

Abstract
Thirty years ago, p53 was discovered as a cellular partner of SV40 Large Tumor Antigen, the oncoprotein of this tumor virus. The first decade of p53 research saw the cloning of p53 DNA and the realization that p53 is not an oncogene but a tumor suppressor that is very frequently mutated in human cancer. In the second decade, the function of p53, a transcription factor induced by stress, resulting in cell cycle arrest, apoptosis and senescence, was uncovered. In its third decade new functions were revealed, including regulation of metabolic pathways and cytokines required for embryo implantation. The fourth decade may see new p53-based drugs to treat cancer. What is next is anybody’s guess.
By now, anybody with interest in cancer research is already well aware of the existence of p53 and its relevance to practically every aspect of tumor biology 1. It is impossible to overlook the prominence of p53: with nearly 50,000 PubMed-listed publications so far and a steady flow of new ones hitting the cyberspace every week, p53 is undoubtedly one of the most extensively studied genes and proteins. Every other year hundreds of scientists gather for an International p53 Workshop to discuss a single gene and protein. However, the notion that p53 is a pivotal tumor suppressor and a major mainstay in our body’s natural anti-cancer defense, now taken for granted, did not come easily. When discovered 30 years ago, p53 was little more than just another “interesting” protein that most cancer researchers did not consider worthy of much attention, let alone of investment of research time and resources. Unlike well-behaved oncogenes, which were often brought into the main stage shortly after their discovery, p53 received relatively little attention in its first years. The road leading to p53’s eventual rise to prominence and its recognition as the most frequently altered gene in human cancer was rather long and winding, with concepts being repeatedly revised, extensively modified and sometimes even totally turned upside down. The history of p53 research over the last 30 years provides a rich example of how knowledge evolves in unexpected ways and how both research “fashions” and new methodological breakthroughs make us perceive the same facts in totally different ways as time progresses. It also teaches us how extensive delving into a single protein can lead to the discovery of new fundamental and general principles that apply to much broader areas of biology and biochemistry.
Prelude: tumor viruses, oncogenes and the road to p53
In the 1970’s, much of the attention of “modern” cancer researchers focused on cancer-causing viruses. In particular, it became evident that such viruses carried oncogenes. The bigger picture was first resolved for RNA tumor viruses; there, it was shown that the virus “hijacks” a cellular gene, which it subsequently reintroduces into the cell that it infects 2. This leads to the vast overexpression of the encoded cellular protein, sometimes in modified form, and eventually causes transformation. Similarly, oncogenes were uncovered by examining the genes adjacent to the integration sites of retroviruses that resulted in the overexpression of those genes and the formation of tumors in animals.
Over the next fifteen years a long list of oncogenes were identified and it became clear that oncogenes were the cause of cancers in animals. It was thus not at all far-fetched to expect that DNA tumor viruses might operate by essentially the same principle – that they had picked up oncogenes from the cell or encoded their own viral oncogenes. It became rapidly clear that the DNA tumor viruses contained oncogenes not related to the cellular oncogenes of the RNA tumor viruses. But how did these viral oncogenes act to transform cells and produce tumors in animals? It was proposed that the DNA tumor virus oncogenes encode viral proteins that lead indirectly to the excessive induction of putative cellular oncoproteins. It was on that fertile conceptual soil that p53 first emerged.
Tumors induced in experimental animals by small DNA tumor viruses, such as SV40, typically express a limited number of viral encoded proteins. These are recognized by the immune system of the host, leading to the production of antibodies against these proteins. By the mid-1970’s, such antibodies started to gain popularity as tools to identify and monitor proteins encoded by the viral genome and expressed in transformed cells. Based on their mode of detection, these proteins were dubbed viral tumor antigens. Subsequent genetic analysis revealed that the genes encoding these viral tumor antigens were often those also responsible for the transforming activity of the virus, namely the viral oncoproteins. In the case of SV40, the two viral proteins identified in this manner were called large T-antigen and small t-antigen, respectively.
It was while studying these SV40-derived tumor antigens that several groups independently stumbled on p53. This happened in 1979, thirty years ago (Timeline). Working at the ICRF (now London Institute for Cancer Research), David Lane and Lionel Crawford realized that when sera from animals bearing SV40-induced tumors were employed to immunoprecipitate SV40 large T-antigen, a non-viral protein with an apparent molecular mass of about 53 kDa came along for the ride 3. Further analysis established that this cellular protein was physically complexed with SV40 large T-antigen. Thus the viral protein, previously shown to be largely responsible for the transforming and tumorigenic activity of the SV40 virus, had selected this hitherto unknown cellular protein as its partner for an intimate, specific interaction. At the same time, Daniel Linzer and Arnold Levine applied a similar immunological approach to SV40 transformed cells, and came up with essentially the same observations, namely that such cells harbored a complex between the SV40 large T-antigen and the cellular 53 kDa protein 4. Three other groups, those of Alan Smith in the UK, Robert Carroll in New York and Pierre May in France, simultaneously made very similar findings, all published in 1979 5-7.
Interestingly, Linzer and Levine also found that their antisera precipitated the same 53 kDa protein from teratocarcinoma (a germ cell tumor)-derived cells, despite the fact that the latter did not harbor any SV40 proteins; this indicated that a subset of the antibodies raised against the viral-induced tumor were capable of interacting directly with this cellular protein 4. In parallel, Lloyd Old and coworkers demonstrated that animals immunized with non-virally transformed cells produced antibodies to the same 53 kDa protein 8, rightfully qualifying it as a cellular tumor antigen. Moreover, Varda Rotter, working in the lab of David Baltimore, was able to identify the same protein being produced in excess in cells transformed by a retrovirus, the Abelson murine leukemia virus 9. Hence, very high levels of this new cellular protein were present not only in SV40-transformed cells but also in other types of cancer cells, but little or no p53 protein could be detected in non-transformed cells.
As is often the case with independent discoveries of the same protein, each lab gave it a different name and continued to publish subsequent papers using their favorite name, creating quite a bit of confusion in this very young field. It was only in 1983, during the first p53 Workshop in Oxted, UK (Fig. 1), that representatives of the different p53 groups got together to discuss a common nomenclature. After a reasonable deal of inevitable debate, the term “p53” emerged as the winner and has stayed with us ever since. Ironically, p53 is actually a misnomer. When coined, it purportedly related to the molecular mass of the protein, which on the basis of its migration in SDS-polyacrylamide gels was estimated to be about 53 kDa. As realized later, this was a gross overestimate, presumably due to the presence of a proline-rich region that slows down the migration of the protein in such gels. In fact, the correct molecular mass of the human p53 protein is only 43.7 kDa, and that of the mouse protein is even less. But who would dare change a winning name?
Fig. 1. The first p53 workshop.

The first p53 workshop took place at The Marie Curie Research Institute at The Chart in Oxted, Surrey, UK from 7-10 May 1983. The program did not include any presentation titles, but the hottest issued on the agenda were the first disclosures of the cloned murine p53 cDNA sequences, as well as reports on ongoing attempts to clone human p53 cDNA and genomic DNA. The table below shows the list of participants in the workshop. With some notable exceptions (David Lane), this was practically almost the entire p53 community in 1983. Program details and participant list courtesy of Varda Rotter.
The early years: p53 is an oncogene?
