

Abstract
Post-traumatic stress disorder (PTSD) is characterized by recurrent distressing memories of an emotionally traumatic event. In this review, we present neuroscientific data highlighting the function of two brain areas—the amygdala and ventromedial prefrontal cortex (vmPFC)—in PTSD and related emotional processes. A convergent body of human and non-human studies suggests that the amygdala mediates the acquisition and expression of conditioned fear and the enhancement of emotional memory, whereas the vmPFC mediates the extinction of conditioned fear and the volitional regulation of negative emotion. It has been theorized that the vmPFC exerts inhibition on the amygdala, and that a defect in this inhibition could account for the symptoms of PTSD. This theory is supported by functional imaging studies of PTSD patients, who exhibit hypoactivity in vmPFC but hyperactivity in amygdala. A recent study of brain-injured and trauma-exposed combat veterans confirms that amygdala damage reduces the likelihood of developing PTSD. But contrary to the prediction of the top-down inhibition model, vmPFC damage also reduces the likelihood of developing PTSD. The putative roles of amygdala and vmPFC in the pathophysiology of PTSD, as well as implications for potential treatments, are discussed in light of these results.
Post-traumatic stress disorder (PTSD) is an anxiety disorder that can develop following exposure to a traumatic event. Events precipitating PTSD include terrifying or life-threatening ordeals such as military combat, traffic accidents, rape, assault, or natural disasters. PTSD is characterized by three clusters of symptoms: 1) re-experience (e.g. flashbacks or nightmares of the traumatic event); 2) emotional numbing and avoidance (e.g. loss of interest, feelings of detachment, avoiding reminders of the traumatic event); and 3) hyperarousal (e.g. excessive vigilance, exaggerated startle) (American Psychiatric Association 2000). Over half the U.S. population has experienced a serious traumatic event, and around 1 in 12 has suffered from PTSD at some point in their lifetime (Kessler and others 1995; Stein and others 2000). Of those afflicted with PTSD, more than a third fail to recover even after many years (Kessler and others 1995). A deeper understanding of the neurobiological basis of PTSD may explain individual differences in susceptibility to the disorder and aid in the development of more effective treatments. In this review, we describe neuroscientific evidence that implicates two areas of the brain—the amygdala and medial prefrontal cortex—in the pathophysiology of PTSD.
Before delving into the function of each of these brain areas, we will first provide some background on their structural anatomy. The amygdala is a subcortical collection of nuclei within the anterior temporal lobe of each hemisphere (Figure 1). The amygdala projects to brainstem and hypothalamic regions that execute the physiological, autonomic, and musculoskeletal components of an emotional response (LeDoux and others 1988). The medial prefrontal cortex (mPFC) refers to the medial wall of the anterior frontal lobes. There are dense white matter connections between the ventral region of mPFC (vmPFC) (Figure 2) and amygdala, facilitating bidirectional communication between these areas (Aggleton and others 1980; Stefanacci and Amaral 2002).
Figure 1.
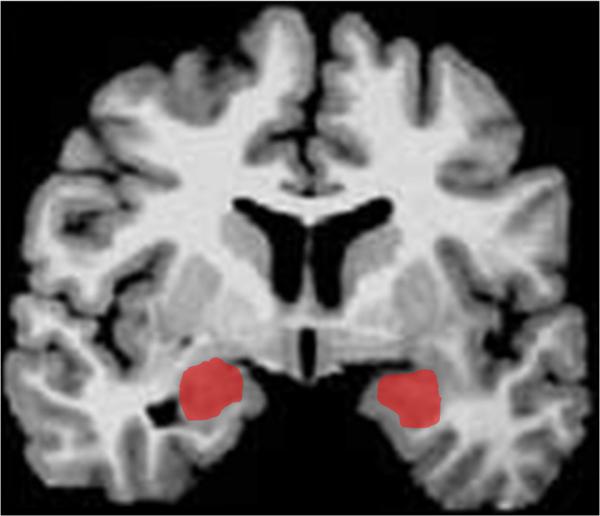
Depiction of amygdala (in red) in a coronal slice.
Figure 2.
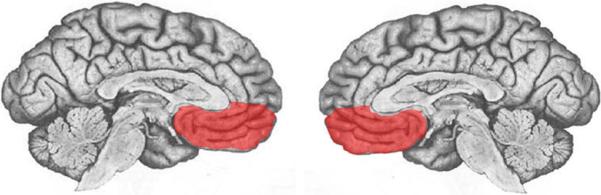
Depiction of vmPFC (in red) in midline views of each hemisphere.
A wealth of neuroscientific data supports the assertion that vmPFC exerts inhibitory control over the amygdala, and that this vmPFC-mediated inhibition of the amygdala is critically involved in the pathogenesis of PTSD. Although the empirical support is detailed in the following the sections, we first provide a brief overview here. The model's underlying supposition is that amygdala activity plays a causal role in the experience of negative affect such as fear, anxiety, and distress. In healthy brains, amygdala activity is thought to be dampened via top-down inhibition by vmPFC, yielding a reduction in subjective distress. However, in PTSD (the model holds), a defect in mPFC function impairs the amygdala inhibition, resulting in unchecked amygdala activity and pathological distress (Milad and others 2006; Rauch and others 2006; Shin and others 2006). Support for this model has been derived through a variety of experimental approaches.
