

Abstract
We report structure-activity studies of a large number of dialkyl imidazoles as inhibitors of Trypanosoma cruzi lanosterol-14α-demethylase (L14DM). The compounds have a simple structure compared to posaconazole, another L14DM inhibitor that is an anti-Chagas drug candidate. Several compounds display potency for killing T. cruzi amastigotes in vitro with values of EC50 in the 0.4–10 nM range. Two compounds were selected for efficacy studies in a mouse model of acute Chagas disease. At oral doses of 20–50 mg/kg given after establishment of parasite infection, the compounds reduced parasitemia in the blood to undetectable levels, and analysis of remaining parasites by PCR revealed a lack of parasites in the majority of animals. These dialkyl imidazoles are substantially less expensive to produce than posaconazole and are appropriate for further development toward an anti-Chagas disease clinical candidate.
Introduction
Chagas Disease causes the third largest parasitic disease burden in the world and the largest in the Western hemisphere, currently affecting 16–18 million people throughout Central and South America1. The disease is caused by the parasite Trypanosoma cruzi (T. cruzi)a, which is able to invade a wide variety of host cells. The vaccine prospects for preventing Chagas disease are not promising because the pathogen has developed complex immune evasion techniques to allow for persistent infection2. Drug therapy options for Chagas disease are limited. The principal drugs are benznidazole and nifurtimox, which have modest efficacy during the acute phase of the disease and are not effective for treatment of the chronic, life-threatening stage. Both of these nitroheterocyclic drugs are poorly tolerated by adults. In short, more effective and better-tolerated anti Chagas disease drugs are greatly needed3.
Sterol biosynthesis is a complex enzymatic pathway, which produces membrane lipids for many eukaryotic organisms. Mammals produce cholesterol as their primary sterol, fungi produce ergosterol, and T. cruzi produces a mixture of ergosterol-like sterols that contain various alkyl substituents at C244. Sterol synthesis in mammals, yeast, and Trypanosoma cruzi go through the common intermediate lanosterol, which is formed in several steps from acetyl-CoA. The first of the post-lanosterol processing steps is the removal of the methyl group at C14 by lanosterol 14α-demethylase (cytochrome P450 subfamily 51) (L14DM). Since the parasite apparently cannot survive soley on cholesterol salvaged from its host, enzymes of the parasite sterol biosynthetic pathway offer potential targets for the development of drugs. Urbina and others have shown that a number of inhibitors of fungal L14DM, including the recently developed anti-fungal drug posaconazole, are potent inhibitors of T. cruzi L14DM, and are able to cure mice suffering from acute and chronic Chagas disease5. The use of posaconazole to treat Chagas disease is being considered, however its manufacturing costs may limit widespread use especially in the long-term treatment of the chronic disease6.
In our studies of inhibitors of protein farnesyltransferase as anti-parasite agents such as 1 (Figure 1), we found that compounds such as 2, which lack the methionyl group, do not inhibit parasite protein farnesyltransferase and yet display potent activity in blocking the growth of T. cruzi amastigote stage (intracellular) parasites 7. Further studies showed that compounds in this class led to the accumulation of lanosterol in amastigotes and to the formation of unusual sterols that are predicted to result from the blockade of the lanosterol 14α-demethylation step 7. Compounds in this class were also found to bind to recombinant T. cruzi L14DM, causing a spectral shift that is consistent with coordination of the unsubstituted imidazole nitrogen to the heme iron of this cytochrome P4507. Since the methyl ester of 2 is rapidly hydrolyzed by enzymes in serum, we went on to prepare derivatives including 3 (Figure 1) that displayed desirable pharmacokinetic properties in mice7. Administration of a single dose of 3 to mice at 30 mg/kg by oral gavage led to a maximal drug concentration in the plasma of 16 µM after 1 hr, and 3 was lost with a serum half-life of ~4 hr. Administration of 3 at 50 mg/kg, twice per day for 14 days to T. cruzi-infected mice reduced parasitemia in blood to ~1% of the level seen in untreated mice7. These encouraging results along with the low cost of goods anticipated for this new class of L14DM inhibitors, which lack stereogenic centers, prompted us to carry out extensive structure-activity studies of 3 in an effort to maximize potency against T. cruzi amastigotes while maintaining respectable pharmacokinetic properties. The synthesis of analogs of 3 was guided using a model of the structure of T. cruzi L14DM templated on the x-ray structure of L14DM from M. tuberculosis8. The present article discloses the synthesis and structure activity relationship studies of a series of dialkyl substituted imidazole derivatives as potent L14DM inhibitors for Chagas chemotherapy.
Figure 1.

Dialkyl imidazole-based L14DM inhibitors.
Results and Discussions
Chemistry
Synthetic procedures are illustrated in Scheme 1 – Scheme 7. Scheme 1 shows the synthesis of dialkyl imidazoles containing a methoxycarbonyl group and also shows the synthetic steps used to make most of the compounds reported in this study. The first of which is alkylation of the N-tritylated imidazole carboxaldehyde 4 to give the N-alkylated imidazole 5.9,10 The second piece, the substituted aniline 9, contains a phenyl-phenyl bond, whch is made using the Suzuki cross coupling, followed by standard functional group transformation11. The two inhibitor pieces are joined by reductive amination to give 10a-l 10.
Scheme 1. aSynthesis of the dialkylimidazoles containing the methoxycarbonyl group.

aReagents and conditions: (a) TrtCl, Et3N, DMF, r.t, 90–94%; (b) RC6H4CH2Br, CH3CN, 60 °C, 24 h,70–75%; (c) PhB(OH)2, Pd(OAc)2, K2CO3, acetone/H2O, 85–89%; (d) KMnO4, pyr/H2O, 100%; (e)SOCl2, MeOH, reflux, 80%; (f) SnCl2. 2H2O, EtOAc, reflux, 6 h, 86%; (g) AcOH, 5, MeOH, 4Å mol. seives, NaCNBH3, overnight, r.t, 60–70%.
Scheme 7. aSynthesis of dialkylimidazoles with an ortho amino group.

a Reagents and conditions: (a) PhB(OH)2, Ba(OH)2.8H2O, Pd(PPh3)4, DME/H2O (5:1), reflux, 90%; (b) NBS, CCl4, overnight, reflux, 60%; (c) 1-Trityl-1H-imidazole-4-carbaldehyde, CH3CN, 60 °C, overnight, 55%; (d) AcOH, R = 23b, 23c, 26a, 26d, 14a, 4Å mol. seives, MeOH, NaCNBH3, overnight, 60%; (e) SnCl2.2H2O, EtOAc, reflux, 2 h, 80–82%.
Scheme 2 shows the preparation of dialkyl imidazoles containing additional acyl groups (esters and amides). It is based on standard functional group transformations and the general methods outlined in Scheme 1. Dialkyl imidazoles containing a ketone functional group are shown in Scheme 3. After several unsuccessful attempts, we found that the Fries rearrangement worked well to install the ketone group12,13. The phenolic hydroxyl group was conveniently activated as a triflate ester to generate 18a-c followed by the usual Suzuki cross coupling to produce 19a-c.
Scheme 2. aPreparation of dialkylimidazoles containing esters and amides.

aReagents and conditions: (a) SOCl2, ROH or ROH, EDCI, HOBt, CH2Cl2, r.t, 24 h, 90 % or NH2R, Et3N, CH2Cl2,0 °C - r.t, 2–4 h, 85%; (b) SnCl2.2H2O, EtOAc, reflux, 86–90%; (c) AcOH, 5, MeOH, 4Å mol. sieves, NaBH3CN, overnight, r.t , 60%.
Scheme 3. aSynthesis of dialkylimidazoles containing ketones.

aReagents and conditions: (a) i) (RCO)2O, pyridine, 1.5 h, r.t; ii) AlCl3, 120 °C, overnight, 40%; (b) Tf2O, pyridine, r.t, overnight, 96%; (c) PhB(OH)2, Pd(PPh3)4, K2CO3, DME/EtOH/H2O, 54 %; (d) i) NaOH, i-PrOH/H2O, reflux, overnight, 81%; ii) AcOH, 5, MeOH, 4Å mol. sieves, NaBH3CN, overnight, r.t, 20–40%.
Non acyl-containing functional groups were introduced into the inhibitor scaffold in place of the methoxycarbonyl group using a Sandmeyer reaction14,15,16 (22c, Scheme 4a), by heating the aryl halide with the secondary amine (22d,e) or reductive amination with formaldehyde (22f). Incorporation of small heterocycles into the dialkyl imidazole scaffold is shown in Scheme 4b using the appropriate aryl bromide in the presence of KOAc and catalytic Pd (PPh3)4 in DMAC 17–19.
Scheme 4. aaSynthesis of dialkylimidazoles containing non-acyl functional groups.

baSynthesis of dialkylimidazoles containing heterocycles.
aReagents and conditions: (a) i) Br2, AcOH, r.t, 1h, 76%; ii) PhB(OH)2, Pd(OAc)2, K2CO3, acetone/H2O, reflux, 86% (b) NaNO2, H2SO4, AcOH i) CuBr2, HCl, 71%; ii) CuCl2, HCl, 75%; (c) Zn(CN)2, Pd(PPh3)4, DMF, 100 °C, overnight, 60–65%; (d) piperidine, 100 °C, 2 h, 90%; (e) pyrrolidine, 80 °C, 2–3 h, 80–90%; (f) HCHO, H2SO4, NaBH4 THF, 10–30°C, 0.5h, 65–70%; (g) SnCl2.2H2O , EtOAc, reflux, 6 h, 76 –80%; (h) AcOH, 5, MeOH, 4Å mol. sieves, NaBH3CN, overnight, r.t, 0–65% (i) Heterocycle, pd(PPh3)4, KOAc, DMAC, 160°C, overnight, 32–35%.