As outlined above, in 1979 retroviruses were already well known to promote neoplastic transformation by overexpressing “hijacked” cancer-promoting cellular proteins. In that light, the observation that SV40 drives the overproduction of p53 in transformed cells seemed to lead very logically to the conclusion that p53, too, was a positive effector of transformation. Of note, a temperature sensitive mutant of the SV40 large T-antigen gene was found to regulate p53 levels in a temperature dependent fashion; cellular p53 levels were very high when T-antigen was functional, but much lower when T-antigen was non-functional, seemingly implying that more p53 means more transformation 10. In short, p53 looked like a cellular oncogene. Additional findings only appeared to lend further support to this conjecture. For instance, work of Peter Sarnow in the Levine lab revealed that the E1B 55 kDa viral tumor antigen encoded by another small DNA tumor virus, adenovirus, also binds p53 and promotes its excessive cellular accumulation 11. Furthermore, Rotter showed that many tumors produced high amounts of p53, whereas this was not observed in normal tissue 12, lending generality to the early observations of DeLeo and co-workers that transformed, but not non-transformed, cell lines expressed high levels of p53 8.
With that notion in mind, attempts were then undertaken to demonstrate experimentally the oncogenic properties of p53. Key to this goal was the need to have in hand p53-encoding DNA, so that p53 expression might be specifically manipulated and the biological consequences of such manipulation assessed in vitro and in vivo. Thus started the race to clone p53, initiated more or less in parallel in a number of laboratories. In the early 1980’s, gene cloning was not as simple as it is now; in fact, it was a rather tedious trial-and-error (mostly error) exercise, grabbing on thin leads and calling for a lot of improvisation, ingenuity, and - not the least - good luck. Nevertheless, a number of groups managed to overcome the hurdles. Within a relatively short stretch of time, several mouse and human p53 cDNA and genomic clones were isolated, validated and reported 13-20. Of note, given the low efficiency of the gene cloning protocols available at the time, special efforts were made to identify cells in which the protein of interest was relatively abundant, assuming that the corresponding mRNA is also more abundant and thus easier to clone. Since the p53 protein is more copious in cancer-derived cells, it was only natural that the first p53 cDNA cloning attempts, including the ones that eventually met with success, employed RNA from transformed cells rather than from normal tissue. This fact, whose significance was appreciated in full only a number of years later, was largely responsible for the rather unusual course that p53 research took in its first decade.
Using the newly obtained p53 DNA clones, and acting on the assumption that p53 overexpression contributed to tumorigenic processes, the consequences of such overexpression were next assessed in a number of experimental model systems. Sure enough, the results fulfilled the expectations. A series of studies by the labs of John Jenkins, Moshe Oren, Varda Rotter and Robert Weinberg revealed that transfected p53 could cooperate quite efficiently with a number of established oncogenes, most notably H-Ras, to transform primary cells in culture, and could facilitate the immortalization of such cells when overexpressed on its own 21-23. In these assays, p53 acted somewhat similarly to the c-myc oncogene. Furthermore, it could be shown that the cloned p53 augmented the transformed properties of established cell lines 24, and – most notably – could increase the in vivo tumorigenic properties of otherwise p53-null cells 25. In sum, by the mid 1980’s p53 was generally acknowledged as an oncogene, whose significance and mechanism of action still remained to be uncovered.
Grand entry into the tumor suppressor main stage
The realization that p53 is actually not an oncogene but rather the opposite, namely a tumor suppressor, took several more years to crystallize. In fact, clues that p53 might be a tumor suppressor had already been around. In retrospect, the first clue was provided by David Wolf and Rotter, who reported in 1984 that the p53 gene was inactivated by retroviral insertion in an Abelson murine leukemia-transformed mouse cell line 26 Similar observations were made by Sam Benchimol, Alan Bernstein and coworkers, studying leukemias induced in mice by the Friend erythroleukemia virus 27, 28. Furthermore, Rotter and coworkers showed that the p53 gene was extensively rearranged and its coding sequences virtually deleted in the human leukemia-derived cell line HL60, precluding production of p53 protein 29.The simplest way to interpret such striking observations was by concluding that loss of p53 promotes cancer, implying that sustained p53 function is necessary to prevent cancer. However, the implications of these findings were not apparent at the time. In the face of the seemingly unequivocal evidence for p53 being an oncogene, these were viewed as exceptional cases that did not reflect on the role of p53 in most common types of cancer. When a group believes something, evidence to the contrary tends to be more readily dismissed.
The cracks in the p53=oncogene wall widened when yet another p53 cDNA clone was put to work by Cathy Finlay and Phil Hinds in the Levine lab. Surprisingly, this clone was totally unable to reproduce the transforming effects observed with the earlier clones, even though Finlay, Hinds, Levine and colleagues could easily reproduce such effects with a clone obtained from the Oren lab. The enigma was solved when they compared the DNA sequences of the various p53 clones employed by them as well as in the earlier studies of others, and realized that no two clones were identical in sequence, suggesting that at least some if not all of the previously tested clones actually carried mutations in the p53 coding region. This was firmly proven to be the case when the sequence of murine wild type p53 (wtp53), derived from normal tissue, was formally established 30, 31. It then became clear that p53 mutations are often present in tumor-derived murine cell lines 30-32., including the ones employed by a number of labs for cDNA cloning, and that only p53 cDNAs carrying such mutations are capable of exerting transforming activities in experimental settings. Hence, it emerged that while mouse tumor-derived p53 mutants can indeed promote cell transformation, wtp53 clearly can not.
So what does the wtp53 have to do with cancer? The answer was provided through the combined impact of extensive analysis of DNA from human cancer specimens and in vitro functional assays. Thus, Bert Vogelstein and co-workers demonstrated that, in human colorectal tumors, the wild type p53 alleles were frequently lost by mutations, deletions or a combination of both, such that the tumor cells did not retain any wtp53 33; this is a hallmark of a tumor suppressor gene. In parallel, functional analysis performed independently in the Levine and Oren labs revealed that, in stark contrast to the transforming activities of tumor-derived p53 mutants, overexpression of wtp53 effectively repressed the transformation of cultured cells by a combination of the potent oncogenes c-myc and H-Ras 34, 35. Together with the earlier observations of loss of p53 in murine tumors and human cancer-derived cells and the subsequent investigation of humans afflicted with the Li-Fraumeni syndrome and of p53 knockout mice (see later), these data finally firmly established p53 as a bona fide tumor suppressor.
It did not take long to show that p53 mutations are frequent not only in colorectal cancer but in most of the common types of human tumors. In at least some types of cancer, p53 mutations were found to be primarily a late-occurring event, presumably playing a role in progression to advanced, invasive and metastatic disease 36, An extensive body of data, encompassing thousands of studies, has revealed p53 mutations in about half of all tumor specimens studied, making it arguably the most frequently mutated gene in human cancers, and the most frequently analyzed gene in many human tumor specimens. Dedicated p53 mutation databases and p53 knowledge bases have been established (http://www-p53.iarc.fr/, http://p53.free.fr/, and http://p53.bii.a-star.edu.sg/index.php). First of their kind for a single cancer-related gene and unprecedented in size, these databases have rapidly become a valuable resource to the p53 research community and to cancer researchers at large. Of note, once it had been figured out that the wtp53 protein was a tumor suppressor and that p53 mutations occurred frequently in cancer cells, the earlier observations that led to the suggestion that p53 is oncogenic could now be explained and placed in the right context. Thus, it became apparent that all the studies demonstrating oncogenic activity of p53 had employed mutant alleles of murine or human p53, typically derived from a cancer cell line overexpressing that particular mutant. Such mutants can exert cancer-promoting effects, by dominant negative inactivation of the endogenous wtp53 as well as by authentic oncogenic gain of function activities (see the Review by Brosh and Rotter, also in this issue 37) Hence, whereas wtp53 is a potent tumor suppressor, cancer-associated p53 mutants indeed possess attributes of oncogenes.