Fear conditioning and extinction
Fear conditioning is a widespread and longstanding experimental paradigm for investigating the neural circuitry involved in processing fear in humans as well as non-human animals. In a typical fear-conditioning paradigm (Figure 3), a neutral event such as a tone (the conditioned stimulus, or CS) is paired with an aversive event, such as a shock (the unconditioned stimulus, or US), which normally elicits a measurable fear response such as freezing, startle, or autonomic/neuroendocrine changes (the unconditioned response, or UR). After several pairings of the tone and shock, the presentation of the tone itself leads to the fear response (the conditioned response, or CR). Extinction occurs when a CS is presented alone, without the US, for a number of trials and eventually the CR is diminished or eliminated. The recall of extinction can be assessed with re-exposure to the CS after a subsequent delay. Given the ostensible similarity between conditioned fear and PTSD symptomotology (in both cases the circumstances related to a previous threat are sufficient to induce a fear-related response), researchers have conceptualized PTSD as a defect in the extinction of conditioned fear (Hamner and others 1999; Milad and others 2006; Rauch and others 2006). Animal and human studies converge to implicate the amygdala and mPFC as the core neural substrates for the acquisition and extinction of conditioned fear.
Figure 3.
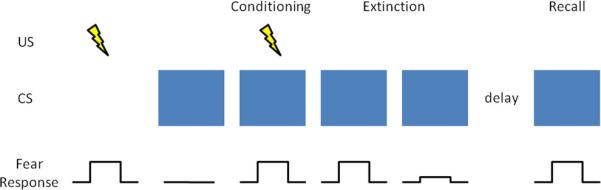
Fear conditioning and extinction. In a typical fear-conditioning paradigm, a neutral event such as a blue light (the CS), which normally does not elicit a fear response, is paired with an aversive event, such as a shock (the US), which normally does elicit a fear response (the UR). After several pairings of the blue light and shock (conditioning phase), the presentation of the blue light itself leads to a fear response (the CR). Extinction occurs when a CS is presented alone, without the US, for a number of trials and eventually the CR is diminished or eliminated. The recall of extinction can be assessed with re-exposure to the CS after a subsequent delay.
Non-human research
Animal data demonstrating the importance of the amygdala in the acquisition and expression of conditioned fear is well chronicled (Davis 2000; LeDoux 2000). In brief, rodent studies employing a range of techniques (electrical stimulation, lesion/ablation, electrophysiological recording, and pharmacological manipulation) reveal that the central nucleus of the amygdala mediates the physiological and behavioral expression of conditioned fear (e.g. endocrine responses, freezing, and startle), while the basolateral nucleus of the amygdala mediates the acquisition (learning) of the conditioned response. Several lines of evidence demonstrate that vmPFC is critically involved in the extinction of the CR. Early primate studies and subsequent rodent studies show that vmPFC lesions impair extinction of the CR (Butter and others 1963; Morgan and LeDoux 1995; Morgan and others 1993), particularly the recall of extinction after a long delay (Lebron and others 2004; Quirk and others 2000). Neurophysiological recordings from rodents indicate that the mPFC neurons signal extinction recall (Milad and Quirk 2002) and that mPFC stimulation decreases the responsiveness of amygdala neurons (Quirk and others 2003; Rosenkranz and others 2003), suggesting the mPFC mediates CR extinction through inhibition of the amygdala.
Human research
Human data on fear conditioning corroborate the conclusions from the animal research. Neurological patients with focal brain lesions involving the amygdala exhibit impairment in the acquisition of conditioned fear responses (Bechara and others 1995; LaBar and others 1995), while fMRI studies associate amygdala activity with conditioned fear responses (Knight and others 2005; LaBar and others 1998; Phelps and others 2004) and vmPFC activity with their extinction (Phelps and others 2004). Furthermore, human vmPFC thickness is correlated with the retention of extinction in healthy adults (Milad and others 2005; Rauch and others 2005). Together, the human and non-human fear conditioning data strongly implicate vmPFC-mediated inhibition of the amygdala as the neural mechanism for the extinction of conditioned fear. If PTSD symptomotology is related to a defect in fear extinction processes, as has been proposed (Hamner and others 1999; Milad and others 2006; Rauch and others 2006), there is strong reason to suspect dysfunction in the vmPFC-amygdala circuit as the underlying cause.
