

Abstract
Introduction
Under specific conditions, a weak lead stimulus, or “prepulse”, can inhibit the startling effects of a subsequent intense abrupt stimulus. This startle-inhibiting effect of the prepulse, termed “prepulse inhibition” (PPI), is widely used in translational models to understand the biology of brain based inhibitory mechanisms and their deficiency in neuropsychiatric disorders. In 1981, four published reports with “prepulse inhibition” as an index term were listed on Medline; over the past 5 years, new published Medline reports with “prepulse inhibition” as an index term have appeared at a rate exceeding once every 2.7 days (n = 678). Most of these reports focus on the use of PPI in translational models of impaired sensorimotor gating in schizophrenia. This rapid expansion and broad application of PPI as a tool for understanding schizophrenia has, at times, outpaced critical thinking and falsifiable hypotheses about the relative strengths vs. limitations of this measure.
Objectives
This review enumerates the realistic expectations for PPI in translational models for schizophrenia research, and provides cautionary notes for the future applications of this important research tool.
Conclusion
In humans, PPI is not “diagnostic”; levels of PPI do not predict clinical course, specific symptoms, or individual medication responses. In preclinical studies, PPI is valuable for evaluating models or model organisms relevant to schizophrenia, “mapping” neural substrates of deficient PPI in schizophrenia, and advancing the discovery and development of novel therapeutics. Across species, PPI is a reliable, robust quantitative phenotype that is useful for probing the neurobiology and genetics of gating deficits in schizophrenia.
Keywords: Animal models, Antipsychotic, Dopamine, Prepulse inhibition, Schizophrenia, Sensorimotor gating, Startle
Introduction
Among the paths to understanding the neurobiology of schizophrenia, one heavily traveled, has been the study through preclinical and clinical models of sensorimotor gating and its neural and genetic substrates. A laboratory paradigm frequently used to operationally measure sensorimotor gating is prepulse inhibition of the startle reflex (PPI). Medline lists over 1400 published reports utilizing the key word “prepulse inhibition” and over 580 that also include the key word “schizophrenia”. Research using PPI to probe the neural and genetic bases of schizophrenia has crossed every level of the “top down” and “bottom up” investigations of this disorder—from studies of the psychological implications of PPI to those assessing the control of PPI by signal transduction pathways and the genes that regulate them. Arising implicitly and explicitly from such a broad application of the PPI paradigm have been assumptions and expectations that we hope to examine critically in this review. In so doing, we hope to offer some perspectives on both potentially productive directions of this work, and the degree to which some assumptions and expectations may, or may not, be reasonable.
Historical overview
The popularity of PPI as an experimental paradigm for understanding schizophrenia comes from its conceptual linkage to clinical observations that schizophrenia patients are unable to optimally filter or “gate” irrelevant, intrusive sensory stimuli (Bleuler 1911; Kraepelin and Robertson 1919; McGhie and Chapman 1961; Venables 1964). These clinical observations led to the formulation of a construct—“gating deficits” in schizophrenia—that has been extended to refer to deficient inhibition of both sensory and cognitive information. The PPI paradigm was developed as a measure of automatic or preconscious inhibition in normal comparison subjects, as one variant of numerous paired-pulse paradigms in which the presentation of a lead stimulus led to the reduced perceptual or motor response to a second stimulus (Peak 1939; Graham 1975) (Fig. 1). Braff et al. (1978) first merged the construct and its operational measurement by identifying PPI deficits in schizophrenia patients, a finding that has since been replicated by many independent groups and [as reviewed previously (Braff et al. 2001b) and below], has become among the most influential paradigms in the field of schizophrenia psychophysiology. A comprehensive review through the year 2000 of all reports linking PPI deficits to schizophrenia in clinical populations is found in Braff et al. (2001b); reports subsequent to this date are listed in Table 1. Animal studies first linked this finding to a neurochemical (DA) and anatomical (ventral striatum) substrate (Sorenson and Swerdlow 1982; Swerdlow et al. 1986), and subsequent reports centered these substrates within an extended forebrain and pontine circuit that regulates PPI in rodents (Koch and Schnitzler 1997; Swerdlow et al. 1992, 2000a; see Table 4). Animal studies have identified developmental (Geyer et al. 1993; Lipska et al. 1995; see Table 3) and genetic (Carter et al. 1999; Ralph et al. 1999; Geyer et al. 2002; see Table 3) influences on PPI and have led to predictive models for antipsychotic development (Swerdlow et al. 1994) that have been modified and widely applied towards antipsychotic discovery. A comprehensive review through the year 2000 of all reports using PPI in models predicting antipsychotic properties is found in Geyer et al. (2001); reports subsequent to this date are listed in Table 2.
Fig. 1.
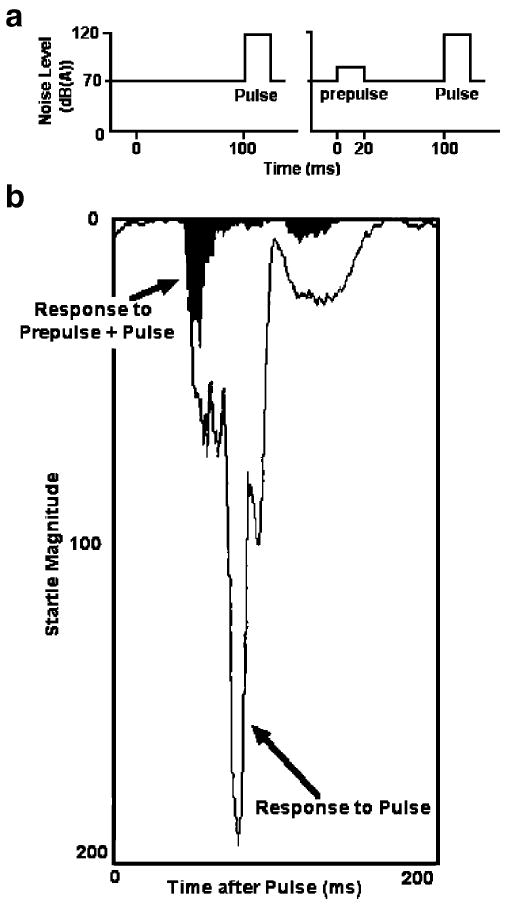
Schematic representation, adapted from Swerdlow et al. (1994), of stimuli used to elicit PPI in laboratory measures (a). b shows superimposed tracings of electromyography of the right orbicularis oculi in an adult male subject, from sequential trials that included either a prepulse [20 ms noise burst 4 dB over a 70-dB(A) background] followed 100 ms later by a 118-dB(A) 40 ms startle noise pulse (solid black area), or the startle pulse alone (open area). Tracings in (b) begin at pulse onset. The amount of inhibition generated by the prepulse can be appreciated visually by subtracting the solid area from the open area
Table 1. Studies of PPI in schizophrenia patients and related groups, ca. 2001-2007a.
- Studies reporting PPI deficits in schizophrenia patients
- Studies reporting PPI deficits in subgroups of schizophrenia patients
- Studies reporting PPI deficits in schizophrenia patients under specific experimental conditions
- Studies reporting PPI deficits in schizophrenia patients
- Studies reporting PPI deficits in populations conceptually linked to schizophrenia
| Reference | Sex, medications, n, other characteristics | PPI deficits compared to normal comparison subjects (NCS)? | Other measures or factors examined in relation to PPIb | Eye side | Background DBc | Startle stimuli | Prepulse stimuli | Prepulse interval (ms) |
|---|---|---|---|---|---|---|---|---|
| I. Studies reporting PPI deficits in schizophrenia patients | ||||||||
| Braff et al. 2005 | F, MED (n=25) | Yes | R | 70 | 40 ms 115 dB WN | 20ms 78 and 86 dB WN | 30, 120 | |
| Cadenhead et al. 2002 | M/F 10/11, MED (n = 4), UNMED (n = 17) | 33% of PTS<1 SD of PPI of NCS | Medication, clinical characteristics, P50, AS | R,L | 70 | 40 ms 115 dB WN | 20 ms 86 dB WN | 30, 120 |
| Duncan et al. 2003a | M, MED (n = 27), UNMED (n = 14) | Yes | Medication | R | 70 | 40 ms 116 dB 1 KHz | 20 ms 85 dB 1 KHz | 30–120 |
| Duncan et al. 2003b | M, study 1: pre- and post-medication (n = 16); study 2: MED (n = 43), UNMED (n = 21) | Yes, independent of medication status | Medication | R | 70 | 40 ms 116 dB 1 KHz | 20 ms dB 1 KHz | 30–120 |
| Heresco-Levy et al. 2007 | M/F 18/12, MED | Yes | Clinical characteristics, serum glycine and glutamate levels | R | 70 | 40 ms 115 dB 1 KHz | 20 ms 84 dB 1 KHz | 30–120 |
| Hong et al. 2007 | M/F 46/13, MED | Yes | Medication, P50 | R | 70 | 40 ms 116 dB WN | 20 ms 80 dB WN | 30–500 |
| Kumari et al. 2003a | M, MED (n = 7). | Trend towards lower PPI | fMRI | R | None | 40 ms airpuff 30 psi | 20 ms airpuff 10 psi | 100 |
| Kumari et al. 2005a | M, MED (n = 23) | Yes | Medication, violence ratings, clinical characteristics, illness duration | R | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 30–150 |
| Kumari et al. 2005b | Study 1: M, MED (n = 35–39); study 2: M/F 23/12, MED | Yes | Medication, clinical characteristics, AS | R | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 30–120 |
| Kumari et al. 2007B | M/F 17/3, UNMED | Yes | R,L | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 30–120 | |
| Ludewig and Vollenweider 2002 | M/F 49/18, MED | Yes | Medication, clinical characteristics | R | 70 | 40 ms 115 dB WN | 20 ms 86 dB WN | 30–2000 |
| Ludewig et al. 2002 | M/F 15/4, MED | Yes | R | 70 | 40 ms 115 dB | 20 ms 86 dB | 30–2000 | |
| Ludewig et al. 2003 | M, UNMED (n = 24) | Yes | Clinical characteristics | R | 70 | 40 ms 115 dB WN | 20 ms 86 dB WN | 30–2000 |
| Mackeprang et al. 2002 | M/F 14/6, MED | Yes independent of medication status | Medication | R | 70 | 40 ms 116 dB | 85 dB | 30–120 |
| Perry et al. 2002 | M and F (n = 41); M/F 25/16, MED (n = 20), UNMED (n = 21) | Yes | Medication | R | 70 | 40 ms 115 dB WN | 20 ms 95 dB WN | 30–120 |
| Perry et al. 2004 | M/F 8/6, MED | Yes | Sex | R | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 30–120 |
| Swerdlow et al. 2006f | M/F 72/31, MED (n = 94), UNMED (n = 9) | Yes | Medication, sex, clinical characteristics, neurocognitive and functional measures, smoking | R,L | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 20–120 |
| II. Studies reporting PPI deficits in subgroups of schizophrenia patients | ||||||||
| Kumari et al. 2004 | M/F 27/15, MED | Yes in men, but not in women | Medication, sex, clinical characteristics, PPF | R | 70 | 40 ms 115 dB WN | 20 ms 78 or 85 dB WN | 30–150 |
| Leumann et al. 2002 | M/F 25/8, MED | Yes with typical but not atypical APs | Medication, LI | R | 70 | 40 ms 115 dB | 20 ms 86 dB | 30–2000 |
| Meincke et al. 2004 | M/F 22/14, MED | Yes during acute, but not remitted clinical state | Clinical characteristics, psychopathological symptoms | R | 65 | 20 ms 115 dB WN | 20 ms 73 dB WN | 30, 100 |
| Minassian et al. 2007 | M/F 16/7, admission: MED (n = 15), UNMED (n = 8), 2 weeks later: MED (n = 23), UNMED (n = 1) | Yes at hospital admission, but not 2 weeks later | Medication, clinical characteristics | R | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 30–120 |
| Oranje et al. 2002 | M/F 31/13, MED | Yes in PTS with typical but not with atypical APs | Medication | R | NS | 30 ms 115 dB 1 KHz | 30 ms 80 dB 1.5 KHz | 120 |
| Quednow et al. 2006 | M/F 19/9, pre-study: MED (n = 9), UNMED (n = 16), post-randomization: typical APs (n = 12), atypical APs (n = 16) | Yes during baseline session in first week of treatment, but not after prolonged treatment | Number of previous episodes, clinical characteristics, therapeutic success | R | 70 | 40 ms 116 dB | 20 ms 86 dB | 120 |
| III. Studies reporting PPI deficits in schizophrenia patients under specific experimental conditions | ||||||||
| George et al. 2006 | M/F 9/6, smokers, MED | Smoking abstinence: ↓PPI; smoking reinstatement: ↑PPI | Smoking | NS | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 30–120 |
| Hazlett et al. 2003 | M/F 14/4, UNMED PTS with schizotypical personality disorder | Greater PPI during attended vs. ignored prepulses in NCS, but not in PTS | PPF | R | 45 | 40 ms 104 dB WN | 5 or 8 s 70 dB 0.8 or 1 KHz | 120, 240 |
| Kedzior and Martin-Iverson 2007 | M/F 7/1, MED | deficits in “attend” condition only | Smoking | Ld | 60 | 50 ms 100 dB WN | 20 ms 70 dB 5 KHz | 20–200 |
| Kumari et al. 2002 | M, MED (n = 30) | Yes in PTS treated with typical APs but not with RIS | Medication, clinical characteristics, duration of illness | R | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 30–120 |
| Kumari et al. 2003b | M/F 7/4, MED (n = 11) | ↓ PPI in response to procyclidine | R | 70 | 40 ms 115 dB WN | 20 ms 78 or 85 dB WN | 30–120 | |
| Wynn et al. 2004 | PTS: M/F 74/2, MED (typical APs (n = 22), atypical APs (n = 43), mixed or unknown (n = 11), unaffected siblings: M/F 17/19 | No PPI deficits in PTS or unaffected siblings | Medication, PPF, sex, clinical characteristics | L | None | 50 ms 105 dB WN | 20 ms 75 dB WN | 120 |
| IV. Studies reporting no PPI deficitsins PPI deficits in schizophrenia patients | ||||||||
| Duncan et al. 2006a | M, MED (n = 52), UNMED (n = 21) | No | Medication, clinical characteristics | R | 70 | 40 ms 116 dB 1 KHz | 20 ms 85 dB 1 KHz | 30–120 |
| Postma et al. 2006 | M, MED (n = 9) | No. Smoking enhanced PPI in PTS and NCS | fMRI | R | None | 40 ms airpuff 30 psi | 20 ms air airpuff 10 psi | 30–120 |
| Volz et al. 2003 | M/F 23/26, MED (n = 42), UNMED (n = 7) | No | L | NS | 50 ms 100 dB WN | Pictures presented for 6 s | 150–3,800 | |
| V. Studies reporting PPI deficits in populations conceptually linked to schizophrenia | ||||||||
| Kumari et al. 2005d | M/F 4/15, unaffected siblings of SZ PTS | Reduced PPI in siblings of SZ PTS with binaural stimulus presentation | Schizotypy ratings | R | 70 | 40 ms 115 dB WN | 20 ms 85 dB WN | 30–120 |
| Sobin et al. 2005a | M/F 11/14, children with 22q11 DS | Yes | Sex, age, clinical characteristics, latency reduction, attention network test, reaction time | R | 50 | 50 ms 104 dB WN | 40 ms 70 dB WN | 100 |
| Sobin et al. 2005b | M/F 13/12, children with 22q11 DS | Yes | Sex, age, clinical characteristics, symptom severity, subsyndromal symptoms of other disorders | R | 56 | 50 ms 104 dB WN | 30 ms 70 dB WN | 100 |
| Weike et al. 2001 | Ss “believe in extraordinary phenomena” (n = 16, M/F = 5/11) or not (n = 16, M/F = 10/6) | PPI not different between believers and non-believers | Sex, age, schizotypal personality, magical ideation/perceptual aberration scales | L | NS | 50 ms 105 dB WN | 20 ms 1000 Hz | 30–480 |
APs Antipsychotics, AS anti-saccade measures, F female, fMRI functional magnetic resonance imaging, L left, LI latent inhibition, M male, MED medicated, NCS normal comparison subjects, NS not specified, P50 P50 event-related potential suppression, PPF prepulse facilitation, PPI prepulse inhibition, PTS patients, R right, RIS risperidone, Ss subjects, SZ schizophrenia, UNMED unmedicated, WN white noise, ↓ reduced, ↑ increased
All tables are preceded by outlines describing their organizational structure. In distilling this substantial literature into tabular form, a substantial amount of information is lost. The abbreviated descriptions herein cannot do justice to the wealth of data and interpretations found in the original reports. References are provided to guide readers to the source material.
Demographics reported as independent measures in most studies
All dB A scale unless not specified in text; stimuli described in KHz are pure tones.
Right eye, n = 1
Table 4. Examples of studies providing anatomically-specific information regarding the neural substrates of PPI, ca. 2001–2007.
- Nucleus accumbens
- Hippocampus
- Prefrontal cortex
- Entorhinal cortex
- Amygdala
- Dorsomedial thalamus
- Habenula
- Medial septum
- Nucleus Basalis of Meynert
- Inferior Colliculus
- Pedunculopontine nucleus
- Laterodorsal tegmental nucleus
- Raphe complex
- Brainstem
| Reference | Rat strain, sex | Brain regions | Manipulation | Effect on PPI | |
|---|---|---|---|---|---|
| I. NAC | |||||
| Adults | |||||
| Caceda et al. 2005 | LE, M | Virally mediated increase in NT1 receptor | Blocked AMP & DIZ-induced PPI deficits | ||
| Culm et al. 2003 | SD, M | Infusion of PTX | Blocked QUIN-induced PPI deficit | ||
| Culm et al. 2004 | SD, M | Infusion of Sp-cAMP | Blocked QUIN-induced PPI deficit | ||
| Mohr et al. 2007 | Mice, C3H, F | Infusion of DIH or QUIN | ↑PPI after QUIN, but Ø PPI after DIH | ||
| Nagel et al. 2003 | SD, M | Infusion of MSX-3 (A2 antagonist) | ↓PPI | ||
| Pothuizen et al. 2005 | WI, M | Core, shell | Infusion of muscimol | Loss of PP intensity dependency after infusion into NAC core but not shell | |
| Pothuizen et al. 2006 | WI, M | Core | NMDA-lesion of the NAC core | enhanced PPI disruption by DIZ but not APO | |
| Powell et al. 2003 | LE, F | Intra-NAC 6-OHDA in SR & IR rats | blocked ↓ in PPI in IR rats | ||
| Schwienbacher et al. 2002 | SD, M | + VTA | Infusion of DAergic, adenosinergic, or GABAergic compounds into NAC and/or VTA | ↑PPI after combined VTA PTX + NAC SCH23390 | |
| Swerdlow et al. 2006d | SD, LE and F1 (SDxLE), M | + Striatum | Measured DA-stimulated [35S]GTPγS-binding in NAC, striatum | PPI-APO sensitivity: SD>F1>LE. [35S]GTPγS-binding in NAC, striatum: LE>F1>SD | |
| II. HPC | |||||
| Adults | |||||
| Ellenbroek et al. 2002,b | WI, M | CA1 | Infusion of AMP, SKF81297 or QUIN | ↓PPI after AMP, SKF81297 or QUIN; AMP-induced PPI deficits blocked by intra-NAC SCH23390 but not sulpiride | |
| Ma and Leung 2004 | LE, M | CA1 | Electrical kindling | ↓PPI (and ↓PA) | |
| Finamore et al. 2001 | Rats | Infusion of KA or NMDA antagonists | ↓PPI with infusion of NMDA antagonists | ||
| Fitting et al. 2006a | SD, M | Infusion of viral toxin TAT | ↑PPI (and ↓PA) | ||
| Inada et al. 2003 | WI, M | Antisense NR1 knockdown | ↓PPI with knockdown 6, but not 14d pre-testing | ||
| Neonates | |||||
| Fitting et al. 2006c | SD, M, F | Neonatal infusion of viral toxin gp120 | ↓PPI (and ↑PA); + Vehicle: ↓PPI with increasing gp120 doses. + APO: ↑PPI with increasing gp120 doses | ||
| Fitting et al. 2006b | SD, M, F | Neonatal TAT infusion | Males: ↓PPI at d 30 and 60, but not d 90 | ||
| vHPC | |||||
| Adults | |||||
| Howland et al. 2004b | LE, M | + dHPC | Electrical stimulation VHPC vs. DHPC combined with NAC microdialysis | ↓PPI after VHPC but not DHPC stim.; ↑DA efflux: ipsi-but not contralateral NAC after unilateral stim. VHPC but not DHPC | |
| Klamer et al. 2005b | SD, M | Microdialysis of the VHPC after systemic (or local) PCP | ↓PPI and ↑cAMP after PCP; blocked by NO-synthase inhibitor L-NAME | ||
| Kusljic and van den Buuse 2004 | SD, M | + dHPC | 5,7-DHT lesion | ↓PPI for DHPC lesioned rats, and partially for VHPC lesioned rats | |
| Zhang et al. 2002a | WI, M | + dHPC | Infusion of NMDA | ↓PPI after intra-VHPC but not-DHPC infusion | |
| Swerdlow et al. 2004b | SD, M | + FX | Infusion of NMDA into the VHPC in rats with EL lesions of the FX | ↓PPI after NMDA infusion into VHPC, unaffected by FX lesion; IA lesion of the VHPC but not EL FX lesion enhanced ↓PPI by APO | |
| Neonates | |||||
| Laplante et al. 2005 | SD, M | IA-neonatal lesion | ↓PPI; blocked by biperiden | ||
| Zhang et al. 2002b | WI, M | + dHPC | Muscimol or TTX infusion | ↓PPI, not blocked by HAL or CLO | |
| Risterucci et al. 2005 | SD, M | IA-neonatal lesion | ↓PPI, ↓blood flow in NAC, BLA,VP, BNST, entorhinal–piriform and orbital CTX | ||
| Adults | |||||
| Caine et al. 2001 | LH, M | dSUB or vSUB | QA-lesions | ↓PPI after vSUB lesions. ↓PPI to AMP (not APO) after vSUB lesions. | |
| III. PFC | |||||
| Adults | |||||
| de Jong and van den Buuse 2006 | SD, M | Infusion of SCH23390 | enhanced PPI deficits to APO but not DIZ | ||
| Neonates | |||||
| Grobin et al. 2006 | M, F | + MD | Neonatal elevation of allopregnanolone | ↓PPI in castrates before and after puberty (PD20 and 80), but Ø PPI during puberty (PD40 and 60) | |
| Rajakumar et al. 2004 | SD, M | Neonatal infusion of antibody to the p75 neurtrophin receptor | ↓PPI at age 10 wks, but not 5 wks | ||
| mPFC | |||||
| Adults | |||||
| Afonso et al. 2007 | SD, F | NMDA-lesion | ↓PPI | ||
| Bast et al. 2001 | WI, M | Infusion of NMDA | ↓PPI, not blocked by HAL or CLO | ||
| Day-Wilson et al. 2006 | LH, M | IR (associated with ↓mPFC volume) | ↓PPI | ||
| Schwabe and Koch 2004 | WI, M | IA-lesion | lesion blocked DIZ-induced ↓PPI but not APO-induced ↓PPI | ||
| Shoemaker et al. 2005 | SD, M | Infusion of SCH23390 into the mPFC; infusion of NMDA into the VHPC in rats with IA mPFC lesion | ↓PPI after infusion of SCH23390 in mPFC; mPFC lesions block ↓PPI after intra-VHPC NMDA infusion | ||
| Swerdlow et al. 2006c | SD, M | + NAC | Systemic SCH23390, IA lesion of mPFC, 6-OHDA DA depletion of mPFC or NAC | ↓PPI after SCH23390, not blocked by either NAC DA depletion or mPFC lesion; ↓PPI after mPFC DA depletion | |
| Neonates | |||||
| Schneider and Koch 2005 | WI, M | IA-neonatal lesion | ↑PPI in juveniles; enhanced PPI deficits to APO in adults | ||
| Schwabe et al. 2004 | WI, M | IA-neonatal lesion | ↑PPI after neonatal lesions; ↓PPI in both lesioned and intact rats after APO | ||
| IV. eCTX | |||||
| Adults | |||||
| Goto et al. 2002 | WI, M | IA lesion | ↓PPI, partially blocked by HAL | ||
| Goto et al. 2004 | WI, M | + NAC | eCTX lesion with IA, microdialysis of NAC | ↓PPI, ↑DA concentration in NAC | |
| Uehara et al. 2007 | WI, M | + mPFC | eCTX lesion with QA, mPFC lidocaine infusion | ↓PPI after eCTX lesion or mPFC lidocaine | |
| V. AMY | |||||
| Neonatal | |||||
| Daenen et al. 2003 | F1 of WI/ UWU, M | AMY (or vHPC) | Neonatal AMY or VHPC lesions with IA | ↓PPI in rats lesioned in the AMY or VHPC on d 7, but not on d 21 | |
| BLA | |||||
| Adults | |||||
| Howland et al. 2007 | LE, M | + eCTX, + vHPC | Electrical kindling | ↓PPI shortly after kindling of BLA, but not of eCTX or VHPC | |
| Kusljic and van den Buuse 2006 | SD, M | + CnA | 5,7-DHT lesion | ↓PPI with lesions of CnA but not BLA | |
| Shoemaker et al. 2003 | SD, M | QA lesion of the BLA | ↓PPI, blocked by quetiapine | ||
| Stevenson and Gratton 2004 | LE, M | + Striatum | Infusion of SCH23390 or raclopride | ↑PPI after intra-BLA SCH23390, ↓PPI after intra-BLA raclopride | |
| VI. MD | |||||
| Adults | |||||
| Swerdlow et al. 2002c | SD, M | Infusion of QUIN or TTX | ↓PPI after TTX but not QUIN, not blocked by quetiapine | ||
| VII. Habenula | |||||
| Adults | |||||
| Heldt andRessler 2006 | Mice, C57, M | Electrolytic lesion | Ø PPI in the absence of stress; but ↓PPI after stress in habenula lesioned rats; blocked by CLO | ||
| VIII. mS | |||||
| Adults | |||||
| Ma and Leung 2007 | LE, M | + SUM | Infusion of muscimol | Muscimol into mS or SUM blocked ketamine-or DIZ-induced PPI deficits | |
| Ma et al. 2004 | LE, M | Infusion of muscimol | Infusion of muscimol into mS blocked PCP-induced PPI deficits | ||
| IX. NBM | |||||
| Ballmaier et al. 2002 | SD, M | Immunolesion of cholinergic NBM neurons | ↓PPI, blocked by single or repeated admin. of rivastigmine | ||
| X. IC | |||||
| Adults | |||||
| Silva et al. 2005 | LE, M | Electrical stimulation | ↓PPI | ||
| Sandner et al. 2002 | SD, M | + PnC | Evoked potentials from IC or PnC | ↓PPI by ketamine and ↑evoked potentials | |
| Yeomans et al. 2006 | WI, M | SC, + intercollicular nuc. or PPTg | Electrical PP and pulses via electrodes to the SC, IC, intercollicular nucleus, or PPTg | PPI after electrical PP to most SC sites. Longer PPI latencies for electrical PP to the SC than IC, intercollicular nuc. or PPTg | |
| XI. PPTg | |||||
| Adults | |||||
| Diederich and Koch 2005 | WI, M | Infusion of muscimol | ↓PPI at intervals≥120 ms | ||
| Takahashi et al. 2007 | Mice, ICR, M | + lGP, ssCTX | Infusion of phaclofen into the PPTg or lidocain into the lGP, c-fos labeling of brain regions after acoustic pulses or prepulses | ↓PPI after intra-PPTg phaclofen or intra-lGP lidocaine; ↑c-fos in lGP after prepulses; ↑c-fos in NAC shell, PnC, and ssCTX after pulses, blocked in NAC and PnC by prepulses | |
| XII. LDTN, SN | |||||
| Adults | |||||
| Jones and Shannon 2004 | SD, M | IA-lesion of the LDTN or SN | ↓PPI after lesion of LDTN but not SN | ||
| XIII. DRN or MRN | |||||
| Adults | |||||
| Kusljic et al. 2006 | SD, M | 5,7-DHT lesion | ↓PPI in MRN but not DRN lesioned rats, blocked by HAL or CLO | ||
| Kusljic et al. 2003 | SD, M | 5,7-DHT lesion | ↓PPI at all PP intensities for MRN-lesioned rats and for some PP intensities for DRN lesioned rats | ||
| XIV. Brainstem | |||||
| Neonates | |||||
| Shishkina et al. 2004 | WI, M | Neonatal infusion of antisense oligonucleotide complementary to the α2 adrenoceptor | ↓PPI at PD34, associated with ↑α2 adrenoceptors in HPC, AMY |
AMP Amphetamine, AMY amygdala, APO apomorphine, BG background, BLA basolateral amygdala, BNST bed nucleus of the stria terminalis, C57 C57BL/6J, CLO clozapine, CnA central nucleus of the amygdala, CPA N(6)-cyclopentanyladenosine, CTX cortex, d dorsal, 5,7-DHT 5,7 dihydroxytryptamine, DIH dihydrexidine, DIZ dizocilpine, DRN dorsal raphe nucleus, e entorhinal, EL electrolytic, F females, FX fornix, HAL haloperdidol, HPC hippocampus, IA ibotenic acid, IC inferior colliculus, IR isolation rearing, KA kainic acid, l lateral, LDTN laterodorsal tegmental nucleus, LE Long Evans, LH Lister Hooded, M males, m medial, MD dorsomedial thalamus, MET methamphetamine, MRN median raphe nucleus, NAC nucleus accumbens, NBM nucleus basalis of Meynert, NMDA N-methyl-d-aspartate, NO nitric oxide, OVX ovariectomized, NT neurotensin, 6-OHDA 6-hydroxydopamine, PD postnatal day, PA pulse alone trial, PCP phencyclidine, PFC prefrontal cortex, PnC nucleus reticularis pontis caudalis, PPI prepulse inhibition, PPTg pendunculopontine nucleus, PTX pertussis toxin, QA quinolinic acid, QUIN quinpirole, S septum, SD Sprague–Dawley, SC superior colliculus, SN substantia nigra, Sp-cAMP cyclic adenosine monophosphate analogue, SR socially reared, ss somatosensory, SUB subiculum, SUM supramamillary area, v ventral, VP ventral pallidum, VTA ventral tegmental area, WI Wistar, ↓decreased, ↑ increased, Ø unchanged
Table 3. Model organisms, ca. 2001–2007.