For a gene to be unequivocally accepted as a tumor suppressor, two additional criteria are expected to be met: humans carrying germline mutations in that gene should exhibit increased cancer susceptibility, and its loss should confer a cancer-prone phenotype in experimental animal models. Gratifyingly, both criteria were fully met by p53. Indeed, germline p53 mutations are largely responsible for the devastating hereditary Li-Fraumeni syndrome, characterized by early onset cancers of diverse types 38, 39. Furthermore, p53 knock-out mice, first described by Donehower and co-workers in 1992, develop cancer (mostly lymphomas) with a very high penetrance 40 (see Timeline by Donehower and Lozano, this issue)
These results also called for a reinterpretation of how the viral oncogenes of the DNA tumor viruses functioned. It could be shown that the SV40 T-antigen and the adenovirus E1A protein from many different adenovirus serotypes combined with the retinoblastoma protein, pRb, and this liberated E2F transcription factors to send the cell into S-phase 41-44. The small DNA tumor viruses do this so as to obtain the enzymes and substrates to support their own DNA replication. However this unusual S-phase event is recognized by p53, which then attempts to kill the cell via induction of apoptosis, preventing further viral replication. These viruses counter that move by binding the p53 protein with the large T-antigen or the E1B 55 kDa protein, events that inactivate p53 function just as mutations do in human cancers45, and lead to accumulation of the inactivated p53 in the transformed cells. Importantly another group of DNA tumor viruses, the human papilloma viruses (HPV), including HPV16 and HPV18, which cause most cervical cancers and are therefore of high clinical relevance, encode two proteins that also target the same cellular proteins: E7, which binds to pRb and liberates E2F transcription factors, and E6, which binds p53 and helps promote its degradation and inactivation45. Incidentally, HPV E6 provided the first clue to the importance of the ubiquitin-proteasome pathway in controlling cellular p53 levels 46.
Thus, in the early part of the last decade of the 20th century, p53 was ushered in as a major tumor suppressor and a most fashionable gene to study. That fashion still remains in effect to this date.
How does p53 do it?
With the appreciation of the centrality of p53 in both the life cycle of the DNA tumor viruses and in human cancers, came the quest for elucidating its mode of action. This quest took two main directions: on the one hand, delineating the biological processes that allow p53 to suppress tumors, and on the other hand elaborating the molecular mechanisms that underlie such processes.
Biological activities of p53
Substantial progress was made when p53 function was reconstituted in transformed cells. This was achieved by a variety of means and tricks, taking advantage of the progress in gene manipulation methodology in the late 80’s and early 90’s. One particularly useful tool was a temperature sensitive mutant of p53, discovered by accident owing to a misadjusted incubator; this mutant exerts wtp53 activity at 32°C, but loses this activity at 37°C or above 47. Through the use of this mutant, it was found that reconstitution of wtp53 activity can impose growth arrest, both at G1 and G2/M 47. Similar conclusions were reached by employing other methodologies 48 49, 50. Remarkably, in other types of transformed cells, such as the M1 leukemia cell line, reactivation of temperature sensitive p53 had a rather different and striking outcome: within a couple of days, all cells in the culture died; a closer look revealed that this death exhibited typical features of apoptosis 51. This study, along with similar findings by Shaw and co-workers 52, established a novel role for p53 as a mediator of apoptosis, and provided the first evidence that apoptosis can serve as a mechanism of tumor suppression. More recently, induction of cellular senescence was identified as an additional mechanism whereby p53 acts to curb neoplastic processes 53 54. This is now emerging as a major mechanism for tumor suppression by p53 55, 56. Overall, the common feature shared by apoptosis and replicative senescence is that cells undergoing those processes are prevented from giving rise to like malignant progeny. Consequently, induction of either process by p53 in an aspiring cancer cell will prevent that cell from spawning a full-blown tumor.
Molecular mechanisms behind p53’s functions
A huge body of information has been accumulated on how p53 works biochemically, which is beyond the scope of this review. Yet, the most prominent property of p53 as a protein is its action as a transcription factor. A number of seminal studies revealed that p53 possesses a functional transactivation domain 57, 58 (actually, we now know that there are two59), and that it can bind tightly to specific DNA sequences 60, 61,62-64- In fact, the ability to bind to specific sequences and transactivate genes is the property that distinguishes wtp53 from virtually all cancer-associated mutant forms.
These studies provided the first description of a p53 binding consensus sequence, eventually enabling genome-wide computational searches for putative p53 binding sites. Through these and many subsequent studies (e.g. 65-68), p53 was firmly canonized as a sequence-specific transcription factor (see review by Resnick, this issue) 69.
Many dozens of p53 target genes have been identified, and shown to be transactivated by p53 upon its binding to p53 response elements upstream to or within these genes. Many of those genes encode proteins that are intimately involved in apoptosis or in control of cell cycle progression (see review by Resnick, this issue). Together, this palette of genes provides plausible mechanistic explanations for the ability of p53, when activated, to promote cell death and/or growth arrest, as realized early on when the genes encoding the cyclin-dependent kinase inhibitor p21 and the proapoptotic BAX were found to be directly transactivated by p5370, 71. It is also noteworthy that the first expression microarrays used to examine the genes regulated by a specific and inducible (by DNA damage) transcription factor were obtained using p53 as the initiating event 72. The complexity of the p53 response to different stimuli is still being digested. The quest to identify additional p53 target genes is steadily ongoing; as induction of many of these genes by p53 is turning out to be cell type-restricted or context restricted, it is likely that the list is yet far from being complete. A recent addition to this growing list is microRNAs: a multitude of studies has identified several microRNAs, most notably members of the miR-34 family, as being subject to transcriptional regulation by p53 (73-76). In parallel, p53 was found to also act as a transcriptional repressor 77. The mechanisms underlying p53-mediated transcriptional repression are many and diverse, mostly not involving direct binding of p53 to consensus sequences within the gene. The transcriptional activities of p53 imply that it can simultaneously alter the expression of hundreds of genes, thereby explaining how it can exert such profound effects on cell fate.
Of note, p53 was subsequently found to also possess non-transcriptional biochemical activities. These are again quite diverse, and can be exerted both in the cell nucleus and in the cytoplasm. A detailed account of those additional p53 activities is not in the scope of this review; yet, most notable among them is the ability of p53 to interact in the cytoplasm with members of the Bcl2 family of apoptosis-regulatory proteins, thereby directly contributing to mitochondrial outer membrane permeabilization, release of cytochrome C and apoptosis (reviewed in 78, 79).
p53 and MDM2: in and out of loops
A common means to explore the biochemical properties of a protein of interest is by identifying other proteins with which it interacts. This has been performed quite exhaustively for p53, yielding many tens of proven and putative interaction partners. Curiously, p53 was one of the first mammalian proteins subjected to the then novel yeast-two-hybrid screen, which yielded a couple of previously unknown p53 interactors, denoted 53BP1 and 53BP2 for lack of better functional descriptions 80.