Emotion regulation
The volitional control of negative emotion is another affective function that is germane to PTSD, and one in which vmPFC and amygdala again play prominent roles. Human functional imaging studies have employed paradigms in which subjects are instructed to reappraise upsetting or anxiety-provoking stimuli in order to reduce the intensity of negative affect. A subset of such studies have found that the volitional suppression of negative emotion in healthy adults is associated with an inverse relationship between vmPFC and amygdala activity (Delgado and others 2008; Johnstone and others 2007; Urry and others 2006). That is, during emotion regulation, an increase in vmPFC activity is associated with a decrease in amygdala activity as well as a decrease in the experience of negative affect. Furthermore, this normative vmPFC-amygdala inverse coupling during emotion regulation is disrupted in patients with major depressive disorder (Johnstone and others 2007), which is characterized by pathologically high levels of negative affect.
It is noteworthy that the inverse coupling between vmPFC and amygdala activity during volitional emotion regulation (unique to humans) bears remarkable resemblance to the inverse coupling between vmPFC and amygdala activity during the extinction of conditioned fear (across species) (Delgado and others 2008). These convergent results lend compelling support to a neurocircuitry model in which vmPFC diminishes negative emotion through top-down inhibition of the amygdala. For the remainder of the article, we turn to the question of whether this mPFC-amgydala circuit may be a critical neural substrate in the pathophysiology of PTSD.
Functional imaging studies of PTSD
Functional imaging techniques have the capability of localizing differences in brain function between healthy individuals and those with a particular mental illness. Researchers have used this approach to identify areas of PTSD patients’ brains that exhibit abnormally high or low levels of activity during exposure to trauma-related stimuli. Several such studies report diminished activation within mPFC (Bremner and others 1999a; Bremner and others 1999b; Shin and others 2004), but elevated activation in the amygdala (Driessen and others 2004; Hendler and others 2003; Liberzon and others 1999; Shin and others 2004). A recent meta-analysis of fifteen functional imaging studies investigating negative emotion processing in PTSD patients demonstrates significant hypoactivation within vmPFC but significant hyperactivation of the amygdala (Etkin and Wager 2007). Coupled with the aforementioned data from fear conditioning and emotion regulation studies, the PTSD neuroimaging data support a model of PTSD pathogenesis which proposes two key elements: 1) the emotional distress characterizing PTSD arises from hyperactivity in the amygdala, and 2) the amygdala hyperactivity is caused by defective inhibition from a hypoactive vmPFC (Milad and others 2006; Rauch and others 2006; Shin and others 2006).
Despite the compelling empirical support for this model of PTSD pathophysiology, there are strong reasons to seek additional corroborative evidence. Whereas conditioned fear can be measured across species with relatively simple autonomic or behavioral responses that may even occur non-consciously in humans (Bechara and others 1995; Ohman and Soares 1998), PTSD is a uniquely human disorder that involves explicit re-experience of the traumatic event, subjective distress, and complex emotions such as guilt and social detachment. Thus, it is fair to say that fear conditioning is an informative but ultimately insufficient experimental model for PTSD. Likewise, the emotion regulation studies described above do not begin to approximate the extreme negative affect and behavioral changes that characterize PTSD. So despite the theoretical connections among PTSD, conditioned fear, and emotion regulation, the substantial differences between PTSD and fear conditioning/emotion regulation paradigms make the direct study of PTSD in humans an indispensible means for revealing the neuropathological basis of the disorder. In this regard, human functional imaging studies of PTSD patients have provided key evidence. However, functional imaging data, which is correlative by nature, is not sufficient evidence for a definitive attribution of causation. In other words, functional imaging studies can associate PTSD with abnormal activity in certain brain areas, but they cannot determine whether the abnormal activity actually reflects the underlying cause of the disorder (Table 1) (Pitman 1997). In sum, these limitations on the aforementioned research warrant complementary data that directly address the causal relationship between brain function and PTSD.
Table 1.
Possible Origins of an Association between a Biological Abnormality and Posttraumatic Stress Disorder
| 1. | The abnormality was preexisting and increased the risk of the individual's being exposed to a traumatic event. |
| 2. | The abnormality was preexisting and increased the individual's vulnerability to develop PTSD upon the traumatic exposure. |
| 3. | The traumatic exposure caused the abnormality, and the abnormality caused the PTSD. |
| 4. | The traumatic exposure caused the PTSD, and the PTSD caused the abnormality. |
| 5. | The traumatic event independently caused both the abnormality and the PTSD. |
| 6. | The traumatic exposure caused PTSD, the PTSD caused a sequel or complication, and the sequel or complication caused the abnormality. |
Lesion study of PTSD
The lesion method is a unique and powerful means of determining the importance of a particular brain area for a particular function. The lesion method refers to an approach whereby a focal area of brain damage is associated with the development of a defect in some aspect of cognition or behavior, and then an inference is made that the damaged brain region is a critical part of the neural substrate for the impaired function. That is, unlike functional imaging data, lesion data reveal causality—damage to a particular brain area causes a change in a particular function. In principle, lesion studies could elucidate the causal contribution of vmPFC and amygdala to PTSD by determining if damage to these brain areas changes the likelihood of developing PTSD. However, in an illness such as PTSD that is not amenable to animal lesion studies, this requires the standardized clinical evaluation of a large group of people who suffered the unlikely coincidence of a localizable focal brain lesion as well as emotionally traumatic events. In addition, the lesions would need to adequately sample various areas of the brain, including the vmPFC and amygdala. This unique resource is in fact available in the Vietnam Head Injury Study (VHIS) (Koenigs and others 2008b; Raymont and others 2008).