Preparation of dialkyl imidazoles containing the benzothiazole appendage is shown in Scheme 5. The key reaction is treatment of the aryl bromide with benzothiazole in the presence of Pd(PPh3)4 and KOAc in DMAC (Heck reaction). The latter proceeded in poor yield so we developed a second method starting with benzoic acid derivative 7, which was converted to the acid chloride and then condensed with 2-amino-thiophenol using pyridine in xylene20 (not shown). The addition of PTSA to the reaction mixture considerably improved the overall yield up to 70%.
Scheme 5. aSynthesis of dialkylimidazoles containing a benzothiazole group.

aReagents and conditions: (a) PhR1B(OH)2, Ba(OH)2.8H2O, Pd(PPh3)4, DME-H2O (5:1), reflux, 90%; (b) NaNO2, H2SO4, AcOH, CuBr2, HCl, 71%; (c) benzothiazole, Pd(PPh3)4, KOAc, DMAC, 160 °C, overnight, 32–35%; (d) SnCl2.2H2O, EtOAc, reflux, 6 h, 76–80%; (e) AcOH, 5, MeOH, 4Å mol. sieves, NaBH3CN, overnight, r.t, 60–65%.
Scheme 6a shows the synthesis of additional dialkyl imidazoles containing different substituents on each of the phenyl groups using methods derived mainly from Scheme 1. Scheme 6b shows the route to a dialkyl imidazole containing a 3,4-diphenyl unit, which was installed via Suzuki cross coupling.
Scheme 6. aaSynthesis of dialkylmidazoles with various substitutions on the two phenyl groups.

a Reagents and conditions: (a) R1B(OH)2 Pd(OAc)2, K2CO3, acetone/H2O, 89%; (b) SnCl2.2H2O, EtOAc, reflux, 2 h 90%; (c) AcOH, 5, MeOH, 4Å mol. sieves, NaBH3CN, r.t, overnight, 80%.
ba Synthesis of a dialkylimidazole bearing a 3,4-diphenyl unit.
a Reagents and conditions: (a) Tf2O, pyridine, r.t, overnight, 96%; (b) PhB(OH)2, Pd(PPh3)4, K2CO3, DME/EtOH/H2O, 82%; (c) SnCl2.2H2O, EtOAc, reflux, 2 h, 90%; (d) AcOH, 5, MeOH, 4Å mol. sieves, NaBH3CN, r.t, overnight, 40%.
Finally, Scheme 7 shows the route for preparing dialkyl imidazoles with an ortho-amino group on the benzyl unit attached to the imidazole nitrogen. Several compounds in this series were prepared as we found that the addition of this amino group improved anti-parasite potency. The route is derived from Scheme 1. We also developed a large scale synthesis of 44a and 44d. In this case, all reaction products were purified by recrystallization except for 41 and 42.
Molecular modeling and structure activity relationship studies
We made use of the homology model of the T. cruzi L14DM in complex with tipifarnib, based on the Mycobacterium tuberculosis enzyme structure, earlier described21. Design and docking studies were carried out with the FLO/QXP program suite, version 060221. In each case amino acid residues within 11 Å of tipifarnib were included in the binding site model for Metropolis Monte Carlo searches and energy minimization procedures. Details of the procedures were earlier described21.
In order to understand the structure-activity relationship of the various modifications of the dialkyl imidazoles, we docked the various compounds into the homology model of the T. cruzi L14DM. The scaffold of all compounds in this paper consists of a dialkyl imidazole, where one substituent is benzyl or biphenyl and the other is biphenylamine. The imidazole nitrogen binds to the heme iron, and the two substituents occupy mainly hydrophobic clefts. The anilino fragment of the scaffold is surrounded by several hydrophobic residues, Tyr 77, Phe 84 and Ala 265, but the amino group does not interact directly with the enzyme, thus acting as a spacer.
To study the effect of substitutions on biological activity we explored the possibility with various functional groups by varying the size, polarity and position on the phenyl ring (Table 1). Compounds described in this report were generated from previous compounds shown to have potent activity against T. cruzi cells. The benzyl substituent on the imidazole fits in a hydrophobic cleft created by Phe 264, Leu 330 and Val 435, and allows for small substituents in the ortho and meta positions only on one side of the aromatic ring and for larger substituents in the para position. Small meta substituents (10c, 10d 10e) also contact Val 435. Various hydrophobic para substituents are tolerated (10c, 10d, 10e, 10h) consistent with the lipophilic character of the cleft’s extension defined by Tyr 77, Met 80, Ile 183 and Met 434. Not surprisingly, the p-phenyl substituent (10h) is the most potent, with and EC50 of 0.5 nM because of its size. The t-butyl substitution (10k) leads to poor activity as it is too bulky for the narrow cleft. Hydrophilic substituents in the para position (10b, 10i) are incompatible with the lipophilic character of the cleft. Only one ortho substituent appears to be tolerated (10i), projecting an amino group towards His 268 and making a hydrogen bond with the imidazole. Hydrogen bond acceptors (10b) or hydrophobic groups (10c, 10d, 10e) in the ortho position would desolvate the imidazole and hence are expectd to have very poor activity.
Table 1.
Activities of compounds showing general structure I against T. cruzi amastigotes (EC50) and Murine fibroblast cells (Scheme 1).
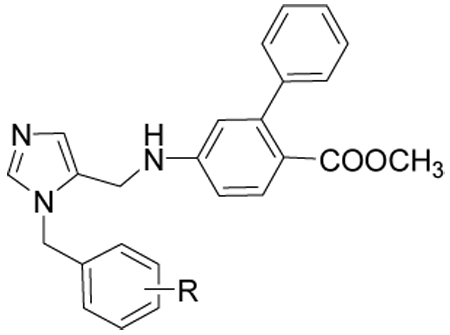 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R | T-cruzi EC50 (nM) | Fibroblasts EC50 (nM) | T-cruzi EC50 (nM) | Fibroblasts EC50 (nM) | T-cruzi EC50 (nM) | Fibroblasts EC50 (nM) |
| Para | meta | ortho | |||||
| 10a | H | 80 | >1000 | – | – | – | – |
| 10b | NO2 | 100 | >10000 | 30 | >10000 | 100–1000 | 10000 |
| 10c | CH3 | 5 | 50000 | 20 | >10000 | 1000 | 10000 |
| 10d | Cl | 5 | 25000 | 20 | >10000 | 100–1000 | 10000 |
| 10e | Br | 20 | >10000 | 20 | >10000 | 100 | 10000 |
| 10f | OMe | 25 | ND | ND | ND | ND | ND |
| 10g | CN | 40 | >10000 | 100 | >10000 | 200 | 25000 |
| 10h | Ph | 0.5, 0.9, 1.0 | >1000 | ND | ND | ND | ND |
| 10i | NH2 | 250 | >10000 | 130 | >10000 | 5 | 25000 |
| 10j | F | ND | ND | 150 | >1000 | ND | ND |
| 10k | C(CH3)3 | ND | ND | 475 | >1000 | ND | ND |
| 10l | 2,4-F2 | 21, 25 | >1000 | – | – | – | – |
We achieved considerable potency in the ester series of analogs (Table 1), but these compounds lack metabolic stability in rodents because of hydrolases that are prevalent both extra- and intracellularly. Schematic changes have been made on the basic skeleton by docking into the homology model. Based on the previous data (Table 1), we decided to maintain the biphenyl group on the imidazole and modify the ester functionality. Our first approach involved the synthesis of various esters and amides in anticipation of reducing the risk of plasma hydrolysis. We further explored the series and undertook a systematic survey of structure-activity relationships.
Various substituents coming off the para position of the anilino fragment in the scaffold (Table 2) can make limited contacts with Tyr 77 and Phe 84 but mainly point into the solvent surrounding the enzyme. This explains why almost all substitutions are tolerated in this position. While the esters (15a, 15b, 15c) have good activity in vitro they are liable to hydrolysis in vivo, hence we began efforts to find non-hydrolysable replacements such as halogens, ketones, amides, and various ring systems. In particular we investigated ester replacements of our previous compounds and assessed their viability by molecular modeling. We observed that there was sufficient room in the hydrophobic binding pocket, and we introduced a variety of heterocycles at the para position to the biphenyl aniline system. We determined that substituting the ester with other functional groups, such as benzothiazole, chloro, or methoxy, which can fit into the hydrophobic binding pocket, was needed for potency as well as stability. Pharmacokinetic stability was examined on selected analogs from Table 2. Compounds 27a and 27d showed good pharmacokinetic profile in mice7, but these compounds are relatively ineffective against cultured T. cruzi (Table 2). In the hope of maintaining these improved pharmacokinetic properties we focused on further modifications to increase potency. In particular, the benzothiazole from the structure-activity study in Table 2 was retained and the biphenyl was replaced with substituted benzyl groups on the imidazole. Branched p-substituents on the benzyl, isopropyl (32a) or methylsulfonyl (32e) do not fit in the narrow hydrophobic cleft (Table 3). As to the m-phenyl substituent on the anilino fragment of the scaffold, various residues lining the pocket are flexible and can easily adopt alternative rotamers as they are not packed by the other residues Arg 96 and Met 97. It is not surprising that the methyl group of 32g and 32h can make extra contacts and afford a slightly better activity. These structural differences do not significantly affect the binding behaviour, as shown in Table 3.
Table 2.