Yet, probably the most important protein-protein interaction of p53 was discovered in 1992, when the MDM2 protein, previously described as a putative oncoprotein, was shown to bind tightly to p53 and inhibit its biochemical activity 81. Since then, MDM2 (the human protein is often called HDM2) has emerged as perhaps the key cellular regulator of p53, effectively serving as the p53 gatekeeper. MDM2 can inhibit p53 activity by a variety of means. First, by binding to the transactivation domain of p53, it sterically blocks the function of that domain 82. Moreover, by acting as a p53-specific E3 ubiquitin ligase, MDM2 promotes the ubiquitylation and subsequent proteasomal degradation of p53 83-85.
A further twist to the p53-MDM2 story came when it was realized that the Mdm2 gene is a direct transcriptional target of p53 86, 87. Thus, p53 and MDM2 form a negative feedback loop, wherein p53 induces the expression of MDM2, which in turn promotes the degradation of p53 and quenches cellular p53 activity 86-88. Extensive work done over the years has taught us that in non-stressed cells, p53 function is kept at a low basal state. This is largely achieved through the constant action of the MDM2-p53 loop, which effectively eliminates excessive p53 activity by triggering the production of MDM2. In contrast, in cells exposed to a variety of stress conditions, most notably those that put the cellular genome at risk, p53 is rapidly activated 89. The first demonstration of the inducible nature of p53 in response to genomic stress was by Warren Maltzman, who showed that UV exposure increases cellular p53 concentrations 90. This and subsequent pivotal findings by Michael Kastan 91, 92, led Lane to dub the p53 as “guardian of the genome” 93, a notion that was only strengthened over the years and is believed to represent much of p53’s raison d’etre in multicellular organisms. The stress-induced switch in p53 levels and activity lies at the heart of p53’s performance as a multifaceted, omnipotent tumor suppressor 89 (Fig. 2). To a large measure, it is MDM2 that guards this switch and ensures that it is not triggered inappropriately. Furthermore, activation of the p53 response in cells experiencing oncogenic stress calls for disengagement of MDM2 and abrogation of its inhibitory effects (Fig. 2), often through a combination of reduced MDM2 levels, reduced MDM2 binding to p53, and reduced E3 activity of MDM2 towards p53, as well as inhibition of post-ubiquitylation functions of MDM2 in promoting p53 degradation. Just as one illustration, seminal work by Chuck Sherr and coworkers revealed that ARF, a respectable tumor suppressor in its own right, acts largely through binding MDM2 and thereby augmenting p53 levels and function 94.
Fig. 2. Simplified scheme of the p53 pathway.
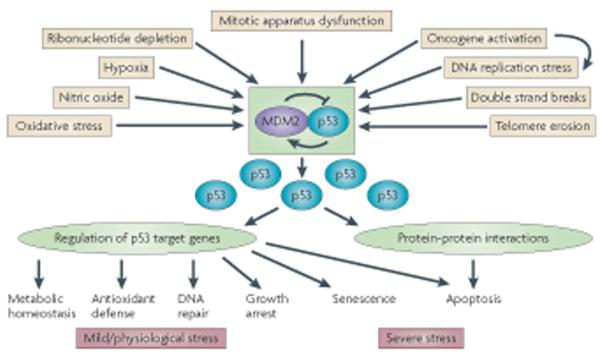
The p53-MDM2 feedback loop is the “heart” of the p53 pathway. Under normal conditions, it maintains p53 levels and activity at constantly low steady state levels. A variety of stress signals (only a representative subset of p53-activating signals are depicted), related in many ways to carcinogenesis, impinge on this central loop to release p53 from MDM2-mediated inhibition. This increases p53 protein levels and activity, inducing various phenotypic changes. Many p53-activating signals are closely interrelated, as exemplified here for oncogenes, whose impact on p53 is partly due to their propensity to induce DNA replication stress. The downstream effects of p53 are largely due to its ability to transactivate and repress various subsets of target genes; however, at least in the case of apoptosis, protein-protein interactions (primarily with Bcl2 family members) also play an important role. It is generally believed that the nature of the phenotypic response to p53 activation is, at least partially, proportionate to the amplitude, duration and nature of the activating signal. Severe stress induces more extreme, usually irreversible responses, namely apoptosis and senescence, whereas milder stress would lead to a transient growth arrest coupled with an attempt to deal with the cause of stress and repair the damage caused by it. Recent evidence indicates that p53 also has an important role in enabling the cell to adjust its metabolism in response to mild normal physiological fluctuations, including those in glucose and other nutrient levels, oxygen availability, and reactive oxygen species levels (see Focus by Vousden).
In 1996, MDM2 was joined by a new cousin, MDMX (MDM4 in the mouse) 95. Like MDM2, MDMX too binds to the N terminal region of p53 and inhibits its activity. While MDMX does not possess measurable E3 activity, it does contribute to p53 degradation; apparently, dimerization with MDMX augments the E3 ligase activity of MDM2 96, 97. Remarkably, inactivation of either MDM2 or MDMX in the mouse results in early embryonic lethality, owing to rampant p53 activation; this dramatic consequence of loss of function of either MDM2 or MDMX can be completely eliminated by concomitant knockout of p53, demonstrating that both MDM2 and MDMX are critical negative regulators of p53 in real life 98-100. In view of the above, it was not surprising to find that excessive expression of MDMX is contributory to human cancer, very much like what had been previously reported for MDM2 (reviewed in 101).The clinical relevance of the intricate MDM2-p53 interplay was further brought into focus by the discovery of a single nucleotide polymorphism (SNP) in the human MDM2 gene 102. Remarkably, individuals carrying a particular MDM2 allele that confers higher expression levels of this gene are predisposed to early-onset cancer, suggesting that subtle differences in basal p53 activity might be enough to affect individual cancer risk 103. While association of this SNP with cancer has been confirmed by many independent studies, contradictory results have also been reported, particularly with breast cancers103 This may stem from the fact that this SNP operates largely in conjunction with the estrogen receptor (ER) in premenopausal women104 ; consequently, association studies that do not separate ER+ from ER− tumors may greatly underestimate the impact of this SNP.
Not surprisingly, the MDM2-p53 loop has caught the attention of many systems biologists and computational biologists, and has since become a favorite model for regulatory interactions within a pathway (e.g. 105). On a more practical level, this loop is being targeted by a number of promising experimental anti-cancer drugs (discussed below). Since the discovery of the MDM2-p53 loop, many additional regulatory loops that involve p53 have been uncovered, and the list is constantly expanding, serving as further testimony to the elaborate networking that p53 is engaged in.
What is this good for? p53 and cancer therapy
The centrality of p53 in human cancer makes it a potentially very lucrative target for cancer therapy development. It is thus no wonder that many efforts have been undertaken over the years, both in industry and in academia, to develop novel p53-based anti-cancer treatments. This is no simple task: p53 is neither a cell surface protein nor a typical enzyme. Hence, antibodies and low molecular weight enzyme inhibitors, which have served as the basis for almost all of the recently developed targeted anti-cancer therapies, are not pertinent options in the case of p53. Researchers and developers have therefore had to resort to less standard approaches (see Review by Lane, this issue).
p53 Gene therapy
Early efforts focused largely on various types of gene therapy. The most intuitive strategy was to extend the gene transfer experiments, which served to establish p53 as a tumor suppressor, to human cancer patients. Indeed, a number of organizations took up this approach, and promising data from clinical trials were reported by Introgen Therapeutics, Texas, as early as 1996 106.