- Low and high baseline PPI levels
- Sub-strains selected by drug sensitivity
- Genetically engineered organisms, based on genes related to:
- Vulnerability for schizophrenia
- Dopamine
- Glutamate
- Noradrenaline
- Histamine
- Catecholamines (general)
- Acetylcholine
- GABA
- Second Messenger Systems
- Neuropeptides
- Other
- Models for specific disorders
- Developmental models
- Isolation/Deprivation/Stress-related
- Isolation rearing
- Maternal deprivation
- Developmental stressors
- Immune-related
- Developmental drug exposure
- Developmental hypoxia
- Developmental nutritional deprivation
- Neonatal lesions
- Drug-related models
- Drug withdrawal
- Toxin exposure
- Other
| Superscript designates study-specific findings | |||||
|---|---|---|---|---|---|
| References | Species, Strain, Sex | Model description/background/rationale | Basal PPI | Effects of drugs typically used to induce PPI deficits | Effects of (presumed) antipsychotics/other treatments |
| I. Low and high baseline PPI levels | |||||
| Humans | |||||
| Bitsios et al. 20051; Swerdlow et al. 2006a2; Vollenweider et al. 20063 | M | Basal PPI differences between subgroups (“low vs. high gaters”) | ØPER, ØAMA1 in low gaters ↓PER1, ↓AMA1 in high gaters | QUE (↑PPI2), CLO (↑PPI3), both at short PP intervals and in low gaters; ØPPI3 in high gaters | |
| Rats | |||||
| Feifel and Priebe 20011; Feifel et al. 20042 | BB, M | Basal PPI deficits | ↓PPI1,2 | CLO and PD 149 163 (a neurotensin mimetic; both ↑PPI), but HAL (ØPPI)1,2; subchronic HAL (↑PPI)1 | |
| Ferguson and Cada 20041; van den Buuse 20042 | SHR vs. SD and WKY, M, F | SHR rats display behavioral abnormalities thought to model clinical symptoms | ↓PA1,2, ↓PPI relative to SD and WKY1; ↓PPI relative to SD (trend only)2, but ØPPI relative to WKY rats2 | AMP (↓PPI in SHR and WKY, but ØPPI in SD rats)2; APO (↓PPI in SD, but ØPPI in SHR and WKY rats)2; DIZ, 8-OHDPAT (both ↓PPI in SHR, WKY, and SD)2 | |
| Freudenberg et al. 2007 | Former WI, M | Selective breeding of rats with high vs. low PPI | |||
| Fujiwara et al. 2006 | LEC and WI, M | A putative animal model of WD | ↓PPI in LEC rats | CU (↓PPI in both LEC and WI rats) | |
| II. Sub-strains selected by drug sensitivity | |||||
| Rats | |||||
| APO Susceptibility | |||||
| Sontag et al. 20031; van der Elst et al. 20062, 20073 | APO-SUS and APO-UNSUS, M | APO-SUS and APO-UNSUS rats were selectively bred to achieve high (SUS) vs. low APO (UNSUS) susceptibility | ↓PPI in APO-SUS vs. APO-UNSUS rats1,3 (not apparent in2) | Sensitivity to the PPI-disruptive effects of COC2 or AMP3 (APO-SUS > APO-UNSUS)2,3 | Removal of isolation stress: ↑PPI in APO-SUS, but ↓PPI in APO-UNSUS rats1; REMO (↓AMP in APO-SUS, but ØAMP in APO-UNSUS)3; aMpT (depleted cytosolic DA, ØPPI in both strains, ØAMP in APO-SUS, ↓AMP in APO-UNSUS)3, RES (ØPPI in both strains, ØAMP in both strains)3; Tests in APO SUS only: REMO (ØPPI, ↓COC)2, PRAZ (↑PPI, ↓COC)2, KETS (↑PPI, ØCOC)2, aMpT + RES (↓AMP)3 |
| Alcohol preference | |||||
| Bell et al. 20031; Ehlers et al. 20072 | F, M | Selective breeding of female rats or selection of male rats with high (P) vs. low (NP) alcohol preference | ØPPI after selective breeding; ↑PPIinPrats2; ↓PPI in P rats housed in isolation2 | Adult rats: AMP (↓PPI in P, but ↑PPI in NP rats); Adolescent rats: AMP (↓PPI in P, but ØPPI in NP rats) | |
| III. Genetically engineered organisms, based on genes related to: | |||||
| A. Vulnerability for schizophrenia | |||||
| Mice | |||||
| Clapcote et al. 2007 | Missense mutations in exon 2 of the DISC1 gene | DISC1 is a proposed schizophrenia susceptibility gene | ↓PPI in mice with missense mutation at residues 31L or 100P | CLO, HAL, BUP, Rolipram (PDE4 inhibitor; all ↑PPI; reversal of PPI was dependent on the specific type of missense mutation) | |
| Barr et al. 2007 | KO for reelin receptors VLDLR or APOER2, M, F | Reelin is reportedly reduced in brains of schizophrenia patients | Ø acoustic PPI in both KO, ↓crossmodal PPI in VLDLR mice, ↑crossmodal PPI in APOER2 mice | PPI-disruptive effects of PCP: KO>WT | |
| Podhorna and Didriksen 2004 | Heterozygous, reeler mutants, M, F | Reeler mice have a mutation in the gene for reelin and have been suggested as an animal model for schizophrenia | ↓PPI only in fully adult F (trend only) | ||
| Boucher et al. 2007 | Heterozygous NRG1 KO, M | NRG1 is a proposed schizophrenia susceptibility gene | ØPPI | PPI-disruptive effects of THC: KO>WT | |
| Mukai et al. 2004 | ZDHH8-KO, M, F | ZDHH8 is a proposed schizophrenia susceptibility gene | ↓PPI in F, but ØPPI in M | ||
| B. Dopamine | |||||
| DA receptors | |||||
| Mice | |||||
| Ralph-Williams et al. 2002 | D1-KO, or D2-KO, M, F | ØPPI in D1-KO, but ↓PPI in D2-KO | APO, SKF82958 (both ØPPI in D1-KO, but ↓PPI in WT; ↓PPI in D2-KO and WT); AMP (↓PPI in D1-KO and WT, ØPPI in D2-KO, but ↓PPI in WT); DIZ (↓PPI in D1-KO, D2-KO, and WT) | ||
| Holmes et al. 2001 | DA D5 null mutants, M, F | ØPPI | SKF 81297 (ØPPI in mutants, but ↓PPI in WT) | ||
| DAT | |||||
| Mice | |||||
| Barr et al. 20041; Yamashita et al. 20062 | DAT-KO, M | Increase dopamine activity has been proposed in schizophrenia | ↓PPI1,2 | COC, METP (both ↑PPI in KO, but ↓PPI in WT)2 | M100907 (5-HT2A antagonist, ↑PPI in KO, but ØPPI in WT)1; FLX, NSX (a NET inhibitor, both ↑PPI in KO, but ØPPI in WT)2, CIT (ØPPI in KO and WT)2 |
| Ralph-Williams et al. 2003b | DAT-knock-downs, M, F | ØPPI | |||
| Other Dopamine related | |||||
| Eells et al. 2006 | Mice, nuclear receptor Nurr1 null mutants | Nurr1 is important for development of DA neurons; early postnatal isolation | ↓PPI after postnatal isolation in Nurr1+/− mice | ||
| C. Glutamate | |||||
| NR1 | |||||
| Rats | |||||
| Inada et al. 2003 | WI, M | Antisense knock-down of HPC NR1 by HPJ-liposome vector | ↓PPI | ||
| Mice | |||||
| Bickel et al. 20081; Duncan et al. 20042, 2006a3; Moy et al. 20064 | TG with reduced expression of NR1, M, F | NMDA receptor signaling may be reduced in schizophrenia. Microtubule stabilization in neurons depends on STOP | ↑PA1,2,3,4, ↓PPI1,2,3,4 | Sensitivity to PPI-disruptive effects of AMP: TG>WT4 | HAL, CLO, RIS (all ↑PPI in both TG and controls)3 |
| Fradley et al. 2005 | TG with reduced expression of NR1 or STOP-KO, M, F | ↓PPI (in both mouse types) | CLO (ØPPI in both mouse types) | ||
| NR2 | |||||
| Mice | |||||
| Boyce-Rustay and Holmes 20061; Spooren et al. 20042 | NR2A-KO, M, F | ØPPI1,2 | Ro 63-1908 (a selective NR2B receptor antagonist,↓PPI)2 | ||
| Takeuchi et al. 2001 | NR2A, NR2B, N2C, N2D, or GLURδ2 mutants | NR2A-D are known subunits of the NMDA receptor channel. GLURδ2 is a relatively novel GLU receptor subunit | ↑PA in NR2A, B, C and D mutants, ØPAin GLURδ2, ↑PPI in NR2B and GLURδ2, ØPPI in NR2A, C and D | ||
| NR3 | |||||
| Brody et al. 2005 | Mice NR3A-KO or TG NR3A overexpressors, M, F | ↑PPI at 3–4 weeks old M, but ØPPI in FKO; ØPPI in TG | |||
| AMPA | |||||
| Wiedholz et al. 2008 | Mice, AMPA GLUR1-KO, M, F | The gene encoding GLUR1 lies within a chromosomal region that is associated with schizophrenia | ↓PPI | ||
| mGLU | |||||
| Mice | |||||
| Brody et al. 2003a, b | mGLU1-KO, M | Reduced glutamate function has been proposed in schizophrenia | ↓PPI1,2,3 | PCP (↓PPI in KO and WT) | RAC (ØPPI in KO and WT), LAM (↑PPI in KO and WT) |
| Brody and Geyer 2004b1; Brody et al. 2004a2; Lipina et al. 20073 | mGLU5-KO, M, F | ↓PPI | PPI deficit of KO mice could not be mimicked in WT mice with the mGLU5 antagonist MPEP1, no further disruption of PPI by DIZ in KO3 | RAC, CLO, LAM (all ØPPI)2, CX546 and ARIR (positive modulators of AMPA, both ↑PPI (less pronounced with ARIR))3 | |
| Other glutamate related | |||||
| Mice | |||||
| Szumlinski et al. 2005 | Homer1-KO or Homer2-KO, M, F | Homer proteins interact with mGLU, and modify the morphology of GLU synapses. A SNP in Homer1 was associated with schizophrenia | ↓PPI in Homer1-KO, but ØPPI in Homer2-KO | HAL (↑PPI in Homer1-KO) | |
| Tsai et al. 2004 | Heterozygous GLYT-KO, M | GLY is a co-agonist at the NMDA-receptor with presumed sub-saturating concentrations at the receptor | ØPPI | Sensitivity to the PPI-disrupting effects of AMP (KO < WT) or DIZ (KO > WT) | |
| Wolf et al. 2007 | CPB-K vs. Balb, M | CPB-K mice display low levels of NMDA receptors | ↑PA, ↓PPI relative to BalbC mice | Acute or subchronic CLO (ØPPI) | |
| D. Noradrenaline | |||||
| Lahdesmaki et al. 2004 | Mice, adrenergic α2A-KO, M, F | Adrenergic α2A receptors modulate transmitter release of DA and 5-HT neurons | ↑PPI | AMP (↓PPI in KO, and WT; greater sensitivity to AMP in KO), DEXM (an α2 agonist, ØPPI, ↓AMP in KO, but not WT) | ATI (an α2 antagonist, ØPPI, ØAMP) |
| E. Histamine | |||||
| Dai et al. 2005 | Mice, H1-KO, M | Histaminic abnormalities have been implicated in the pathophysiology of schizophrenia | ↓PPI in WT, but ØPPI in KO after IR | Sensitization to METH enhanced effects of IR on PPI in WT, but not in KO mice | |
| F. Cathecholamines (General) | |||||
| Klejbor et al. 2006 | Mice, FGFR1-TG, M, F | FGFR1-TG express a dominant-negative mutant from the catecholaminergic, neuron-specific TH promoter | ↑PA, ↓PPI | FLUP (a DA antagonist, ↑PPI) | |
| G. Acethylcholine (ACh) | |||||
| Nicotinic | |||||
| Mice | |||||
| Bowers et al. 2005 | Nicotinic α7-KO, M | Evidence suggests reduced α7 expression in schizophrenia patients | ØPPI | PPI-disruptive effects of EtOH: KO = WT | |
| Cui et al. 2003 | Nicotinic β3-KO, M, F | β3-subunit of the nACH is highly expressed in DA neurons of the SN and VTA | ↓PPI | ||
| Muscarinic | |||||
| Thomsen et al. 2007 | Mice, M5-KO, M, F | The M5 muscarinic ACh receptor has been implicated in susceptibility to schizophrenia | ↓PPI | AMP (↓PPI in KO and WT) | CLO (↑PPI in KO, but ØPPI in WT; ØAMP in both KO and WT), HAL (↑PPI in KO and WT; ↓APO in both KO and WT) |
| H. GABA | |||||
| GABAA | |||||
| Mice | |||||
| Hauser et al. 2005 | GABAA α5 mutants, M, F | The α5 subunit of the GABAA channel is strongly expressed in the HPC | ↓PPI | ||
| Yee et al. 2005 | GABAA α3-KO, M, F | The α3-GABAA receptor is the main receptor subtype expressed by GABA-ergic neurons involved in controlling monoaminergic Neurons | ↓PPI | HAL (↑PPI) | |
| Other GABA related | |||||
| Mice | |||||
| Chiu et al. 2005 | GAT1-KO, M, F | GAT1-KOs display behavioral abnormalities proposed to model some aspects of psychopathology | ↓PPI | ||
| Heldt et al. 2004 | GAD65-KO, M, F | GAD65 is a GABA synthesizing enzyme | ↓PPI | CLO reversed the PPI deficit of KO | |
| I. Second messenger systems | |||||
| Mice | |||||
| Gould et al. 20041; Kelly et al. 20072 | TG with a constituitively active Gsα or TG with R(AB), or Gsα x PKA double TG mice, M, F | R(AB) TG express a PKA type inhibitor. G-protein signaling related to the cAMP/PKA pathway may be abnormal in schizophrenia | ↓PPI in Gsα TG, but ØPPI in PKA TG1,2 and Gsα × R(AB) double TG2 | HAL (↑PPI in Gsα TG, but ØPPI in R(AB) TG), ROL (↑PPI in Gsα TG)2 | |
| Harrison et al. 2003 | LAP1, M, F | LAP1 is a G-protein coupled receptor with developmental expression suggesting a role in psychopathology | ↓PPI | ||
| Koh et al. 2008 | PLCβ1-KO, M, F | PLCβ1 may be altered in brains of schizophrenia patients | ↓PPI | HAL (↑PPI in KO, but ØPPI in WT) | |
| Shum et al. 2005 | CaMKIV-KO, M | CaMKIV is thought to be involved in neuroplasticity and aspects emotional behavior | ↓PPI (and ↓PA) | ||
| van den Buuse et al. 2005a | Gzα-KO, M | Gzα is a G-protein of the Gi type and associated with DA D2-receptors | ØPPI | Sensitivity to the PPI disruptive effects of AMP, APO (both: KO>WT) or DIZ (KO = WT) | |
| J. Neuropeptides | |||||
| Neurotensin | |||||
| Rats | |||||
| Caceda et al. 2005 | M | Virally mediated over expression of NT1 in the NAC | ØPPI | ↓AMP, ↓DIZ | |
| Mice | |||||
| Kinkead et al. 2005 | NT null mutants, M, F | NT is proposed to have “endogenous antipsychotic” properties | ↑PA, ↓PPI | AMP (ØPPI in mutants, but ↓PPI in WT) | HAL, QUET (both ØPPI in mutants, but ↑PPI in WT), CLO (↑PPI in mutants and WT), OLA (ØPPI in mutants amd WT) |
| CRF | |||||
| Mice | |||||
| Dirks et al. 20021, 20032; Groenink et al. 20083 | TG CRF1 overexpressors, M | CRF abnormalities may play a role in psychopathology | ↓PPI1,2,3 | CRF1 antagonists (↑PPI in TG, but ØPPI in WT), GR antagonists (ØPPI in TG and WT), adrenelectomy (ØPPI in TG and WT)3; HAL, CLO, RIS, but not CDP all reduce PPI deficit of TG relative to WT | |
| Risbrough et al. 2004 | CRF1-KO, M | ØPPI | CRF (↑PPI in KO, but ↓PPI (and ↑PA) in WT) | ||
| Arginine Vasopressin | |||||
| Egashira et al. 2005 | Mice, V1b-KO, M | V1b plays a role in regulation of the physiological response to stress | ↑PA, ↓PPI | CLO, RIS (both ↑PPI), but HAL (ØPPI) | |
| Gastrin | |||||
| van den Buuse et al. 2005b | Gastrin-KO, M | Gastrin is a peptide hormone. It is also produced in the brain and binds to the CCK receptor. CCK interacts with DA in the brain | ØPPI | Sensitivity to the PPI-disruptive effects of AMP (KO<WT), but for APO, DIZ, 8-OHDPAT (all KO = WT) | |
| Neurexin | |||||
| Beglopoulos et al. 2005 | Mice, Nxph3-KO, M | Nxph3 is a ligand of synaptic α-neurexins | ↑PA, ↓PPI | ||
| PACAP | |||||
| Tanaka et al. 2006 | Mice, Adcyap1-mutants | Adcyap1 mutants lack the gene encoding for PACAP and display marked behavioral abnormalities including hyperlocomotion and jumping behavior | ↓PPI | AMP (↑PPI) | HAL (ØPPI) |
| K. Other | |||||
| Wang et al. 2003a, b | Mice, adenosine A2-KO, M | Adenosine may influence PPI by interacting with the DA system of the brain | ↓PA, ↓PPI | AMP (ØPPI in KO and WT (slight trend towards ↓PPI in KO, but ↑PPI in WT); DIZ (↓PPI in KO and somewhat more pronounced in WT) | |
| Wolinsky et al. 2007 | Mice, TA1-KO, M | Trace amines have been implicated in schizophrenia | ↓PPI | ||
| Mice | |||||
| Gogos et al. 20061; van den Buuse et al. 20032 | Aro-KO, M, M (castrated), F | Gender differences in psychiatric disease; aromatase converts testosterone into estrogen | ØPPI in M, castrated M, and F1; age-dependent ↓PPI in M, but ØPPI in F (slight trends only)2 | PPI-disruptive effect of 8-OHDPAT (F K0 = F WT; M KO>M WT; enhanced in castrated M WT, but not in KO)1 | |
| Weil et al. 2006 | MT1-KO, M, F | Melatonin has been implicated in psychiatric disease | ↓PPI | ||
| Mice | |||||
| Petitto et al. 2002a | Double deletion of IL2/IL15Rβ-double-KO, M, F | IL2 and IL15 are cytokines that share a common β-receptor subunit, which is essential for intracellular signaling. Expression levels are high in HPC and limbic regions | ↓PA, ↓PPI | ||
| Petitto et al. 2002b | MRL-lpr substrain, M | MRL-lpr mice develop lupus-like autoimmune disease, but also reduced IL2 production. Schizophrenia may involve immune processes of the CNS | ØPPI in pre-disease MRL-lpr, but ↓PPI with evidence of autoimmune disease | ||
| Mice | |||||
| Irintchev et al. 2004 | L1 or CHL1, M, F | The cell-adhesion molecule L1 and its close homologue CHL1 may be linked to schizophrenia | ↓PPI | ||
| Jaworski et al. 2005 | TIMP-2-KO or knock-down, M, F | TIMP influence extracellular matrix molecules and may be involved in brain plasticity and possibly brain development | ↓PPI in KO when comparedto heterozygous,butnot knock-downorWT animals | ||
| Pillai-Nair et al. 2005 | NCAM-TG, M, F | NCAM is elevated in brains from schizophrenia patients | ↓PPI | CLO (↑PPI), HAL (ØPPI) | |
| Mice | |||||
| Brunskill et al. 20051; Erbel-Sieler et al. 20042 | Npas1-KO, Npas3-KO, M, F | Npas is a transcription factor highly expressed in developing neuroepithelium | ↓PPI in Npas12 and Npas31,2-KO | ||
| Burne et al. 2005 | Vitamin D receptor KO, M, F | Vitamin D contributes to normal brain development. | ↓PPI at long PP intervals | ||
| Cao and Li 2002 | Emx1 mutants, M, F | Emx1 is implicated in forebrain development and behavioral processes. | (↓PPI; trend only) | ||
| McDonald et al. 2001 | FGFR3-null mutants, M, F | FGFR may contribute to neuronal growth, angiogenesis, mitagenesis and skeletal development | ↓PPI | ||
| Miyakawa et al. 2003 | CN-mutants, M | CN is involved in neurite extension and neuronal plasticity. CN-mutants display behavioral abnormalities | ↓PPI | ||
| Park et al. 2002 | CDF mutants and Catna2-TG (of CDF mutants) | CDF-mutants show morphological abnormalities in the HPC and cerebellum as well as behavioral abnormalities. Catna2-TG have partially restored CDF regions and normal HPC and cerebellum morphology | ↓PPI in CDF-mutants; ØPPI in Catna2-TG | ||
| Porras-Garcia et al. 2005 | Heterozygous Lurcher mutants, M | Lurcher mutants display a progressive loss of Purkinje neurons | ↓PPI | ||