Activities of compounds showing general structure II against T. cruzi amastigotes (EC50) and Murine fibroblast cells (nM) (Scheme-2,Scheme-3,Scheme-4).
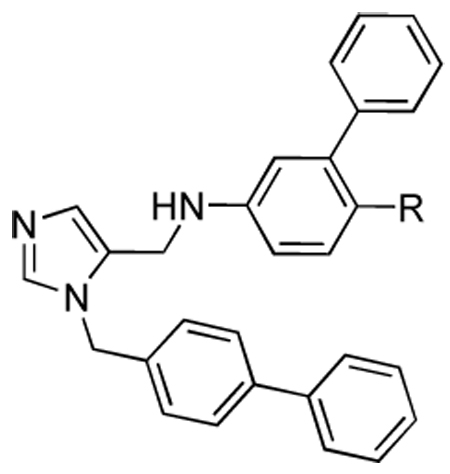 | |||
|---|---|---|---|
| Compound | R | EC50T. cruzi (nM) | EC50 Fibroblasts (nM) |
| 15a | COOEt | 10 | >10,000 |
| 15b | COOiPr | 50 | >10,000 |
| 15c | COOCy | 10 | >10,000 |
| 16a | C(=O)NHCH3 | 2.4, 3.0, 3.8, 5.1 | >1000 |
| 16b | C(=O)N(CH3)2 | 9.3, 14 | >1000 |
| 16c | C(=O)N(CH2)5 | 24, 39 | >1000 |
| 20a | C(=O)CH3 | 40 | >10,000 |
| 20b | C(=O)Et | 40 | >10,000 |
| 20c | C(=O)Pr | 40 | >10,000 |
| 24b | Cl | 6, 7 | >1000 |
| 24c | CN | 1.3, 1.8 | >1000 |
| 24d | 1-piperidine | 23, 35 | >1000 |
| 24e | 1-pyrrolidine | 22, 41 | >1000 |
| 24f | N(CH3)2 | 20, 26 | >1000 |
| 24g | OCH3 | 7, 9 | >1000 |
| 27a | 5-thiazole | 9, 9, 22, 28, 28 | >1,000 |
| 27b | 2-pyrrole | 100 | >10,000 |
| 27c | 2-benzofuran | 40 | >10,000 |
| 27d | 2-benzothiazole | 19,7,7,15,14,22,21 | >1,000 |
| 27e | 2-benzoxazole | 100 | >10,000 |
| 27f | 3-benzisoxazole | 10 | >10,000 |
| 27g | 3-anthranil | 50 | >10,000 |
Table 3.
Activities of compounds showing general structure III against T. cruzi amastigotes (EC50) and Murine fibroblast cells (nM) (Scheme-5).
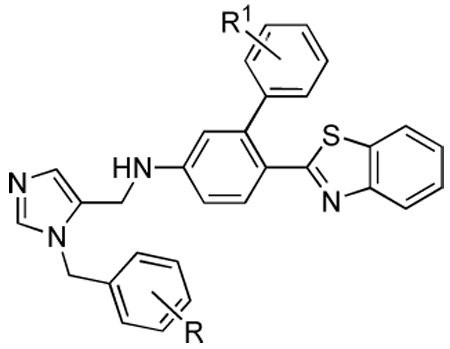 | ||||
|---|---|---|---|---|
| Compound | R | R1 | EC50T. cruzi (nM) | EC50Fibroblasts (nM) |
| 32a | p-isopropyl | H | 540, 860 | >1000 |
| 32b | p-ethyl | H | 110, 190 | >1000 |
| 32c | p-chloro | H | 250 | >1000 |
| 32d | o, p-difluoro | H | 420, 630 | >1000 |
| 32e | p-methyl sulfonyl | H | >1000 | >1000 |
| 32f | p-(2-toluyl) | H | 120, 180 | >1000 |
| 32g | p-Ph | 2-methyl | 31, 42 | >1000 |
| 32h | p-Ph | 2, 3-dimethyl | 64, 101 | >1000 |
Using the structure-activity from Table 1, we decided to explore further combinations of substitutions of the two-phenyl rings of the scaffold, in the absence of the methyl acetate ester substituent. Para substitutions on the benzyl recapitulate our findings from Table 1, with 35d being the most active. None of the substitutions on the anilino fragment were as active as the m-phenyl. Finally we combined the most active substituents of Table 1 – Table 4. All compounds exhibit EC50s against T. cruzi in the 1–25 nM range (Table 5). We focused our attention towards the ortho amino substituted compound 10i, which is the analog in this series showing the best potency against cultured T. cruzi. The combination of these observations resulted in a series of compounds possessing inhibition the 1–2 nM range against T. cruzi (Table 5) and these structural differences significantly affect the potency. Among these the most active is 44d, which projects the amino-substituted biphenyl in the hydrophobic cleft that has a uniquely placed histidine for forming a hydrogen bond. It also possesses a benzothiazole that makes hydrophobic interactions with Tyr 77 and Phe 84. The predicted binding mode is shown in Figure 2.
Table 4.
Activities of compounds showing general structure IV against T. cruzi amastigotes (EC50) and Murine fibroblast cells (nM) (Scheme-6).
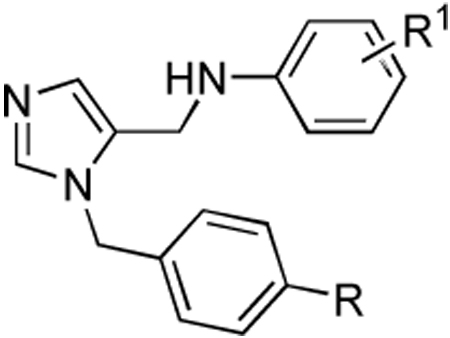 | ||||
|---|---|---|---|---|
| Compound | R | R1 | EC50T. cruzi(nM) | EC50 Fibroblasts (nM) |
| 35a | NO2 | 3-Ph | 100 | 10,000 |
| 35b | CH3 | 3-Ph | 100 | 10,000 |
| 35c | Cl | 3-Ph | 80 | 10,000 |
| 35d | Ph | 3-Ph | 10, 29, 23, 20 | 10,000 |
| 35e | H | 3-Ph | 360, 400 | >750 |
| 35f | H | 3-CH(CH3)2 | >1000 | >1000 |
| 35g | H | 3-(o-toluyl) | 230, 310 | >1000 |
| 35h | H | 3-(m-toluyl) | 550 | >1000 |
| 35i | H | 3-(o,m-dimethyl phenyl) | 260, 300 | >1000 |
| 35j | H | 3-(3’-pyridyl) | 600, 760 | >1000 |
| 35k | H | 3-(4’-pyridyl) | 520, 510 | >1000 |
| 35l | Ph | 4-COOCH3 | 80 | > 10,000 |
| 35m | Ph | 2-Ph | 80 | > 10,000 |
| 35n | Ph | 4-Ph | 200 | > 10,000 |
| 39 | Ph | 3-Ph; 4-Ph | 80 | > 10,000 |
Table 5.
Activities of compounds showing general structure V against T. cruzi amastigotes (EC50) and Murine fibroblast cells (nM) (Scheme-7).
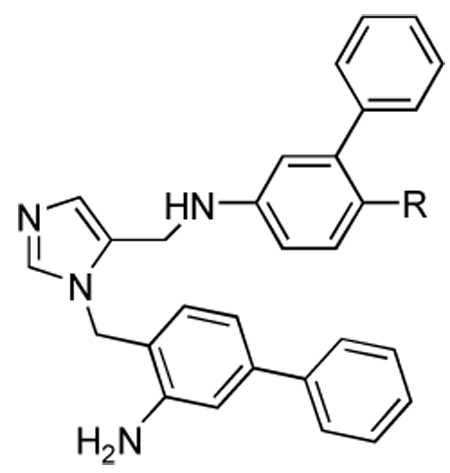 | |||
|---|---|---|---|
| Compound | R | EC50T. cruzi (nM) | EC50 Fibroblasts (nM) |
| 44a | Cl | 1.2, 1.5, 2.1 | >1000 |
| 44b | CN | 1.5, 2.6 | >1000 |
| 44c | OCH3 | 0.8, 2.1 | >1000 |
| 44d | 2-benzothiazole | 0.4, 0.6, 0.7, 1.1, 1.4 | >1000 |
| 44e | 5-thiazole | 10, 13 | >1000 |
| 44f | C(=O)NHCH3 | 12, 26 | >1000 |
Figure 2.

Homology model of T. cruzi L14DM in complex with 44d.
Binding of 44a and 44d to recombinant T. cruzi L14DM in vitro
We tested the binding of key compounds 44a and 44d to recombinant T. cruzi L14DM by monitoring the difference visible spectral change that occurs when the imidazole nitrogen coordinates to the heme iron (Soret band shift) (Figure 3). Both compounds were found to bind tightly to L14DM, with a maximal difference spectrum obtained when the amount of inhibitor approached the total amount of enzyme in solution. This indicates that the equilibrium dissociation constant for the L14DM-inhibitor complex is << 2.2 µM, the concentration of enzyme used in the assay. It was not possible to obtain accurate values of the dissociation constants because the use of lower enzyme concentrations gives rise to spectral signals that are not significantly above the noise. The spectral shift observed (type II difference spectrum7) is consistent with direct coordination of the imidazole nitrogen of the inhibitors to the heme-iron. Thus, it is expected that these compounds compete with lanosterol for binding to L14DM, but this was not established with kinetic studies.
Figure 3. Difference spectra for the binding of 44a (top panel) and 44d (bottom panel) to recombinant T. cruzi L14DM.