Yet, completion of these trials and transfer to the clinic were slow to come. Eventually, p53 gene therapy, delivered with the aid of an adenovirus vector, was approved in 2004 for the treatment of head and neck cancer in China 107. Although one still has to await a critical assessment of the results, it is remarkable that this is the first ever gene therapy protocol approved for routine clinical use in humans. Cancer researchers and patients alike are also eagerly awaiting the outcome of critical phase 3 clinical trials with Advexin, the adenovirus p53 gene therapy agent developed originally in the US by Introgen Therapeutics 108.
An alternative gene therapy strategy was developed by McCormick and coworkers at Onyx, in California. This again employed infection of the tumor with recombinant adenovirus. However, rather than transducing a p53 gene, this virus was deficient in the E1B 55kDa protein, which binds and inactivates p53. Consequently, this oncolytic virus can replicate in tumor cells that lack functional p53 and kill them, whereas it is incapable of replicating in normal, p53-proficient cells 109. Although clinical trials yielded promising results 110, approval for clinical use still needs to be evaluated 111. In the meantime, a related oncolytic virus, operating on similar principles, has also been approved for cancer therapy in China 112, 113; Future will tell whether p53-based gene therapy will indeed be able to make a significant impact on cancer treatment.
Restoring p53 activity
An entirely different set of strategies has been centered on the development of low molecular weight compounds that restore p53 activity in tumor cells. In one approach, such molecules were developed to interact with mutant p53 proteins within tumor cells and thereby alter their conformation and restore their function114 115. The results of clinical trials testing the efficacy of such compounds, exemplified by PRIMA-1 114, are likely to be coming in within the next several years. This type of mutant p53-activating drug is potentially applicable in tumors harboring p53 mutations, comprising in principle about 50% of all cancer patients.
The second approach aims to target the other half of patients, namely those that retain wtp53. Towards that end, efforts have evolved to produce compounds that disrupt MDM2-p53 binding, thereby liberating p53 from its negative inhibitor and enabling elevated p53 activity. The best documented successful attempt was made by a team in Roche; this led to the development of the family of compounds known as Nutlins 116 117. Nutlins interact with the p53-binding pocket of the MDM2 molecule, effectively dislodging p53 from MDM2 and leading to extensive p53 activation and induction of a full blown p53 response, which can trigger tumor shrinkage in experimental animals. A complementary approach led to the identification of RITA; whereas Nutlins bind to MDM2, RITA binds to p53 and prevents it from being attacked by MDM2 118. RITA may be particularly promising because, unlike Nutlins, it has a strong apoptotic impact on tumor cells 119 It is still too early to tell whether and when Nutlins, RITA or more recently identified compounds that target the interaction of p53 with MDMX, rather than MDM2 101, will enter into the clinic. In the meantime, however, these compounds have become popular research tools, serving as a “clean” means to activate p53 without imposing wide-ranging cellular stress. These types of compounds may be most useful in tumor cells that have amplified and overexpressed MDM2 and therefore inactivated p53. Other compounds leading to the accelerated degradation of MDM2 or the inhibition of MDM2 in cells also could be a promising approach 120. It is certain that efforts to create novel p53-based therapies will be with us for quite some time.
Prognosis
The discovery that p53 plays a pivotal role in cell killing by DNA damaging agents, many of which are in routine use for cancer chemotherapy, gave rise to the expectation that p53 mutation status will prove a reliable predictor of therapy response and patient prognosis. To a great measure, these expectations have not been fulfilled, probably reflecting the complex genetic nature and extensive diversity of individual tumors, as well as the fact that the p53 pathway is often disarmed in tumors by mechanisms that do not involve direct p53 gene mutations. Yet, there are cases in which p53 mutations can indeed predict prognosis 121-123. It is likely that the predictive power of p53 mutation status will increase when tumor subsets are better defined and individual cases more rigorously stratified.
The Future
What will the next decade of p53 research bring us? Surely we will need to understand what the various isoforms of p53, p63 and p73 (Text Box A) do in both the developing organism and in adults. In particular there are already clues that the ratios of some of these isoforms, which can antagonize each other by acting as either transcriptional activators or repressors, are critical for cellular function124. There is some evidence that the monomers or dimers of p53 can form hetero-tetramers with p63 or p73 and these three transcription factors can give rise to a combined or new activity, further complicating the possible combinatorics and their functionalities.
Text box 1. The family grows: p63, p73 and isoforms of p53.
For quite a while, p53 was believed to be a unique protein, with no obvious relative. This was changed by the discovery of two additional members of the family, p63 and p73 (reviewed in 130) . Both p63 and p73 play essential roles in development (skin, nervous system, female reproduction, etc) 131-133 and can under some circumstances act as tumor suppressors 134, 135. Curiously p53, p63 and p73 have very closely related DNA binding domains, bind to similar DNA sequences and can induce the transcription of some of the same genes, but can also induce the transcription of very different genes in specific cell types 130.
Furthermore, p53 itself is also not a single protein. Over the past few years it has become clear that, through extensive alternative splicing as well as alternative transcriptional initiation, the TP53 gene produces as many as nine different isoforms, containing various combinations of alternative amino-terminal and carboxy-terminal portions of the protein 136. Of note, deletion of the amino-terminus produces a dominant negative repressor of p53-regulated genes. Some of the p53 isoforms are observed in different tissues and during different stages of development. The precise roles of each of the reported p53 isoforms remain largely unknown. Future unraveling of this story promises to provide both more excitement and more complexity to the ever growing p53 picture.
To make matters even more complicated, each of these proteins can be extensively modified by phosphorylation, acetylation, methylation, ubiquitylation, sumoylation, neddylation and even addition of N-acetyl glucosamine. This often happens in response to a stress signal or a physiological change in the cell. For that reason we might suspect that these modifications have important functional consequences; yet, definitive evidence for what those consequences are is still lacking. For example the Wip1 phosphatase (an oncogene amplified in a number of cancers) acts upstream of p53 to inactivate the ATM protein kinase and prevents p53 phosphorylation by MEK kinase at two specific serine residues125-127. This inactivates the functions of the wtp53 protein in a cell, suggesting that these protein modifications are important. Maybe the next ten years will see a clinical trial with a Wip1 inhibitor that activates p53 in cancers where Wip1 is amplified in the genome.
Although studies in cultured cells have provided a rich body of seemingly impeccable evidence for the pivotal importance of p53 post-translational modifications, many of these conclusions are challenged by data from mutant p53 knock-in mice 128. It is these confusing results that will need to be reconciled over the next ten years.
Some clarification about these modifications, as well as roles for p53 isoforms are expected to come from studies that focus on the development of fruit flies, round worms, zebrafish, mice and even humans. In some of these organisms there is growing evidence that p53, p63 and/or p73 play roles in female fertility, female germ line genomic stability and reproduction100. Over the next ten years we will surely learn more about this new chapter of p53 family functions. Sexual dimorphism (that is a role for p63 and p73 in the female germ line but not the male germ line100) of the p53 sisters is explained in part by p53-controlled genes that are also regulated by estrogen and the estrogen receptor (e.g. MDM2, LIF and WIP1) 104, 125, 129. There will probably be androgen regulated genes and perhaps other nuclear receptors and ligands that will be shown to play a role in the p53 pathway in conjunction with diverse kinds of stress.