| Yukawa et al. 2005 | STAT6-deficient, M | STAT6 is expressed in the CTX, HPC, striatum (developing brain), and basal forebrain (adults). STAT are signaling molecules that mediate cytokine-related mechanisms | ↓PPI | ||
| L. Models for specific disorders | |||||
| Fragile X-syndrome | |||||
| Humans + mice | |||||
| Frankland et al. 20041; Spencer et al. 20062 | Human children with FXS, Fmr1-KO, Fmr2, or Fmr1+2 double KO mice, M | Fmr-KO mice are putative animal models of FXS | ↓PPI in children with FXS1 and in Fmr1-KO1, but ØPPI in Fmr1-KO2 and FMR1+2 double KO2 | ||
| Bontekoe et al. 2002 | FXR2-KO, M | FXR2 is a homolog of the FMRP protein, which is lacking or mutated in FXS. | ↓PPI | ||
| Chen and Toth 2001 | FMR1-KO, M | FMR1-gene encodes the FMRP protein, which is lacking or mutated in FXS. | ↑PPI | ||
| Nasu-Hasu-Hikala disease | |||||
| Kaifu et al. 2003 | Mice, DAP12-deficient, M, F | DAP12 deletions lead to the Nasu-Hikala disease | ↓PA, ↓PPI | ||
| 22q11 deletion syndrome | |||||
| Paylor et al. 2006 | Mice, with chromosomal deletions Df1, 2, 3, 4 or 5, or mutations of genes Tbx1, Gnb1l, or Cdcrel1, M, F | Chromosomal Df1 deletions are a putative animal model of 22q11 deletion syndrome, which is linked to high schizophrenia rates. Df1 deletions were “behaviorally mapped” to mutations of single genes via PPI | ↓PPI in mice with deletions of Df1, Df2, Df3 and mutations of TbX1, Gnb1l. ØPPI in mice with deletions of Df4 or Df5, and mutations of Cdcrel1 | ||
| Huntington’s disease | |||||
| Van Raamsdonk et al. 2005 | Mice, YAC128 | HD patients have motor-, cognitive-and psychiatric disturbances. YAC128 mice express mutant huntingtin and are a presumed animal model for HD | ↓PPI in older mice | ||
| Alzheimer’s disease | |||||
| Mice | |||||
| Ewers et al. 2006 | APP&PS1 double-KO, M, F | AD involves neuropathological changes in the HIP | ØPPI, but correlation between PPI deficits and neuropathological changes | ||
| McCool et al. 2003 | CRND8-TG, M | CRND8-TG show over expression of Swedish/Indiana familial mutations of APP and an age dependent increase of amyloid production | ↓PPI (small effects) | ||
| Taniguchi et al. 2005 | WILD and N279K mutants, M, F | TAU mutations may play a causal role in forms of dementia and PD. N279K and WILD mutants contain a mutation of the human TAU gene | ↓PPI in N279K mutants, ØPPI in WILD mutants | ||
| IV. Developmental models | |||||
| A. Isolation/deprivation/stress-related | |||||
| 1. Isolation Rearing | |||||
| Rats | |||||
| Barr et al. 20061; Cilia et al. 20052; Day-Wilson et al. 20063; Harte et al. 20074; Powell et al. 20025, 20036; Rosa et al. 20057 | SD, FLH, M; LE, M; WI, M | IR | ↓PPI in M and F, and all strains1,2,3,4,5,6,7 | Water deprivation (ØPPI)5 | Iloperidone (broad spectrum DA/5-HT/NE antagonist, maternal dep 3. ØPPI)1; compound A (α7 agonist, ↑PPI2); DA-depletion with 6-OHDA (↑PPI)6; handling (↑PPI)7 |
| Nunes Mamede Rosa et al. 2005 | WI, M | Post-weaning isolation for 10 days | ↑PPI (not reversed by resocialization) | ||
| Mice | |||||
| Dai et al. 20041, 20052; Sakaue et al. 20033; Varty et al. 20064 | C57, ddY, 129 and H1-KO, all M | IR | ↓PPI in WT mice1,2,3; ↓PPI (C57 and 129 mice in atleast one of the two test sessions)4 ØPPI in H1-KO2 | Sensitization to AMP enhanced effects of IR on PPI in WT1,2 but not in H1-KO2, RIS and MKC-242 (a 5HT1a agonist, both ↑PPI)3 | |
| 2. Maternal deprivation | |||||
| Rats | |||||
| Choy and van den Buuse 2007 | Early stress: MD. Later stress: implantation of CORT pellets | ↓PPI (trend only in MD rats) | APO (↓PPI in CTR and rats treated with either MD or CORT, but ØPPI in rats exposed to MD and CORT); AMP (↓PPI in CTR, but ØPPI in rats exposed to MD); 8-OHDPAT (↓PPI in all groups) | ||
| Ellenbroek and Cools 2002 | WI, M, nulliparous F | IR, MD, rearing by MD mother | ↓PPI in IR rats; ↓PPI in MD rats; ØPPI in MD +IR rats; ↓PPI in pups reared by a MD mother; ↓PPI in MD pups reared by a non-MD mother | ||
| Garner et al. 2007 | WI, F | Early stress: MD. Later stress: Implantation of CORT pellets | ↓PPI in MD rats, ØPPI in CORT treated rats | ||
| Husum et al. 2002 | WI, M | MD | ↓PPI | ||
| 3. Developmental stressors | |||||
| Rats | |||||
| Hauser et al. 2006 | WI, M, F | Prenatal DEX exposure | ↑PPI in M (not replicated in a second study) | ||
| Koenig et al. 2005 | SD, M | Exposure of pregnant females to stressors | ↓PPI | ||
| Burton et al. 20061; Lovic and Fleming 20042 | SD, M, F | Exposure of pregnant females to restraint stress or exposure of offspring to AR with or without mechanical stimulation | ØPPI in response to restraint condition1. ↓PPI after AR with minimal stimulation1,2, but ØPPI after AR with maximal stimulation2 | ||
| Iso et al. 2007 | Mice, C57, M | Animals were exposed to enriched or impoverished conditions during development | ↓PPI in mice continuously kept in impoverished conditions | ||
| 4. Immune-related | |||||
| Rats | |||||
| Borrell et al. 20021; Romero et al. 20072 | WI, M, F | Prenatal bacterial immune challenge with LPS | ↓acoustic PPI in M rats1,2, ↓visual PPI in F rats2 | HAL, CLO (both ↑PPI in M and F)1; chronic HAL (↑PPI)2 | |
| Fortier et al. 2007 | SD, M | Prenatal (or postnatal) systemic bacterial (LPS), viral (poly I: C) or local (TUR) immune challenge | LPS (↓PPI at E15-16 and E18-19); poly I:C (ØPPI); TUR (↓PPI at E15-16) | ||
| Pletnikov et al. 2002 | Lewis or Fisher, M | IC-infusion of BDV on PND0 | ↓PPI in Fisher rats, but ØPPI in Lewis rats | ||
| Mice | |||||
| Nyffeler et al. 20061; Ozawa et al. 20062 | C57, M, F; Balb, M, F | Prenatal viral (poly I:C) immune challenge | ↓PPI in adults, but ØPPI in Balb juveniles2. Correlation between immunoreactivity for α2 GABAA immunoreactivity in the ventral dentate gyrus and PPI in CTR, but not in immune-challenged C557 mice1 | ||
| Rajakumar et al. 2004 | SD, M | IC-injection of antibody against the p75 neurotrophin receptor at PND0 to suppress neurotrophin activity | ↓PPI | ||
| Shi et al. 2003 | Balb or C57, M, F | Prenatal systemic immune challenge with influenza or poly I:C virus | ↓PPI under both conditions | CLO, CHLO (both ↑PPI following challenge with influenza virus)1 | |
| B. Developmental drug exposure | |||||
| Rats | |||||
| Tan 2003 | WI, M, F | Exposure to AMP or vehicle during pregnancy (GD8 to parturition) followed by an acute AMP or vehicle exposure challenge on the day of testing | ↓PPI and ↑PA after prenatal AMP treatment | ||
| Harris et al. 2003 | SD, M, F | Neonatal DIZ exposure | ↓PPI in F, but ØPPI in M | ||
| Rasmussen et al. 2007 | SD, M, F | Neonatal PCP or vehicle exposure on PND 7, 9 and 11 followed by a single PCP or vehicle exposure at PND45. Rats were tested at PND32-34 and 1,4 and 6 weeks after the PND45 treatment | ØPPI after neonatal PCP treatment only; transient ↓PPI after neonatal + adolescent PCP; ↑PPI in F, but ØPPI in M after adolescent PCP only | ||
| Takahashi et al. 2006 | WI, M, F | Daily exposure to PCP over 2 weeks in neonatal vs. adult rats | Persistent ↓PPI after neonatal PCP treatment, in M and F, transient ↓PPI after adult PCP administration in M | ||
| Wang et al. 2003a | SD | Neonatal PCP or vehicle exposure on PND 7, 9 and 11 | ↓PPI | M40403 (a SOD mimetic, ØPCP after short term treatment, but ↓PCP after long term treatment) | |
| Slawecki and Ehlers 2005 | SD, M | Alcohol exposure during adolescence or adulthood | ↑PPI after adolescent exposure, but ØPPI after adult exposure (↓PA for both groups) | ||
| Schneider and Koch 20031; Schneider et al. 20052 | WI, M | Chronic prepubertal, pubertal, or adult exposure to the CB agonist WIN 55,212-2 | ↓PPI in prepubertal2 and pubertal1 rats, but ØPPI in adult1 rats | HAL (↑PPI)1,2 | |
| Schneider et al. 2006 | WI, M | Prenatal valproate exposure | ↓PPI | Environmental enrichment (↑PPI) | |
| Gizerian et al. 20061; Grobin et al. 20062 | SD, M, F | Neonatal ALO administration on PND2 or PND5 or PND1 and PND5 | ↓PPI at PND 801,2 and 202, but not at PND 402 and 602 | CLO (↑PPI in the ALO PND2 group; ØPPI in the PND5 group)1 | |
| Watanabe et al. 2004 | SD, M | Cytokines have been implicated in the pathophysiology of schizophrenia. Neonatal challenge with the cytokine LIF from PND2 to PND10 | ↓PPI during and after adolescence | ||
| Futamura et al. 20031; Sotoyama et al. 20072 | SD, M, F | Neonatal perturbation of neurotrophic signaling via EGF administration | ↑PA1,2, ↓PPI1,2 | Sensitivity to the PPI-disruptive effects of subthreshold APO or QUIN in EGF-treated rats > controls; SKF38393 (ØPPI)2 | Subchronic CLO (↑PPI), but subchronic HAL (ØPPI)1 |
| Henck et al. 2001 | WI, M, F | Neonatal exposure to supraphysiological doses of the mitogen EGF | ↓PPI in F, but ØPPI in males | ||
| Mice | |||||
| Thomsen et al. 2007 | DBA, C57, C3H, ddyY, M, F | Neonatal EGF administration | ↑PA for all strains, ↓PPI in DBA and C57, but ØPPI in C3H and ddyY mice | ||
| Rats | |||||
| Elmer et al. 2004 | SD, M | Prenatal challenge with antimitotic Ara-C. | ↓PA, ↓PPI in post-adolescent rats | APO (ØPPI) | |
| Jongen-Relo et al. 20041; Le Pen et al. 20062 | WI, F, M; SD, M | Prenatal challenge with antimitotic MAM (at different time points during pregnancy) | ↓PPI in M SD rats2, (↓PPI for specific PP and PND of MAM challenge in WI rats, trend only)1 | ||
| Shishkina et al. 2004 | WI, M | Neonatal short-term reduction of brainstem α2 adrenergic receptors via injection of antisense oligonucleotides | ↓PPI at PND 34, but ØPPI at PND 22 and 80 | ||
| Howland et al. 2004a | Rats, LE, M | Neonatal i.p. injections of KA | ↓PPI | Sensitivity to the PPI-disruptive effects of APO in KA-treated rats = controls | |
| C. Developmental hypoxia | |||||
| Guinea pigs | |||||
| Rehn et al. 2004 | DH, Dunkin-Hartley, F | Reduction in utero-placental blood flow via unilateral ligation of the uterine artery | ↓PPI | ||
| Rats | |||||
| Tejkalova et al. 2007 | WI, M | Hypoxia on PND12 via bilateral carotid arterial occlusion | ↓PPI | ||
| Sandager-Nielsen et al. 2004 | SPF-WI, M | Anoxia on PND9 | (↓PPI in 1 of 2 experiments only) | AMP(↓PPItolowdose,trend only) | |
| Schmitt et al. 2007 | SD, M | Repeated mild hypoxia from PND4-8 | ↓PPI | ||
| D. Developmental nutritional deprivation | |||||
| Rats | |||||
| Burne et al. 2004 | SD, M, F | Pre-and postnatal vitamin D deprivation. | ↓PPI only after combined Pre-and chronic postnatal vitamin D deficiency | ||
| Palmer et al. 2004 | WKY, M, F | Prenatal protein deprivation. Prenatal malnutrition may increase the risk for schizophrenia | ↓PPI in Fat PND 56,but ØPPI at PND 35; ØPPI in M | ||
| E. Neonatal lesions | |||||
| Rats | |||||
| Daenen et al. 2003 | WI, M | Neonatal IA-lesion of the vHPC or AMY | ↓PPI in adult rats lesioned at PND7, ØPPI in adult rats lesioned at PND21 | ||
| Laplante et al. 20051; Powell et al. 2006; Le Pen and Moreau 20022; Le Pen et al. 20032; Rueter et al. 20044; Zhang et al. 20065 | SD, M | Neonatal IA-lesion of the vHPC | ↓PPI in lesioned post-pubertal rats1,2,3,4,5 | OXO (a muscarinic agonist, ØPPI in lesioned rats, but ↓PPI in non-lesioned rats)1 | HAL (ØPPI in lesioned rats and ↓PPI in non-lesioned rats2, but ↑PPI in lesioned rats and ØPPI in non-lesioned rats)5, CLO and OLA (↑PPI in lesioned rats, ↓PPI in non-lesioned rats)2, RIS2, BIP1 (a muscarinic antagonist), GLY3, and ORG 245983 (a NMDA co-agonist, all ↑PPI in lesioned rats, ØPPI in non-lesioned rats)3; chronic CLO or RIS (↑PPI); BP 897, AVE 5997, A-437203 (all preferential D3 antagonists, ØPPI)5 |
| Schneider and Koch 2005 | Rats, WI, M | Neonatal IA-lesion of the mPFC. Morphological changes in the mPFC in schizophrenia patients have been reported | ↑PPI after neonatal lesion in juvenile rats, but ØPPI in adult rats | Sensitivity to the PPI-disruptive effects of APO in adults: lesioned > non-lesioned | |
| Schwabe et al. 2004 | Rats, WI, M | Neonatal or adult IA-lesion of the mPFC | ↑PPI in adult rats after neonatal lesion, but ØPPI after adult lesions | APO (↓PPI) | |
| V. Drug-related models | |||||
| A. Drug withdrawal | |||||
| Rats | |||||
| Peleg-Raibstein et al. 2006a1, b2; Tenn et al. 20033 | WI, M, SD, M | Withdrawal from repeated, escalating AMP administration schedules (up to 5 mg/kg, 8 mg/kg, or 10 mg/kg). The endogenous DA system of unmedicated schizophrenia patients has been hypothesized to be “sensitized” | ØPPI with up to 5mg/kg AMP in WI1, but ↓PPI in SD3; ↓PPI under all other conditions | ||
| Wilmouth and Spear 2006 | SD, M | Withdrawal from nicotine (7 days of exposure). Withdrawal was induced by mecamylamine after nicotine treatment | ↓PPI in adolescents on day 1, but ØPPI on day 4 of withdrawal. ØPPI at either day in adults | ||
| B. Toxin exposure | |||||
| Rats | |||||
| Terry et al. 2007 | SD, M | Chronic, intermittent exposure to the organophosphate pesticide chlorpyrifos | ↓PPI | ||
| Tadros et al. 2005 | WI, M | Repeated injection of the mitochondrial toxin 3-NP leads to selective striatal lesions and behavioral changes linked to HD | ↓PPI (↓PA) | TAUR (a semi-essential β-amino acid, when administered prior to 3-NP: ↓3-NP) | |
| VI. Other | |||||
| Rats | |||||
| Pijlman et al. 2003 | WI, M | Exposure to physical stress (PS, foot shock) or emotional stress (ES, witness of foot shock to PS rat) | ↑PPI in PS, but ØPPI in ES rats | ||
| Mice | |||||
| van den Buuse et al. 2004 | C57, M | ADX. CORT replacement. Stress is a risk factor in psychiatric disease | ØPPI (for ADX, ADX+ CORT) | HAL (↑PPI in ADX+CORT and CTRL mice, but ØPPI in ADX mice) | |
| Rats | |||||
| Byrnes et al. 2007 | SD, F postpartum rats | Postpartum female rats | ØPPI | Sensitivity to the PPI-disrupting effects of QUIN: Postpartum rats < controls | |
| Mice | |||||
| Tremolizzo et al. 2005 | BtC3Fe, M | Hypermethylation may be related to downregulation of Reelin and GAD67 in schizophrenia patients. Methionine exposure for 2 weeks is used as an epigenetic model for schizophrenia | ↓PPI | Chronic VAL (↓Methionine), acute IMID (↓Methionine) | |
ACH Acetylcholine (receptor), AD Alzheimer’s disease, ADX adrenalectomy, ALO allopregnanolone, AMA amantadine, AMP amphetamine, aMpT alpha-methyl-para-tyrosine, AMY amygdala, APP amyloid precursor protein, AR artificial rearing, ARIR ariracepam, APO apomorphine, ATI atipamezole, BB Brattleboro, BDV Borna Disease virus, BIP biperiden, BUP bupropion, CaMKIV Calcium–calmodulin-dependent protein kinease IV, CCK cholecystokinin, CB cannabinoid (receptor), CDP chlordiazepoxide, CHLO chlorpromazine, CIT citalopram, CLO clozapine, CNS central nervous system, COC cocaine, CORT corticosterone, CRF corticotropin releasing factor, CTR controls, CU copper, DA dopamine (receptor), DAT dopamine transporter, DEX dexamethasone, DEXM dexmedetomidene, DIZ dizocilpine, E embryonic day, EGF epidermal growth factor, F female, FGFR fibroblast growth factor receptor, Fmr1 fragile X mental retardation 1 gene, FLUP flupenthixol, FLX fluoxetine, FXS Fragile X syndrome, GAD glutamic acid decarboxylase, GAT GABA transporter, GD gestation day, GR glucocorticoid receptor, GLU glutamate, GLY glycine, H histamine (receptor), HAL haloperidol, HD Huntington’s disease, HPC hippocampus, IMID imidazenil, KA kainic acid, KETS ketanserin, KO knock-out, LAM lamotrigine, LAP lysophosphatidic acid receptor, LE Long–Evans rat, LEC Long–Evans Cinnamon rat, LIF leukemia inhibitory factor, LPS lipopolysaccharide, M male, m metabotropic, MAM metholazoxymethanol acetate, MD maternal deprivation/maternally deprived, MT melatonin (receptor), n nicotinic, METH metamphetamine, METP methylphenidate, NAC nucleus accumbens, NBM nucleus basalis magnocellularis, NCAM neural cell adhesion molecule, NET norepinephrine transporter, 3-NP 3-nitropropionic acid, NR NMDA receptor subunit, NRG neuregulin, NSX nisoxetine, NT neurotensin, Nxph neurexophilin, OLA olanzapine, OXO oxotremorine, PA response to pulse alone, PACAP pituitary adenylate-cyclase-activating polypeptide, PD Parkinson’s disease, PND postnatal day, PER pergolide, PLC phospholipase C, poly I:C polyinosinic:polycytidylic acid, PRAZ prazosin, PS1 presinilin1, QUET quetiapine, QUIN quinpirole, RAC raclopride, REMO remoxipride, RIS risperidone, ROL rolipram, ROP ropinirole, SD Sprague–Dawley rat, SHR spontaneously hypertensive rat, SN substantia nigra, SOD superoxide dismutase, STAT signal transducers and activators of transcription, SUS susceptible, TA trace amine (receptor), TAUR taurine, TG transgenic, TH tyrosine hydroxylase, THC tetrahydrocannabinol, TIMP tissue inhibitor of metalloprotease, TUR turpentine, UNSUS unsusceptible, VAL valproate, VTA ventral tegmental area, V1b Vasopressin receptor 1b, WD Wilson’s Disease, WI Wistar rat, WKY Wistar–Kyoto rat, WT wild-type, ↓decreased, ↑increased, Ø unchanged
Table 2. Examples of studies using PPI to assess or predict antipsychotic properties, ca. 2001–2007.