Compounds were added to 2.2 µM enzyme in increments of 0.2 µM. Maximum difference spectra were obtained with equimolar enzyme and compound. The absorbance around 360 nm in the spectra with 44d is attributed to the compound alone. Binding of compound to enzyme caused an increase in absorbance at ~440 nm and a decrease at ~390 nm.
Pharmacokinetics and activity of compounds in the murine model of Chagas disease
Two compounds with potent activity against T. cruzi in vitro, 44a and 44d, were subjected to single dose (50 mg/kg by oral gavage) pharmacokinetic studies on Balb/c mice in groups of three. Table 6 gives the data summary, and plasma concentration-versus-time plots for each mouse are provided as Supplemental Material. For 44a, the peak average blood level of 2.4 µg/mL (Cmax) was obtained in 0.8 hr (Tmax) (average from 3 mice). Values for 44d are Cmax = 7.2 µg/mL and Tmax = 1.7 hr. We collected plasma drug concentration data out to 5 hr and obtained AUC0–5hr of 6.3 and 28.7 µg-hr/mL for 44a and 44d, respectively. Accurate terminal elimination half-lifes were not obtained, but the data show that the half-life is in excess of 3 hr for 44a (Supplemental Material). For 44d, significant drug loss was not observed out to 5 hr (Supplemental Material) showing that this compound is more stable than 44a in mice. These values are not very different from the published terminal phase half-life of posaconazole of 7–9 hours in mice and rats22. A long elimination half-life, as exhibited by these compounds, is believed to be important for successful elimination of the slowly dividing T. cruzi during chronic infection23.
Table-6.
Single oral dose pharmacokinetics of T. cruzi L14DM inhibitors
| Compound | 44a | 44d | ||
|---|---|---|---|---|
| Mouse 1, 2, 3 | Avg ± SD | Mouse 1, 2, 3 | Avg ± SD | |
| Cmax (µg/mL) | 2.8, 2.4, 1.9 | 2.4 ± 0.5 | 5.8, 8.6, 7.2 | 7.2 ± 1.4 |
| Tmax (hr) | 0.5, 1.0, 1.0 | 0.8 ± 0.3 | 1.0, 2.0, 2.0 | 1.7 ± 0.6 |
| AUC 0_5hr (µg-hr/mL) | 6.7, 6.7, 5.6 | 6.3 ± 0.6 | 24.7, 32.4, 29.0 | 28.7 ± 3.9 |
The efficacies of the 44a and 44d in the murine model of Chagas disease were compared to vehicle and posaconazole (Figure 4). The compounds or vehicle were administered to a group of 6 mice at the indicated doses twice per day for 21 consecutive days to mice by oral gavage from days 7–27 post-infection. Parasitemia was monitored by microscopic examination of blood through 97 days post-infection. All mice in the vehicle group developed overwhelming parasitemia and were dead by day 16 post-infection. All drug-treated groups manifested a low level of parasitemia in the post-treatment period that gradually declined to levels that were microscopically undetectable by the end of the experiment (Figure 4). The differences in parasitemia between the four compound-treated groups were not statistically significant at any if the time points. At 100 days, the mice were sacrificed, and 200 µL of whole blood was subjected to PCR for detection of T. cruzi. Parasitemia was suppressed below PCR detectability in all the posaconazole treated mice and in 4 of 6 mice treated with 44d at 50 mg/kg. Although parasitemia was dramatically suppressed by 44d at 20 mg/kg and by 44a at 50 mg/kg, most or all of these mice had detectable parasites by PCR (Table 7). It is uncertain whether or not the PCR negative mice are absolutely cured of the T. cruzi infection due to the intermittent nature of parasitemia in chronically infected animals. Future studies will also employ the approach of giving immunosuppression to the mice at the termination of the experiment to more definitively determine if the mice are parasitologically cured24.
Figure 4. Efficacy of compounds in mice infected with T. cruzi.

Mice in groups of 6 were given compounds or vehicle by oral gavage twice per day from days 7–27 post-infection. Parasitemia was quantified on wet mounts of fresh blood. All vehicle treated mice were dead by day 16. Dramatic suppression of parasitemia was observed in all compound treated groups, with 44d at 50 mg/kg showing the most suppression along with posacanzole. The mice tolerated all of the treatments without apparent side effects.
Table-7.
Mice parasitemia by PCR
| Compound | Dose | PCR parasite detection after 100 days post infection |
|---|---|---|
| 44d | 50 mg/kg | 2 of 6 mice |
| 44d | 20 mg/kg | 5 of 6 mice |
| 44a | 50 mg/kg | 6 of 6 mice |
| Posaconazole | 20 mg/kg | 0 of 6 mice |
The mice appeared to tolerate the drug treatments without apparent side effects. All mice in the four drug-treated groups survived to the termination of the experiment at day 100. Weights were monitored on a weekly basis, and all treated animals gradually gained weight from the completion of drug treatment until the end of the experiment (data shown in Supplemental Material).
Conclusions
From this study have come several new inhibitors of T. cruzi L14DM that are among the most potent inhibitors of in vitro T. cruzi amastigote growth known to date (sub-nanomolar to low-nanomolar potency). Two of the most promising compounds, 44a and 44d, were chosen for studies in mice and display excellent pharmacokinetic properties including oral activity and reasonable stability in mouse plasma. These two compounds are efficacious in a mouse model of acute Chagas disease with efficacy comparable to that of posaconazole. This new class of L14DM inhibitors should be much cheaper to produce than posaconazole. Further studies are in progress to explore the animal toxicology profile of these compounds as well as their efficacy in a chronic model of Chagas disease in mice as well as efficacy studies in larger animals.
Experimental Section
Synthesis of compounds
Unless otherwise indicated, all anhydrous solvents were commercially obtained and stored under nitrogen. Reactions were performed under an atmosphere of dry nitrogen in oven dried glassware and were monitored for completeness by thin layer chromatography (TLC) using silica gel 60 F-254 (0.25 mm) plates with detection with UV light. 1H-NMR spectra were recorded on dilute solutions in CDCl3, CD3OD or DMSO-d6 at 300 MHz. Chemical shifts are reported in parts per million (δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Electron spray ionization mass spectra were acquired on an Bruker Esquire LC00066.
Flash chromatography was carried out with silica gel (40–63 µm). Preparative reverse phase HPLC was performed on an automated Varian Prep star system using a gradient of 20% MeOH to 100% MeOH (with 0.1% trifluoroacetic acid) at 12 ml/min over 30 min using a YMC S5 ODS column (20×100 mm, Waters Inc.). All compounds tested on parasites were purified to a single peak by HPLC as described above. HPLC purified compounds were submitted to 1H NMR analysis, and compounds were submitted to testing on parasites if the 1H NMR spectrum was free of detectable non-compound peaks (estimated purity at least 95%).
1-Triphenylmethyl-4-imidazole carboxaldehyde (4)
To a 1L three-necked round-bottomed flask with an addition funnel was added 3H-imidazole-4-carbaldehyde (12 g, 0.125 mol), trityl chloride (38.3 g, 0.137 mol), and acetonitrile (400 mL). The mixture was stirred at r.t. to give a slurry. Triethylamine (30 mL, 0.215 mol) was added dropwise over 20 min. After the addition was complete, the reaction mixture was stirred at r.t. for 20 h. Hexane (40 mL) and water (400 mL) were added. The slurry was stirred for 30 min and filtered. The cake was washed with water (3 × 100 mL) and dried in a vacuum oven at 50 °C, for 20 h to give 4 as a white solid (39.8 g, 94%). 1H NMR (CDCl3) δ 9.81 (s, 1H), 7.54 (s, 1H), 7.46 (s, 1H), 7.29 (m, 10H), 7.04 (m, 5H); MS m/z 339.4 (M+H)+.
General Procedure for the synthesis of 3-Alkyl -3H-imidazole-4-carbaldehyde (5)
A solution of compound 4 (1.5 mmol) in acetonitrile (10 mL) was treated with alkyl bromide (1.5 mmol) at r.t. and heated to 60 °C and stirred overnight under nitrogen. The reaction mixture was cooled and concentrated under vacuum, and the resulting paste was triturated with acetone (20 mL) and stirred for 2–3 h. The resulting solid was isolated by filtration and extracted with CH2Cl2 (2 × 25 mL) and washed with saturated NaHCO3. The organic layers were combined, dried over Na2SO4 and concentrated to give 5 as a yellow solid6.
2-Methyl-5-nitro-biphenyl (6)
To a solution of 4-nitro-2-bromotoluene (6.48 g, 30 mmol) and phenyl boronic acid (3.84 g, 31.5 mmol) in 70 mL of acetone was added 85 mL of water, potassium carbonate hydrate (12.4 g, 75 mmol, 2.5 equiv) and Pd(OAc)2 (0.33 g, 5% equiv). The mixture was refluxed for 10 h and then cooled. The deep black solution was extracted with ether and 3 N HCl. The ether fraction was passed through a layer of Celite. After evaporating solvent, the residue was dried and then recrystallized from methanol to give flake crystals7 (4.60 g, 88%). mp 77–78 C. 1H NMR (CDCl3) δ 8.09–8.11 (m, 2H, aryl), 7.40–7.49 (m, 4H, aryl), 7.30–7.33 (m, 2H, aryl), 2.37 (s, 3H). MS m/z 213.3 (M+H)+.