The VHIS (Phase 3) includes 193 Vietnam veterans with lesions distributed throughout the brain (as a result of penetrating head injuries sustained during combat) and 52 veterans with combat exposure but no brain injury. During the last several years, each of these 245 individuals has been evaluated for PTSD using the Structured Clinical Interview for DSM-IV-TR Axis I disorders, Non-Patient edition (SCID-N/P) (First 2002). In addition, all brain-injured VHIS veterans have undergone a CT scan, allowing the accurate determination of lesion location. Thus each veteran could be classified as either having developed PTSD at some point in their lifetime (PTSD-positive) or having never developed PTSD (PTSD-negative). In a recent study of these VHIS patients (Koenigs and others 2008b), we sought to test the two main predictions of the conventional mPFC-amygdala model of PTSD pathogenesis. Specifically, if PTSD symptomotology is caused by amygdala hyperactivity due to defective inhibition by vmPFC, then veterans with amygdala damage will have a lower-than-normal likelihood of developing PTSD, whereas veterans with vmPFC damage (but intact amygdala) will have a greater-than-normal likelihood of developing PTSD.
PTSD prevalence following vmPFC or amygdala damage
To test these hypotheses directly, we divided the VHIS participants into four groups based on lesion location: 1) significant damage to vmPFC in either hemisphere (vmPFC lesion group; n=40; Figure 4), 2) damage to amygdala in either hemisphere (amygdala lesion group; n=15; Figure 5), 3) damage not involving vmPFC or amygdala (non-vmPFC /non-amygdala lesion group; n=133), and 4) no brain damage (non-brain damaged group; n=52). Brain-injured groups did not significantly differ from each other on basic demographic variables (age, race, sex, and education) nor did they differ in combat exposure, tour duration, or intellectual decline following brain injury, thereby ruling out a host of variables as potential explanations for differences in PTSD occurrence among the groups of brain-injured veterans.
Figure 4.

The color indicates the number of veterans in the vmPFC group (n=40) with damage to a given voxel. The greatest lesion overlap (red) occurred in the anterior vmPFC bilaterally. Top row, sagittal views of the vmPFC group lesion overlap. The left hemisphere (x=-8) is on the left and the right hemisphere (x=6) is on the right. Middle row, coronal views of a healthy adult brain. Slices are arranged with the anterior-most slice on the left (y=66; y=56; y=46; y=36; respectively). Bottom row, coronal views of the vmPFC group lesion overlap, corresponding to the slices in the middle row. From Koenigs and others 2008b.
Figure 5.
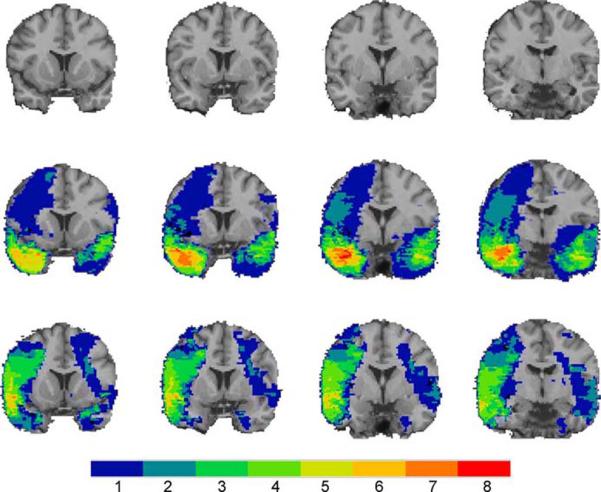
The color indicates the number of veterans with damage to a given voxel. Top row, coronal views of a healthy adult brain. Slices are arranged with the anterior-most slice on the left (y=14; y=8; y=2; y=-4; respectively). Middle row, coronal views of the amygdala group lesion overlap. Bottom row, coronal views of the temporal lobe comparison group lesion overlap. Slices in the middle and bottom rows correspond to the top row. The overlap maps were similar, except for the medial anterior temporal area containing the amygdala, which was damaged in the amygdala group, but intact in the temporal lobe comparison group. From Koenigs and others 2008b.
The prevalence of PTSD in the non-brain damaged group (48%) and the non-vmPFC/non-amygdala lesion group (40%) was similar to published estimates of PTSD prevalence among Vietnam veterans exposed to intense combat (Dohrenwend and others 2006). Remarkably, none of the veterans in the amygdala lesion group (0%) developed PTSD. To rule out the possibility that the absence of PTSD in the amygdala group was due to accompanying damage in anterior temporal cortex or medial temporal lobe structures, rather than damage to the amygdala per se, we selected from the non-vmPFC/non-amygdala group those veterans who had anterior temporal and/or medial temporal lobe damage, but no amygdala damage (n=28) (“temporal lobe comparison group”, Figure 5). The PTSD prevalence among these veterans (32%) was significantly greater than the amygdala group, but not significantly different than the non-brain damaged group or the rest of non-vmPFC/non-amygdala group. These data confirm the prediction that the amygdala damage would result in a lower-than-normal likelihood of developing PTSD, and thereby support the hypothesis that amygdala hyperactivity plays a causal role in the pathophysiology of PTSD.
