of each tRNA (Thiolated and Nonthiolated, respectively). The hybridization probes and the numbers of the RNA samples are indicated at the top of the autoradiographs; the numbers correspond to the samples described under the diagram at the bottom. The quantification of the modification is presented at the bottom panel and is expressed as a percentage of the thiolated signal from the thiolated + nonthiolated signals (as presented in the -APM gel at the middle panel), normalized against the control fibroblasts. The deviations are indicated as a result of two to three independent measures (for the control fibroblasts, the deviation was quasi null and is therefore not indicated).
To study the effect of the hypomodification on tRNA stability, we performed RNA hybridization of total RNA extracted from the patients' fibroblasts. This analysis disclosed slightly lower levels of several tRNAs, which was nonspecific for the thio-modified tRNAs (Figure 5). We therefore conclude that the TRMU mutations did not affect either the transcription level or the stability of the hypo-modified tRNAs to a significant extent.
Figure 5.
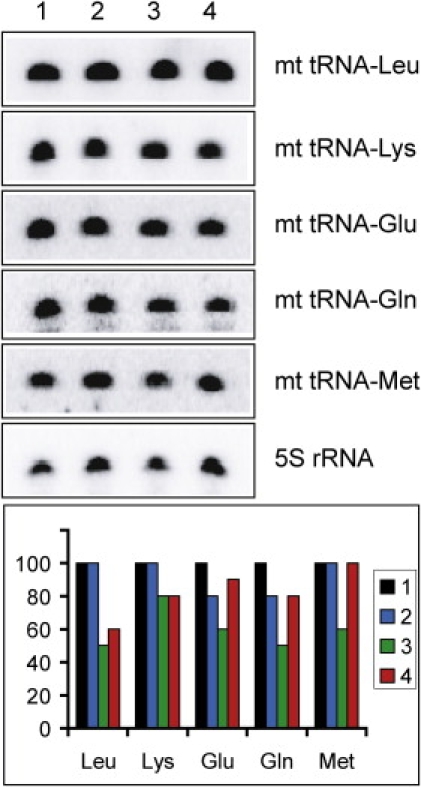
Quantification of Mitochondrial tRNAs by RNA Hybridization
RNA was isolated from the fibroblasts of a control (1) and three patients (2–4 for patients 2624, 2859, and 1910, respectively). Relative values normalized to the 5S rRNA signal are presented in the diagram below the autoradiographs (the various tRNAs are indicated on the x axis only by their respective amino acid abbreviation; thus, Leu stands for mitochondrial tRNA-Leu transcript). Average values of two to three independent experiments are presented. The error was never higher than 10%.
In view of these findings, we propose that the mitochondrial translation defect in our patients is the result of reduced modification of several mitochondrial tRNAs. In E. coli, the 2-thiouridylation stabilizes the codon-anticodon interaction and confers the tRNA an efficient ribosome binding.11,12 Until now, only one mutation in the human TRMU gene, A10S, had been reported. Homozygosity for this mutation had aggravated the deafness phenotype of patients who harbored the homoplasmic A1555G mutation in the mitochondrial gene encoding the 12S rRNA, MTRNR1 (MIM 561000). The combination of TRMU and MTRNR1 mutations was associated with reduced 2-thiouridylation and low content of the mitochondrial tRNAs, which led to impaired mitochondrial protein synthesis.13
The TRMU protein requires sulfur for its activity; cysteine desulfurase, which transfers sulfur from cysteine to the TRMU ortholog, has been shown to be essential for the thio-modification of bacterial tRNAs.14 The availability of cysteine in the neonatal period is limited because its endogenous synthesis from methionine by the transsulfuration pathway is markedly attenuated. The activity of the rate-limiting enzyme in the pathway, cystathionase, is very low at birth and increases slowly during the first few months of life.15 For this reason, cysteine is considered a conditionally essential amino acid, at least in preterm infants. Furthermore, metallothionein, a source of cysteine, is at its peak at birth and declines rapidly during the first month of life.16 We propose that there is a window of time, during 1–4 months of age, whereby patients with TRMU mutations are at an increased risk of developing liver failure. Dietary- and metallothionein-derived cysteine may provide some protection during the first month of life, and the rising activity of cystathionase serves a similar purpose after 3–4 months of age. Nonetheless, an intercurrent illness combined with reduced dietary (cysteine) intake at 1–4 months of age may further compromise TRMU activity in these patients. This may account for the timing of the clinical presentation, mostly at 2–4 months of age, and the lack of recurrence in patients who survive the neonatal episode. Sequence determination of the TRMU gene is warranted in patients with acute liver failure in the first year of life, predominantly when the onset is at 1–4 months of age.
Acknowledgments
We are grateful to the patients and their families, to Mrs. Noa Cohen and Mrs. Corinne Belaiche for their dedicated assistance, to Prof. Shoshy Altuvia for fruitful discussions, to Prof. Michael Wilschanski for sharing of patient 1116 data, and to Dr. Israela Lerer and Prof. Elon Pras for provision of anonymous control samples. This work was supported in part by funding from the Joint Research Fund of the Hebrew University and Hadassah Medical Organization to N.O.; the Israel Science Foundation (1354-2005) to A.S and O.E; the Israeli Ministry of Health and Association Française contre les Myopathies to A.S, I.T., and O.K.; and the Fondation pour la Recherche Médicale and Agence Nationale de la Recherche Scientifique to I.T. and A.M.M.H.