5-Nitro-biphenyl-2-carboxylic acid (7)
2-Methyl-5-nitro-biphenyl (3.2 g, 15 mmol) was dissolved in 15 mL of pyridine and 30 mL of water. The mixture was heated to 90 °C and KMnO4 (14.2 g, 90 mmol) was added in portions. The mixture was refluxed for 5 h. The black solid was filtered off, and the filtrate was acidified with 6 N HCl. The mixture was cooled in an ice bath and the white precipitate was collected 7 (3.1 g, 85%). mp 175–176 °C. 1H NMR CCDCl3) δ 8.25–8.33 (m, 2H, aryl), 8.08 (d, J = 8.9 Hz, 1H, aryl), 7.41–7.51 (m, 3H, aryl), 7.31–7.39 (m, 2H, aryl). MS m/z 243.2 (M+H)+.
5-Nitro-biphenyl-2-carboxylic acid methyl ester (8)
A solution of compound 7 (2.84 g, 11.7 mmol) in methanol (35 mL) was treated with SOCl2 (4.16 g, 35 mmol) dropwise at 0 °C. The mixture was heated to reflux for 6 h, then cooled to r.t.. Upon transfer, spontaneous crystallization occurred. The residual solvent was then filtered off, and the resulting solid was left to dry overnight to give 8 (2.28 g, 76%). 1H NMR CCDCl3) δ 8.25 (m, 2H), 7.92 (d, J = 8.4 Hz, 1H), 7.43 (m, 3H), 7.32 (dd, 2H), 3.67 (s, 3H, COOCH3). MS m/z 258.1 (M+H)+.
5-Amino-biphenyl-2-carboxylic acid methyl ester (9)
A mixture of 5-nitro-biphenyl-2-carboxylic acid methyl ester 8 (2.0 g, 7.8 mmol) and SnCl2.2H2O (8.8 g, 39 mmol) in EtOAc (75 mL) was stirred at reflux under nitrogen for 2.5 h. Upon cooling, saturated NaHCO3 (150 mL) was added. The organic layer was removed, and the aqueous layer washed with EtOAc (2 ×100 mL). The combined organic layers were dried (Na2SO4) and concentrated to dryness to give 9 as a white solid (1.53 g, 86%). 1H NMR CCDCl3) δ 7.80 (d, J = 8.4 Hz, 1H), 7.27–7.37 (m, 5H, aryl), 6.63 (dd, J = 8.4, 2.1 Hz, 1H), 6.56 (d, J = 8.4 Hz), 3.59 (s, 3H, COOCH3). MS m/z 228.3 (M+H)+.
General procedure for reductive amination10
To a solution of 9a-l (0.32 mmol) and 5 (0.32 mmol) in MeOH (6 mL) was added 0.4 g of 4 angstrom molecular sieves. The solution was stirred at r.t. under nitrogen for 1 h, after which acetic acid (4.4 mmol) was added. After 5 min, NaCNBH3 (0.64 mmol) was added in portions. The mixture was stirred at r.t. under nitrogen overnight. The reaction mixture was mixed with CH2Cl2 and saturated NaHCO3. The aqueous layer was extracted with CH2Cl2, after which the combined organic layers were washed with brine. The organic layer was dried with Na2SO4, and the solvent was removed by evaporation. The crude product was purified by preparative HPLC to give 10 a-l.
See Supporing Information for p-10b, p-10c, p-10d, p-10e, p-10f, p-10h, p-10i, p-10k, m-10b, m-10c, m-10d, m-10e, m-10g, 10l, m-10i, m-10j, o-10b, o-10c, o-10d, o-10e, o-10g, and o-10i.
Compounds 11a-b were prepared by following the procedure described for the synthesis of 8 (Supporting Information).
Representative amidation
A solution of compound 7 (1.75 g, 7.2 mmol) in SOCl2 (3 mL) was heated to reflux for 2 h, then cooled to r.t. and concentrated under vacuo to give a pale white solid. The crude acid chloride (1.87 g, 6.7 mmol) was dissolved in CH2Cl2 (25 mL) and was added to a stirred solution of the amine (6.7 mmol) and triethylamine (15.4 mmol) in CH2Cl2 at 0 °C, and the mixture was stirred for 4 h at r.t., diluted with H2O and extracted with CH2Cl2 (2 × 30 mL) and concentrated under vacuo to provide 12a-c. Compounds were submitted to nitro reduction conditions as described for the synthesis of 9.
Compounds 13a-c were prepared by following the procedure described for the synthesis of 9 (Supporting Information).
Compounds 15a-c were prepared from 13 and 5 by following the procedure described for the synthesis of 10 (Supporting Information).
Compounds 16a-c were prepared via reductive amination of 14a-c with 5 following the procedure described previously (Supporting Information).
Representative acylation and Fries rearrangement
A mixture of 3-aminophenol (1.09g, 10 mmol) and butyryl anhydride (4.1 mL, 25 mmol) in pyridine (10 mL) was stirred at r.t. for 1.5 h. The reaction mixture was diluted with EtOAc (60 mL), and the organic layer washed with sat. NaHCO3 (2 × 60 mL), 10% HCl (2 × 60 mL), dried (Na2SO4) and concentrated, and to the resulting oil was added 1.5 mL of 1,2-dichlorobenzene and AlCl3 (2.67g, 20 mmol) and the solution was stirred at 120 °C overnight under nitrogen. The resulting tar was added to EtOAc (60 mL), and washed with water (3 × 60 mL) and brine (60 mL). The organic layers combined and dried over Na2SO4 and concentrated. The resulting tan solid was recrystallized from EtOAc to yield 17a-c as a brown solid (1.04 g, 40%) (Supporting Information).
Representative triflation
To solution of compound 17a-c (249 mg, 1.0 mmol) in pyridine (3 mL) was added Tf2O (0.20 mL, 1.2 mmol) dropwise at 0 °C and the mixture was stirred at r.t. overnight. The solution was diluted with EtOAc (10 mL) and washed with water and brine. The organic layers were combined, dried (Na2SO4) and concentrated under vacuo to get 18a-c as brown oil (365 mg, 96%) (Supporting Information).
Representative Suzuki cross coupling
To a mixture of 18 (193 mg), phenylboronic acid (101 mg, 0.83 mmol, 1.4 equiv) and Pd(PPh3)4 (69 mg, 10 mol%) in dimethoxyethane (5 mL) was added ethanol (0.5 mL) and K2CO3 (0.59 mL of 2 M in H2O, 2 equiv), and the solution was stirred at reflux overnight under nitrogen. The solvent was removed under vacuum and the resulting solid was taken up into CH2Cl2 (30 mL) and washed with brine (30 mL). The solvent was removed by evaporation, and the resulting black solid was purified by column chromatography using a gradient of 1:1 Hex:EtOAc yielded a white solid (81 mg, 54%).
See Supporting Information for 19a-c.
Representative N-acyl deprotection and reductive amination
Biphenyl 19 (180 mg, 0.58 mmol) was dissolved in 5 mL i-PrOH, and NaOH (232 mg, 5.8 mmol in 0.5 mL H2O) was added. The solution was heated to reflux overnight, after which the solvent was removed under vacuum. The resulting solid was taken up into EtOAc (60 mL) and washed with sat. NaHCO3 (60 mL), water (60 mL), and brine (2 × 60 mL). The organic layer was dried over sodium sulfate to yield an orange oil, (112 mg, 81%). The resulting aniline was coupled to 5 as described for compound 10.
See Supporting Information for 20a-c.
2-Phenyl-4-nitrobromobenzene (22a)
Sodium nitrite (2.66g, 38.5 mmol, 1.1 equiv.) was added in portions to 21 mL of concentrated sulfuric acid at r.t.. The suspension was cooled to 10 °C and acetic acid (22 mL) was added dropwise. The mixture was stirred for 20 min. at 10 °C, and 2-phenyl-4-nitroaniline (7.56 g, 35 mmol) was added in portions over 30 minutes. The solution was stirred for 2h. at 10 °C, and water (15 mL) was added to clear the suspension. The solution was stirred for 1 h at r.t. and cupric bromide (13.07 g, 56 mmol, 1.6 equiv.) in 27 mL of 2M HCl was added slowly. The resulting black sludge was stirred for 20 min at r.t. and 1h at 60 °C. The solution was added to ether (200 mL) and washed with water (3 × 100 mL) and brine (150 mL). The organic layer was dried with sodium sulfate and concentrated to give an orange solid which was purified by recrystallization from methanol to yield a red solid (6.94 g, 71%). 1H NMR (CDCl3) δ 8.20 (d, J = 2H, 1H, ortho to NO2), 8.06 (dd, J = 2 Hz and 9 Hz, 1H, ortho to NO2), 7.86 (d, J = 9 Hz, 1H, ortho to Br), 7.41–7.49 (m, 5H, Ar).
Synthesis of 2-chloro-5-nitro-biphenyl(22b)
Synthesis of 22b performed as described for 22a. The procedure for the large-scale synthesis of 22b is as follows. A mixture of phenylboronic acid (1.51 g, 10.05 mmol), tetrakis(triphenylphosphine)palladium(0) (0.35 g, 0.30 mmol, 3 mol%) and K2CO3 (2.76 g, 20.0 mmol) in 10 mL (4:1) dry toluene/EtOH was stirred at room temperature under argon for 15 min. To this mixture was added 3-bromo-4-chloronitrobenzene 16 (2.37 g, 10.0 mmol), and the mixture refluxed for 24 h. The contents were cooled and filtered. The filtrate was diluted with toluene and washed with 10% citric acid (2 × 20 mL), 1M NaHCO3 (2 × 20 mL) and brine (2 × 20 mL). The combined organic layers were evaporated, to give a brown oil, which was recrystallized from hot hexane to give 22b as a pale yellow solid (1.56 g, 88%).