With regard to the other predicted result, we did not find that the prevalence of PTSD in the vmPFC lesion group was greater-than-normal. To the contrary, PTSD prevalence in the vmPFC lesion group (18%) was significantly lower than that of the non-brain damaged and the non-vmPFC/non-amygdala lesion comparison groups. The markedly low levels of PTSD among veterans vmPFC damage calls into question the veracity of a model in which defective vmPFC-mediated inhibition is the basis of the amygdala hyperactivity in PTSD. If this were the case, then vmPFC damage would presumably diminish the inhibition of the amygdala, resulting in greater susceptibility to PTSD.
Discussion of the role of vmPFC in PTSD
The seemingly discrepant results between functional imaging studies of PTSD (which associate PTSD with vmPFC hypoactivity) and our lesion study (which associates vmPFC damage with resistance to PTSD) warrant further discussion. On one hand, the disparate findings highlight the difference in inferential strength between functional imaging and lesion methods. As illustrated in Table 1, the finding of vmPFC hypoactivity in functional imaging studies of PTSD does not necessarily reflect a causal contribution to the disorder. It is entirely possible that the vmPFC hypoactivity observed in PTSD develops as a consequence of chronic distress associated with PTSD, or perhaps as a downstream effect of primary dysfunction of the amygdala. The latter possibility is supported by human functional imaging data demonstrating abnormal vmPFC activity in patients with amygdala lesions (Hampton and others 2007). But still, it is important to explain why vmPFC lesions result in decreased susceptibility to PTSD. In other words, if the key function of vmPFC (with respect to PTSD) is not inhibition of the amygdala, then what is it? We propose that the causal role of vmPFC in PTSD may be related to its function in self-insight and self-reflection. PTSD is characterized by the experience of distress and anxiety associated with past autobiographical events. Thus a loss of self-insight or self-reflection may diminish the core symptoms of the disorder. Multiple lines of evidence support the plausibility of this interpretation. Previous studies of vmPFC lesion patients document diminished self-insight and self-monitoring (Barrash and others 2000; Beer and others 2006). In addition, vmPFC patients exhibit diminished levels of shame, guilt, embarrassment, and regret (Beer and others 2006; Camille and others 2004; Koenigs and others 2007), all of which are emotions involving some element of self-awareness or self-reflection. Furthermore, data from another recent study of ours (Koenigs and others 2008a) indicate that vmPFC lesion patients experience normal levels of the “somatic” symptoms of depression (e.g. fatigue and changes in appetite), but markedly diminished levels of the “cognitive/affective” symptoms (e.g. self-dislike and guilt), which presumably involve a greater degree of self-reflection and rumination. Taken together, these results hint that the function of vmPFC may extend far beyond inhibition of the amygdala. Future studies will be needed to specify the extent of vmPFC function, and its importance for mood and anxiety disorders such as PTSD.
Discussion of the role of amygdala in PTSD
Unlike the findings for vmPFC, the lesion results regarding the amygdala are precisely as predicted from the functional imaging data, which associate PTSD with amygdala hyperactivity. The finding that amygdala damage abolishes the development of PTSD among combat veterans supports the assertion that amygdala hyperactivity plays a causal role in the pathophysiology of PTSD. What is not entirely clear, however, is the neurocognitive or neurobehavioral mechanism by which the amygdala mediates PTSD. As detailed above, ample human and non-human data demonstrate that the amygdala is critically involved in the expression of negative affect and emotion-related behavior, such as conditioned fear responses. Thus one possibility is that amygdala damage could confer resistance to PTSD through the diminished expression of fear- or anxiety-related responses. A second possibility is that the amygdala could underlie PTSD by virtue of its role in the consolidation of emotional memories. A robust literature indicates that emotionally arousing stimuli are generally better remembered than emotionally neutral stimuli, and that the amgydala is responsible for this emotional memory enhancement (McGaugh 2004). In some sense, PTSD can be conceived as a disorder of extreme emotional memory enhancement, in which emotionally traumatic events are consolidated and recalled to an excessive, pathological degree. Perhaps amygdala damage impairs the obtrusive recall of emotionally traumatic events that defines the disorder. A third possibility is that the amygdala's role in PTSD is related to its function in the detection and evaluation of biologically relevant stimuli, such as threat (Sander and others 2003). It is important to note that these proposed mechanisms are not mutually exclusive; amygdala damage could confer resistance to PTSD through impairment in threat detection, fear expression, and/or emotional memory enhancement.