- Anti-dopaminergics
- D2/mixed receptor antagonists
- D3-preferential antagonists
- D4-preferential antagonists
- Glutamatergic mechanisms
- mGLUR
- NMDA
- GLY
- Serotonergic mechanisms
- Noradrenergic mechanisms
- Cholinergic mechanisms
- Nicotinic agonists
- Muscarinic agonists
- AChE inhibitors
- Histaminergic mechanisms
- Cannabinoid mechanism
- CB1-antagonists
- Endocannabinoid transport inhibitor
- Cannabidiol
- Neuropeptide mechanisms
- Neurotensin agonists
- Opioids
- CCK
- Adenosine mechanisms
- GABA agonists
- GABA agonists
- Hormones
- Second-messenger inhibitors
- Nitric oxide synthase inhibitors
- Guanylate cyclase+NOS inhibitors
- PDE-inhibitors
- Miscellaneous
| References | Species, strain, sex | PPI deficit induced by | Primarydrug/mechanismtested | Effects | Other drug types tested |
|---|---|---|---|---|---|
| I. Anti-dopaminergics | |||||
| A. D2/mixed receptor antagonists | |||||
| Rats | |||||
| Bast et al. 2001 | WI, M | Intra-VHPC NMDA infusion | HAL D2 family antagonist | ØNMDA | CLO (ØNMDA) |
| Cilia et al. 2007 | SD, M | KET | HAL | ØKET, not potentiated by mGLUR5 antagonist MPEP | CLO, RIS, lamotrigine, SB-271046-A (all similar effects to HAL) |
| Conti et al. 2005 | WKY, M; BN, M | ICV CRF infusion | HAL | ↓CRF in WKY rats, ØCRF in BN rats | CLO (↓CRF in WKY rats, overcompensation of PPI-deficit in BN rats) |
| van den Buuse and Gogos 2007 | SD, M | 8 OHDPAT | HAL | ↓8 OHDPAT (at 100 ms PP interval) | RAC, ARI, BUS (all ↓8-OHDPAT), CLO, OLA, RIS, AMI, MDL73,005EF (partial 5-HT1A agonist) (all Ø8-OHDPAT) all at 100 ms PP interval |
| Rats + mice | |||||
| Metzger et al. 2007 | Mice, C57; Rats, SD, M | AMP or APO | HAL (implanted HAL polymer, or acute HAL) | HAL implants: ↓AMP in mice, ↓APO in rats Acute HAL: ↓APO in rats | |
| Mice | |||||
| Russig et al. 2004 | C57, M | APO | HAL | ↓APO | CLO (ØAPO) |
| Human subgroups (+rats) | |||||
| Vollenweider et al. 2006 | Humans (“low vs. high gaters”) | Basal PPI, differences between subgroups | CLO D1 - 4/5-HT2/α1/muscarinic antagonist | ↑PPI in “low gaters” (at short PP intervals), ØPPI in “high gaters” | |
| Swerdlow et al. 2006a | Humans (“low vs. high gaters”), M; rats, SD, M; rats, BN, M | Basal PPI, differences between human subgroups or rat strains | Quetiapine D1 - 2/5-HT2/α1/H1/muscarinic antagonist | ↑PPI in human low gaters and SD rats (at short PP intervals), ↑PPI in BN rats | CLO (↑PPI at short PP intervals), HAL (ØPPI) in SD rats |
| Primates | |||||
| Linn et al. 2003 | Capuchin monkeys, F | PCP | CLO | ↓PCP | HAL (ØPCP) |
| Rats | |||||
| Erhardt et al. 2004 | SD, M | ↑Endogenous KYNA by kynurine or PNU 156561A | CLO | ↓KYNA | HAL (↓KYNA) |
| Le Pen and Moreau 2002 | SD, M | nHPC lesion | CLO | ↑PPI | OLA (↑PPI), RIS (↑PPI), HAL (ØPPI) |
| Depoortere et al. 2007b | SD, M | APO | F15063 D2/D3-Antagonist, D4-partial agonist, 5-HT1A-agonist | ↓APO | |
| Depoortere et al. 2007a | SD, M | Basal PPI | F15063 | ØPPI | |
| Barr et al. 2006 | SD, M | IR vs. induction of PPI deficits by APO, PCP, or CIR in SR rats | Iloperidone DA/5-HAT/NA antagonist | ØPPI in IR rats, but ↓APO, ↓PCP, ↓CIR in SR rats | |
| Ellenbroek et al. 2001 | WI, M | Basal PPI, APO, or AMP | JL13 Predominant D1/5-HT2 binding | ↑PPI (basal), ↓APO, ↓AMP | HAL, CLO (both, ØPPI (basal), ↓APO, ↓AMP) |
| Ojima et al. 2004 | SD, M | Basal PPI | Perospirone D2/5-HT2A/5-HT1A antagonist | ↑PPI | HAL (ØPPI), RIS (↑PPI (relative to HAL)) |
| Rueter et al. 2004 | SD, M | nVHPC lesion | Risperidone D2/5-HT2/α antagonist (chronic low-dose treatment) | ↑PPI | CLO (↑PPI) |
| Bubenikova et al. 2005 | WI, M | DIZ | Zotepine D1/D2/D3/5-HT2A/5-HT2C5-HT6/5-HT7/α1/H1/NET-affinity | ØDIZ | RIS (ØDIZ), CLO, OLA (both ↓DIZ, but ↓PPI relative to vehicle (no DIZ)) |
| Mice | |||||
| Fejgin et al. 2007 | NMRI, M | Basal PPI or PCP | Aripiprazole partial agonist at D2/5-HT1A and antagonist at 5-HT2A | ↑PPI (basal), ↓PCP | CLO (↑PPI (trend), ØPCP), OLA, (ØPPI, ØPCP), HAL (↑PPI, ØPCP) |
| Brea et al. 2006 | Swiss, M | APO or DOI | QF2004B D1 - 4/5-HT1A,2A,2C/α1,2/M1,2/H1-binding | ↓APO, ↓DOI | CLO, HAL (both ↓APO, ↓DOI) |
| Flood et al. 2008 | DBA/2NCrl, DBA/2J, 2NHsd, 2NTac1, 2NTac2, C57BL/6Tac, 129S6/SvEvTac | Basal PPI | Olanzapine D1/D2/5-HT2/α1/muscarinic/H1 antagonist | Reversal of PPI deficit (tested only in DBA/2NCrl mice) | ARI (reversal of PPI deficit), β-CD (reversal of PPI deficit compared to H2O) in DBA/2NCrl mice; both drugs were not tested in other strains |
| B. D3-preferential antagonists | |||||
| Rats | |||||
| Zhang et al. 2007b | WI, M | PD128907 (D3 agonist), or APO | A-691990 | ↓PD128907, ØAPO | HAL (ØPD128907, ↓APO), RAC (ØPD128907), CLO, RIS (both: ↓PD128907, ↓APO), SB 277011 (↓PD128907, ØAPO) |
| Mice | |||||
| Zhang et al. 2006 | DBA, M | Basal PPI or nVHPC lesion | A-437203 | ↑PPI in unlesioned animals, but ØPPI after nVHPC lesion) | Intact mice: HAL (↑PPI), RIS (↑PPI), SB277011 (D3 antagonist, ↑PPI), AVE 5997 (D3 antagonist, ØPPI); nVHPC lesion: HAL (↑PPI), AVE 5997 (ØPPI); BP897 (preferential D3/D2 antagonist, ↑PPI in lesioned and intact mice) |
| Park et al. 2005 | ICR, M | APO | KKHA 761 | ↓APO | |
| C. D4-preferential antagonists | |||||
| Boeckler et al. 2004 | Rats, WI, M | FAUC 213 | ↓APO | ||
| II. Glutamatergic mechanisms | |||||
| A. mGLUR | |||||
| Rats | |||||
| Kinney et al. 2005 | SD, M | AMP | CDPPB Metabotropic GLU 5 allosteric potentiator | ↓AMP | |
| Mice | |||||
| Galici et al. 2005 | C57, M | AMP or PCP | LY487379 Metabotropic GLU 2 allosteric potentiator | ↓AMP, ØPCP | LY379268 (GLU 2/3 agonist; ØAMP, ØPCP) |
| B. NMDA | |||||
| Rats | |||||
| Zajaczkowski et al. 2003 | WI, M | DIZ | CGP 40116 Competitive NMDA antagonist | ↓DIZ | |
| C. GLY | |||||
| Rats | |||||
| Le Pen et al. 2003 | SD, M | nVHPC lesion | Glycine | ↑PPI | ORG 24598 (GLYT1 inhibitor, ↑PPI) |
| Mice | |||||
| Adage et al. 2007 | C57, M | PCP | AS057278 DAAO inhibitor; DAAO is the enzyme which oxidizes D-serine (→see below) | ↓PCP | CLO (↓PCP) |
| Depoortere et al. 2005 | DBA, M | Basal PPI | SSR5504734 GLYT antagonist | ↑PPI | |
| Kinney et al. 2003 | DBA, M | Basal PPI | NFPS GLYT1 antagonist | ↑PPI | CLO (↑PPI) |
| Lipina et al. 2005 | C57, M | Basal PPI or DIZ | d-Serine modulator of the GLY site of the NMDA receptor | ↑PPI (basal PPI), ØDIZ | l-Serine (ØPPI), ALX 5407 (GLYT1 inhibitor, ↓PPI, ↓DIZ), CLO (↑PPI, ↓DIZ) |
| III. Serotonergic mechanisms | |||||
| 5-HT1 | |||||
| Rats | |||||
| Auclair et al. 2006 | SD, M | APO | SSR181507 5-HT1A agonist, partial D2 agonist | ØAPO ↓APO (when co-administered with WAY100635) | SLV313 (similar to SSR81507), sarizotan (ØAPO), bifeprunox, HAL, ARI, RIS, OLA, QUE, ZIP (all ↓APO) |
| Auclair et al. 2007 | SD, M | Basal PPI | SSR181507 | ↓PPI (reversed by WAY100,635) | Sarizotan, bifeprunox, 8-OHDPAT, (all ↓PPI), HAL, ARI, RIS, OLA, QUE, ZIP (all ØPPI) |
| Krebs-Thomson et al. 2006 | 5-MeO-DMT (hallucinogen) | Way100,635 5-HT1A antagonist | ↓5 MeO-DMT | M100907 (5-HT2A antagonist, Ø5-MeO-DMT), SER-082 (5-HT2C antagonist ↓5-MeO-DMT) | |
| Mice | |||||
| Sakaue et al. 2003 | ddY, M | IR, APO or DIZ | MC-242 5-HT1A agonist | ↑PPI (in IR mice, antagonized by Way100,635), ØAPO (in SR mice), ØDIZ (in SR mice) | RIS (↑PPI in IR mice, ↓APO in SR mice) |
| 5-HT2 | |||||
| Rats | |||||
| Vanover et al. 2006 | SD, M | DOI | ACP-103 5-HT2A inverse agonist | ↓DOI | |
| Siuciak et al. 2007 | WI, M | APO | CP-809,101 5-HT2C agonist | ↓APO | HAL (↓APO) |
| Ouagazzal et al. 2001a, b | SD, M | LSD (hallucinogen) | M100907 | ↓LSD | SB 242084 (5-HT2C antagonist), SDZ SER 082 (5-HT2b/2C antagonist), RO 04-6790 (5-HT6 antagonist), HAL (all ØLSD) |
| Mice | |||||
| Barr et al. 2004 | DAT-KO, M | Basal PPI | M100907 5-HT2A antagonist | ↑PPI | |
| Marquis et al. 2007 | DBA/2N, M | Basal PPI, DIZ, or DOI | WAY 163909 5-HT2C agonist | ↑PPI, ↓DIZ, ↓DOI, ↑AMP | |
| 5-HT6 | |||||
| Pouzet et al. 2002a | Rats, WI, M | AMP or PCP | SB-271046 5-HT6 antagonist | ↓AMP, ØPCP | CLO (↓AMP, ØPCP) |
| 5-HT7 | |||||
| Rats (+ mice) | |||||
| Pouzet et al. 2002b | WI, M | AMP or PCP | SB-258741 5-HT7 antagonist | ØAMP, ↓PCP | RIS (↓AMP, ↓PCP) |
| Rats + mice | |||||
| Semenova et al. 2008 | 5-HT7KO, M; Mice, C57, M; Rats, SD, M | APO, AMP, or PCP | SB-269970 5-HT7 antagonist | No SB-269970: ↓PCP in KO vs. WT mice; ↓APO and ↓AMP in both KO and WT. SB-269970: ØPCP in C57 mice and SD rats | |
| IV. Noradrenergic mechanisms | |||||
| Rats | |||||
| Ballmaier et al. 2001a | SD, M | Coapplication of Idazoxan α2 antagonist) + RAC (D2/D3 antagonist) | ↓APO, but no additional impact of idazoxan over RAC | ||
| Sallinen et al. 2007 | SD, M; WI, M | PCP | JP-1302 α2C antagonist | ↓PCP in both strains | Atipamezole (α2 antagonist, ØPCP) |
| V. Cholinergic mechanisms | |||||
| A. Nicotinic agonists | |||||
| Rats | |||||
| Cilia et al. 2005 | LH, M | IR | Compound A α7-agonist | ↑PPI | |
| Suemaru et al. 2004 | WI, M | APO or PCP | Nicotine | ØPPI, ↓APO (eliminated by mecamylamine, but not hexamethonium), ØPCP | Methyllycaconitine (α7 antagonist), dihydro-beta-erthoidine (α4β2 antagonist), both ØPPI, HAL (↓APO, ØPCP), CLO (↓PCP) |
| Mice | |||||
| Andreasen et al. 2006 | BALB, M; NMRI, M | PCP | Nicotine | ↓PCP in BALB mice, ØPCP in NMRI mice | CLO (similar pattern than nicotine), RIS (ØPCP in either strain) |
| Spielewoy and Markou 2004 | DBA, C3H, C57BL or 129, all M | PCP | Nicotine | ØPPI in all strains, ↓PCP in DBA and C3H (trend), ØPCP in C57 and 129 mice | |
| B. Muscarinic agonists | |||||
| Rats | |||||
| Jones et al. 2005 | SD, M | APO or SCO | Xanomeline M1/M4 muscarinic agonist | ↓APO, ↓SCO | BuTAC (M2/4-preferring agonist, ↓APO), oxotremorine, RS86, pilocarpine, milameline,sabcomeline (all muscarinic agonists, all ↓APO), HAL (↓APO, ↓SCO), OLA (↓APO), SCH23390 (ØCLO) |
| Stanhope et al. 2001 | SD, M | APO | Xanomeline | ØPPI, ↓APO | HAL (ØPPI, ↓APO), pilocarpine (↓PPI, ØAPO), MUS (↓PPI), SCO (↓PPI), methyoscopolamine (ØPPI) |
| C. AChE-inhibitors | |||||
| Rats | |||||
| Hohnadel et al. 2007 | WI, M | APO, DIZ, or SCO | Donepezil | ↓APO, ØDIZ, ↓SCO | Galantamine (↓APO, ↓DIZ, ↓SCO) |
| Ballmaier et al. 2002 | SD, M | Immunolesioning of cholinergic neurons in nucleus basalis | Rivastigmine | ↑PPI | |
| VI. Histaminergic mechanisms | |||||
| Roegge et al. 2007 | Rats, SD, M | DIZ | Pyrilamine H1 antagonist | ↓DIZ | |
| Mice | |||||
| Fox et al. 2005 | DBA, M | Basal PPI | ABT-239 H3 receptor antagonist | ↑PPI | RIS (↑PPI) |
| Ligneau et al. 2007 | Swiss, M | APO | BF2.649 H3 receptor antagonist/inverse agonist | ↓APO | |
| Browman et al. 2004 | DBA, M; C57, M | Basal PPIa,(b) | Thioperamide H3 receptor antagonist/inverse agonist | ↑PPI in DBA, ØPPI in C57 mice | Ciproxifan (↑PPI in DBA and C57 (trend), RIS (↑PPI in both strains) |
| VII. Cannabinoid mechanisms | |||||
| A. CB1 antagonists | |||||
| Rats | |||||
| Ballmaier et al. 2007 | SD, M | PCP, DIZ, APO | AM251 | ↓PCP, ↓DIZ, ↓APO | Rimonabant (↓APO, ↓DIZ, ↓PCP), CLO (↓PCP) |
| Mice | |||||
| Malone et al. 2004 | Swiss, M | APO | SR 141716 | ↓APO | |
| Nagai et al. 2006 | ddY, M | Δ9-THC | SR 141716 | ↓THC | HAL (↓THC), RIS (↓THC) |
| B. Endocannabinoid transport inhibitor | |||||
| Bortolato et al. 2006 | Rats, SD, M | Basal PPI | AM404 | ØPPI | Win55,212 (ØPPI), APO (↓PPI), DIZ (↓PPI) |
| C. Cannabidiol | |||||
| Long et al. 2006 | Mice, Swiss, M | DIZ | Non-psychoactive constituent of the Cannabis sativa plant, agonist of the TRP receptor VAN1, inhibitor of anandamide-uptake | ↓DIZ, ØDIZ (if pretreated with TRP agonist capsazepine) | CLO (↓DIZ) |
| VIII. Neuropeptide mechanisms | |||||
| A. Neurotensin agonists | |||||
| Rats | |||||
| Shilling et al. 2004 | SD, M | DOI or CIR | NT69L | ↓DOI, ↓CIR | PD149163 (NT antagonist, ↓CIR) |
| Shilling et al. 2003 | SD, M | AMP or DIZ | NT69L | ↑PPI, ↓AMP, ↓DIZ | |
| B. Opioids | |||||
| Bortolato et al. 2005 | Rats, SD, M | U50488 kappa-opioid agonist | Nor-BNI Kappa-opioid antagonist | ↓U50488, but ØAPO, ØDIZ | CLO (↓U50488), HAL (ØU50488) |
| Ukai and Okuda 2003 | Mice, ddY, M | APO | Endomorphin-1 Endogenous mu opioid agonist (ICV-infusion) | ØPPI, ↓APO (antagonized by the mu1 antagonist naloxonazine, but not by ICV-infusion of the mu antagonist β-funaltrexamine) | Naloxonazine (ØAPO) |
| C. CCK | |||||
| Shilling and Feifel 2002 | Rats, SD, M | AMP, DIZ or DOI | SR146131 CCKA antagonist | ØAMP, ↓DIZ, ↓DOI | |
| IX. Adenosine | |||||
| Wardas et al. 2003 | Rats, WI, M | PCP | CGS 21680 Adenosine A2 agonist | ↓PCP | |
| X. GABA agonists | |||||
| Bortolato et al. 2004 | Rats, SD, M | PPI, APO or DIZ | Baclofen | ØPPI, ØAPO,↓DIZ, (prevented by SCH50911) | |
| Bortolato et al. 2007 | Juvenile mice: DBA, M; C57, M | Basal PPI in DBA (and C57) | Baclofen | ↑PPI (prevented by SCH50911) in DBA, ØPPI in C57 mice | CLO (↑PPI in DBA, ØPPI in C57), HAL (ØPPI in both strains) |
| XI. Anticonvulsants/mood stabilizers | |||||
| Rats | |||||
| Frau et al. 2007 | SD, M | Basal PPI, APO or DIZ | Topiramate GABAA agonist, Voltage-gated Na-channel, AMPA/Kainate blocker | ↑PPI, ↓APO, potentiation of HAL (↓APO) and CLO (↓APO) effects, ØDIZ, attenuation of CLO (↓DIZ) | HAL (↓APO), CLO (↓APO, ↓DIZ) |
| Mice | |||||
| Brody et al. 2003a, b | 129, M; C57, M | AMP, KET | Lamotrigine Na-channel blocker | ↓KET in 129 mice, ØAMP in both strains, ↑PPI in KET and ctrl mice (C57) | |
| Mice | |||||
| Ong et al. 2005 | 129, M; C57, M | AMP or KET | Lithium | ØPPI1,2, ↓AMP1,2, ØKET1 | Carbamazepine (ØPPI, ↓KET, ØAMP), Phenytoin (↑↓PPI (dose-dependent), ØKET, ØAMP), valproate (ØPPI, ØKET, ØAMP); all in 129 mice |
| Umeda et al. 2006 | ddY, M | APO or DIZ | Valproate | ØPPI, ↓APO, ØDIZ | Lithium (ØPPI, ↓APO, DIZ), carbamazepine (ØPPI, ↓APO, ↑DIZ) |
| XII. Hormones | |||||
| Rats | |||||
| Czyrak et al. 2003 | WI, M | 8-OHDPAT | Corticosterone hormone | ↓8-OHDPAT (repeated CORT), Ø8-OHDPAT (acute CORT) | Way100,135 (↓8OHDPAT) |
| Gogos and Van den Buuse 2004 | SD, F, OVX | 8-OHDPAT | Estrogen (implant, 2 weeks) sex hormone | ↓8-OHDPAT, ↓8-OHDPAT (in cotreatment with progesterone) | Progesterone (implant, Ø8-OHDPAT) |
| Myers et al. 2005 | SD, M | PCP | Secretin peptide functional in gut and brain | ↓PCP | |
| XIII. Second-messenger inhibitors | |||||
| A. Nitric oxide synthase inhibitors | |||||
| Rats | |||||
| Salum et al. 2006 | WI, M | AMP, APO, BRO, QUI | l-NOARG (two injections) | ↓AMP, but ØAPO, ØBRO, (↓QUI (trend)) | SKF38393 (ØPPI-independent of pretreatment with l-NOARG) |
| Mice | |||||
| Klamer et al. 2005b | Mice deficient of neuronal NOS vs. B6129SF2 (ctrl), M | PCP or DIZ | l-NAME | ↓PCP | |
| Klamer et al. 2004b | NMRI, M | PCP | N-propyl-arginine | ↓PCP | |
| B. Guanylate cyclase + nos inhibitors | |||||
| Klamer et al. 2004a | Mice, NMRI, M | PCP | Methylene blue | ↓PCP | |
| C. PDE-inhibitors | |||||
| Kanes et al. 2007 | Mice, C57, M | PPI or AMP | Rolipram Phosphodiesterase (PDE)4 inhibitor | ↑PPI, ↓AMP | HAL (↑PPI) |
| XIV. Miscellaneous | |||||
| Rats | |||||
| Wang et al. 2003a | SD, | Perinatal PCP (3 applications) | M40403 Superoxide Dismustase Mimetic | ↓PCP (by both short and long-term treatment with M40403) | |
| Mice | |||||
| Palsson et al. 2007 | NMRI, M | PCP | l-Lysine (subchronic; l-artinine transport inhibitor) | ↓PCP | |
| Zhang et al. 2007a | Std:ddy, M | DIZ | Minocycline Second generation antibiotic | ↓DIZ | |
AChE acetylcholinesterase, AMP amphetamine, APO apomorphine, ARI aripipazole, BN Brown Norway, BRO bromocriptine, CB cannabinoid receptor, β-CD (2-hdroxypropyl)-beta-cyclodextrin, CIR cirazoline, CLO clozapine, CORT corticosterone, DAAO d-aminoacid oxidase, DA dopamine, DAT dopamine transporter, DIZ dizocilpine, F female, GLU glutamate, GLY glycine, GLYT glycine transporter, HAL haloperidol, ICV intracerebroventricular, IR isolation rearing, KET ketamine, KYNA kynuric acid, LE Long Evans, LH Lister hooded, LSD lysergic acid dyethylamide, M male, MPEP 2-methyl-t-(phenylethnyl)-pyridine, MUS muscarine, NE norepinephrine, NET norepinephrine transporter, nHPC neonatal hippocampus, NOS nitric oxyde synthase, NT neurotensin, OLA olanzapine, OVX ovariectomized, ppm parts per million, PND postnatal day, PP prepulse, QUE quetiapine, QUI quinpirole, RAC raclopride, RIS risperidone, Rx treatment, SCO scopolamine, SD Sprague Dawley, SR social rearing, THC tetrahydracannabinol, TRP transient receptor potential channel, V ventral, VAN vanilloid, WI Wistar, WKYs Wistar Kyoto, WT wild type, ZIP ziprasidone, ↓XYZ reduction of effect XYZ, ↑XYZ enhancement of effect XYZ, ØXYZ no change of effect XYZ
This quantitative physiological abnormality in schizophrenia patients, conceptually linked to an intuitive clinical construct and neurochemical, anatomical, developmental, and genetic substrates, has provided a powerful focus for scientific developments. With the rapid expansion and broad application of variations of PPI measures, new expectations for its use to inform us about the biology of schizophrenia have at times outpaced critical thinking and falsifiable hypotheses about the relative strengths vs limitations of these complex studies. Here, we hope to enumerate some of these expectations and the future promises and potential limitations of PPI studies.
Human studies: What can our field realistically expect to learn about schizophrenia based on studies of PPI in humans?
Diagnosis
As an isolated measure, PPI is not a “diagnostic instrument”. There is substantial variability and significant overlap in PPI distributions among normal and disordered populations. In addition, there are many different disorders in which affected individuals are characterized by reduced PPI, on average, compared to a normal comparison population (cf. Braff et al. 2001b). The reason for the “non-pathognomonic” nature of PPI deficits is simple: the amount of PPI exhibited by any organism at any given moment reflects activity at many different levels of integrated cortico–striato–pallido–thalamic (CSPT) circuitry and its output via the pontine tegmentum. Low levels of PPI can result from normal variations at several levels of this circuitry; alternatively, disease processes can impact different levels of this circuit, with synergistic effects on pontine activity that mediates PPI. Conceivably, disease processes might even impact this circuitry in such a way as to bias it towards elevated levels of PPI, and compensatory or allostatic changes within feedback or downstream elements of the circuitry might offset the effects of otherwise PPI-disruptive disease processes. Thus, absolute levels of PPI—either low or high—are neither diagnostically nor neurophysiologically specific.
A corollary of this fact—that PPI is not “diagnostic”—is that no simple qualitative value of “normal” or “deficient” can accurately be applied to any particular level of PPI, particularly among clinically normal individuals. It is common in the literature (including our own reports) to describe relatively low levels of PPI as “deficient”, “impaired”, or “poor”. In fact, we know of no clear adaptive or functional advantage of higher vs. lower levels of PPI among clinically normal individuals. Perhaps, this idea is most easily conveyed in the comparison between clinically normal men and women: on average, under specific stimulus conditions (e.g., 20 ms white noise prepulses, 10 dB over a 70-dB(A) white nose background, 100 ms before a 115-dB(A) 40 white noise pulse), men exhibit more PPI than do women (Swerdlow et al. 1993b, 2006f; Kumari et al. 2004; Aasen et al. 2005). Furthermore, there is some evidence that among normal women, PPI shifts across the menstrual cycle (Swerdlow et al. 1997; Jovanovic et al. 2004). Clearly, there is no basis for describing PPI in women vs. men as “deficient”, nor for describing luteal- vs. follicular-phase PPI as “impaired”. Similarly, drugs that increase PPI in normals cannot be accurately claimed to “improve” PPI.
At a more basic level, at any given moment in time, individuals are not characterized by a single “PPI” value, in the same manner in which they might be characterized by other quantitative traits such as height, Q–T interval, or fasting glucose level. One of PPI’s strengths as an experimental measure is its exquisite sensitivity to stimulus parameters and test conditions [as described for the startle reflex by Davis 1984]. The inhibition generated by prepulses under different stimulus conditions likely reflects different underlying physiological substrates. Thus, under a variety of test/stimulus conditions, the same clinical population might conceivably exhibit PPI levels that are reduced, equal to, or elevated, compared to normal comparison subjects. An instructive example from preclinical studies of PPI is found in the report that inbred Brown Norway (BN) rats exhibit “deficient” PPI compared to outbred Sprague Dawley (SD) rats, based on measurements with 100 ms prepulse intervals (Palmer et al. 2000). Subsequent studies reproduced this finding, but also demonstrated that at shorter prepulse intervals, the opposite relationship existed: BN rats exhibited significantly more PPI compared to SD rats (Swerdlow et al. 2006a, 2008). Thus, depending on the stimulus parameters, populations can exhibit either relatively reduced or excessive PPI.
PPI is also highly sensitive to state variables and influences, such as medications (Table 1), cigarette smoking (Table 1), fatigue (van der Linden et al. 2006), stress (Grillon et al. 1998), and hormonal status (Swerdlow et al. 1997; Jovanovic et al. 2004). While some of these variables and influences can be controlled under experimental conditions, the notion of using such a sensitive measure in isolation as a diagnostic tool is not realistic. This being said, one potentially valuable strategy in the characterization of clinical populations is the use of PPI in combination with multiple other measures of forebrain inhibitory function, such as P50 event-related potential (ERP) suppression (“P50 gating”; Adler et al. 1982) and antisaccade deficits (Radant et al. 2007), to identify multiple measures and patterns of normal vs. deficient function (Cadenhead et al. 2002; Braff et al. 2008; Sugar et al. 2007). PPI and P50 gating are both deficient but correlate weakly, if at all, in schizophrenia patients (Braff et al. 2007b); similarly, PPI and antisaccade performance are both deficient but do not correlate significantly in schizophrenia patients (Kumari et al. 2005b). Thus, these measures apparently assess forebrain inhibitory processes that are dissociable and nonredundant. More importantly, there are patients who exhibit normal levels of some but not other gating measures (and presumably normal function within brain circuitry regulating some but not other measures), and subpopulations of patients who exhibit different profiles in these deficits (Kumari et al. 2005b; Swerdlow et al. 2006f; Braff et al. 2007b). These subpopulations may reflect different patterns of brain dysfunction and conceivably distinct genetic substrates and treatment sensitivities (Braff et al. 2007a).
Symptoms, course, and outcome
Can we predict the clinical course or even clinical features of schizophrenia based on PPI levels? There is no compelling data to suggest that among schizophrenia patients, levels of PPI predict clinical course, nor are there consistent robust relationships between lower levels of PPI and higher levels of specific symptoms of schizophrenia, or cumulative positive or negative symptoms scores (Table 1). Certainly, there is much interest in determining whether, with repeated or longitudinal measures, a change in PPI predicts or accompanies clinical deterioration or improvement, including the prediction of illness onset in prodromal subjects (Cadenhead 2002; Addington et al. 2007; Cannon et al. 2008). Very few studies have collected longitudinal measures of PPI in schizophrenia populations with adequate sample size and duration to be informative, although some are in progress. One might predict a relationship between PPI and psychosis in extreme conditions, such as the shift from euthymic to manic bipolar disorder, but even in this case, studies have been limited to cross-sectional comparisons, and results across studies have not been consistent (Perry et al. 2001; Rich et al. 2005; Barrett et al. 2005; Carroll et al. 2007). Duncan et al. (2006a, b) did detect an association between lower levels of PPI, and greater levels of psychotic symptoms and psychological discomfort among unmedicated schizophrenia patients.