5-Nitro-biphenyl-2-carbonitrile 22c
To a solution of 22a (0.2 g, 0.72 mmol) in DMF (5 mL) was placed under high vacuum for 15 min. The solution was purged with Ar for 15 min. While purging was continued, ZnCN2 (101 mg, 0.86 mmol) and Pd(PPh3)4 (83 mg, 0.072 mmol) were added. The reaction mixture was heated at 100 °C under Ar for 18 h and cooled to r.t. and added to H2O. The mixture was extracted with EtOAc and then washed with brine, dried (Na2SO4), filtered and concentrated. The residue was purified by flash column chromatography using 30% EtOAc/hexane to give 97 mg of cyano compound 22c (60%).
Preparation of compounds 22d-e
Compounds 22d-e were prepared from 22a (1.0 g, 3.5 mmol) and 4.3 mmol of the corresponding amine. Starting materials were mixed together and heated at 100 °C for 2–3 h followed by usual workup to give the required nitro intermediate compounds.
Dimethyl-(5-nitro-biphenyl-2-yl)-amine (22f)
A solution of 21 (0.5g, 2.33 mmol) and sodium borohydride (0.43 g, 11.6 mmol) in THF (25 ml) was added drop wise to an efficiently stirred solution of 3M sulfuric acid (11.5 mL, 35 mmol) and 35% aqueous formaldehyde (9.3 mmol) in THF (5 ml) at 10–15 °C. After the first half of the addition, the mixture is acidified with 3M H2SO4 (11.5 ml, 35 mmol) and then stirred for 1 h. To the resultant mixture, water (20 ml) was added followed by the addition of aqueous KOH to raise the pH to about 9-–0. The organic phase was separated and the aqueous phase was extracted with ether (2 × 100 ml). The combined organic layers were washed with saturated NaCl and dried over sodium sulfate. The ether layer was evaporated to dryness, and the crude material was purified by flash chromatography eluting at 10% EtOAc to afford 22f, 0.45 g (80%).
Synthesis of 23a-g
Compounds were prepared as described for compound 9.
Synthesis of 24b-f
Compounds were prepared as described for compound 10. See Supporting Information for 24b-g.
Representative heterocyclic Heck reaction: 3-(5-Nitro-biphenyl-2-yl)-benzo[d]isoxazole (25f)
A mixture of 2-phenyl-4-nitrobromobenzene (22a, 556 mg, 2.0 mmol), Pd(PPh3)4 (116 mg, 0.1 mmol), and potassium acetate (294 mg, 3.0 mmol, 1.5 equiv.) were flushed with nitrogen for 5 min. after which N,N-dimethylacetamide (5 mL) was added. The solution was flushed with nitrogen for an additional 5 min. and 1,2-benzisoxazole (0.24 mL, 286 mg, 2.4 mmol, 1.2 equiv.) was added. The solution was stirred at 160 °C overnight. The solution was added to ether (150 mL) and washed with water (3 × 100 mL). The organic layer was dried with sodium sulfate and concentrated to yield a black oily solid which was purified by column chromatography 4:1 (Hexane:EtOAc) to obtain the crude product (206 mg), which was used without further purification.
See Supporting Information for 25a-c and 25e-g.
2-(5-Nitro-biphenyl-2-yl)-benzothiazole (25d)
A large scale synthesis of this intermediate was carried out as follows. A mixture of 7 (2 g, 8.23 mmol) and SOCl2 (20 mL) was refluxed for 3h. SOCl2 was removed under reduced pressure to give the acid chloride. The resulting 5-nitro-biphenyl-2-carbonyl chloride (2 g, 7.6 mmol) was added to a mixture of 2-aminothiophenol (0.82 mL, 7.6 mmol) and pyridine (0.61 mL, 7.6 mmol) in p-xylene (60 mL). The contents were stirred at room temperature for 1 h, then p-TsOH·H2O (7.2 g, 38.0 mmol) was added, and the reaction mixture was stirred at reflux. After 12 h, the reaction was cooled, extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were washed with saturated NaHCO3 (2 × 50 mL), brine (100 mL) and dried over Na2SO4. Evaporation of organics resulted in a green solid which was recrystallized from EtOAc to obtain 2-(5-nitro-biphenyl-2-yl)-benzothiazole 25d as a white solid (1.7 g, 70%). 1H NMR (CDCl3) δ 8.30–8.35 (m, 2H), 8.28 (d, 1H, J = 2 Hz), 8.07 (d, 1H, J =7 Hz), 7.72 (d, 1H, J = 9 Hz), 7.33–7.49 (m, 7 H).
Synthesis of compounds 26a-g
Compounds were prepared from 25a-g as described for the synthesis of 9.
Synthesis of compounds 27a-g
Reductive amination with 26 and 5 was performed as for compound 10. See Supporting Information for 27a-g.
Synthesis of compounds 32a-h
Preparation of compounds 32a-h were performed as described for 10. See Supporting Information for 32a-h.
Preparation of compounds 35a-n
Compounds were prepared from 34a-n and 5 under reductive amination conditions as performed previously. See Supporting Information for 35a-k.
3,4-diphenylnitrobenzene (37)
The synthesis was carried out according to the procedure for 19. 1H NMR (acetone-d6): δ 7.58 (dd, 1 H, J=3 and 9 Hz), 7.51 (d, 1 H, J=3 Hz), 7.00 (d, 1 H, J=9 Hz), 6.54–6.58 (m, 5 H), 6.48–6.51 (m, 4 H).
3,4-diphenylaniline (38)
The synthesis was carried out using 37 according to the procedure described for 9 1H NMR (acetone-d6) δ 7.02–7.20 (m, 11 H), 7.71–7.75 (m, 2 H).
(3-Biphenyl-4-ylmethyl-3H-imidazol-4-ylmethyl)-[1,1';2',1"]terphenyl-4'-yl-amine (39)
The reaction was carried out using 38 and 5 according to the procedure described for 10. Yellow solid, 40%. mp 195–199 °C. 1H NMR (acetone-d6) δ 7.68 (s, 1 H), 7.59–7.62 (m, 4 H), 7.44 (t, 2 H, J=7 Hz), 7.35 (tt, 2 H, J=2 and 8 Hz), 7.26 (d, 2 H, J=8 Hz), 7.121–7.17 (m, 5 H), 7.06–7.12 (m, 4 H), 7.01–7.04 (m, 2 H), 6.98 (s, 1 H), 6.72 (dd, 1 H, J=2 and 8 Hz), 6.64 (m, 1 H), 5.40 (s, 2 H), 4.31 (s, 2 H). 13C NMR (CDCl3) δ 146.65, 141.79, 141.47, 141.20, 138.87, 135.03, 131.58, 129.86, 129.71, 129.25, 128.85, 128.72, 127.81, 127.73, 127.59, 127.24, 127.03, 126.45, 125.74, 115.19, 112.34, 48.68, 38.29. HRMS [FAB M+H]+ (C35H30N3); calcd., 492.2439; found, 492.2440.
4-Methyl-3-nitro-biphenyl (40)
A large scale synthesis of 40 was carried out as follows. A mixture of 4-bromo-2-nitrotoluene 3 (1.0 g, 4.6 mmol), phenyl boronic acid (0.6 g, 5.09 mmol) and Ba(OH)2. 8H2O (3.2 g, 9.2 mmol) in 15 mL of DME:H2O (5:1) was stirred under Ar for 15 min. To this, Pd (PPh3)4 (0.53 g, 0.46 mmol) was added and the resulting solution was refluxed for overnight. The reaction was cooled and diluted with EtOAc (30 mL) and washed with NaHCO3 and brine. The resulting organic layer was filtered through celite, dried over (Na2SO4) and evaporated to give a brown solid, which was recrystallized from hexane to obtain a white solid (0.89 g, 90%). 1H NMR (CDCl3) δ 8.23 (d, J = 1.8 Hz, 1H), 7.75 (dd, J = 7.8, 1.8 Hz, 1H), 7.63 (m, 1H), 7.60 (d, J = 1.2 Hz, 1H), 7.53–7.45 (m, 2H), 7.44–7.39 (m, 2H) 2.65 (s, 3H) . MS m/z 214.2 (M +H+).
4-Bromomethyl-3-nitro-biphenyl (41)
A large scale synthesis of 41 was carried out as follows. A solution of 2-methyl-3-nitrobipyhenyl (40, 0.382 g, 1.79 mmol), NBS (0.333 g, 1.87 mmol), and a few crystals of benzoyl peroxide in CCl4 (15 mL) was refluxed for 36 h. The mixture was cooled and treated with benzene (50 mL). The resulting solution was filtered and evaporated to dryness in vacuo. The crude material was chromatographed (hexane/ EtOAc, 20:1) to give 41 (0.31 g, 60%). 1H NMR (CDCl3) δ 8.30 (d, J = 2.0 Hz, 1H), 7.86 (dd, J = 8.0, 2.0 Hz, 1H), 7.69–7.64 (m, 4H), 7.53 (d, J = 1.8 Hz,1H), 7.48 (m, 1H), 4.91 (s, 2H). MS m/z 293.1 (M+H+).