Implications for treatment
Despite the incomplete understanding of PTSD pathophysiology, the research to date has yielded important implications for the development of more effective treatments for PTSD. The parallel observations of amygdala hyperactivity in PTSD (Etkin and Wager 2007) and lack of PTSD in Vietnam vets with amygdala damage (Koenigs and others 2008b) suggest that a selective deactivation of the amygdala may be an effective treatment strategy for PTSD. There are several potential means for disrupting amygdala function. Perhaps the most drastic approach would be surgical resection of one or both amygdala. Unilateral amygdalectomy is a relatively common occurrence during anterior temporal lobe resections performed to treat pharmacologically intractable epilepsy. Cases of severe, intractable PTSD (which involve extreme debilitation and suicidality) may warrant similarly aggressive treatment. However, the hypothetical efficacy of this surgical approach is debatable; there are at least two documented cases of PTSD developing in individuals who had undergone unilateral removal of the left amygdala as part of treatment for epilepsy (Adami and others 2006; Smith and others 2008). A more elegant (and reversible) approach would be deep brain stimulation (DBS), which involves the surgical implantation of electrodes to manipulate neuronal activity. DBS is a rapidly advancing technique that has been employed to treat a host of neurological and psychiatric illnesses, including Parkinson's disease, depression, and obsessive-compulsive disorder. In principle, electrodes implanted in or around one or both amygdala could effectively inhibit amygdala output, and thereby diminish the experience of anxiety and distress. As a third approach, recent advances in cellular and molecular neuroscience hold promise for the development of amygdala-focused pharmacological treatments. For example, rodent research indicates that inhibition of certain ion channels expressed preferentially in the amygdala suppresses innate and conditioned fear and anxiety (Coryell and others 2007; Wemmie and others 2003).
In addition, vmPFC may be an appropriate neuroanatomical target for PTSD treatments. Our recent lesion studies indicate that vmPFC damage is associated with a conspicuous absence of both PTSD (Koenigs and others 2008b) and depression (Koenigs and others 2008a). These findings suggest that inhibition of vmPFC may diminish the subjective distress and negative affect characterizing PTSD. Indeed, this proposal is consistent with outcomes of surgical interventions used to treat affective disorders. Subcaudate tractotomy, a surgical procedure that interrupts white matter tracts connecting the vmPFC to subcortical structures, has shown efficacy in reducing symptoms of depression and anxiety (Cosgrove 2000; Shields and others 2008). Similarly, a pioneering DBS study reports antidepressant effects of inhibiting subgenual PFC white matter (Mayberg and others 2005), which is adjacent to and interconnected with vmPFC. Perhaps DBS-mediated inhibition of vmPFC could be an effective treatment for PTSD.
Summary
In sum, the mPFC and amygdala are critical nodes in the neurocircuitry underlying emotion processing, and play a fundamental role in the pathophysiology of PTSD. Neuroscientific investigation into the functional interaction between these areas, as in fear conditioning and emotion regulation paradigms, has yielded important insight into the neural mechanisms of emotion-related behavior. Building on this framework, neuroscientific studies of PTSD have underscored the importance of mPFC and amygdala in the development of the disorder, and suggested avenues for more effective treatments. For PTSD, the translation of neuroscientific study into clinical application appears close at hand.
Acknowledgements
This work was supported in part by the National Institute of Neurological Disorders and Stroke intramural research program, DAMD17-01-1-0675 (J.G.)
References
- Adami P, Konig P, Vetter Z, Hausmann A, Conca A. Post-traumatic stress disorder and amygdalahippocampectomy. Acta Psychiatr Scand. 2006;113:360–363. doi: 10.1111/j.1600-0447.2005.00737.x. discussion 363-364. [DOI] [PubMed] [Google Scholar]
- Aggleton JP, Burton MJ, Passingham RE. Cortical and subcortical afferents to the amygdala of the rhesus monkey (Macaca mulatta). Brain Res. 1980;190:347–368. doi: 10.1016/0006-8993(80)90279-6. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition. American Psychiatric Association; Washington DC: 2000. Text Revision (DSM-IV-TR) [Google Scholar]
- Barrash J, Tranel D, Anderson SW. Acquired personality disturbances associated with bilateral damage to the ventromedial prefrontal region. Dev Neuropsychol. 2000;18:355–381. doi: 10.1207/S1532694205Barrash. [DOI] [PubMed] [Google Scholar]
- Bechara A, Tranel D, Damasio H, Adolphs R, Rockland C, Damasio AR. Double dissociation of conditioning and declarative knowledge relative to the amygdala and hippocampus in humans. Science. 1995;269:1115–1118. doi: 10.1126/science.7652558. [DOI] [PubMed] [Google Scholar]