Web Resources
The URLs for data presented herein are as follows:
NCBI Conserved Domains, http://www.ncbi.nlm.nih.gov/sites/entrez?db=cdd
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Mandel H., Szargel R., Labay V., Elpeleg O., Saada A., Shalata A., Anbinder Y., Berkowitz D., Hartman C., Barak M. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat. Genet. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- 2.Naviaux R.K., Nguyen K.V. POLG mutations associated with Alpers' syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004;55:706–712. doi: 10.1002/ana.20079. [DOI] [PubMed] [Google Scholar]
- 3.Spinazzola A., Viscomi C., Fernandez-Vizarra E., Carrara F., D'Adamo P., Calvo S., Marsano R.M., Donnini C., Weiher H., Strisciuglio P. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 4.Saada A., Shaag A., Elpeleg O. mtDNA depletion myopathy: elucidation of the tissue specificity in the mitochondrial thymidine kinase (TK2) deficiency. Mol. Genet. Metab. 2003;79:1–5. doi: 10.1016/s1096-7192(03)00063-5. [DOI] [PubMed] [Google Scholar]
- 5.Miller C., Saada A., Shaul N., Shabtai N., Ben-Shalom E., Shaag A., Hershkovitz E., Elpeleg O. Defective mitochondrial translation due to a ribosomal protein (MRPS16) mutation. Ann. Neurol. 2004;56:734–738. doi: 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- 6.Kolesnikova O.A., Entelis N.S., Jacquin-Becker C., Goltzene F., Chrzanowska Lightowlers Z.M., Lightowlers R.N., Martin R.P., Tarassov I. Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum. Mol. Genet. 2004;13:2519–2534. doi: 10.1093/hmg/ddh267. [DOI] [PubMed] [Google Scholar]
- 7.Enriquez J.A., Cabezas-Herrera J., Bayona-Bafaluy M.P., Attardi G. Very rare complementation between mitochondria carrying different mitochondrial DNA mutations points to intrinsic genetic autonomy of the organelles in cultured human cells. J. Biol. Chem. 2000;275:11207–11215. doi: 10.1074/jbc.275.15.11207. [DOI] [PubMed] [Google Scholar]
- 8.Edvardson S., Shaag S., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-tRNA synthetase gene is associated with ponto-cerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Igloi G.L. Interaction of tRNAs and of phosphorothioate-substituted nucleic acids with an organomercurial. Probing the chemical environment of thiolated residues by affinity electrophoresis. Biochemistry. 1988;27:3842–3849. doi: 10.1021/bi00410a048. [DOI] [PubMed] [Google Scholar]
- 10.Shigi N., Suzuki T., Tamakoshi M., Oshima T., Watanabe K. Conserved bases in the TPsi C loop of tRNA are determinants for thermophile-specific 2-thiouridylation at position 54. J. Biol. Chem. 2002;277 doi: 10.1074/jbc.M207323200. 39128–3913. [DOI] [PubMed] [Google Scholar]
- 11.Ashraf S.S., Sochacka E., Cain R., Guenther R., Malkiewicz A., Agris P.F. Single atom modification (O→S) of tRNA confers ribosome binding. RNA. 1999;5:188–194. doi: 10.1017/s1355838299981529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yarian C., Marszalek M., Sochacka E., Malkiewicz A., Guenther R., Miskiewicz A., Agris P.F. Modified nucleoside dependent Watson-Crick and wobble codon binding by tRNALysUUU species. Biochemistry. 2000;39:13390–13395. doi: 10.1021/bi001302g. [DOI] [PubMed] [Google Scholar]
- 13.Guan M.X., Yan Q., Li X., Bykhovskaya Y., Gallo-Teran J., Hajek P., Umeda N., Zhao H., Garrido G., Mengesha E. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 2006;79:291–302. doi: 10.1086/506389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Umeda N., Suzuki T., Yukawa M., Ohya Y., Shindo H., Watanabe K., Suzuki T. Mitochondria-specific RNA-modifying enzymes responsible for the biosynthesis of the wobble base in mitochondrial tRNAs. Implications for the molecular pathogenesis of human mitochondrial diseases. J. Biol. Chem. 2005;280:1613–1624. doi: 10.1074/jbc.M409306200. [DOI] [PubMed] [Google Scholar]
- 15.Zlotkin S.H., Anderson G.H. The development of cystathionase activity during the first year of life. Pediatr. Res. 1982;16:65–68. doi: 10.1203/00006450-198201001-00013. [DOI] [PubMed] [Google Scholar]
- 16.Zlotkin S.H., Cherian M.G. Hepatic metallothionein as a source of zinc and cysteine during the first year of life. Pediatr. Res. 1988;24:326–329. doi: 10.1203/00006450-198809000-00010. [DOI] [PubMed] [Google Scholar]