3-(3-Nitro-biphenyl-4-ylmethyl)-3H-imidazole-4-carbaldehyde (42)
A large scale synthesis of 42 was carried out as follows. 1-Trityl-4-imidazole carboxaldehyde (0.5 g, 1.5 mmol) and bromo compound 41 (0.43 g, 1.5 mmol) were stirred in acetonitrile (10 mL) at 60 °C under nitrogen overnight. The solvent was removed by evaporation, and the resulting paste was triturated with acetone (20 mL). The resulting solid was isolated by filtration and extracted with CH2Cl2 and saturated NaHCO3. The organic layer was dried over Na2SO4 and evaporated to dryness. The residue was purified by column chromatography (EtOAc/MeOH, 9.5 : 0.5) to give 42 as a solid 0.2 g. 1H NMR (CDCl3) δ 9.75 (s, 1H, -CHO), 8.36 (d, J = 1.8 Hz, 1H), 7.95 (s, 1H), 7.89 (s, 1H), 7.51 (dd, J= 8.1, 1.8 Hz, 1H), 7.59–7.55 (m, 2H), ), 7.50–7.41 (m, 3H), 6.84 (d, J = 8.1 Hz,1H), 5.95 (s, 2H). MS m/z 308.3 (M+H+).
Preparation of compounds 43a-f
The compounds were prepared by treating 23b,c, 26a,d and 14a with 42 under reductive amination conditions as performed previously for 10.
(6-Chloro-biphenyl-3-yl)-[3-(3-nitro-biphenyl-4-ylmethyl)-3H-imidazol-4-ylmethyl]-amine (43a)
A large scale synthesis was carried out as follows. To a mixture of 6-chloro-biphenyl-3-ylamine 26a (0.5 g, 2.46 mmol), 3-(3-Nitro-biphenyl-4-ylmethyl)-3H-imidazole-4-carbaldehyde 42 (0.72, 2.46 mmol) in methanol (25 mL), was added acetic acid (0.35 mL, 4.92 mmol) and 4Å molecular sieves. The resulting mixture was stirred at room temperature under argon for 0.5 h. To this, NaCNBH3 (0.4 g, 6.15 mmol) was added and then stirring continued overnight. The resulting solid was separated and dissolved in EtOAc (100 mL). The undissolved material was filtered off, and the organic layer was evaporated to give 43a as a yellow solid (0.715 g, 70%). 1H NMR 300 MHz (CD3OD) δ 9.05 (s, 1H), 8.41 (d, J = 2.1 Hz, 1H), 7.80 (dd, J = 1.8, 8.1 Hz, 1H), 7.70 (s, 1H), 7.45–7.65 (m, 5H), 7.25–7.35 (m, 5H), 7.15 (d, J = 8.4 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 6.52 (dd, J = 2.4, 8.4 Hz, 1H), 6.35 (d, , J = 2.4 Hz, 1H), 6.0 (s, 2H), 4.42 (s, 2H)). MS m/z 495.5 (M+H+).
See Supporting Information for 43b, 43c, 43e, and 43f.
(6-Benzothiazol-2-yl-biphenyl-3-yl)-[3-(3-nitro-biphenyl-4-ylmethyl)-3H-imidazol-4-ylmethyl]-amine (43d)
A large scale synthesis based on previous work25 was carried out as follows. To a solution of 26d (1.1 g, 3.67 mmol) and 42 (1.12 g, 3.67 mmol) in CH2Cl2 (40 mL) was added TiCl4 (1M solution in dichloromethane) (1.83 mL, 1.83 mmol) dropwise at r.t., and the mixture was stirred for 30 min. To this was added NaCNBH3 (276 mg, 4.4 mmol) in MeOH (6 mL), the resulting mixture was stirred overnight and the solvents was removed by evaporation. The resulting crude material was dissolved in CH2Cl2 (150 mL) and sat. NaHCO3 (50 mL) was added. The organic layer was separated and dried (Na2SO4) to obtain a yellow solid which was recrystallized from EtOH to afford 43d (1.52 g, 70%). 1H NMR 300 MHz (CD3OD) δ 9.1 (s, 1H), 8.42 (d, J = 2.1 Hz 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.71–7.81 (m, 4H), 7.57–7.62 (m, 2H), 7.24–7.49 (m, 8H), 7.13–7.18 (m, 2H), 6.95 (d, J = 8.4 Hz, 1H), 6.65 (dd, J = 2.4, 8.4 Hz, 1H), 6.40 (d, J = 2.4 Hz, 1H), 6.0 (s, 2H), 4.6 (s, 2H). MS m/z 594.4 (M+H+).
Preparation of compounds 44a-e
Compounds were synthesized by following the procedure described for 9.
(6-Chloro-biphenyl-3-yl)-[3-(3-amino-biphenyl-4-ylmethyl)-3H-imidazol-4-ylmethyl]-amine (44a)
A large scale synthesis was carried out as follows. To a solution of 43a (0.5 g, 1.01 mmol) in EtOAc (75 mL) was added SnCl2.2H2O (1.13 g, 5.05 mmol), and the mixture was stirred at reflux for 3 h. Upon, cooling saturated NaHCO3 100 mL) was added until the pH was neutral, and the liquid was filtered through a pad of celite. The organic layer was separated, and the aqueous layer was extracted with EtOAc (2 × 100 mL). The combined organic layers were washed with brine and dried over Na2SO4. Evaporation of the solvent resulted in crude material 44a, which was recrystallized from isopropanol (0.374 g, 80%). Off-white solid, m.p. 188–190 °C; 1H NMR (CD3OD) δ 8.70 (s, 1H), 7.51–7.58 (m, 3H), 7.32–7.47 (m, 8H), 7.23 (d, J = 8.1 Hz 1H), 7.14 (d, J = 1.8 Hz 1H), 7.10 (d, J = 8.1 Hz 1H), 6.98 (dd, J = 1.8, 7.8 Hz, 1H), 6.67 (dd, J = 2.4, 8.7 Hz, 1H), 6.59 (d, , J = 2.4 Hz, 1H), 5.45 (s, 2H), 4.50 (s, 2H). MS m/z 465.5 (M+H+).
See Supporting Information for 44b-f.
Preparation of posaconazole
Noxafil (Schering-Plough), was purchased from a pharmacy, and the active compound was purified from the suspension by organic extraction and flash chromatography. The liquid formulation (12.5 mL) was added to a 1 L separating funnel and diluted with water (300 mL) and then extracted with EtOAc (2 × 500 mL), and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (3 × 300 mL). The combined organic layers were evaporated to dryness and the solid obtained was purified by flash column chromatography eluting with 3% methanol in CH2Cl2 to yield 425 mg (85%).
T. cruzi and murine fibroblast growth inhibition assays
Compounds were screened against the β-galactosidase expressing the Tulahuen strain of T. cruzi in 96 well tissue culture plates as described previously.25, 26 The Tulahuen strain originated in Chile, and is grouped with the more common TCII phylogenetic lineage of T. Cruzi. In this assay T. cruzi proliferate as intracellular amastigotes withint murine 3T3 fibroblasts.
Compounds were screened in triplicate to determned values of EC50. Standard errors within assays were consistently less than 15%. Compounds were separately screened against murine 3T3 fibroblast cells to determine the EC50 values against these host cells. Growth of the 3T3 fibroblasts was quantified by the resazurin assay as previously described.25 Although some compounds were yellow-colored as solids, there was no effect of the diluted compounds on the colorimetric readouts.
Pharmacokinetic studies in mice
Compounds were suspended at 10 mg/ml in 20% (w/v) Trappsol™ hydroxypropyl beta-cyclodextrin (pharmaceutical grade) (CTD, Inc.) and administered to BALB/c mice (7–8 week females weighing approximately 20 g) by oral gavage in a volume of 100 µL. Thus, the mice received a dose of 50 mg/kg. At timed intervals, 40 µL of tail blood was collected in heparinized capillary tubes. Plasma was separated and frozen for later analysis according to the method previously reported27. Individual traces of plasma drug concentration versus time for each mouse are provided as Supporting Information.
Efficacy studies in mice
BALB/c mice (7–8 week females) were infected with 1 × 104 T. cruzi trypomastigotes (Tulahuen strain) by subcutaneous injection. By 7 days post-infection, every mouse had microscopically observable parasites on slides of peripheral blood. On day 7 post-infection, mice (in groups of 6) began receiving treatments by oral gavage twice per day for 21 days. Compounds were administered in a volume of 100 µL per dose using the vehicle, 20% (w/v) Trappsol™ hydroxypropyl beta-cyclodextrin (pharmaceutical grade) (CTD, Inc.). Parasitemia was monitored by placing 5 µL of tail blood under a cover slip and counting 50 high-powered fields. Mice that were pre-morbid from progressive infection were euthanized. All surviving mice were sacrificed on day 100 post-infection, and ~200 µL of blood from cardiac puncture was taken for PCR analysis of parasitemia.
L14DM binding studies
Expression of T. cruzi L14DM in E. coli and purification will be reported elsewhere. Binding reactions contained 2.2 µM T. cruzi L14DM (concentration determined from the Soret peak, absorbance at 420 nm minus absorbance at 490 nm using an extinction coefficient of 111 mM−1 cm−1) in 1 mL of 50 mM sodium phosphate, pH 7.5, 300 mM NaCl, 10% glycerol. Binding of inhibitors was monitored by difference spectroscopy (340–600 nm) in which 1 mL of the above protein solution was placed in the sample and reference cuvettes. After setting the difference spectrum to zero, 1 µL aliquots of inhibitor stock solution (200 µM in DMSO) was added to the sample compartment, and 1 µL of DMSO only was added to the reference cuvette. The inhibitor concentration was varied from 200 to 2000 nM.