The next ten years should see a more detailed description of the roles of the p53 protein in metabolism (see Focus by Vousden), reproduction and fertility, genomic instability in the germ line as well as in cancers, longevity of the organism (see the Focus by Donehower), and the use of p53 mutational spectra to suggest a therapy or predict an outcome (see Focus by Brosh and Rotter 37). There may even be other diseases, besides cancer, where p53 could be shown to play a role. While we can see and even imagine the exploration of these areas of research, there are other surprises surely in store for us. If there is one thing we have learned from the first thirty years of p53 research, it is that something we could not have guessed or thought of, will become clear and obvious over the next decade. What we believe today will surely be modified and this central gene and its protein, its sisters and its ancestors, will continue to teach us and challenge us and perplex us well into 2019.
Acknowledgements
We wish to thank the thousands of researchers whose outstanding work over the last 30 years has made p53 research so exciting. We apologize to all our colleagues whose important contributions could not be cited for lack of space. Work in the authors’ laboratories is supported by grant R37 CA40099 from the National Cancer Institute, a Center of Excellence grant from the Flight Attendant Medical Research Institute, grants from the European Commission (Mutp53, FP6 Contract 502983 and OncomiRs, FP7 Contract 201102), and the Robert Bosch Foundation (to MO), and by grant PO1 CA 87497 from the National Institutes of Health, grant W81XWH-06-1-0514 from the Department of Defense, and grants from the Breast Cancer Research Foundation (to AJL) and general support to the Simons Center for Systems Biology at the Institute for Advanced Study from the Simons Foundation. MO is the incumbent of the Andre Lwoff Chair in Molecular Biology.
Timeline- timeline
- 1979
- 1983
- 1984
- 1984
- 1988
- 1989
- 1990
- 1990
- 1990-1992
- 1991
- 1992
MDM2 negatively regulates p5381
- 1992
p53 knock-out mice are cancer-prone40
- 1992
- 1993
p21 is a p53 target gene70
- 1993
- 1994
first p53-DNA complex structure139
- 1997
- 1997
- 1997
ARF-p53 connection143
- 1997
p53 implicated in senescence54
- 1998
- 2000
- 2001
- 2002
p53 implicated in aging150
- 2003
- 2004
p53 gene therapy approved in China 107
- 2004
MDM2 polymorphism accelerates cancer102
- 2004
Nutlins activate p53116
- 2005
multiple p53 isoforms 153
- 2005
p53 has antioxidant function154
- 2005
- 2007
p53 is required for embryo implantation 157
- 2007
- 2007
- 2007
p53 inhibits the IGF-1/mTOR pathway164
References
- 1.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–3. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 3.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–3. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 4.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 5.Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol. 1979;31:472–83. doi: 10.1128/jvi.31.2.472-483.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melero JA, Stitt DT, Mangel WF, Carroll RB. Identification of new polypeptide species (48-55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology. 1979;93:466–80. doi: 10.1016/0042-6822(79)90250-2. [DOI] [PubMed] [Google Scholar]
- 7.Smith AE, Smith R, Paucha E. Characterization of different tumor antigens present in cells transformed by simian virus 40. Cell. 1979;18:335–46. doi: 10.1016/0092-8674(79)90053-9. [DOI] [PubMed] [Google Scholar]
- 8.DeLeo AB, et al. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci U S A. 1979;76:2420–4. doi: 10.1073/pnas.76.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rotter V, Witte ON, Coffman R, Baltimore D. Abelson murine leukemia virus-induced tumors elicit antibodies against a host cell protein, P50. J Virol. 1980;36:547–55. doi: 10.1128/jvi.36.2.547-555.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linzer DI, Maltzman W, Levine AJ. The SV40 A gene product is required for the production of a 54,000 MW cellular tumor antigen. Virology. 1979;98:308–18. doi: 10.1016/0042-6822(79)90554-3. [DOI] [PubMed] [Google Scholar]
- 11.Sarnow P, Ho YS, Williams J, Levine AJ. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell. 1982;28:387–94. doi: 10.1016/0092-8674(82)90356-7. [DOI] [PubMed] [Google Scholar]
- 12.Rotter V. p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells. Proc Natl Acad Sci U S A. 1983;80:2613–7. doi: 10.1073/pnas.80.9.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chumakov PM, Iotsova VS, Georgiev GP. Isolation of a plasmid clone containing the mRNA sequence for mouse nonviral T-antigen. Dokl Akad Nauk SSSR. 1982;267:1272–5. [PubMed] [Google Scholar]
- 14.Oren M, Levine AJ. Molecular cloning of a cDNA specific for the murine p53 cellular tumor antigen. Proc Natl Acad Sci U S A. 1983;80:56–9. doi: 10.1073/pnas.80.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matlashewski G, et al. Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene. Embo J. 1984;3:3257–62. doi: 10.1002/j.1460-2075.1984.tb02287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harlow E, Williamson NM, Ralston R, Helfman DM, Adams TE. Molecular cloning and in vitro expression of a cDNA clone for human cellular tumor antigen p53. Mol Cell Biol. 1985;5:1601–10. doi: 10.1128/mcb.5.7.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pennica D, et al. The amino acid sequence of murine p53 determined from a c-DNA clone. Virology. 1984;134:477–82. doi: 10.1016/0042-6822(84)90316-7. [DOI] [PubMed] [Google Scholar]
- 18.Leppard K, et al. Purification and partial amino acid sequence analysis of the cellular tumour antigen, p53, from mouse SV40-transformed cells. Embo J. 1983;2:1993–9. doi: 10.1002/j.1460-2075.1983.tb01690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zakut-Houri R, Bienz-Tadmor B, Givol D, Oren M. Human p53 cellular tumor antigen: cDNA sequence and expression in COS cells. Embo J. 1985;4:1251–5. doi: 10.1002/j.1460-2075.1985.tb03768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf D, Harris N, Goldfinger N, Rotter V. Isolation of a full-length mouse cDNA clone coding for an immunologically distinct p53 molecule. Mol Cell Biol. 1985;5:127–32. doi: 10.1128/mcb.5.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eliyahu D, Raz A, Gruss P, Givol D, Oren M. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature. 1984;312:646–9. doi: 10.1038/312646a0. [DOI] [PubMed] [Google Scholar]
- 22.Jenkins JR, Rudge K, Currie GA. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature. 1984;312:651–4. doi: 10.1038/312651a0. [DOI] [PubMed] [Google Scholar]
- 23.Parada LF, Land H, Weinberg RA, Wolf D, Rotter V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature. 1984;312:649–51. doi: 10.1038/312649a0. [DOI] [PubMed] [Google Scholar]
- 24.Eliyahu D, Michalovitz D, Oren M. Overproduction of p53 antigen makes established cells highly tumorigenic. Nature. 1985;316:158–60. doi: 10.1038/316158a0. [DOI] [PubMed] [Google Scholar]
- 25.Wolf D, Harris N, Rotter V. Reconstitution of p53 expression in a nonproducer Ab-MuLV-transformed cell line by transfection of a functional p53 gene. Cell. 1984;38:119–26. doi: 10.1016/0092-8674(84)90532-4. [DOI] [PubMed] [Google Scholar]
- 26.Wolf D, Rotter V. Inactivation of p53 gene expression by an insertion of Moloney murine leukemia virus-like DNA sequences. Mol Cell Biol. 1984;4:1402–10. doi: 10.1128/mcb.4.7.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.David Y. Ben, Prideaux VR, Chow V, Benchimol S, Bernstein A. Inactivation of the p53 oncogene by internal deletion or retroviral integration in erythroleukemic cell lines induced by Friend leukemia virus. Oncogene. 1988;3:179–85. [PubMed] [Google Scholar]
- 28.Mowat M, Cheng A, Kimura N, Bernstein A, Benchimol S. Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature. 1985;314:633–6. doi: 10.1038/314633a0. [DOI] [PubMed] [Google Scholar]
- 29.Wolf D, Rotter V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc Natl Acad Sci U S A. 1985;82:790–4. doi: 10.1073/pnas.82.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eliyahu D, et al. Meth A fibrosarcoma cells express two transforming mutant p53 species. Oncogene. 1988;3:313–21. [PubMed] [Google Scholar]
- 31.Finlay CA, et al. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol Cell Biol. 1988;8:531–9. doi: 10.1128/mcb.8.2.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halevy O, Rodel J, Peled A, Oren M. Frequent p53 mutations in chemically induced murine fibrosarcoma. Oncogene. 1991;6:1593–600. [PubMed] [Google Scholar]