Interestingly, while robust relationships between PPI and the most common clinical indices of schizophrenia have been hard to detect, reports have identified significant correlations between PPI and a number of relatively complex clinical measures, ranging from quantitative Rorschach ink blot indices of thought disturbance (Perry and Braff 1994) to scales of distractibility and attention (Karper et al. 1996). One report (Swerdlow et al. 2006f) identified a significant positive correlation between PPI and global functioning levels (GAF score) in schizophrenia patients, but this relationship was evident only among male patients, and the correlation—while highly significant (p<0.005)—accounted for a relatively modest amount of the total PPI variance. In addition, PPI levels were associated with levels of independent living, also perhaps reflecting its relationship to global functioning. As a result, more sophisticated and sensitive analyses of PPI, related gating measures, and function in schizophrenia patients are being pursued (Light et al. 2007a; Braff et al. 2007a). Studies have detected modest but statistically significant relationships between PPI and measures of executive function in some patient groups [e.g., children with 22q11DS (Sobin et al. 2005a, b)]. A preliminary qualitative article by Butler et al. (1991) noted a nonsignificant trend toward greater tactile (but not acoustic) PPI among six (predominantly male) patients with schizophrenia and low levels of Wisconsin Card Sorting Test perseverative responses than among nine (predominantly female) patients distinguished by high levels of Wisconsin Card Sorting Test (WCST) perseverative responses. Kumari et al. (2007a) recently reported a significant (p<0.03) correlation between tactile PPI and WCST perseverative responses in male schizophrenia patients. Significant positive relationships between acoustic PPI and working memory as well as other formal indices of neurocognitive function have been detected among clinically normal individuals (Bitsios et al. 2006; Light et al. 2007b, 2008; Csomor et al. 2008), although no such relationships have been reported for schizophrenia patients.
The relative insensitivity of PPI to clinical state speaks of the importance of trait features of this measure, which may reflect more “hard-wired” anatomical and genetic determinants. The fact that some relationships can be detected between PPI and relatively global measures of function in schizophrenia patients, but not between PPI and clinical state per se, is consistent with the hypothesis that the causal link between genes and functional outcome in schizophrenia reflects the impact of forebrain circuits that regulate basic gating mechanisms, more than those that control the expression of specific symptom states (Light et al. 2004; Braff and Light 2004; Light and Braff 2005). Thus, while diagnosis in schizophrenia will remain symptom-based for the foreseeable future, it could be argued that studies of the biology of schizophrenia and its relationship to functional outcome may be best advanced through quantitative measures of forebrain inhibitory function such as PPI.
Treatment
As PPI deficits in schizophrenia reflect dysfunction in forebrain circuitry and are linked to both cognitive and functional deficits in schizophrenia patients, can PPI or its potentiation by drugs in patients be used to predict individualized treatment for this disorder? Certainly, in terms of preclinical predictive models, PPI has been quite powerful, as discussed below. In schizophrenia patients, cross-sectional data and some longitudinal findings demonstrate that antipsychotic treatment is associated with elevated (i.e., “normalized”) PPI and that this association is most robust with atypical antipsychotics as a class, compared to first generation antipsychotics (Table 1). Of course, interpreting medication effects in most of these reports is difficult because patients are uniformly being treated with complex multidrug regimens across a range of doses, and medication compliance is known to be poor among schizophrenia outpatients (Lieberman et al. 2005). A recent controlled study with a multidrug cross-over design detected PPI-increasing effects of olanzapine (but not risperidone or haloperidol) in chronically ill schizophrenia patients (Wynn et al. 2007). Findings of PPI-increasing effects of both quetiapine and clozapine in clinically normal, “low-gating” subjects suggests that the PPI-increasing effects of these drugs in schizophrenia patients may not reflect disorder-specific processes (Swerdlow et al. 2006a; Vollenweider et al. 2006). We do not know if the PPI-enhancing effects of these drugs, and conceivably some of their clinical benefit, may reflect their ability to optimize function within spared (intact) gating mechanisms, rather than their ability to correct or normalize activity within dysfunctional mechanisms.
Still, it is reasonable to ask whether the ability of drugs to normalize PPI in patients, or to increase PPI in “low-gating” normals, might reflect their impact on brain processes and resulting cognitive abilities that ultimately would have clinical utility and perhaps cognitive-enhancing effects in schizophrenia. While clinically effective antipsychotics (particularly atypical antipsychotics) are associated with increased PPI in patients and low-gating normals (Table 1), PPI is also increased in non-patients by ketamine and methylenedioxymethamphetamine (MDMA; discussed below; Duncan et al. 2001; Abel et al. 2003; Vollenweider et al. 1999), neither of which would be on anyone’s list of likely antipsychotic agents. Nicotine is associated with increased PPI in schizophrenia patients (Kumari et al. 2001; Swerdlow et al. 2006f), but despite the hypothesis that smoking reflects a form of “self-medication” in schizophrenia patients, there is no clear evidence for either antipsychotic or cognitive-enhancing effects of nicotine in these patients. While there is an active quest by many groups to develop cognitively enhancing nicotinic receptor-specific agonists, based on the putative relationship between the alpha-7 nicotinic receptor subtype and schizophrenia (Freedman et al. 1997), there is presently no evidence that such compounds either increase PPI or enhance cognition in patients. Thus, screening compounds as effective antipsychotics based on their PPI-enhancing effects in clinical or special populations is likely to yield both true and false positives. At this point, there is an inferential, but not empirical, basis for using PPI enhancement as a basis for predicting the ability of a compound to enhance cognition and real-world daily functioning in schizophrenia. Clearly, this is an area of active investigation, and such empirical evidence might emerge based on these efforts.
A reliable, robust quantitative phenotype
While the realistic expectations for PPI as a clinically useful biomarker may be somewhat limited, it is very realistic to expect that PPI will continue to be a valuable tool for investigating brain functions relevant to several neuropsychiatric disorders, including schizophrenia. The many strengths of PPI as an experimental measure have been reviewed elsewhere (Braff et al. 2001b), and none of the realistic limitations described above detract from its attributes as an objective, quantifiable, reliable, robust, neurochemically and parametrically sensitive cross-species measure of a neurobiologically important process. Nonetheless, even in its use as an investigative experimental tool in humans, there should be a realistic assessment of what we can and cannot expect from PPI.
Two types of studies speak strongly to the general reliability of this quantitative phenotype. First, test–retest reliability has been established for PPI in normal comparison subjects (NCS), across days (Abel et al. 1998; Swerdlow et al. 2001c; Flaten 2002), weeks, and months (Cadenhead et al. 1999; Ludewig et al. 2002). More recently, 1-year retest data collected in 68 schizophrenia patients yielded intra-class correlations of 0.75 (30 ms)–0.89 (120 ms; Light et al. 2007a), suggesting a very high stability of this phenotype in patients. Second, a multisite study of PPI in NCS was conducted, using carefully standardized equipment, test methods, and inclusion/exclusion criteria. No significant differences in PPI were detected across seven geographically dispersed test sites, despite some modest methodological drift that was detected via rigorous quality assurance efforts (Swerdlow et al. 2007). Thus, within individuals, and across test samples, PPI appears to be a reliable phenotype.
While PPI is a reliable phenotype, at least among NCS, it is not reasonable to expect that every schizophrenia patient will exhibit a “deficient-PPI” phenotype. In fact, as noted above, there is no way to test this possibility because there is no absolute value that defines “deficient” PPI. Under commonly used test conditions, there is substantial overlap in the distribution of PPI values, between schizophrenia patients and community comparison subjects (cf. Braff et al. 2001b). Clearly, there are schizophrenia patients who have higher levels of PPI compared to many NCS. The overlapping group distributions with this measure likely reflect the many influences on PPI, other than schizophrenia-related pathology, such as sex, hormonal status, smoking, withdrawal from caffeine or nicotine, fatigue, and medications. There are also normal interindividual differences in activity within brain circuitry (e.g., in the pallidum, pons, or cerebellum) that regulates PPI, but is not primarily involved in schizophrenia. With typical testing parameters, NCS vs. unmedicated patients or patients receiving only typical antipsychotics, group separation in mean percent PPI might be reasonably expected to reach 1 SD (e.g., Kumari et al. 1999; Ludewig et al. 2003; Swerdlow et al. 2006f), which corresponds to 55% non-overlap. However, when patients taking atypical antipsychotics are included, group separation drops dramatically, to about 0.3 SD (e.g. Swerdlow et al. 2006f)—or 21% nonoverlap. This latter fact is particularly important, given that upwards of 90% of schizophrenia patients in most current open-enrollment studies report taking atypical antipsychotic medications [although true compliance is likely lower (Dolder et al. 2002; Lacro et al. 2002)].
In addition to medication status, studies have reported many other variables in patient selection that influence group separation in comparisons of schizophrenia patients vs. NCS. One issue that may ultimately impact the utility of PPI as a quantitative phenotype is its potential sensitivity to ascertainment bias. As noted above, PPI correlates positively with global function in schizophrenia patients. Thus, on average, studies of lower functioning patients will detect greater separation vs. NCS, and those of higher functioning patients will detect less group separation. For this reason, investigators are considering the impact of study designs that select for higher-vs. lower-functioning schizophrenia patients, such as those that require a proband within an intact family structure (and who thus may be relatively higher functioning) vs. those utilizing patients without intact families, who are often homeless or medically indigent (Calkins et al. 2007).
Perhaps equally important as the selection of patients is the selection of NCS. Comparison samples differ substantially across studies and can range from generally healthy, young college students, to “professional controls”, who are often low-functioning and unemployed, beyond their activities as test subjects in biomedical research. The latter group is more likely to have histories of disorders that are associated with reduced PPI, such as anxiety disorders (OCD, panic disorder or post-traumatic stress disorder) or “cluster A” personality disorders; they may also be more likely to carry vulnerability genes for neuropsychiatric disorders, take psychotropic medications that influence PPI, and have histories of substance use or brain trauma that might impact PPI-regulatory brain circuitry. Much has been written about the considerations in selecting a “matched”, “representative”, “normal” or “supernormal” comparison group in biomedical research (e.g., Roy et al. 1997; Calkins et al. 2004), and without belaboring this point, these same considerations apply to studies of PPI and may greatly impact group separation in comparisons of control vs. schizophrenia populations.
As reviewed in Braff et al. (2001b) and elsewhere, the amount of separation between schizophrenia and NCS populations in PPI is highly dependent on testing conditions, and specifically, on stimulus parameters. Thus, if all else is equal, schizophrenia-linked PPI deficits are most pronounced under conditions in which prepulse salience, often based on its intensity over background, is within a “dynamic range”: not too high, but not too low. For example, most studies find this “sweet spot” of maximal schizophrenia vs. NCS separation using discrete white noise prepulses 8–16 dB over a 70-dB(A) background, with about 60 ms prepulse intervals [or stimulus onset asynchronies (SOAs; Table 1)]. Some studies failing to detect PPI deficits in schizophrenia samples have used prepulses in the absence of a background white noise, effectively creating very large prepulse intensities of 25–40 dB(A; Hazlett et al. 2003, Wynn et al. 2004, 2005). In addition to prepulse intensity relative to background, prepulse frequency (e.g., tone vs. white noise), duration (discrete vs. continuous) and other variables (including the use of binaural vs. mono-aural stimuli) may contribute to maximizing the group separation in PPI between schizophrenia and NCS populations (Braff et al. 2001a; Hsieh et al. 2006; Kumari et al. 2005b, 2007b).
As noted above, the temporal “sweet spot” for detecting automatic (uninstructed) PPI deficits in schizophrenia patients appears to occur with prepulse intervals between 30 and 240 ms, depending somewhat on other stimulus characteristics. The temporal range around 60 ms appears to be most sensitive in several studies (Braff et al. 1978, 1992, 2005; Weike et al. 2000; Leumann et al. 2002; Swerdlow et al. 2006f) and may be the range in which PPI deficits are most resistant to normalization by antipsychotic medications. Interestingly, this interval sits at the juncture between preconscious and conscious information processing, based on perceptual detection thresholds (Libet et al. 1979; Kanabus et al. 2002). The possibility that PPI in this temporal range may be most deficient in schizophrenia suggests that automatic inhibitory mechanisms may be most “porous” at a critical barrier between preconscious processing and conscious awareness. While clearly a point for more systematic analysis, such a notion suggests a biological mechanism that is syntonic with psychological models for the intrusion of unedited, preconscious content into conscious awareness in this disorder (Libet et al. 1979; Gray 1995; Swerdlow 1996; Grobstein 2005).
A useful tool for probing the neurobiology and genetics of gating deficits in schizophrenia
Perhaps the most realistic expectation is that PPI is and will remain a useful tool for studying the neurobiology of information processing abnormalities in schizophrenia. While the PPI deficit “signal” in genetic studies of schizophrenia has been blunted by the widespread use of atypical antipsychotics, investigators are increasingly well informed about the many other factors affecting the measurement of PPI and the detection of schizophrenia-associated deficits, and in this way are better positioned to study the basis for these deficits at the levels of their neurobiological and genetic substrates. These studies will be aided by special populations, including “low-gating” normals (Swerdlow et al. 2006a; Vollenweider et al. 2006) and asymptomatic relatives of schizophrenia probands (Kumari et al. 2005b), and by patients with related disorders, such as 22q11 deletion syndrome and unmedicated “prodromal” individuals (Sobin et al. 2005a, b).
As a relatively robust and reliable quantitative phenotype, PPI will be used to map genes associated with deficient sensorimotor gating in schizophrenia probands and families (Swerdlow et al. 2007; Greenwood et al. 2007). The strength of this “endophenotype” approach to understanding disease genetics has been described by many, including Gottesman and Gould (2003), Gould and Gottesman (2006), and Braff et al. (2007a), and largely reflects the fact that the quantitative laboratory measure (in this case, PPI), is closer to the underlying biology (i.e., aberrant neural circuits and their regulation by disease genes), compared to the more variable clinical phenotype (Braff et al. 2007a). There are a small but growing number of examples in which this strategy has proven successful, in identifying genes that confer risk for colon cancer (Leppert et al. 1990) and Type II diabetes (Scott et al. 2007). Whether this strategy can succeed in identifying vulnerability genes for more complex neuropsychiatric disorders is a question at the core of several large ongoing investigative efforts.
Gains will likely be made through the combined use of PPI with sophisticated neurocognitive, neuroimaging, and genetic/genomic tools in schizophrenia and normal populations. It is realistic to expect that these various applications will converge in a top down or bottom up fashion, i.e., to link: (1) genes with (2) brain substrates that cause (3) gating deficits responsible for (4) neurocognitive disturbances and (5) the resulting daily functional impairment in schizophrenia. Based on the genes and brain substrates identified in these studies, one might reasonably expect that novel treatments will be identified, perhaps acting on intracellular G-protein-coupled signal transduction mechanisms that have already been implicated in the regulation of PPI (van den Buuse et al. 2005a; Kelly et al. 2007; Swerdlow et al. 2006d; Culm et al. 2004; Svenningsson et al. 2003), and which may also be abnormal in some schizophrenia patients (cf. Catapano and Manji 2007). There are also mature lines of research suggesting that novel treatments may target neuropeptides, such as neurotensin (Kinkead et al. 2005; Feifel et al. 2004), that potently regulate PPI and its dopaminergic control, or may target specific dopamine receptors subtypes that regulate PPI via relatively localized effects within mesolimbic and limbic–fronto–striatal circuits (e.g., Zhang et al. 2006). At some stage, it is reasonable to expect that the development of any one of these or other novel treatments might be guided by their effects on PPI in control or clinical populations.
A surrogate measure for neural processes with wide-reaching psychological implications
The frontal, limbic, and mesolimbic circuitry that regulates PPI also regulates many higher-order psychological processes. Thus, PPI can be viewed as a simple surrogate “readout” of activity in this circuitry—an experimentally generated signal from the forebrain, detected through efferents descending through a “pontine portal”. Alternatively, PPI can be viewed as a measure of a fundamental psychological process—sensorimotor gating—with broad-reaching implications for the structure of complex behavior and thoughts. In truth, both views are at least partly accurate, under specific uses of the PPI paradigm.
“Gating” can be a very specific process when operationalized in the laboratory, but is less precisely defined when used as a psychological construct. How broadly can we extrapolate from the laboratory measure of one type of gating—sensorimotor gating—to other forms of automatic inhibition of sensory, cognitive, or motor information? There is credible evidence that PPI correlates significantly with a form of perceptual “gating”, measured by the degree to which the prepulse reduces the perceived intensity of the startling stimulus (Peak 1939; Swerdlow et al. 2005b). On the other hand, PPI does not correlate strongly with the most structurally similar form of “gating”—sensory gating—measured by suppression of the P50 auditory event-related potential (ERP; Light et al. 2006; Hong et al. 2007). Nor does PPI in normal humans correlate strongly with other measures thought to assess inhibitory processes that contribute to forms of “cognitive gating”, such as latent inhibition (Murphy et al. 2001; Leumann et al. 2002; Peleg-Raibstein et al. 2006a, b) or visuospatial or semantic priming (Swerdlow et al. 1995b). Certainly, there is little evidence that PPI assesses processes that are strong determinants of normal personality structure and dimensions (Swerdlow et al. 2003d). At the least, it is important to recognize that the construct of “gating” is applied to many different processes and that it is reasonable to expect PPI to be informative about some, but not all or even most of these processes.
Summary: human studies
Human studies of PPI will continue to provide one important level of information within a top down or bottom up understanding of the biology of schizophrenia. PPI offers great promise as a quantitative phenotype for genetic studies and will be used in combination with other measures to connect an aberrant physiological signal (impaired startle inhibition) with its underlying neural substrates (via neuroimaging studies) and with its consequences in terms of cognitive deficits (via neurocognitive measures) and real-life impairment (via functional measures). It is realistic to expect that as we gain a better understanding of its modulating variables and optimal experimental methods, PPI in humans will continue its evolution, started in 1978 (Braff et al. 1978) from an isolated laboratory-based psychophysiological phenomenon, into a productive clinical research tool for understanding psychopathology. As we learn more about PPI, our scientific approaches to its use will continue to become more sophisticated, and we will be better positioned to take full advantage of what it can tell us about normal and abnormal brain functions.
Animal studies: What can our field realistically expect to learn about schizophrenia based on studies of PPI in laboratory animals?
Etiology
Two general applications of animal studies of PPI will be considered here: (1) the use of PPI to evaluate models or model organisms relevant to the etiology of schizophrenia; and (2) the use of PPI to “map” the neural substrates of deficient PPI in schizophrenia.
Model organisms, created via genetic, developmental, surgical, pharmacological, or immune manipulations, have been a mainstay of studies of the etiology, pathophysiology, and treatment of schizophrenia. Of course, schizophrenia—as defined clinically—is a uniquely human disorder (least we ascribe to rats the ability to have “two or more voices conversing with one another or voices maintaining a running commentary on the [rat’s] thoughts or behavior,” or the ability to conceptualize that “alien thoughts have been put into his or her mind…”, or to have homologous complex social cognitive deficits; APA 2000). However, investigators can apply schizophrenia-linked constructs to these models and test whether the resulting animal reproduces laboratory-based phenotypes exhibited by schizophrenia patients. The degree to which these phenotypes are reproduced in the model organism provides a level of validity to the construct, even if it is specific to the laboratory-based phenotype, rather than the broader clinical disorder.
For example, given a particular schizophrenia candidate gene “X”, it is reasonable to ask whether manipulations of gene “X” produce an animal that exhibits reduced levels of PPI compared to a wild-type animal. If so, then the gene “X” mutant would be a valid model for PPI deficits in schizophrenia. Such an approach has been taken with many different animal models (Table 3). There are obvious limitations to the specificity and sensitivity of this approach, which could be deduced from the above discussions of the PPI findings in humans.
Because deficient PPI is not unique to schizophrenia populations, there is no a priori justification for claiming that such a mutant specifically models the PPI deficits in schizophrenia, rather than OCD (Swerdlow et al. 1993a; Hoenig et al. 2005), Tourette Syndrome (Smith and Lees 1989; Castellanos et al. 1996; Swerdlow et al. 2001b), Blepharospasm (Gomez-Wong et al. 1998), or a number of other conditions. The specificity of the linkage of the model with schizophrenia, and hence with PPI deficits in schizophrenia, must come from the construct. For example, the finding of PPI deficits in a murine model of 22q11 deletion syndrome (22q11DS) links this model to PPI deficits in schizophrenia (Paylor et al. 2006; Sobin et al. 2005a, b), on the basis of the clinical relationship between 22q11DS and schizophrenia. Without this clinical relationship, this would just be a mouse with low PPI, and the model would most likely be a “false positive” for the schizophrenia phenotype.
Certainly, it is unlikely that most genes associated with low vs. high levels of PPI will be related to reduced PPI in schizophrenia or any one other disease states. This is because the most potent influences regulating baseline PPI involve physiological substrates that are probably not relevant to schizophrenia. For example, a very potent determinant of acoustic PPI is hearing threshold, as an organism that cannot hear a prepulse will not exhibit PPI. Thus, many candidate “PPI genes” identified via gene inactivation or mapping strategies of drug-free PPI in inbred and recombinant rodents will likely be associated with hearing threshold. Beyond the level of sensory detection, the most potent neural control of baseline PPI is exerted by the pedunculopontine nucleus (PPTg) (Swerdlow and Geyer 1993a), which mediates PPI via its impact on the nucleus reticularis pontis caudalis (NRPC; Koch et al. 1993). For the same reasons noted for hearing threshold, genetic studies of PPI will likely be influenced strongly by genes coding for the normal function of the PPTg—a structure that does not play a central role in any model for the pathophysiology of schizophrenia. In contrast, the prefrontal cortex (PFC)—which is viewed as a critical substrate for some core symptoms of schizophrenia (e.g., cognitive disorganization, deficient working memory, executive functioning, abstract reasoning, cognitive flexibility and context processing, and negative symptoms)—is likely to be three or four synapses removed from the primary startle circuit; in a normal human or rodent, genes controlling the PFC will likely contribute only weakly to a genetic “signal” based on levels of baseline PPI.
One might argue that a finding of PPI deficits provides additional validation that a particular model reproduces one of the quantitative phenotypes associated with schizophrenia. But as noted above, there is no definitive evidence that PPI deficits—or the neural abnormalities that produce them—are necessary for the expression of the broader schizophrenia phenotype. Rather, it is almost certainly true that there are large numbers of functionally impaired, symptomatic schizophrenia patients who exhibit levels of PPI in the “normal” range. Thus, rejecting animal models on the basis of “normal” PPI levels would likely result in a number of “false-negative” models—i.e., ones in which some features of the model accurately recreate important aspects of the biology of schizophrenia, but do not result in reduced PPI.
Perhaps the most realistic expectation of PPI in the assessment of animal models of schizophrenia is that it can provide validation for specific existing constructs—i.e., that the construct can reproduce PPI deficits exhibited by a significant subgroup of the heterogeneous population of schizophrenia patients. On the other hand, “normal” or unaltered PPI should not be used as the basis for rejecting a model: even in the presence of “normal” (i.e., wild-type, sham lesioned or placebo-treated) PPI levels, it is very possible that a model might be highly informative about the biology of schizophrenia.
Animal studies are also used to explicate the neural regulation of PPI, as a means of understanding the neural basis of PPI deficits in schizophrenia and other disorders. In this case, the manipulations are selected not necessarily based on a “construct” of schizophrenia, but rather based on the extant PPI neural “map”, and the understanding of anatomical and neurochemical properties of that map. In general, the organism used in these studies is not a schizophrenia “model” per se, but is more akin to a canvas on which a neural map can be painted. A reasonably comprehensive understanding of this “map”, ca. 2000, is found in Swerdlow et al. (2001a), and an updated list of studies of “PPI anatomy” is found in Table 4.
Much can be gleaned about PPI and its broader context by considering two facts related to its anatomical substrates. First, PPI remains intact after acute trans-collicular decerebration in the rat (Davis et al. 1982). In other words, the expression of unimodal acoustic PPI in rats does not require any part of the forebrain, and therefore, it must be mediated at or below the pons. The prepulse does not (and by physical and temporal constraints, cannot) “travel” to the forebrain to generate its inhibitory impact on the simple startle reflex (see discussion in Swerdlow et al. 2001a). Second, PPI can be regulated, and even eliminated, by subtle pharmacological manipulations at the most rostral tip of the forebrain [e.g., D1 receptor blockade within the medial prefrontal cortex (Ellenbroek et al. 1996; Shoemaker et al. 2005; Swerdlow et al. 2005c)]. Thus, brain substrates at the furthest point from the PPI “mediating” circuitry in the pons are capable of potently regulating the amount of inhibition generated by the prepulse, presumably via tonic, “thermostat”-like stimulus-independent changes in activity within descending circuitry.
These two facts lead to a simple conclusion: while PPI is mediated via the pons, it can be regulated by the forebrain. A relative loss of PPI in clinical populations, and in the animal models that are used to study them, can be a consequence of aberrant activity within this descending circuitry—somewhere “between” the cortex and pons—or within substrates that impinge upon it. The efforts to “map” this PPI-regulatory circuitry, point-to-point, from cortex to pons, are aimed to help investigators identify candidate substrates that contribute to the loss of PPI in patient populations and candidate targets for therapeutic interventions. Of the many words of caution related to this use of animals to “map PPI”, two will be noted here.