- Beer JS, John OP, Scabini D, Knight RT. Orbitofrontal cortex and social behavior: integrating self-monitoring and emotion-cognition interactions. J Cogn Neurosci. 2006;18:871–879. doi: 10.1162/jocn.2006.18.6.871. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Narayan M, Staib LH, Southwick SM, McGlashan T, Charney DS. Neural correlates of memories of childhood sexual abuse in women with and without posttraumatic stress disorder. Am J Psychiatry. 1999a;156:1787–1795. doi: 10.1176/ajp.156.11.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Staib LH, Kaloupek D, Southwick SM, Soufer R, Charney DS. Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: a positron emission tomography study. Biol Psychiatry. 1999b;45:806–816. doi: 10.1016/s0006-3223(98)00297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butter CM, Mishkin M, Rosvold HE. Conditioning and extinction of a food-rewarded response after selective ablations of frontal cortex in rhesus monkeys. Exp Neurol. 1963;7:65–75. doi: 10.1016/0014-4886(63)90094-3. [DOI] [PubMed] [Google Scholar]
- Camille N, Coricelli G, Sallet J, Pradat-Diehl P, Duhamel JR, Sirigu A. The involvement of the orbitofrontal cortex in the experience of regret. Science. 2004;304:1167–1170. doi: 10.1126/science.1094550. [DOI] [PubMed] [Google Scholar]
- Coryell MW, Ziemann AE, Westmoreland PJ, Haenfler JM, Kurjakovic Z, Zha XM. Targeting ASIC1a reduces innate fear and alters neuronal activity in the fear circuit. Biol Psychiatry. 2007;62:1140–1148. doi: 10.1016/j.biopsych.2007.05.008. others. [DOI] [PubMed] [Google Scholar]
- Cosgrove GR. Surgery for psychiatric disorders. CNS Spectr. 2000;5:43–52. doi: 10.1017/s1092852900007665. [DOI] [PubMed] [Google Scholar]
- Davis MC. The role of the amygdala in conditioned and unconditioned fear and anxiety. In: Aggleton JP, editor. The Amygdala: A Functional Analysis. Oxford University Press; Oxford: 2000. [Google Scholar]
- Delgado MR, Nearing KI, Ledoux JE, Phelps EA. Neural circuitry underlying the regulation of conditioned fear and its relation to extinction. Neuron. 2008;59:829–838. doi: 10.1016/j.neuron.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrenwend BP, Turner JB, Turse NA, Adams BG, Koenen KC, Marshall R. The psychological risks of Vietnam for U.S. veterans: a revisit with new data and methods. Science. 2006;313:979–982. doi: 10.1126/science.1128944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessen M, Beblo T, Mertens M, Piefke M, Rullkoetter N, Silva-Saavedra A. Posttraumatic stress disorder and fMRI activation patterns of traumatic memory in patients with borderline personality disorder. Biol Psychiatry. 2004;55:603–611. doi: 10.1016/j.biopsych.2003.08.018. others. [DOI] [PubMed] [Google Scholar]
- Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164:1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- First MB, editor. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version. Patient Edition. Biometrics Research, New York State Psychiatric Institute; New York: 2002. (SCID-I/P) [Google Scholar]
- Hamner MB, Lorberbaum JP, George MS. Potential role of the anterior cingulate cortex in PTSD: review and hypothesis. Depress Anxiety. 1999;9:1–14. [PubMed] [Google Scholar]
- Hampton AN, Adolphs R, Tyszka MJ, O'Doherty JP. Contributions of the amygdala to reward expectancy and choice signals in human prefrontal cortex. Neuron. 2007;55:545–555. doi: 10.1016/j.neuron.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Hendler T, Rotshtein P, Yeshurun Y, Weizmann T, Kahn I, Ben-Bashat D. Sensing the invisible: differential sensitivity of visual cortex and amygdala to traumatic context. Neuroimage. 2003;19:587–600. doi: 10.1016/s1053-8119(03)00141-1. others. [DOI] [PubMed] [Google Scholar]
- Johnstone T, van Reekum CM, Urry HL, Kalin NH, Davidson RJ. Failure to regulate: counterproductive recruitment of top-down prefrontal-subcortical circuitry in major depression. J Neurosci. 2007;27:8877–8884. doi: 10.1523/JNEUROSCI.2063-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- Knight DC, Nguyen HT, Bandettini PA. The role of the human amygdala in the production of conditioned fear responses. Neuroimage. 2005;26:1193–1200. doi: 10.1016/j.neuroimage.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Koenigs M, Huey ED, Calamia M, Raymont V, Tranel D, Grafman J. Distinct regions of prefrontal cortex mediate resistance and vulnerability to depression. J Neurosci. 2008a;28:12341–12348. doi: 10.1523/JNEUROSCI.2324-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigs M, Huey ED, Raymont V, Cheon B, Solomon J, Wassermann EM. Focal brain damage protects against post-traumatic stress disorder in combat veterans. Nat Neurosci. 2008b;11:232–237. doi: 10.1038/nn2032. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigs M, Young L, Adolphs R, Tranel D, Cushman F, Hauser M. Damage to the prefrontal cortex increases utilitarian moral judgements. Nature. 2007;446:908–911. doi: 10.1038/nature05631. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA. Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron. 1998;20:937–945. doi: 10.1016/s0896-6273(00)80475-4. [DOI] [PubMed] [Google Scholar]