- 33.Baker SJ, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–21. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 34.Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focus formation. Proc Natl Acad Sci U S A. 1989;86:8763–7. doi: 10.1073/pnas.86.22.8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–93. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 36.Baker SJ, et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990;50:7717–22. [PubMed] [Google Scholar]
- 37.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009 doi: 10.1038/nrc2693. (in press) [DOI] [PubMed] [Google Scholar]
- 38.Malkin D, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–8. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 39.Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348:747–9. doi: 10.1038/348747a0. [DOI] [PubMed] [Google Scholar]
- 40.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 41.DeCaprio JA, et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–83. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 42.Shirodkar S, et al. The transcription factor E2F interacts with the retinoblastoma product and a p107-cyclin A complex in a cell cycle-regulated manner. Cell. 1992;68:157–66. doi: 10.1016/0092-8674(92)90214-w. [DOI] [PubMed] [Google Scholar]
- 43.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–30. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 44.Whyte P, et al. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–9. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 45.Levine RL, Carroll M. A common genetic mechanism in malignant bone marrow diseases. N Engl J Med. 2009;360:2355–7. doi: 10.1056/NEJMe0902257. [DOI] [PubMed] [Google Scholar]
- 46.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 47.Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation and of cell proliferation by a temperature-sensitive mutant of p53. Cell. 1990;62:671–80. doi: 10.1016/0092-8674(90)90113-s. [DOI] [PubMed] [Google Scholar]
- 48.Mercer WE, et al. Negative growth regulation in a glioblastoma tumor cell line that conditionally expresses human wild-type p53. Proc Natl Acad Sci U S A. 1990;87:6166–70. doi: 10.1073/pnas.87.16.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science. 1990;249:912–5. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- 50.Diller L, et al. p53 functions as a cell cycle control protein in osteosarcomas. Mol Cell Biol. 1990;10:5772–81. doi: 10.1128/mcb.10.11.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yonish-Rouach E, et al. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–7. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- 52.Shaw P, et al. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci U S A. 1992;89:4495–9. doi: 10.1073/pnas.89.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, Blandino G, Oren M, Givol D. Induced p53 expression in lung cancer cell line promotes cell senescence and differentially modifies the cytotoxicity of anti-cancer drugs. Oncogene. 1998;17:1923–30. doi: 10.1038/sj.onc.1202113. [DOI] [PubMed] [Google Scholar]
- 54.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 55.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 56.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fields S, Jang SK. Presence of a potent transcription activating sequence in the p53 protein. Science. 1990;249:1046–9. doi: 10.1126/science.2144363. [DOI] [PubMed] [Google Scholar]
- 58.Raycroft L, Wu HY, Lozano G. Transcriptional activation by wild-type but not transforming mutants of the p53 anti-oncogene. Science. 1990;249:1049–51. doi: 10.1126/science.2144364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu J, Zhou W, Jiang J, Chen X. Identification of a novel p53 functional domain that is necessary for mediating apoptosis. J Biol Chem. 1998;273:13030–6. doi: 10.1074/jbc.273.21.13030. [DOI] [PubMed] [Google Scholar]
- 60.Bargonetti J, Friedman PN, Kern SE, Vogelstein B, Prives C. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell. 1991;65:1083–91. doi: 10.1016/0092-8674(91)90560-l. [DOI] [PubMed] [Google Scholar]
- 61.Kern SE, et al. Identification of p53 as a sequence-specific DNA-binding protein. Science. 1991;252:1708–11. doi: 10.1126/science.2047879. [DOI] [PubMed] [Google Scholar]
- 62.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–9. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 63.Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol Cell Biol. 1992;12:2866–71. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zauberman A, Barak Y, Ragimov N, Levy N, Oren M. Sequence-specific DNA binding by p53: identification of target sites and lack of binding to p53 - MDM2 complexes. Embo J. 1993;12:2799–808. doi: 10.1002/j.1460-2075.1993.tb05941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farmer G, et al. Wild-type p53 activates transcription in vitro. Nature. 1992;358:83–6. doi: 10.1038/358083a0. [DOI] [PubMed] [Google Scholar]
- 66.Pietenpol JA, et al. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc Natl Acad Sci U S A. 1994;91:1998–2002. doi: 10.1073/pnas.91.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scharer E, Iggo R. Mammalian p53 can function as a transcription factor in yeast. Nucleic Acids Res. 1992;20:1539–45. doi: 10.1093/nar/20.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen X, Farmer G, Zhu H, Prywes R, Prives C. Cooperative DNA binding of p53 with TFIID (TBP): a possible mechanism for transcriptional activation. Genes Dev. 1993;7:1837–49. doi: 10.1101/gad.7.10.1837. [DOI] [PubMed] [Google Scholar]
- 69.Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009 doi: 10.1038/nrc2730. (in press) [DOI] [PubMed] [Google Scholar]
- 70.el-Deiry WS, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 71.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 72.Zhao R, et al. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–93. [PMC free article] [PubMed] [Google Scholar]
- 73.Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12:414–8. doi: 10.1016/j.ccr.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 74.Raver-Shapira N, Oren M. Tiny actors, great roles: microRNAs in p53’s service. Cell Cycle. 2007;6:2656–61. doi: 10.4161/cc.6.21.4915. [DOI] [PubMed] [Google Scholar]
- 75.He X, He L, Hannon GJ. The guardian’s little helper: microRNAs in the p53 tumor suppressor network. Cancer Res. 2007;67:11099–101. doi: 10.1158/0008-5472.CAN-07-2672. [DOI] [PubMed] [Google Scholar]
- 76.Brosh R, et al. p53-Repressed miRNAs are involved with E2F in a feed-forward loop promoting proliferation. Mol Syst Biol. 2008;4:229. doi: 10.1038/msb.2008.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ginsberg D, Mechta F, Yaniv M, Oren M. Wild-type p53 can down-modulate the activity of various promoters. Proc Natl Acad Sci U S A. 1991;88:9979–83. doi: 10.1073/pnas.88.22.9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–20. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–30. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci U S A. 1994;91:6098–102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 82.Oliner JD, et al. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–60. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 83.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–9. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 84.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–7. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 85.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 86.Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. Embo J. 1993;12:461–8. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 88.Picksley SM, Lane DP. The p53-mdm2 autoregulatory feedback loop: a paradigm for the regulation of growth control by p53? Bioessays. 1993;15:689–90. doi: 10.1002/bies.950151008. [DOI] [PubMed] [Google Scholar]
- 89.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 90.Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689–94. doi: 10.1128/mcb.4.9.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–11. [PubMed] [Google Scholar]
- 92.Kastan MB, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–97. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 93.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–6. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 94.Kamijo T, et al. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U S A. 1998;95:8292–7. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shvarts A, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. Embo J. 1996;15:5349–57. [PMC free article] [PubMed] [Google Scholar]