First, rodent brains and human brains are not the same. Thus, a map of neural circuitry regulating PPI in rodents cannot be expected to translate exactly to human brains. Indeed, it is surprising how much overlap is suggested across species, based on neuroimaging findings in humans (Kumari et al. 2003a, 2005, 2007a; Postma et al. 2006), and based on examples of localized neuropathology associated with PPI deficits in brain disorders such as HD and in rat and murine models of this disorder (Swerdlow et al. 1995a; Carter et al. 1999; Van Raamsdonk et al. 2005). These findings notwithstanding, it is clear that species differences will be most pronounced in phylogenetically newest regions, some of which—e.g., frontal cortex—may be of most relevance to schizophrenia. As we attempt to interpret these circuit maps at higher levels of resolution to guide drug development—i.e., beyond simple efferent/afferent patterns, and down to the receptor-and subcellular levels—these cross-species differences may become increasingly important. A number of these differences are already suggested based on simple pharmacological challenge studies, described below.
Second, all rodent brains are not the same. Strain differences in PPI, and in sensitivity to drug effects on PPI, are quite remarkable across inbred and outbred rat strains, and across inbred and outbred mouse strains. These differences must reflect differences in the PPI-regulatory brain circuitry, potentially at any level from the presence of different cell types within a larger circuit organization, down to differences in the activity of specific enzymes within signal transduction pathways. Inbred Brown Norway rats exhibit significantly more PPI at short prepulse intervals and significantly less PPI at long prepulse intervals, compared to outbred Sprague Dawley (SD) rats (Swerdlow et al. 2006a). These differences are heritable (Swerdlow et al. 2008), and must reflect genetically mediated differences in brain organization. Albino SD and hooded Long Evans (LE) rats differ significantly in their sensitivity to the PPI-disruptive effects of dopamine (DA) agonists (e.g., Swerdlow et al. 2004a, 2006d) and in the expression of DA-regulatory enzymes [e.g., catechol-o-methyl transferase (COMT)] and signal transduction enzymes (e.g., protein kinase) within the nucleus accumbens (Shilling et al. 2008). Which of these strains provides an anatomical/neurochemical “map” of PPI that is most informative about human PPI circuitry, and hence, about PPI circuit abnormalities in schizophrenia? The answer is likely to differ, based on the neural systems and levels of resolution being studied, and the models being applied.
Treatment
It is reasonable to expect that studies of PPI in laboratory animals will continue to play a major role in the discovery and development of novel therapeutics for schizophrenia. As noted above, there is no compelling empirically based reason to expect that increased PPI per se might be desirable or functionally enhancing, nor that the ability of a drug to increase PPI in schizophrenia patients should be necessary or sufficient for clinical benefit. Despite this caveat, there is clear empirical evidence that the ability of drugs to “normalize” PPI levels after they have been reduced experimentally by specific drugs or perhaps by other manipulations (e.g., developmental manipulations) strongly predicts clinical utility and even potency of antipsychotic agents (Swerdlow et al. 1994; Swerdlow and Geyer 1998; Fig. 2). Towards this end, PPI has been used in several different types of predictive models, which differ in their sensitivity, specificity, logistical complexity, and even in the types of antipsychotics that they appear to identify. These issues are reviewed in Geyer et al. (2001), and an update of studies using PPI for its predictive validity since 2000 are found in Table 2.
Fig. 2.
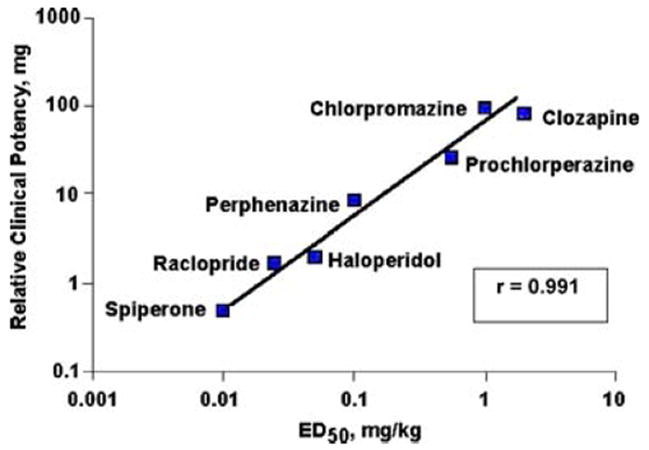
Evidence supporting the predictive validity of one “rapid-throughput” animal model of PPI deficits. In these studies (Swerdlow et al. 1994), PPI was disrupted in adult male Sprague–Dawley rats by the mixed D1/D2 agonist, apomorphine (0.5 mg/kg sc). The ED50 of a number of drugs to reverse this apomorphine effect correlated significantly with their clinical potency. Subsequent studies have identified many other clinically effective antipsychotic agents from different chemical classes that prevent the PPI-disruptive effects of apomorphine in rats [see Table 2 and Geyer et al. (2001)]. A small number of potential “false-positive” compounds have also been detected, primarily in other species or strains. Other predictive models have been developed using PPI as a dependent measure, as described in the text and Table 2, each with different sensitivity, specificity, and logistical complexities
The four most common variations of the PPI paradigm in models predictive of antipsychotic effects involve the use of (1) DA agonists (Fig. 2), (2) NMDA antagonists, (3) isolation rearing (IR), and (4) neonatal ventral hippocampal lesions (NVHLs). While each of these variations is based on a biological “construct” for the etiology of schizophrenia, i.e., hyperdopaminergia, hypoglutamatergia, and specific neurodevelopmental insults, they have all been applied towards predicting antipsychotic properties in novel compounds. In truth, only the former two variants are well suited to traditional “rapid throughput” drug screens, based on the amount of time and resources necessary for the developmental models, and the relatively small (and often strain-or sex-dependent) effects of isolation rearing on PPI (Weiss et al. 1999, 2000; Powell et al. 2002). In each of these variations, the ability of a drug to “normalize” PPI is interpreted as evidence for antipsychotic potential. Some second generation antipsychotics, such as clozapine, quetiapine, and olazapine, tend to increase PPI in otherwise intact animals (Swerdlow and Geyer 1993b; cf. Geyer et al. 2001), particularly in mice, adding some interpretative complexity to their ability to normalize PPI after pharmacological, developmental, or surgical manipulations. In fact, the ability to enhance baseline PPI is a signal that has been used as a predictor of antipsychotic potential in mice, in some normally “low-gating” mouse strains (cf. Ouagazzal et al. 2001a), rat strains (Feifel et al. 2001, 2004), and even in normal “low-gating” humans (Swerdlow et al. 2006a; Vollenweider et al. 2006).
Beyond the dopamine system, some new targets of antipsychotics have emerged in recent years, based in part on studies using variations of PPI paradigms as predictive models. Examples of these targets include (but are not limited to) selective 5-HT2C receptor agonists (Marquis et al. 2007), CB1 cannabinoid receptor antagonists (Nagai et al. 2006), neurotensin-1 receptor agonists (Shilling et al. 2003, 2004; Caceda et al. 2005), selective adenosine A(2A) receptor agonists (Wardas et al. 2003), alpha-7 nicotinic receptor agonists (Suemara et al. 2004), and selective histamine H3 receptor antagonists (Fox et al. 2005; Table 2). It should be emphasized that in some cases, these targets were identified based on PPI assays with less compelling predictive validity, such as the ability of compounds to increase basal PPI levels in mice, or to normalize PPI after its disruption by 5HT agonists or NMDA antagonists. These assays may have strong sensitivity, particularly for identifying compounds with potentially novel mechanisms, but they also may lack specificity for detecting antipsychotic properties, at least in comparison to assays based on the ability to block the PPI-disruptive effects of apomorphine and perhaps other DA agonists (Fig. 2). We will have to await clinical evidence to determine whether these reports reflect “false positives” of these models.
PPI has only more recently begun to be used in models for detecting preventative or neuroprotective interventions, to identify strategies that would prevent the neuropathological and clinical consequences of a vulnerability gene or developmental insult involved in the prodrome and onset of schizophrenia. Some studies are approaching such an application, using early neuroimmune challenges to yield PPI deficits during adulthood (e.g., Borrell et al. 2002), or using sustained early life antipsychotic exposure to blunt the PPI-disruptive effects of developmental insults (Powell et al. 2006a, b). Assuming that these models succeed, it remains to be determined how one would test or apply such interventions in a clinical setting.
A reliable, robust, quantitative phenotype
In any given rodent species and strain, both PPI and its drug sensitivity are quite robust and reliable phenotypes. Within a range of 30–120 ms prepulse intervals, and 2–16 dB noise prepulses over a 65-to 70-dB(A) noise background, and 105–120 dB(A) noise pulses, PPI in rats exhibits a magnitude and parametric sensitivity that are strikingly similar across a number of studies from different laboratories and, conveniently, are also quite similar to those exhibited by humans. Similarly, PPI-disruptive effects of a number of simple manipulations (e.g., administration of a direct DA agonist) have been replicated across laboratories to the point that they have become “standard assays”, in predictive models for antipsychotic development. The PPI-disruptive effects of more complex manipulations, including early developmental lesions or isolation rearing, tend to be more variable across laboratories (discussed above), perhaps due to the complexities (and hence variability) of the methods and uncontrolled sources of variance. Some differences in reports of PPI drug sensitivity and sensitivity to developmental manipulations clearly seem to result from differences in rat strain or even supplier (e.g., Swerdlow et al. 1998, 2000b, 2003a, 2004a), and these differences are being explicated at the levels of heritable differences in neural substrates regulating PPI.
Some disparities in reported drug or other manipulation effects on PPI may also reflect differences in the recording properties of a variety of “home built“ and commercially available startle response acquisition systems. While there is no “gold standard” for such an apparatus, there are a number of characteristics that should be evaluated in interpreting whether response measurements “obey the laws of physiology”, e.g., intensity-and interval-dependence of PPI, and relative insensitivity of PPI to weight differences across animals. These issues are reviewed in Geyer and Swerdlow (1998).
Startle and PPI data can be deceptively complex, and some disparities in reported effects on PPI in rodents undoubtedly reflect these complexities and resulting interpretative differences across studies. Despite the impressive degree of automation in laboratory measures of PPI, one cannot automatically enter startle data into an equation and reasonably expect the calculated percent PPI to be informative. For example, we have previously reviewed the importance of considering the impact of changes in startle magnitude on changes in PPI (Swerdlow et al. 2000a). Simply put, the only unambiguous changes in sensorimotor gating are ones that can be demonstrated in the absence of changes in startle magnitude. In this case, reduced sensorimotor gating reflects a diminished impact of the prepulse on startle magnitude and, hence, an increase in startle magnitude on prepulse + pulse trials only. Any other related pattern of results, involving significantly reduced or increased startle magnitude on pulse-alone trials, must be interpreted in the context of additional supportive evidence. Such evidence might come from the use of low and high pulse intensities or from subgroups of rats that are matched based on comparable levels of startle magnitude.
Another interpretative issue that has been discussed in several recent reports relates to the potential impact of prepulse-induced startle activity on PPI and its modification by drugs or other experimental manipulations (Yee et al. 2004; Swerdlow et al. 2004c). A stimulus is only considered a “prepulse” in relationship to a second stimulus. By any other metric, it is simply a stimulus and can elicit motor activity including a startle reflex, depending on its properties. If the prepulse intensity exceeds the startle threshold, a “prepulse + pulse” configuration is better described as a “paired-pulse” configuration, and the resulting decrement in the startle response elicited by the second pulse is described as “paired-pulse inhibition”, comparable to the phenomenon used to study “blink excitability” (e.g., Kimura and Harada 1976; Valls-Sole et al. 2004). The similarities and differences of PPI and paired-pulse inhibition have been described for a small number of drug effects (e.g. Swerdlow et al. 2002a), but relatively little is known about this relationship for the long list of manipulations that have been applied towards PPI studies.
The interpretative ambiguities created by “prepulse-elicited startle” are most relevant to conditions in which the prepulse exceeds startle threshold. In a rat, for 20 ms noise prepulses over a 70-dB(A) noise background, this threshold is generally between 12 and 15 dB, although the precise value varies with strain, sex, age, and other factors. Other prepulse characteristics, including frequency (pure tone vs. white noise), duration, and configuration (continuous vs. discrete) can impact its motor-inhibiting and activating properties. For the vast majority of published PPI studies, prepulses are used at levels that elicit no or little detectable motor activity; even relatively intense prepulses (e.g., 10–15 dB salience, based on the stimulus conditions described above) might elicit a motor “signal” that is <1% of the total startle signal (Swerdlow et al. 2004c). In fact, this signal is comparable to that detected on “NOSTIM” trials, i.e., when no motor activity is recorded in the absence of stimulus delivery, suggesting that this small signal reflects ongoing motor activity rather than a prepulse-elicited motor response (e.g. Swerdlow et al. 2004c; Weber and Swerdlow 2008). Importantly, only a small fraction of studies utilize prepulses with supra-threshold intensities, and among these, most also utilize much weaker prepulses as internal comparisons. PPI is used to assess many things, and in some cases, a range of prepulse intensities is used to create a complete parametric characterization for purposes unrelated to drug effects (e.g., QTL analyses). Clearly, in these cases, the use of intense prepulses is not a “confound”, but simply a way to fully characterize a phenotype.
It is argued that potentially confounding effects might arise if a drug or other manipulation lowers startle threshold and, hence, transforms a non-startling prepulse into one that elicits a motor response (Yee et al. 2004). Specifically, a potentially confounding interaction might arise if increases in prepulse-evoked motor responses diminished the pre-pulse’s inhibitory effects on a subsequent startle response. In fact, there is no reason to predict such an effect: full startle responses elicited by an S1 in a paired-pulse paradigm do not interfere with the inhibitory impact of S1 on the startle response to S2 (e.g., Swerdlow et al. 2002a), so there is no credible reason to predict that such interference would result from a prepulse-evoked response that is 100-fold less intense. Nonetheless, under drug conditions, a number of control comparisons can be conducted—analogous to those used to understand the impact on PPI of drug-induced changes in startle magnitude—to determine whether drug effects on prepulse-evoked motor activity and PPI can be “dissociated”. We might predict that a common drug receptor (e.g., D1 or D2) might mediate two processes (reduced PPI and increased prepulse-induced motor activity), via effects within different brain substrates. Similar to changes in startle magnitude, a given drug might elicit either increases, decreases, or no change in prepulse-induced motor responses, yet have a consistent effect on PPI (e.g., Weber and Swerdlow 2008); even in cases where drug-induced changes in prepulse-induced activity are detected, they amount to shifts of less than 1% of the total “signal” of the startle response and, as noted above, are comparable to changes observed in “NOSTIM” activity. Thus, while it is a reasonable precaution to consider measuring prepulse-elicited motor activity to ascertain whether it is significantly greater than ongoing background motor activity, and whether it might potentially interact with the startling effects of the startle pulse, in our experience, such an exercise amounts to “much ado about [almost] nothing” (Swerdlow 2005).
A useful tool for modeling the neurobiology and gating and its deficits in humans
The most compelling contribution of animal studies of PPI towards the understanding of the basis for PPI deficits in schizophrenia comes in the ability to directly manipulate neural and genetic substrates and test hypotheses in a controlled experimental setting. The challenges of extrapolating such findings across species are not trivial, as discussed above in relationship to neural circuit maps. Still, for understanding the contribution to PPI deficits in schizophrenia of pathology in medial prefrontal cortex, hippocampus, amygdala or ventral striatum, or of specific candidate genes or early developmental insults, cross-species studies are a unique, powerful tool.
PPI studies have also identified neurobiological bridges across species that may reveal potential limitations of these studies and, perhaps, more generally of animal models of schizophrenia. For example, several drugs potently disrupt PPI in rats and yet increase PPI in normal humans. This is most notable because the drugs in question—ketamine (Abel et al. 2003; Duncan et al. 2001), MDMA (Vollenweider et al. 1999) and under some conditions, DA agonists (Bitsios et al. 2005)—have pharmacological and clinical properties that are central to models for the pathophysiology of schizophrenia. These findings raise both experimental and conceptual issues.
At an experimental level, drug doses, routes of administration, and pharmacokinetic/dynamic properties differ substantially across species. As one example, amphetamine reliably decreases PPI in rats only at doses above 2 mg/kg administered subcutaneously (Mansbach et al. 1988; Sills 1999; Swerdlow et al. 2006d), while the oral dose of amphetamine given to normal humans in PPI studies rarely exceeds 0.29 mg/kg (20 mg total; e.g., Hutchison and Swift 1999; Swerdlow et al. 2003b). Species differences in drug effects might also reflect contextual differences in the test setting. Humans volunteer and are paid for study participation, have the test conditions explained by a supportive research assistant, swallow a pill, and sit in a comfortable chair during testing; by contrast, rats are removed from a cage, injected with a drug, and then placed alone in a plastic tube inside an unfamiliar box where they are exposed to loud, unexpected noises. One might imagine that drug effects on a fight-or-flight reflex (startle) might differ in these two conditions, independent of species. Furthermore, while the parametric properties of PPI (e.g., sensitivity to prepulse intensity and interval) are strikingly similar across species, drug effects might reveal some cross-species differences in these parametric effects. For example, at 120 ms prepulse intervals, ketamine has opposite effects on PPI in rats (disrupts PPI; Mansbach and Geyer 1989) and humans (increases PPI; Abel et al. 2003; Duncan et al. 2001); on the other hand, ketamine can increase PPI in rats at shorter prepulse intervals (e.g., 30 ms; Mansbach and Geyer 1989). Our group has detected similar species-and interval-dependent effects with the NMDA antagonist, memantine (Swerdlow et al. 2003c, 2005a). Conceivably, NMDA-related mechanisms of drug effects on gating at 30 ms in rats might best approximate those at 120 ms in humans.
However, this explanation does not address the conceptual dilemma created by the fact that psychotomimetic drugs increase PPI in normal humans, while schizophrenia is associated with reduced PPI. While PPI deficits in schizophrenia might possibly reflect the consequences of sustained deficiencies in glutamatergic activity in the context of developmentally aberrant neural connections, it does not follow that such effects would be reproduced by an acute challenge of an NMDA antagonist to a normal individual with normal neural connectivity. Furthermore, one might easily imagine that acute drug effects on an intact brain might enhance sensorimotor gating via a mechanism that is very distinct from (e.g., “upstream” or “downstream” from) those responsible for reduced gating in the brain of a schizophrenia patient. Nonetheless, faced with these discrepant effects of psychotomimetic drugs on PPI, it is difficult to know whether the failings lie in the cross-species translation of the PPI model, in the validity of the acute ketamine/ glutamate antagonist model of schizophrenia, or both.
An additional challenge in building neurobiological bridges of PPI studies across species comes from the human side of the bridge—from the observations that drug effects on PPI in humans can differ significantly, depending on basal levels of PPI. A number of drugs—including amphetamine (Swerdlow et al. 2003b), pergolide, amantadine (Bitsios et al. 2005), quetiapine (Swerdlow et al. 2006a), and clozapine (Vollenweider et al. 2006)—have been demonstrated to have effects that differ significantly (and in some cases, are arithmetically opposite) in normal humans with low vs. high PPI levels, relative to the overall test population. Similar findings may be emerging from animal studies, e.g., among inbred strains with low basal levels of PPI (cf. Ouagazzal et al. 2001a). How we interpret this “rate dependency” of drug effects on PPI in humans and laboratory animals and what it means about the many reported drug effects on PPI that have not considered or tested the impact of basal PPI levels, are issues that remain to be resolved.
While this discussion has focused primarily on cross-species comparisons between rodents and humans, and we discussed earlier the strain differences in PPI that have been detected in both rats and mice, it is also worth noting that there are also a number of important cross-species differences in PPI and its parametric and pharmacological sensitivity between rats and mice. Just as one example, while PPI is disrupted by DA agonists in both rats and mice, there is some evidence that this effect primarily reflects activation of D2 receptors in rats (Swerdlow et al. 1994; cf. Geyer et al. 2001), but of D1 receptors in mice (Ralph-Williams et al. 2003a; Ralph and Caine 2005). Within a restricted set of stimulus parameters (particularly prepulse intervals), infusion of D2 agonists into the nucleus accumbens decreases PPI in rats and increases PPI in mice (Mohr et al. 2007). This issue is not yet settled, as mice lacking D2 receptors are insensitive to the PPI-disruptive effects of d-amphetamine (Ralph et al. 1999), and some mouse strains exhibit “rat-like” PPI sensitivity to D2 agonists (Ralph and Caine 2007). Nonetheless, enough data exists that we can be fairly confident that a similar drug effect on PPI in rats and mice does not necessarily reflect a common underlying brain substrate. This raises the dilemma that when modeling the loss of PPI in schizophrenia, we are almost certainly studying very different neurobiological substrates, depending on the model species; this makes it very difficult to identify a clear, a priori rationale for selecting one species over another.
A surrogate measure for neural processes with wide-reaching psychological implications
Models of higher cognitive processes are only now being developed in rodents. Given the limited size and processing capacity of the frontal cortex in mice and rats vs. primates, and its relatively weaker contribution to the organization of behavior, there is reason to be skeptical that rodent models of higher cognitive processes will provide meaningful homology to human cognition. Nonetheless, mice and rats are amenable to complex conditioning schedules and are capable of performing choices and sophisticated behavioral sequences, and it is certain that studies will assess the potential relationship of PPI to these processes (e.g., Roegge et al. 2007; Depoortere et al. 2007a, b; Garner et al. 2007; Paine et al. 2007). Extrapolating these findings to humans will present many challenges. In general, the farther forward one moves in the brain, the greater the anatomical and functional differences between rodents and humans. For example, one might imagine a scenario in which “cognitive” control in rodents involves a prominent role for subcortical (e.g., basal ganglia) functions that overlap with PPI-regulatory circuitry, while in humans, higher cognitive control is “encephalized” to discrete frontal circuits that participate less in the regulation of startle gating.
There is already some evidence for both convergence and divergence of PPI and other operational animal models of “gating”, in terms of their underlying neural substrates. For example, contemporaneous measures of PPI and N40 gating—an animal model of P50 ERP gating in humans—revealed that apomorphine, phencyclidine, and DOI each disrupt PPI and reduce ERP responsivity to the S1 stimulus in the N40-gating paradigm, but do not specifically disrupt N40 gating per se (Swerdlow et al. 2006b). Some overlap has been reported in the pharmacological sensitivity of PPI and [some of the various forms of] latent inhibition to DA agonists and NMDA antagonists (Mansbach and Geyer 1989; Bakshi et al. 1995; Razoux et al. 2007), although many conditions lead to a loss of PPI in rats but leave latent inhibition intact (e.g., amphetamine withdrawal (Peleg-Raibstein et al. 2006a, b) and D2 blockade in the basolateral amygdala (Stevenson and Gratton 2004)). Thus, neurobiological mechanisms of PPI cannot be assumed to be common to experimental measures of either sensory or cognitive gating in rats. The potential overlap in the neurobiology of PPI and higher-order functions in rats, such as working memory, is an area of ongoing investigation. At present, there is no compelling evidence that such an overlap exists or that PPI is informative about higher cognitive functions in rodents.
Summary: animal studies
Animal models will remain an important tool in developing and testing hypotheses for the pathogenesis of brain disorders. As a reliable, quantitative “read out” of relatively well-defined neural circuitry, measures of PPI in laboratory animals will continue to be used to test and validate these hypotheses and to generate important new hypotheses regarding cellular mechanisms and therapeutic strategies. PPI models provide predictive validity in drug discovery and development, both as rapid through-put screens and as components of more biologically sophisticated models involving developmental, immunologic, and genetic manipulations. Areas of convergence and divergence are being identified in the cross-species pharmacology of PPI; areas of convergence will be exploited so that human drug effects can be predicted and understood based on PPI drug effects in rodents and their underlying cellular and molecular substrates. Finally, the relationship of PPI to higher-order learning processes is being explored in rodents, and the findings will be used to generate and test hypotheses regarding the interplay of sensorimotor gating and cognition in normal and disordered humans.
Conclusions
The construct of gating deficits in neuropsychiatric disorders has empirical support and intuitive appeal, and serves as a unifying heuristic for understanding the psychological and neural substrates shared by otherwise apparently unrelated disorders. PPI is an operational measure of basic, brain-based gating processes. It is robust, reliable, easily quantified, and versatile as an experimental tool, and is abnormal in several brain disorders including schizophrenia, that are characterized by clinical evidence of impaired gating of sensory, cognitive, motor of affective information. PPI can be measured across species and is regulated in laboratory animals by neurochemical, anatomical, developmental, and genetic substrates that can be systematically studied and used as the basis for developing and testing hypotheses for the biological basis of PPI deficits in patients.
For all of these reasons, studies of PPI in humans and laboratory animals have multiplied and expanded, and this measure is being used to explore many new questions at many different levels of analysis. While our field does not yet face the floods of the “Sorcerer’s Apprentice” (von Goethe 1779), it is clear that findings have amassed at an exponential rate and are testing our collective ability to critically integrate results, to identify areas of consistency, redundancy, and disagreement. Based on a review of the present literature, we reached several conclusions: (1) in humans, PPI is not “diagnostic”; levels of PPI do not predict clinical course, specific symptoms, or individual medication responses; (2) in preclinical studies, PPI is valuable for evaluating models or model organisms relevant to schizophrenia, “mapping” neural substrates of deficient PPI in schizophrenia, and advancing the discovery and development of novel therapeutics; (3) across species, PPI is a reliable, robust quantitative phenotype that is useful for probing the neurobiology and genetics of gating deficits in schizophrenia. In this review, we also identify some realistic expectations of this paradigm, describing its considerable strengths but also limitations, and stress some interpretative issues for consideration as we move forward with this powerful tool for translational neuropsychiatric research.