- LaBar KS, LeDoux JE, Spencer DD, Phelps EA. Impaired fear conditioning following unilateral temporal lobectomy in humans. J Neurosci. 1995;15:6846–6855. doi: 10.1523/JNEUROSCI.15-10-06846.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebron K, Milad MR, Quirk GJ. Delayed recall of fear extinction in rats with lesions of ventral medial prefrontal cortex. Learn Mem. 2004;11:544–548. doi: 10.1101/lm.78604. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon I, Taylor SF, Amdur R, Jung TD, Chamberlain KR, Minoshima S. Brain activation in PTSD in response to trauma-related stimuli. Biol Psychiatry. 1999;45:817–826. doi: 10.1016/s0006-3223(98)00246-7. others. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Lozano AM, Voon V, McNeely HE, Seminowicz D, Hamani C. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45:651–660. doi: 10.1016/j.neuron.2005.02.014. others. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu Rev Neurosci. 2004;27:1–28. doi: 10.1146/annurev.neuro.27.070203.144157. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quinn BT, Pitman RK, Orr SP, Fischl B, Rauch SL. Thickness of ventromedial prefrontal cortex in humans is correlated with extinction memory. Proc Natl Acad Sci U S A. 2005;102:10706–10711. doi: 10.1073/pnas.0502441102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73:61–71. doi: 10.1016/j.biopsycho.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE. Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci. 1995;109:681–688. doi: 10.1037//0735-7044.109.4.681. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- Ohman A, Soares JJ. Emotional conditioning to masked stimuli: expectancies for aversive outcomes following nonrecognized fear-relevant stimuli. J Exp Psychol Gen. 1998;127:69–82. doi: 10.1037//0096-3445.127.1.69. [DOI] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Pitman RK. Overview of biological themes in PTSD. Ann N Y Acad Sci. 1997;821:1–9. doi: 10.1111/j.1749-6632.1997.tb48264.x. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Pare D. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci. 2003;23:8800–8807. doi: 10.1523/JNEUROSCI.23-25-08800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch SL, Milad MR, Orr SP, Quinn BT, Fischl B, Pitman RK. Orbitofrontal thickness, retention of fear extinction, and extraversion. Neuroreport. 2005;16:1909–1912. doi: 10.1097/01.wnr.0000186599.66243.50. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research--past, present, and future. Biol Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Raymont V, Greathouse A, Reding K, Lipsky R, Salazar A, Grafman J. Demographic, structural and genetic predictors of late cognitive decline after penetrating head injury. Brain. 2008;131:543–558. doi: 10.1093/brain/awm300. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Moore H, Grace AA. The prefrontal cortex regulates lateral amygdala neuronal plasticity and responses to previously conditioned stimuli. J Neurosci. 2003;23:11054–11064. doi: 10.1523/JNEUROSCI.23-35-11054.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander D, Grafman J, Zalla T. The human amygdala: an evolved system for relevance detection. Rev Neurosci. 2003;14:303–316. doi: 10.1515/revneuro.2003.14.4.303. [DOI] [PubMed] [Google Scholar]
- Shields DC, Asaad W, Eskandar EN, Jain FA, Cosgrove GR, Flaherty AW. Prospective Assessment of Stereotactic Ablative Surgery for Intractable Major Depression. Biol Psychiatry. 2008 doi: 10.1016/j.biopsych.2008.04.009. others. [DOI] [PubMed] [Google Scholar]
- Shin LM, Orr SP, Carson MA, Rauch SL, Macklin ML, Lasko NB. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61:168–176. doi: 10.1001/archpsyc.61.2.168. others. [DOI] [PubMed] [Google Scholar]
- Shin LM, Rauch SL, Pitman RK. Amygdala, medial prefrontal cortex, and hippocampal function in PTSD. Ann N Y Acad Sci. 2006;1071:67–79. doi: 10.1196/annals.1364.007. [DOI] [PubMed] [Google Scholar]
- Smith SD, Abou-Khalil B, Zald DH. Posttraumatic stress disorder in a patient with no left amygdala. J Abnorm Psychol. 2008;117:479–484. doi: 10.1037/0021-843X.117.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanacci L, Amaral DG. Some observations on cortical inputs to the macaque monkey amygdala: an anterograde tracing study. J Comp Neurol. 2002;451:301–323. doi: 10.1002/cne.10339. [DOI] [PubMed] [Google Scholar]
- Stein MB, McQuaid JR, Pedrelli P, Lenox R, McCahill ME. Posttraumatic stress disorder in the primary care medical setting. Gen Hosp Psychiatry. 2000;22:261–269. doi: 10.1016/s0163-8343(00)00080-3. [DOI] [PubMed] [Google Scholar]
- Urry HL, van Reekum CM, Johnstone T, Kalin NH, Thurow ME, Schaefer HS. Amygdala and ventromedial prefrontal cortex are inversely coupled during regulation of negative affect and predict the diurnal pattern of cortisol secretion among older adults. J Neurosci. 2006;26:4415–4425. doi: 10.1523/JNEUROSCI.3215-05.2006. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH, Jr., Welsh MJ. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci. 2003;23:5496–5502. doi: 10.1523/JNEUROSCI.23-13-05496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]