PCR detection of T. cruzi parasitemia
Extraction of T. cruzi DNA and PCR amplification of DNA was performed according to previously published methods28–30. Briefly, 200 µl of whole was mixed with 200 µl of 6 M guanidine HCl – 0.2M EDTA and stored at 4°C. Samples were boiled for 15 minutes and extracted with an equal volume of phenol:chloroform (1:1), then extracted with an equal volume of chloroform:isoamyl-alcohol (24:1), followed by ethanol precipitation and resuspension in 20 µl H2O . Precipitated DNA (2 µl of a 1:20 dilution) was subjected to PCR in a volume of 50 µl for 39 cycles using the S35 and S36 primers that amplify a 330-bp minicircle sequence30. Six microliters of the PCR products were separated on 2% agarose gel and visualized with ethidium bromide.
Supplementary Material
Ackowledgement
This research was supported by National Institutes of Health grants AI070218 and CA067771.
Footnotes
Abbreviations: EC50, concentration of compound that causes 50% reduction in parasite growth in vitro; lanosterol 14α-demethylase, L14DM; T. cruzi, Trypanosoma cruzi.
Supporting Information: Pharmacokinetic data for compounds 44a and 44d and mice weight data.
References
- 1.World Health Organization. Control of Chagas disease: Second Report of the Expert Committee (W.H.O., Geneva) W.H.O. Technical Report Series. 2002:905.
- 2.Buckner FS, Van Voorhis WC. Immune response to Trypanosoma cruzi: Control of infection and pathogenesis of Chagas disease. In: Cunningham MW, Fujinami RS, editors. Philadelphia: Lippincott Williams & Wilkins; 2000. pp. 569–591. [Google Scholar]
- 3.Urbina JA, Docampo R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 2003;19:495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 4.Goad LJ, Berens RL, Marr JJ, Beach DH, Holz GG. The activity of ketoconazole and other azoles against Trypanosoma cruzi: Biochemistry and chemotherapeutic action in vitro. Molecular and Biochemical Parasitol. 1989;32:179–190. doi: 10.1016/0166-6851(89)90069-8. [DOI] [PubMed] [Google Scholar]
- 5.Ferraz LM, Gazzinelli TR, Alves OR, Urbina AJ, Romanha JA. The anti-Trypanosoma cruzi activity of posaconazole in a murine model of acute Chagas disease is less dependent on gamma interferon than that of benznidazole. Antimicrobial agents and Chemotherapy. 2007;51:1359–1364. doi: 10.1128/AAC.01170-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buckner FS. Sterol 14-demethylase inhibitors for Trypanosoma cruzi infections. Adv .Exp. Med. Biol. 2008;625:61–80. doi: 10.1007/978-0-387-77570-8_6. [DOI] [PubMed] [Google Scholar]
- 7.Buckner FS, Yokoyama K, Lockman J, Aikenhead K, Ohkanda J, Sadilek M, Sebti MS, Van Voorhis WC, Hamilton AD, Gelb MH. A class of sterol 14-demethylase inhibitors as anti-Trypanosoma cruzi agents. Proc. Natl. Acad. Sci. USA. 2003;100:15149–15153. doi: 10.1073/pnas.2535442100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Podust ML, Poulos LT, Waterman RM. Crystal structure of cytochrome P450 14α-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc. Natl. Acad. Sci. USA. 2001;98:3068–3073. doi: 10.1073/pnas.061562898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bang-Chi C, Amanda PS, Joseph ES, George C, Sarah CT. Efficient molar-scale synthesis of 1-methyl-5-acylimidazole triflic acid salts. Org. Process Research and Development. 2000;4:613–614. [Google Scholar]
- 10.Ohkando J, Lockman JW, Kothare AM, Qian Y, Blaskovich AM, Sebti SA, Hamilton AD. Design and synthesis of potent nonpeptidic farnesyltransferase inhibitors based on a terphenyl scaffold. J. Med. Chem. 2002;45:177–188. doi: 10.1021/jm0103099. [DOI] [PubMed] [Google Scholar]
- 11.Quia Y, Maurgan JJ, Fossum RD, Vogt A, Sebti SM. Probing the hydrophobic pocket of farnesyltransferase: Aromatic substitution of CAAX peptidomimetics lead to highly potent inhibitors. Bio. Med. Chem. 1999;7:3011–3024. doi: 10.1016/s0968-0896(99)00252-7. [DOI] [PubMed] [Google Scholar]
- 12.Martin R. Uses of the Fries rearrangement for the preparation of hydroxyarylketones: A Review. Organic Preparations and Procedures International. 1992;24:373–435. [Google Scholar]
- 13.Gallo R, Gozard JP, Trippitelli S, Rossi P, Portioli R. Method for preparing 4-(alkyl)-3-(alkoxy)-aniline compounds in WO 98/57921: France. 1998 [Google Scholar]
- 14.Kothare AM, Ohkando J, Lockman WJ, Qian Y, Blaskovich AM, Sebti AS, Hamilton AD. Development of a tripeptide mimetic strategy for the inhibition of protein farnesyltransferase. Tetrahedron. 2000;56:9833–9841. [Google Scholar]
- 15.Doyle PM, Siegfried B, Dellaria FJ. Alkyl nitrite-metal halide deamination reactions. J. Org. Chem. 1977;42:2426–2431. [Google Scholar]
- 16.Olepu S, Suryadevara PK, Kasey R, Yokoyama K, Verlinde CLMJV, Chakrabarti D, Van Voorhis WC, Gelb MH. 2-Oxo-tetrahydro-1,8-naphthyridines as selective inhibitors of malarial protein farnesyltransferase and as anti-malarials. Bio. Med. Chem. Lett. 2008;18:494–497. doi: 10.1016/j.bmcl.2007.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pivsa-Art S, Satoh T, Kawamura Y, Miura M, Nomura M. Palladium-catalyzed arylation of azole compounds with aryl halides in the presence of alkali metal carbonates and the use of copper iodide in the reaction. Bulletin of the Chemical Society of Japan. 1998;71:467–473. [Google Scholar]
- 18.Ohta A, Akita Y, Ohkuwa T, Chiba M, Fukunga R. Palladium-catalyzed arylation of furan, thiophene, benzo[b]furan, and benzo[b]thiophene. Heterocycles. 1990;31:1951–1958. [Google Scholar]
- 19.Aoyagi Y, Inoue A, Koizumi I, Hashimoto R, Tokunaga K. Palladium-catalyzed cross-coupling reactions of chloropyrazines with aromatic heterocycles. Heterocycles. 1992;33:257–272. [Google Scholar]
- 20.Johnson SM, Connelly S, Wilson IA, Kelly JW. Biochemical and structural evaluation of highly selective 2-arylbenzoxazole-based transthyretin amyloidogenesis inhibitors. J. Med. Chem. 2008;51:260–270. doi: 10.1021/jm0708735. [DOI] [PubMed] [Google Scholar]
- 21.Hucke O, Gelb MH, Verlinde CLMJ, Buckner FS. The protein farnesyltransferase inhibitor tipifarnib as a new lead for the development of drugs against Chagas disease. J. Med. Chem. 2005;48:5415–5418. doi: 10.1021/jm050441z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rachel C, Sudhakar P, Mark L, Josephine L, Vijay B. Pharmacokinetics, safety, and tolerability of oral posaconazole administered in single and multiple doses in healthy adults. Anti. Agent. Chemo. 2003;47:2788–2795. doi: 10.1128/AAC.47.9.2788-2795.2003. 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urbina JA, Payares G, Sanoja C, Lira R, Romanha AJ. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. International Journal of Antimicrobial Agents. 2003;21:27–38. doi: 10.1016/s0924-8579(02)00273-x. [DOI] [PubMed] [Google Scholar]
- 24.Bustamante JM, Bixby LM, Tarleton RI. Drug-induced cure drives conversion to a stable and protective CD8+ T central memory response in chronic Chagas disease. Nat. Med. 2008;14:542–550. doi: 10.1038/nm1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckner FS, Verlinde CLMJ, La Flamme AC, Van Voorhis WC. Efficient technique for screening drugs for activity against Trypanosoma cruzi using parasites expressing β-galactosidase. Antimicrob. Agents Chemother. 1996;40:2592–2597. doi: 10.1128/aac.40.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brisse S, Barnabe C, Tibayrenc M. Identification of six Trypanosoma cruzi phylogenetic lineages by random amplified polymorphic DNA and multilocus enzyme electrophoresis. Int. J. Parasitol. 2000;30:35–44. doi: 10.1016/s0020-7519(99)00168-x. [DOI] [PubMed] [Google Scholar]
- 27.Kraus JM, Verlinde CLMJ, Karimi M, Lepesheva GI, Gelb MH, Buckner FS. Rational modification of a candidate cancer drug for use against Chagas disease. J. Med. Chem. :2008. doi: 10.1021/jm801313t. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sturm NR, Degrave W, Morel C, Simpson L. Sensitive detection and schizodeme classification of Trypanosoma cruzi cells by amplification of kinetoplast minicircle DNA sequences: Use in diagnosis of Chagas disease. Mol. Biochem. Parasitol. 1989;33:205–214. doi: 10.1016/0166-6851(89)90082-0. [DOI] [PubMed] [Google Scholar]
- 29.Avila HA, Sigman DS, Cohen LM, Millikan RC, Simpson L. Polymerase chain reaction amplification of Trypanosoma cruzi kinetoplast minicircle DNA isolated from whole blood lysates: Diagnosis of chronic Chagas disease. Mol. Biochem. Parasitol. 1991;48:211–221. doi: 10.1016/0166-6851(91)90116-n. [DOI] [PubMed] [Google Scholar]
- 30.Gomes ML, Maced AM, Vago AR, Pena SD, Galvão LM, Chiari E. Trypanosoma cruzi: Optimization of polymerase chain reaction for detection in human blood. Exp. Parasitol. 1998;88:28–33. doi: 10.1006/expr.1998.4191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.