Acknowledgments
The authors of this study were supported by grants from the NIMH (NRS, DLB, GAL: MH 42228, MH 65571; NRS: MH 68366, MH 53484, MH 58903, MH 69589; GAL: MH 79777) and the VISN 22 MIRECC (DLB, GAL). DLB also was supported by a NARSAD Distinguished Investigator Award. The authors have also served as paid consultants to pharmaceutical companies or have had research supported by these companies (NRS, DLB, GAL: Allergan Pharmaceuticals, Pfizer Pharmaceuticals; DLB: Acadia Pharmaceuticals; GAL: Memory Pharmaceuticals, Sepacor and Astra-Zeneca). No funding entity provided any input to, oversight of, or influence over the contents of this review. The authors thank Ms. Maria Bongiovanni and Mr. David Ko for their expert assistance in manuscript preparation.
References
- Aasen I, et al. Sex effects in prepulse inhibition and facilitation of the acoustic startle response: implications for pharmacological and treatment studies. J Psychopharmacol. 2005;19:39–45. doi: 10.1177/0269881105048890. [DOI] [PubMed] [Google Scholar]
- Abel K, et al. Repeated testing of prepulse inhibition and habituation of the startle reflex: a study in healthy human controls. J Psychopharmacol. 1998;12:330–337. doi: 10.1177/026988119801200402. [DOI] [PubMed] [Google Scholar]
- Abel KM, et al. Low dose ketamine increases prepulse inhibition in healthy men. Neuropharmacology. 2003;44:729–737. doi: 10.1016/s0028-3908(03)00073-x. [DOI] [PubMed] [Google Scholar]
- Adage T, et al. In vitro and in vivo pharmacological profile of AS057278, a selective d–amino acid oxidase inhibitor with potential anti-psychotic properties. Eur Neuropsychopharmacol. 2007 doi: 10.1016/j.euroneuro.2007.06.006. in press. [DOI] [PubMed] [Google Scholar]
- Addington J, et al. North American Prodrome Longitudinal Study. North American Prodrome Longitudinal Study: a collaborative multisite approach to prodromal schizophrenia research. Schizophr Bull. 2007;33:665–672. doi: 10.1093/schbul/sbl075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler LE, et al. Neurophysiological evidence for a defect in neuronal mechanisms involved in sensory gating in schizophrenia. Biol Psychiatry. 1982;17:639–654. [PubMed] [Google Scholar]
- Afonso VM, et al. Medial prefrontal cortex lesions in the female rat affect sexual and maternal behavior and their sequential organization. Behav Neurosci. 2007;121:515–526. doi: 10.1037/0735-7044.121.3.515. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (APA) Diagnostic and Statistical Manual of Mental Disorders DSM-IV-TR. Fourth Edition (Text Revision) 2000. [Google Scholar]
- Andreasen JT, et al. Nicotine and clozapine selectively reverse a PCP–induced deficit of PPI in BALB/cByJ but not NMRI mice: comparison with risperidone. Behav Brain Res. 2006;167:118–127. doi: 10.1016/j.bbr.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Auclair AL, et al. Actions of novel antipsychotic agents on apomorphine-induced PPI disruption: influence of combined serotonin 5-HT1A receptor activation and dopamine D2 receptor blockade. Neuropsychopharmacology. 2006;31:1900–1909. doi: 10.1038/sj.npp.1301015. [DOI] [PubMed] [Google Scholar]
- Auclair AL, et al. Putative antipsychotics with pronounced agonism at serotonin 5-HT1A and partial agonist activity at dopamine D2 receptors disrupt basal PPI of the startle reflex in rats. Psychopharmacology (Berl) 2007;193:45–54. doi: 10.1007/s00213-007-0762-7. [DOI] [PubMed] [Google Scholar]
- Bakshi VP, et al. A comparison of the effects of amphetamine, strychnine and caffeine on prepulse inhibition and latent inhibition. Behav Pharmacol. 1995;6:801–809. [PubMed] [Google Scholar]
- Ballmaier M, et al. Combined alpha 2-adrenergic/D2 dopamine receptor blockade fails to reproduce the ability of clozapine to reverse phencyclidine-induced deficits in prepulse inhibition of startle. Psychopharmacology (Berl) 2001a;159:105–110. doi: 10.1007/s002130100905. [DOI] [PubMed] [Google Scholar]
- Ballmaier M, et al. Selective immunolesioning of cholinergic neurons in nucleus basalis magnocellularis impairs prepulse inhibition of acoustic startle. Neuroscience. 2001b;108:299–305. doi: 10.1016/s0306-4522(01)00413-4. [DOI] [PubMed] [Google Scholar]
- Ballmaier M, et al. Rivastigmine antagonizes deficits in prepulse inhibition induced by selective immunolesioning of cholinergic neurons in nucleus basalis magnocellularis. Neuroscience. 2002;114:91–98. doi: 10.1016/s0306-4522(02)00234-8. [DOI] [PubMed] [Google Scholar]
- Ballmaier M, et al. Cannabinoid receptor antagonists counteract sensorimotor gating deficits in the phencyclidine model of psychosis. Neuropsychopharmacology. 2007;32:2098–2107. doi: 10.1038/sj.npp.1301344. [DOI] [PubMed] [Google Scholar]
- Barr AM, et al. The selective serotonin-2A receptor antagonist M100907 reverses behavioral deficits in dopamine transporter knockout mice. Neuropsychopharmacology. 2004;29:221–228. doi: 10.1038/sj.npp.1300343. [DOI] [PubMed] [Google Scholar]
- Barr AM, et al. Iloperidone reduces sensorimotor gating deficits in pharmacological models, but not a developmental model, of disrupted prepulse inhibition in rats. Neuropharmacology. 2006;51:457–465. doi: 10.1016/j.neuropharm.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Barr AM, et al. The reelin receptors VLDLR and ApoER2 regulate sensorimotor gating in mice. Neuropharmacology. 2007;52:1114–1123. doi: 10.1016/j.neuropharm.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett SL, et al. Normal levels of prepulse inhibition in the euthymic phase of bipolar disorder. Psychol Med. 2005;35:1737–1746. doi: 10.1017/S0033291705005702. [DOI] [PubMed] [Google Scholar]
- Bast T, et al. Hyperactivity and disruption of prepulse inhibition induced by N-methyl-d-aspartate stimulation of the ventral hippocampus and the effects of pretreatment with haloperidol and clozapine. Neuroscience. 2001;103:325–335. doi: 10.1016/s0306-4522(00)00589-3. [DOI] [PubMed] [Google Scholar]
- Beglopoulos V, et al. Neurexophilin 3 is highly localized in cortical and cerebellar regions and is functionally important for sensorimotor gating and motor coordination. Mol Cell Biol. 2005;25:7278–7288. doi: 10.1128/MCB.25.16.7278-7288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RL, et al. Amphetamine-modified acoustic startle responding and prepulse inhibition in adult and adolescent alcohol-preferring and-nonpreferring rats. Pharmacol Biochem Behav. 2003;75:163–171. doi: 10.1016/s0091-3057(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Bickel S, et al. Early auditory sensory processing deficits in mouse mutants with reduced NMDA receptor function. Neuropsychopharmacology. 2008 doi: 10.1038/sj.npp.1301536. in press. [DOI] [PubMed] [Google Scholar]
- Bitsios P, et al. The effects of dopamine agonists on prepulse inhibition in healthy men depend on baseline PPI values. Psychopharmacology (Berl) 2005;182:144–152. doi: 10.1007/s00213-005-0056-x. [DOI] [PubMed] [Google Scholar]
- Bitsios P, et al. Increased prepulse inhibition of the acoustic startle response is associated with better strategy formation and execution times in healthy males. Neuropsychologia. 2006;44:2494–2499. doi: 10.1016/j.neuropsychologia.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Bleuler E. Dementia praecox; or, the group of schizophrenias. International Universities Press; New York: 19111950. [Google Scholar]
- Boeckler F, et al. FAUC 213, a highly selective dopamine D4 receptor full antagonist, exhibits atypical antipsychotic properties in behavioural and neurochemical models of schizophrenia. Psychopharmacology (Berl) 2004;175:7–17. doi: 10.1007/s00213-004-1782-1. [DOI] [PubMed] [Google Scholar]
- Bontekoe CJ, et al. Knockout mouse model for Fxr2: a model for mental retardation. Hum Mol Genet. 2002;11:487–498. doi: 10.1093/hmg/11.5.487. [DOI] [PubMed] [Google Scholar]
- Borrell J, et al. Prenatal immune challenge disrupts sensorimotor gating in adult rats. Implications for the etiopathogenesis of schizophrenia. Neuropsychopharmacology. 2002;26:204–215. doi: 10.1016/S0893-133X(01)00360-8. [DOI] [PubMed] [Google Scholar]
- Bortolato M, et al. Baclofen reverses the reduction in prepulse inhibition of the acoustic startle response induced by dizocilpine, but not by apomorphine. Psychopharmacology (Berl) 2004;171:322–330. doi: 10.1007/s00213-003-1589-5. [DOI] [PubMed] [Google Scholar]
- Bortolato M, et al. Kappa opioid receptor activation disrupts prepulse inhibition of the acoustic startle in rats. Biol Psychiatry. 2005;57:1550–1558. doi: 10.1016/j.biopsych.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Bortolato M, et al. Anxiolytic-like properties of the anandamide transport inhibitor AM404. Neuropsychopharmacology. 2006;31:2652–2659. doi: 10.1038/sj.npp.1301061. [DOI] [PubMed] [Google Scholar]
- Bortolato M, et al. Activation of GABA(B) receptors reverses spontaneous gating deficits in juvenile DBA/2J mice. Psychopharmacology (Berl) 2007;194:361–369. doi: 10.1007/s00213-007-0845-5. [DOI] [PubMed] [Google Scholar]
- Boucher AA, et al. Heterozygous neuregulin 1 mice are more sensitive to the behavioural effects of Delta9-tetrahydrocannabinol. Psychopharmacology (Berl) 2007;192:325–336. doi: 10.1007/s00213-007-0721-3. [DOI] [PubMed] [Google Scholar]
- Bowers BJ, et al. Deletion of the alpha7 nicotinic receptor subunit gene results in increased sensitivity to several behavioral effects produced by alcohol. Alcohol Clin Exp Res. 2005;29:295–302. doi: 10.1097/01.alc.0000156116.40817.a2. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Holmes A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic-and antidepressant-like effects in mice. Neuropsychopharmacology. 2006;31:2405–2414. doi: 10.1038/sj.npp.1301039. [DOI] [PubMed] [Google Scholar]
- Braff DL, Light GA. Preattentional and attentional cognitive deficits as targets for treating schizophrenia. Psychopharmacology (Berl) 2004;174:75–85. doi: 10.1007/s00213-004-1848-0. [DOI] [PubMed] [Google Scholar]
- Braff D, et al. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. 1978;15:339–343. doi: 10.1111/j.1469-8986.1978.tb01390.x. [DOI] [PubMed] [Google Scholar]
- Braff DL, et al. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49:206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- Braff DL, et al. Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res. 2001a;49:171–178. doi: 10.1016/s0920-9964(00)00139-0. [DOI] [PubMed] [Google Scholar]
- Braff DL, et al. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology. 2001b;156:234–258. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- Braff DL, et al. Female schizophrenia patients have prepulse inhibition deficits. Biol Psychiatry. 2005;57:817–820. doi: 10.1016/j.biopsych.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Braff DL, et al. Deconstructing schizophrenia: an overview of the use of endophenotypes in order to understand a complex disorder. Schizophr Bull. 2007a;33:21–32. doi: 10.1093/schbul/sbl049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braff DL, et al. Prepulse Inhibition and P50 Suppression are both deficient but not correlated in schizophrenia patients. Biol Psychiatry. 2007b;61:1204–1207. doi: 10.1016/j.biopsych.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Braff, et al. One-year longitudinal study of 200 schizophrenia patients: no clinical, cognitive, neurophysiological or functional differences at intake in retested vs. non-retested patients. Abstr Society for Neuroscience. 2008 in press. [Google Scholar]
- Brea J, et al. QF2004B, a potential antipsychotic butyrophenone derivative with similar pharmacological properties to clozapine. Neuropharmacology. 2006;51:251–262. doi: 10.1016/j.neuropharm.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Brody SA, Geyer MA. Interactions of the mGluR5 gene with breeding and maternal factors on startle and prepulse inhibition in mice. Neurotox Res. 2004b;6:79–90. doi: 10.1007/BF03033300. [DOI] [PubMed] [Google Scholar]
- Brody SA, et al. Disruption of prepulse inhibition in mice lacking mGluR1. Eur J Neurosci. 2003a;18:3361–3366. doi: 10.1111/j.1460-9568.2003.03073.x. [DOI] [PubMed] [Google Scholar]
- Brody SA, et al. Lamotrigine prevents ketamine but not amphetamine-induced deficits in prepulse inhibition in mice. Psychopharmacology (Berl) 2003b;169:240–246. doi: 10.1007/s00213-003-1421-2. [DOI] [PubMed] [Google Scholar]
- Brody SA, et al. Effect of antipsychotic treatment on the prepulse inhibition deficit of mGluR5 knockout mice. Psychopharmacology (Berl) 2004a;172:187–195. doi: 10.1007/s00213-003-1635-3. [DOI] [PubMed] [Google Scholar]
- Brody SA, et al. A developmental influence of the N-methyl-d-aspartate receptor NR3A subunit on prepulse inhibition of startle. Biol Psychiatry. 2005;57:1147–1152. doi: 10.1016/j.biopsych.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Browman KE, et al. Enhancement of prepulse inhibition of startle in mice by the H3 receptor antagonists thioperamide and ciproxifan. Behav Brain Res. 2004;153:69–76. doi: 10.1016/j.bbr.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Brunskill EW, et al. Abnormal neurodevelopment, neurosignaling and behaviour in Npas3-deficient mice. Eur J Neurosci. 2005;22:1265–1276. doi: 10.1111/j.1460-9568.2005.04291.x. [DOI] [PubMed] [Google Scholar]
- Bubenikova V, et al. The effect of zotepine, risperidone, clozapine and olanzapine on MK-801-disrupted sensorimotor gating. Pharmacol Biochem Behav. 2005;80:591–596. doi: 10.1016/j.pbb.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Burne TH, et al. Combined prenatal and chronic postnatal vitamin D deficiency in rats impairs prepulse inhibition of acoustic startle. Physiol Behav. 2004;81:651–655. doi: 10.1016/j.physbeh.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Burne TH, et al. Behavioural characterization of vitamin D receptor knockout mice. Behav Brain Res. 2005;157:299–308. doi: 10.1016/j.bbr.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Burton C, et al. Early adversity alters attention and locomotion in adult Sprague–Dawley rats. Behav Neurosci. 2006;120:665–675. doi: 10.1037/0735-7044.120.3.665. [DOI] [PubMed] [Google Scholar]
- Butler RW, Jenkins MA, Geyer MA, Braff D. Wisconsin card sorting deficits and diminished sensorimotor gating in a discrete subgroup of schizophrenic patients. In: Tamminga CA, Schulz SC, editors. Advances in neuropsychiatry and psychopharmacology. New York, NY: Raven Press; 1991. pp. 163–168. [Google Scholar]
- Byrnes EM, et al. Sensorimotor gating and dopamine function in postpartum rats. Neuropsychopharmacology. 2007;32:1021–1031. doi: 10.1038/sj.npp.1301222. [DOI] [PubMed] [Google Scholar]
- Caceda R, et al. Virally mediated increased neurotensin 1 receptor in the nucleus accumbens decreases behavioral effects of mesolimbic system activation. J Neurosci. 2005;25:11748–11756. doi: 10.1523/JNEUROSCI.4282-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenhead KS. Vulnerability markers in the schizophrenia spectrum: implications for phenomenology, genetics, and the identification of the schizophrenia prodrome. Psychiatr Clin North Am. 2002;25:837–853. doi: 10.1016/s0193-953x(02)00021-7. [DOI] [PubMed] [Google Scholar]
- Cadenhead KS, et al. Prepulse inhibition and habituation of the startle response are stable neurobiological measures in a normal male population. Biol Psychiatry. 1999;45:360–364. doi: 10.1016/s0006-3223(98)00294-7. [DOI] [PubMed] [Google Scholar]
- Cadenhead KS, et al. Neurobiological measures of schizotypal personality disorder: defining an inhibitory endophenotype. Am J Psychiatry. 2002;159:869–871. doi: 10.1176/appi.ajp.159.5.869. [DOI] [PubMed] [Google Scholar]
- Caine SB, et al. Behavioral effects of psychomotor stimulants in rats with dorsal or ventral subiculum lesions: locomotion, cocaine self-administration, and prepulse inhibition of startle. Behav Neurosci. 2001;115:880–894. doi: 10.1037//0735-7044.115.4.880. [DOI] [PubMed] [Google Scholar]
- Calkins ME, et al. Antisaccade performance is impaired in medically and psychiatrically healthy biological relatives of schizophrenia patients. Schizophr Res. 2004;71:167–178. doi: 10.1016/j.schres.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Calkins ME, et al. The consortium on the genetics of endophenotypes in schizophrenia: model recruitment, assessment, and endophenotyping methods for a multi-site collaboration. Schiz Bull. 2007;33:33–48. doi: 10.1093/schbul/sbl044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, et al. Prediction of psychosis in youth at high clinical risk: a multi-site longitudinal study in North America. Arch Gen Psychiatry. 2008;65:28–37. doi: 10.1001/archgenpsychiatry.2007.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao BJ, Li Y. Reduced anxiety-and depression-like behaviors in Emx1 homozygous mutant mice. Brain Res. 2002;937:32–40. doi: 10.1016/s0006-8993(02)02461-7. [DOI] [PubMed] [Google Scholar]
- Carroll CA, et al. Sensorimotor gating in manic and mixed episode bipolar disorder. Bipolar Disord. 2007;9:221–229. doi: 10.1111/j.1399-5618.2007.00415.x. [DOI] [PubMed] [Google Scholar]
- Carter RJ, et al. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos FX, et al. Sensorimotor gating in boys with Tourette’s syndrome and ADHD: preliminary results. Biol Psychiatry. 1996;39:33–41. doi: 10.1016/0006-3223(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Catapano LA, Manji HK. G protein-coupled receptors in major psychiatric disorders. Biochim Biophys Acta. 2007;1768:976–993. doi: 10.1016/j.bbamem.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/s0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- Chiu CS, et al. GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J Neurosci. 2005;25:3234–3245. doi: 10.1523/JNEUROSCI.3364-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy KH, van den Buuse M. Attenuated disruption of prepulse inhibition by dopaminergic stimulation after maternal deprivation and adolescent corticosterone treatment in rats. Eur Neuropsychopharmacol. 2007;18:1–13. doi: 10.1016/j.euroneuro.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Cilia J, et al. Reversal of isolation-rearing-induced PPI deficits by an alpha7 nicotinic receptor agonist. Psychopharmacology (Berl) 2005;182:214–219. doi: 10.1007/s00213-005-0069-5. [DOI] [PubMed] [Google Scholar]
- Cilia J, et al. (+/−) Ketamine-induced prepulse inhibition deficits of an acoustic startle response in rats are not reversed by antipsychotics. J Psychopharmacol. 2007;21:302–311. doi: 10.1177/0269881107077718. [DOI] [PubMed] [Google Scholar]
- Clapcote SJ, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Conti LH, et al. Effects of a typical and an atypical antipsychotic on the disruption of prepulse inhibition caused by corticotropin-releasing factor and by rat strain. Behav Neurosci. 2005;119:1052–1060. doi: 10.1037/0735-7044.119.4.1052. [DOI] [PubMed] [Google Scholar]
- Csomor PA, et al. Haloperidol differentially modulates prepulse inhibition and P50 suppression in healthy humans stratified for low and high gating levels. Neuropsychopharmacology. 2008;33:497–512. doi: 10.1038/sj.npp.1301421. [DOI] [PubMed] [Google Scholar]
- Cui C, et al. The beta3 nicotinic receptor subunit: a component of alpha-conotoxin MII-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J Neurosci. 2003;23:11045–11053. doi: 10.1523/JNEUROSCI.23-35-11045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culm KE, et al. Reduced G(i) and G(o) protein function in the rat nucleus accumbens attenuates sensorimotor gating deficits. Brain Res. 2003;982:12–18. doi: 10.1016/s0006-8993(03)02880-4. [DOI] [PubMed] [Google Scholar]
- Culm KE, et al. Repeated quinpirole treatment increases cAMP-dependent protein kinase activity and CREB phosphorylation in nucleus accumbens and reverses quinpirole-induced sensorimotor gating deficits in rats. Neuropsychopharmacology. 2004;29:1823–1830. doi: 10.1038/sj.npp.1300483. [DOI] [PubMed] [Google Scholar]
- Czyrak A, et al. 8-OHDPAT-induced disruption of prepulse inhibition in rats is attenuated by prolonged corticosterone treatment. Neuropsychopharmacology. 2003;28:1300–1310. doi: 10.1038/sj.npp.1300165. [DOI] [PubMed] [Google Scholar]
- Daenen EW, et al. Neonatal lesions in the amygdala or ventral hippocampus disrupt prepulse inhibition of the acoustic startle response; implications for an animal model of neurodevelopmental disorders like schizophrenia. Eur Neuropsychopharmacol. 2003;13:187–197. doi: 10.1016/s0924-977x(03)00007-5. [DOI] [PubMed] [Google Scholar]
- Dai H, et al. Social isolation stress significantly enhanced the disruption of prepulse inhibition in mice repeatedly treated with methamphetamine. Ann NY Acad Sci. 2004;1025:257–266. doi: 10.1196/annals.1316.032. [DOI] [PubMed] [Google Scholar]
- Dai H, et al. Blockage of histamine H1 receptor attenuates social isolation-induced disruption of prepulse inhibition: a study in H1 receptor gene knockout mice. Psychopharmacology (Berl) 2005;183:285–293. doi: 10.1007/s00213-005-0203-4. [DOI] [PubMed] [Google Scholar]
- Davis M. The mammalian startle response. In: Eaton RC, editor. Neural mechanisms of startle behavior. Plenum Press; New York: 1984. pp. 287–342. [Google Scholar]
- Davis M, et al. A primary acoustic startle circuit: lesion and stimulation studies. J Neurosci. 1982;2:791–805. doi: 10.1523/JNEUROSCI.02-06-00791.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day-Wilson KM, et al. Medial prefrontal cortex volume loss in rats with isolation rearing-induced deficits in prepulse inhibition of acoustic startle. Neuroscience. 2006;141:1113–1121. doi: 10.1016/j.neuroscience.2006.04.048. [DOI] [PubMed] [Google Scholar]
- de Jong IE, van den Buuse M. SCH 23390 in the prefrontal cortex enhances the effect of apomorphine on prepulse inhibition of rats. Neuropharmacology. 2006;51:438–446. doi: 10.1016/j.neuropharm.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Depoortere R, et al. Neurochemical, electrophysiological and pharmacological profiles of the selective inhibitor of the glycine transporter-1 SSR504734, a potential new type of antipsychotic. Neuropsychopharmacology. 2005;30:1963–1985. doi: 10.1038/sj.npp.1300772. [DOI] [PubMed] [Google Scholar]
- Depoortere R, et al. F15063, a compound with D2/D3 antagonist, 5-HT 1A agonist and D4 partial agonist properties. III. Activity in models of cognition and negative symptoms. Br J Pharmacol. 2007a;151:266–277. doi: 10.1038/sj.bjp.0707160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depoortere R, et al. F15063, a compound with D(2)/D(3) antagonist, 5-HT(1A) agonist and D(4) partial agonist properties: (II) Activity in models of positive symptoms of schizophrenia. Br J Pharmacol. 2007b;151:253–265. doi: 10.1038/sj.bjp.0707159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederich K, Koch M. Role of the pedunculopontine tegmental nucleus in sensorimotor gating and reward-related behavior in rats. Psychopharmacology. 2005;179:402–408. doi: 10.1007/s00213-004-2052-y. [DOI] [PubMed] [Google Scholar]
- Dirks A, et al. Reduced startle reactivity and plasticity in transgenic mice overexpressing corticotrophin-releasing hormone. Biol Psychiatry. 2002;51:583–590. doi: 10.1016/s0006-3223(01)01323-3. [DOI] [PubMed] [Google Scholar]
- Dirks A, et al. Reversal of startle gating deficits in transgenic mice overexpressing corticotrophin-releasing factor by antipsychotic drugs. Neuropsychopharmacology. 2003;28:1790–1708. doi: 10.1038/sj.npp.1300256. [DOI] [PubMed] [Google Scholar]
- Dolder CR, et al. Antipsychotic medication adherence: is there a difference between typical and atypical agents. Am J Psychiatry. 2002;159:103–108. doi: 10.1176/appi.ajp.159.1.103. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, et al. Clinical and sensorimotor gating effects of ketamin in normals. Neuropsychopharmacology. 2001;25:72–83. doi: 10.1016/S0893-133X(00)00240-2. [DOI] [PubMed] [Google Scholar]
- Duncan E, et al. Prepulse inhibition of acoustic startle in subjects with schizophrenia treated with olanzapine or haloperidol. Psychiatry Res. 2003a;120:1–12. doi: 10.1016/s0165-1781(03)00161-6. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, et al. Effect of treatment status on prepulse inhibition of acoustic startle in schizophrenia. Psychopharmacology (Berl) 2003b;167:63–71. doi: 10.1007/s00213-002-1372-z. [DOI] [PubMed] [Google Scholar]