- 96.Kostic M, Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE. Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J Mol Biol. 2006;363:433–50. doi: 10.1016/j.jmb.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 97.Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci U S A. 2003;100:12009–14. doi: 10.1073/pnas.2030930100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 99.de Oca Luna R. Montes, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 100.Parant J, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–5. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- 101.Wade M, Wahl GM. Targeting Mdm2 and Mdmx in cancer therapy: better living through medicinal chemistry? Mol Cancer Res. 2009;7:1–11. doi: 10.1158/1541-7786.MCR-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bond GL, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 103.Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9:95–107. doi: 10.1038/nrc2584. [DOI] [PubMed] [Google Scholar]
- 104.Hu W, et al. A single nucleotide polymorphism in the MDM2 gene disrupts the oscillation of p53 and MDM2 levels in cells. Cancer Res. 2007;67:2757–65. doi: 10.1158/0008-5472.CAN-06-2656. [DOI] [PubMed] [Google Scholar]
- 105.Lahav G, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–50. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 106.Roth JA, et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat Med. 1996;2:985–91. doi: 10.1038/nm0996-985. [DOI] [PubMed] [Google Scholar]
- 107.Peng Z. Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Hum Gene Ther. 2005;16:1016–27. doi: 10.1089/hum.2005.16.1016. [DOI] [PubMed] [Google Scholar]
- 108.Senzer N, Nemunaitis J. A review of contusugene ladenovec (Advexin) p53 therapy. Curr Opin Mol Ther. 2009;11:54–61. [PubMed] [Google Scholar]
- 109.Heise C, et al. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3:639–45. doi: 10.1038/nm0697-639. [DOI] [PubMed] [Google Scholar]
- 110.Kirn D, Hermiston T, McCormick F. ONYX-015: clinical data are encouraging. Nat Med. 1998;4:1341–2. doi: 10.1038/3902. [DOI] [PubMed] [Google Scholar]
- 111.Alemany R. Cancer selective adenoviruses. Mol Aspects Med. 2007;28:42–58. doi: 10.1016/j.mam.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 112.Yu W, Fang H. Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets. 2007;7:141–8. doi: 10.2174/156800907780058817. [DOI] [PubMed] [Google Scholar]
- 113.Zhang H, et al. Enhanced therapeutic efficacy by simultaneously targeting two genetic defects in tumors. Mol Ther. 2009;17:57–64. doi: 10.1038/mt.2008.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bykov VJ, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–8. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 115.Boeckler FM, et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A. 2008;105:10360–5. doi: 10.1073/pnas.0805326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vassilev LT, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 117.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 118.Issaeva N, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–8. doi: 10.1038/nm1146. [DOI] [PubMed] [Google Scholar]
- 119.Grinkevich VV, et al. Ablation of key oncogenic pathways by RITA-reactivated p53 is required for efficient apoptosis. Cancer Cell. 2009;15:441–53. doi: 10.1016/j.ccr.2009.03.021. [DOI] [PubMed] [Google Scholar]
- 120.Yang Y, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7:547–59. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 121.Aas T, et al. Specific P53 mutations are associated with de novo resistance to doxorubicin in breast cancer patients. Nat Med. 1996;2:811–4. doi: 10.1038/nm0796-811. [DOI] [PubMed] [Google Scholar]
- 122.O’Shea D, et al. The presence of TP53 mutation at diagnosis of follicular lymphoma identifies a high-risk group of patients with shortened time to disease progression and poorer overall survival. Blood. 2008;112:3126–9. doi: 10.1182/blood-2008-05-154013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Young KH, et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood. 2008;112:3088–98. doi: 10.1182/blood-2008-01-129783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tomasini R, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–91. doi: 10.1101/gad.1695308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Han HS, et al. The estrogen receptor alpha pathway induces oncogenic Wip1 phosphatase gene expression. Mol Cancer Res. 2009;7:713–23. doi: 10.1158/1541-7786.MCR-08-0247. [DOI] [PubMed] [Google Scholar]
- 126.Lu X, et al. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell. 2007;12:342–54. doi: 10.1016/j.ccr.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 127.Shreeram S, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–64. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 128.Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006;6:909–23. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 129.Kang HJ, et al. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0904280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stiewe T. The p53 family in differentiation and tumorigenesis. Nat Rev Cancer. 2007;7:165–8. doi: 10.1038/nrc2072. [DOI] [PubMed] [Google Scholar]
- 131.Celli J, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999;99:143–53. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- 132.Yang A, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–8. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 133.Yang A, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 134.Finlan LE, Hupp TR. p63: the phantom of the tumor suppressor. Cell Cycle. 2007;6:1062–71. doi: 10.4161/cc.6.9.4162. [DOI] [PubMed] [Google Scholar]
- 135.Rosenbluth JM, Pietenpol JA. The jury is in: p73 is a tumor suppressor after all. Genes Dev. 2008;22:2591–5. doi: 10.1101/gad.1727408. [DOI] [PubMed] [Google Scholar]
- 136.Bourdon JC. p53 Family isoforms. Curr Pharm Biotechnol. 2007;8:332–6. doi: 10.2174/138920107783018444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Livingstone LR, et al. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992;70:923–35. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 138.Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–48. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- 139.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 140.Kaghad M, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–19. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- 141.Schmale H, Bamberger C. A novel protein with strong homology to the tumor suppressor p53. Oncogene. 1997;15:1363–7. doi: 10.1038/sj.onc.1201500. [DOI] [PubMed] [Google Scholar]
- 142.Yang A, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–16. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 143.Kamijo T, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–59. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 144.Banin S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 145.Canman CE, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 146.Brodsky MH, et al. Drosophila p53 binds a damage response element at the reaper locus. Cell. 2000;101:103–13. doi: 10.1016/S0092-8674(00)80627-3. [DOI] [PubMed] [Google Scholar]
- 147.Ollmann M, et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell. 2000;101:91–101. doi: 10.1016/S0092-8674(00)80626-1. [DOI] [PubMed] [Google Scholar]
- 148.Derry WB, Putzke AP, Rothman JH. Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science. 2001;294:591–5. doi: 10.1126/science.1065486. [DOI] [PubMed] [Google Scholar]
- 149.Schumacher B, Hofmann K, Boulton S, Gartner A. The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol. 2001;11:1722–7. doi: 10.1016/s0960-9822(01)00534-6. [DOI] [PubMed] [Google Scholar]
- 150.Tyner SD, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 151.Chipuk JE, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 152.Mihara M, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–90. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 153.Bourdon JC, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–37. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sablina AA, et al. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–13. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–9. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jones RG, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 157.Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007;450:721–4. doi: 10.1038/nature05993. [DOI] [PubMed] [Google Scholar]
- 158.Bommer GT, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 159.Chang TC, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.He L, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Raver-Shapira N, et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 162.Tarasov V, et al. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–93. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 163.Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A. 2007;104:15472–7. doi: 10.1073/pnas.0707351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Feng Z, et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–53. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]