- Duncan GE, et al. Deficits in sensorimotor gating and tests of social behavior in a genetic model of reduced NMDA receptor function. Behav Brain Res. 2004;153:507–519. doi: 10.1016/j.bbr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Duncan EJ, et al. Medication status affects the relationship of symptoms to prepulse inhibition of acoustic startle in schizophrenia. Psychiatry Res. 2006a;145:137–145. doi: 10.1016/j.psychres.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Duncan GE, et al. Typical and atypical antipsychotic drug effects on locomotor hyperactivity and deficits in sensorimotor gating in a genetic model of NMDA receptor hypofunction. Pharmacol Biochem Behav. 2006b;85:481–491. doi: 10.1016/j.pbb.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eells JB, et al. Early postnatal isolation reduces dopamine levels, elevates dopamine turnover and specifically disrupts prepulse inhibition in Nurr1-null heterozygous mice. Neuroscience. 2006;140:1117–1126. doi: 10.1016/j.neuroscience.2005.12.065. [DOI] [PubMed] [Google Scholar]
- Egashira N, et al. Disruption of the prepulse inhibition of the startle reflex in vasopressin V1b receptor knockout mice: reversal by antipsychotic drugs. Neuropsychopharmacology. 2005;30:1996–2005. doi: 10.1038/sj.npp.1300784. [DOI] [PubMed] [Google Scholar]
- Ehlers CL, et al. Increased alcohol drinking in isolate-housed alcohol-preferring rats. Behav Neurosci. 2007;121:111–119. doi: 10.1037/0735-7044.121.1.111. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, Cools AR. Early maternal deprivation and prepulse inhibition: the role of the postdeprivation environment. Pharmacol Biochem Behav. 2002;73:177–184. doi: 10.1016/s0091-3057(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, et al. Prepulse inhibition and latent inhibition: the role of dopamine in the medial prefrontal cortex. Neuroscience. 1996;75:535–542. doi: 10.1016/0306-4522(96)00307-7. [DOI] [PubMed] [Google Scholar]
- Ellenbroek BA, et al. Effects of JL13, a pyridobenzoxazepine with potential atypical antipsychotic activity, in animal models for schizophrenia. J Pharmacol Exp Ther. 2001;298:386–391. [PubMed] [Google Scholar]
- Ellenbroek BA, et al. The role of hippocampal dopamine receptors in prepulse inhibition. Eur J Neurosci. 2002b;15:1237–1243. doi: 10.1046/j.1460-9568.2002.01948.x. [DOI] [PubMed] [Google Scholar]
- Elmer GI, et al. Altered prepulse inhibition in rats treated prenatally with the antimitotic Ara-C: an animal model for sensorimotor gating deficits in schizophrenia. Psychopharmacology (Berl) 2004;174:177–189. doi: 10.1007/s00213-003-1757-7. [DOI] [PubMed] [Google Scholar]
- Erbel-Sieler C, et al. Behavioral and regulatory abnormalities in mice deficient in the NPAS1 and NPAS3 transcription factors. Proc Natl Acad Sci USA. 2004;101:13648–13653. doi: 10.1073/pnas.0405310101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt S, et al. Endogenous kynurenic acid disrupts prepulse inhibition. Biol Psychiatry. 2004;56:255–260. doi: 10.1016/j.biopsych.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Ewers M, et al. Associative and motor learning in 12-month-old transgenic APP+PS1 mice. Neurobiol Aging. 2006;27:1118–1128. doi: 10.1016/j.neurobiolaging.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Feifel D, Priebe K. Vasopressin-deficient rats exhibit sensorimotor gating deficits that are reversed by subchronic haloperidol. Biol Psychiatry. 2001;50:425–433. doi: 10.1016/s0006-3223(01)01100-3. [DOI] [PubMed] [Google Scholar]
- Feifel D, et al. Startle and sensorimotor gating in rats lacking CCK-A receptors. Neuropsychopharmacology. 2001;24:663–670. doi: 10.1016/S0893-133X(00)00235-9. [DOI] [PubMed] [Google Scholar]
- Feifel D, et al. Reversal of sensorimotor gating deficits in Brattleboro rats by acute administration of clozapine and a neurotensin agonist, but not haloperidol: a potential predictive model for novel antipsychotic effects. Neuropsychopharmacology. 2004;29:731–738. doi: 10.1038/sj.npp.1300378. [DOI] [PubMed] [Google Scholar]
- Fejgin K, et al. The atypical antipsychotic, aripiprazole, blocks phencyclidine-induced disruption of prepulse inhibition in mice. Psychopharmacology (Berl) 2007;191:377–385. doi: 10.1007/s00213-006-0658-y. [DOI] [PubMed] [Google Scholar]
- Ferguson SA, Cada AM. Spatial learning/memory and social and nonsocial behaviors in the spontaneously hypertensive, Wistar–Kyoto and Sprague–Dawley rat strains. Pharmacol Biochem Behav. 2004;77:583–594. doi: 10.1016/j.pbb.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Finamore TL, et al. Contributions of hippocampal cellular damage and NMDA receptor dysfunction to behavioral markers of schizophrenia. Int J Neurosci. 2001;109:61–70. doi: 10.3109/00207450108986525. [DOI] [PubMed] [Google Scholar]
- Fitting S, et al. Intrahippocampal injections of Tat: effects on prepulse inhibition of the auditory startle response in adult male rats. Pharmacol Biochem Behav. 2006a;84:189–196. doi: 10.1016/j.pbb.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Fitting S, et al. Neonatal hippocampal Tat injections: developmental effects on prepulse inhibition (PPI) of the auditory startle response. Int J Dev Neurosci. 2006b;24:275–283. doi: 10.1016/j.ijdevneu.2006.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, et al. Neonatal intrahippocampal glycoprotein 120 injection: the role of dopaminergic alterations in prepulse inhibition in adult rats. J Pharmacol Exp Ther. 2006c;318:1352–1358. doi: 10.1124/jpet.106.105742. [DOI] [PubMed] [Google Scholar]
- Flaten MA. Test-retest reliability of the somatosensory blink reflex and its inhibition. Int J Psychophysiol. 2002;45:261–265. doi: 10.1016/s0167-8760(02)00034-x. [DOI] [PubMed] [Google Scholar]
- Flood DG, et al. Variables affecting prepulse inhibition of the startle reflex and the response to antipsychotics in DBA/2NCrl mice. Psychopharmacology (Berl) 2008;195:203–211. doi: 10.1007/s00213-007-0894-9. [DOI] [PubMed] [Google Scholar]
- Fortier ME, et al. Effects of prenatal infection on prepulse inhibition in the rat depend on the nature of the infectious agent and the stage of pregnancy. Behav Brain Res. 2007;181:270–277. doi: 10.1016/j.bbr.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Fox GB, et al. Pharmacological properties of ABT-239 [4-(2-{2-[(2R)-2-Methylpyrrolidinyl]ethyl}-benzofuran-5-yl)benzonitrile]: II. Neurophysiological characterization and broad preclinical efficacy in cognition and schizophrenia of a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther. 2005;313:176–190. doi: 10.1124/jpet.104.078402. [DOI] [PubMed] [Google Scholar]
- Fradley RL, et al. STOP knockout and NMDA NR1 hypomorphic mice exhibit deficits in sensorimotor gating. Behav Brain Res. 2005;163:257–264. doi: 10.1016/j.bbr.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Frankland PW, et al. Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol Psychiatry. 2004;9:417–425. doi: 10.1038/sj.mp.4001432. [DOI] [PubMed] [Google Scholar]
- Frau R, et al. Effects of topiramate on the prepulse inhibition of the acoustic startle in rats. Neuropsychopharmacology. 2007;32:320–331. doi: 10.1038/sj.npp.1301115. [DOI] [PubMed] [Google Scholar]
- Freedman R, et al. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci USA. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg F, et al. Selective breeding for deficient sensorimotor gating is accompanied by increased perseveration in rats. Neuroscience. 2007;148:612–622. doi: 10.1016/j.neuroscience.2007.06.034. [DOI] [PubMed] [Google Scholar]
- Fujiwara N, et al. Effects of copper metabolism on neurological functions in Wistar and Wilson’s disease model rats. Biochem Biophys Res Commun. 2006;349:1079–1086. doi: 10.1016/j.bbrc.2006.08.139. [DOI] [PubMed] [Google Scholar]
- Futamura T, et al. Neonatal perturbation of neurotrophic signaling results in abnormal sensorimotor gating and social interaction in adults: implication for epidermal growth factor in cognitive development. Mol Psychiatry. 2003;8:19–29. doi: 10.1038/sj.mp.4001138. [DOI] [PubMed] [Google Scholar]
- Galici R, et al. A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J Pharmacol Exp Ther. 2005;315:1181–1187. doi: 10.1124/jpet.105.091074. [DOI] [PubMed] [Google Scholar]
- Garner B, et al. Early maternal deprivation reduces prepulse inhibition and impairs spatial learning ability in adulthood: no further effect of post-pubertal chronic corticosterone treatment. Behav Brain Res. 2007;176:323–332. doi: 10.1016/j.bbr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- George TP, et al. A preliminary study of the effects of cigarette smoking on prepulse inhibition in schizophrenia: involvement of nicotinic receptor mechanisms. Schizophr Res. 2006;87:307–315. doi: 10.1016/j.schres.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Swerdlow NR. Measurement of startle response, prepulse inhibition, and habituation. In: Crawley JN, Skolnick P, editors. Current protocols in neuroscience. Unit 8.7. Wiley; New York: 1998. pp. 1–15. [DOI] [PubMed] [Google Scholar]
- Geyer MA, et al. Isolation rearing of rats produces a deficit in prepulse inhibition of acoustic startle similar to that in schizophrenia. Biol Psychiatry. 1993;34:361–372. doi: 10.1016/0006-3223(93)90180-l. [DOI] [PubMed] [Google Scholar]
- Geyer MA, et al. Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology. 2001;156:117–154. doi: 10.1007/s002130100811. [DOI] [PubMed] [Google Scholar]
- Geyer MA, et al. Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry. 2002;7:1039–1053. doi: 10.1038/sj.mp.4001159. [DOI] [PubMed] [Google Scholar]
- Gizerian SS, et al. Neonatal neurosteroid administration results in development-specific alterations in prepulse inhibition and locomotor activity: neurosteroids alter prepulse inhibition and locomotor activity. Psychopharmacology (Berl) 2006;186:334–342. doi: 10.1007/s00213-006-0360-0. [DOI] [PubMed] [Google Scholar]
- Gogos A, Van den Buuse M. Estrogen and progesterone prevent disruption of prepulse inhibition by the serotonin-1A receptor agonist 8-hydroxy-2-dipropylaminotetralin. J Pharmacol Exp Ther. 2004;309:267–274. doi: 10.1124/jpet.103.061432. [DOI] [PubMed] [Google Scholar]
- Gogos A, et al. Oestrogen modulation of the effect of 8-OH-DPAT on prepulse inhibition: effects of aromatase deficiency and castration in mice. Psychopharmacology (Berl) 2006;188:100–110. doi: 10.1007/s00213-006-0472-6. [DOI] [PubMed] [Google Scholar]
- Gomez-Wong E, et al. Sensory modulation of the blink reflex in patients with blepharospasm. Arch Neurol. 1998;55:1233–1237. doi: 10.1001/archneur.55.9.1233. [DOI] [PubMed] [Google Scholar]
- Goto K, et al. Reduced prepulse inhibition in rats with entorhinal cortex lesions. Behav Brain Res. 2002;134:201–207. doi: 10.1016/s0166-4328(02)00039-6. [DOI] [PubMed] [Google Scholar]
- Goto K, et al. Involvement of nucleus accumbens dopaminergic transmission in acoustic startle: observations concerning prepulse inhibition in rats with entorhinal cortex lesions. Psychiatry Clin Neurosci. 2004;58:441–445. doi: 10.1111/j.1440-1819.2004.01281.x. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gould TD, Gottesman II. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav. 2006;5:113–119. doi: 10.1111/j.1601-183X.2005.00186.x. [DOI] [PubMed] [Google Scholar]
- Gould TJ, et al. Sensorimotor gating deficits in transgenic mice expressing a constitutively active form of Gs alpha. Neuropsychopharmacology. 2004;29:494–501. doi: 10.1038/sj.npp.1300309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham F. The more or less startling effects of weak prestimuli. Psychophysiology. 1975;12:238–248. doi: 10.1111/j.1469-8986.1975.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Gray JA. The contents of consciousness: a neuropsychological conjecture. Behav Brain Sci. 1995;18:659–722. [Google Scholar]
- Greenwood TA, et al. The Consortium on the Genetics of Schizophrenia (COGS): Preliminary heritability analyses of endophenotypic measures for schizophrenia. Arch Gen Psychiatry. 2007;33:33–48. doi: 10.1001/archpsyc.64.11.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, et al. Effects of experimental context and explicit threat cues on acoustic startle in Vietnam veterans with posttraumatic stress disorder. Biol Psychiatry. 1998;44:1027–1036. doi: 10.1016/s0006-3223(98)00034-1. [DOI] [PubMed] [Google Scholar]
- Grobin AC, et al. Perinatal allopregnanolone influences prefrontal cortex structure, connectivity and behavior in adult rats. Neuroscience. 2006;138:809–819. doi: 10.1016/j.neuroscience.2005.12.026. [DOI] [PubMed] [Google Scholar]
- Grobstein P. Making the unconscious conscious: a bidirectional bridge between neuroscience/ cognitive science and psychotherapy. Cortex. 2005;41:663–668. doi: 10.1016/s0010-9452(08)70283-1. [DOI] [PubMed] [Google Scholar]
- Groenink L, et al. CRF(1) not glucocorticoid receptors mediate prepulse inhibition deficits in mice overexpressing CRF. Biol Psychiatry. 2008;63:360–368. doi: 10.1016/j.biopsych.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Harris LW, et al. Long-term behavioural, molecular and morphological effects of neonatal NMDA receptor antagonism. Eur J Neurosci. 2003;18:1706–1710. doi: 10.1046/j.1460-9568.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- Harrison SM, et al. LPA1 receptor-deficient mice have phenotypic changes observed in psychiatric disease. Mol Cell Neurosci. 2003;24:1170–1179. doi: 10.1016/j.mcn.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Harte MK, et al. Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm. 2007;114:893–898. doi: 10.1007/s00702-007-0627-6. [DOI] [PubMed] [Google Scholar]
- Hauser J, et al. Hippocampal alpha5 subunit-containing GABAA receptors modulate the expression of prepulse inhibition. Mol Psychiatry. 2005;10:201–207. doi: 10.1038/sj.mp.4001554. [DOI] [PubMed] [Google Scholar]
- Hauser J, et al. Prenatal dexamethasone exposure, postnatal development, and adulthood prepulse inhibition and latent inhibition in Wistar rats. Behav Brain Res. 2006;175:51–61. doi: 10.1016/j.bbr.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Hazlett EA, et al. Deficient attentional modulation of the startle response in patients with schizotypal personality disorder. Am J Psychiatry. 2003;160:1621–1626. doi: 10.1176/appi.ajp.160.9.1621. [DOI] [PubMed] [Google Scholar]
- Heldt SA, Ressler KJ. Lesions of the habenula produce stress-and dopamine-dependent alterations in prepulse inhibition and locomotion. Brain Res. 2006;1073–1074:229–239. doi: 10.1016/j.brainres.2005.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt SA, et al. Prepulse inhibition deficits in GAD65 knockout mice and the effect of antipsychotic treatment. Neuropsychopharmacology. 2004;29:1610–1619. doi: 10.1038/sj.npp.1300468. [DOI] [PubMed] [Google Scholar]
- Henck JW, et al. Growth and development in rats given recombinant human epidermal growth factor(1–48) as neonates. Toxicol Sci. 2001;62:80–91. doi: 10.1093/toxsci/62.1.80. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, et al. High glycine levels are associated with prepulse inhibition deficits in chronic schizophrenia patients. Schizophr Res. 2007;91:14–21. doi: 10.1016/j.schres.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Hoenig K, et al. Impaired prepulse inhibition of acoustic startle in obsessive-compulsive disorder. Biol Psychiatry. 2005;57:1153–1158. doi: 10.1016/j.biopsych.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Hohnadel E, et al. Galantamine and donepezil attenuate pharmacologically induced deficits in prepulse inhibition in rats. Neuropharmacology. 2007;52:542–551. doi: 10.1016/j.neuropharm.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, et al. Behavioral characterization of dopamine D5 receptor null mutant mice. Behav Neurosci. 2001;115:1129–1144. [PubMed] [Google Scholar]
- Hong LE, et al. Independent domains of inhibitory gating in schizophrenia and the effect of stimulus interval. Am J Psychiatry. 2007;164:61–65. doi: 10.1176/ajp.2007.164.1.61. [DOI] [PubMed] [Google Scholar]
- Howland JG, et al. Delayed onset of prepulse inhibition deficits following kainic acid treatment on postnatal day 7 in rats. Eur J Neurosci. 2004a;20:2639–2648. doi: 10.1111/j.1460-9568.2004.03731.x. [DOI] [PubMed] [Google Scholar]
- Howland JG, et al. Electrical stimulation of the hippocampus disrupts prepulse inhibition in rats: frequency-and site-dependent effects. Behav Brain Res. 2004b;152:187–197. doi: 10.1016/j.bbr.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Howland JG, et al. Kindling of basolateral amygdala but not ventral hippocampus or perirhinal cortex disrupts sensorimotor gating in rats. Behav Brain Res. 2007;177:30–36. doi: 10.1016/j.bbr.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Hsieh MH, et al. Effects of background and prepulse characteristics on prepulse inhibition and prepulse facilitation: implications for neuropsychiatric research. Biol Psychiatry. 2006;59(6):555–559. doi: 10.1016/j.biopsych.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Husum H, et al. Early maternal deprivation alters hippocampal levels of neuropeptide Y and calcitonin-gene related peptide in adult rats. Neuropharmacology. 2002;42:798–806. doi: 10.1016/s0028-3908(02)00038-2. [DOI] [PubMed] [Google Scholar]
- Hutchison KE, Swift R. Effect of d-amphetamine on prepulse inhibition of the startle reflex in humans. Psychopharmacology (Berl) 1999;143:394–400. doi: 10.1007/s002130050964. [DOI] [PubMed] [Google Scholar]
- Inada K, et al. Antisense hippocampal knockdown of NMDA-NR1 by HVJ-liposome vector induces deficit of prepulse inhibition but not of spatial memory. Neurosci Res. 2003;45:473–481. doi: 10.1016/s0168-0102(03)00012-9. [DOI] [PubMed] [Google Scholar]
- Irintchev A, et al. Impairment of sensorimotor gating in mice deficient in the cell adhesion molecule L1 or its close homologue, CHL1. Brain Res. 2004;1029:131–134. doi: 10.1016/j.brainres.2004.09.042. [DOI] [PubMed] [Google Scholar]
- Iso H, et al. Environmental change during postnatal development alters behaviour, cognitions and neurogenesis of mice. Behav Brain Res. 2007;179:90–98. doi: 10.1016/j.bbr.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Jaworski DM, et al. Prepulse inhibition and fear-potentiated startle are altered in tissue inhibitor of metalloproteinase-2 (TIMP-2) knockout mice. Brain Res. 2005;1051:81–89. doi: 10.1016/j.brainres.2005.05.057. [DOI] [PubMed] [Google Scholar]
- Jones CK, Shannon HE. Lesions of the laterodorsal tegmental nucleus disrupt prepulse inhibition of the acoustic startle reflex. Pharmacol Biochem Behav. 2004;78:229–237. doi: 10.1016/j.pbb.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Jones CK, et al. Pharmacologic interactions between the muscarinic cholinergic and dopaminergic systems in the modulation of prepulse inhibition in rats. J Pharmacol Exp Ther. 2005;312:1055–1063. doi: 10.1124/jpet.104.075887. [DOI] [PubMed] [Google Scholar]
- Jongen-Relo AL, et al. The prenatal methylazoxymethanol acetate treatment: a neurodevelopmental animal model for schizophrenia. Behav Brain Res. 2004;149:159–181. doi: 10.1016/s0166-4328(03)00228-6. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, et al. Menstrual cycle phase effects on prepulse inhibition of acoustic startle. Psychophysiology. 2004;41:401–406. doi: 10.1111/1469-8986.2004.00166.x. [DOI] [PubMed] [Google Scholar]
- Kaifu T, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest. 2003;111:323–332. doi: 10.1172/JCI16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanabus M, et al. Temporal order judgement for auditory and visual stimuli. Acta Neurobiol Exp. 2002;62:263–270. doi: 10.55782/ane-2002-1443. [DOI] [PubMed] [Google Scholar]
- Kanes SJ, et al. Rolipram: a specific phosphodiesterase 4 inhibitor with potential antipsychotic activity. Neuroscience. 2007;144:239–246. doi: 10.1016/j.neuroscience.2006.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karper LP, et al. Preliminary evidence of an association between sensorimotor gating and distractibility in psychosis. J Neuropsychiatry Clin Neurosci. 1996;8:60–66. doi: 10.1176/jnp.8.1.60. [DOI] [PubMed] [Google Scholar]
- Kedzior KK, Martin-Iverson MT. Attention-dependent reduction in prepulse inhibition of the startle reflex in cannabis users and schizophrenia patients—a pilot study. Eur J Pharmacol. 2007;560:176–182. doi: 10.1016/j.ejphar.2007.01.032. [DOI] [PubMed] [Google Scholar]
- Kelly MP, et al. Constitutive activation of Galphas within forebrain neurons causes deficits in sensorimotor gating because of PKA-dependent decreases in cAMP. Neuropsychopharmacology. 2007;32:577–588. doi: 10.1038/sj.npp.1301099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura J, Harada O. Recovery curves of the blink reflex during wakefulness and sleep. J Neurol. 1976;213:189–198. doi: 10.1007/BF00312869. [DOI] [PubMed] [Google Scholar]
- Kinkead B, et al. Neurotensin-deficient mice have deficits in prepulse inhibition: restoration by clozapine but not haloperidol, olanzapine, or quetiapine. J Pharmacol Exp Ther. 2005;315:256–264. doi: 10.1124/jpet.105.087437. [DOI] [PubMed] [Google Scholar]
- Kinney GG, et al. The glycine transporter type 1 inhibitor N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine potentiates NMDA receptor-mediated responses in vivo and produces an antipsychotic profile in rodent behavior. J Neurosci. 2003;23:7586–7591. doi: 10.1523/JNEUROSCI.23-20-07586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney GG, et al. A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther. 2005;313:199–206. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- Klamer D, et al. Phencyclidine-induced behaviour in mice prevented by methylene blue. Basic Clin Pharmacol Toxicol. 2004a;94:65–72. doi: 10.1111/j.1742-7843.2004.pto940203.x. [DOI] [PubMed] [Google Scholar]
- Klamer D, et al. The neuronal selective nitric oxide synthase inhibitor, Nomega-propyl-l-arginine, blocks the effects of phencyclidine on prepulse inhibition and locomotor activity in mice. Eur J Pharmacol. 2004b;503:103–107. doi: 10.1016/j.ejphar.2004.09.042. [DOI] [PubMed] [Google Scholar]
- Klamer D, et al. Activation of a nitric-oxide-sensitive cAMP pathway with phencyclidine: elevated hippocampal cAMP levels are temporally associated with deficits in prepulse inhibition. Psychopharmacology (Berl) 2005a;179:479–488. doi: 10.1007/s00213-004-2051-z. [DOI] [PubMed] [Google Scholar]
- Klamer D, et al. Effects of phencyclidine on acoustic startle and prepulse inhibition in neuronal nitric oxide synthase deficient mice. Eur Neuropsychopharmacol. 2005b;15:587–590. doi: 10.1016/j.euroneuro.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Klejbor I, et al. Fibroblast growth factor receptor signaling affects development and function of dopamine neurons—inhibition results in a schizophrenia-like syndrome in transgenic mice. J Neurochem. 2006;97:1243–1258. doi: 10.1111/j.1471-4159.2006.03754.x. [DOI] [PubMed] [Google Scholar]
- Koch M, Schnitzler HU. The acoustic startle response in rats—circuits mediating evocation, inhibition and potentiation. Behav Brain Res. 1997;89:35–49. doi: 10.1016/s0166-4328(97)02296-1. [DOI] [PubMed] [Google Scholar]
- Koch M, et al. Cholinergic neurons in the pedunculopontine tegmental nucleus are involved in the mediation of prepulse inhibition of the acoustic startle response in the rat. Exp Brain Res. 1993;97:71–82. doi: 10.1007/BF00228818. [DOI] [PubMed] [Google Scholar]
- Koenig JI, et al. Prenatal exposure to a repeated variable stress paradigm elicits behavioral and neuroendocrinological changes in the adult offspring: potential relevance to schizophrenia. Behav Brain Res. 2005;156:251–261. doi: 10.1016/j.bbr.2004.05.030. [DOI] [PubMed] [Google Scholar]
- Koh HY, et al. Deficits in social behavior and sensorimotor gating in mice lacking phospholipase Cbeta1. Genes Brain Behav. 2008;7:120–128. doi: 10.1111/j.1601-183X.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- Kraepelin E, Robertson GM. Dementia praecox and paraphrenia. Livingstone Edinburgh: 1919. [Google Scholar]
- Krebs-Thomson K, et al. The roles of 5-HT1A and 5-HT2 receptors in the effects of 5-MeO-DMT on locomotor activity and prepulse inhibition in rats. Psychopharmacology (Berl) 2006;189:319–329. doi: 10.1007/s00213-006-0566-1. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Normalization of information processing deficits in schizophrenia with clozapine. Am J Psychiatry. 1999;156:1046–1051. doi: 10.1176/ajp.156.7.1046. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Influence of cigarette smoking on prepulse inhibition of the acoustic startle response in schizophrenia. Hum Psychopharmacol. 2001;16:321–326. doi: 10.1002/hup.286. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Prepulse inhibition of the startle response in risperidone-treated patients: comparison with typical antipsychotics. Schizophr Res. 2002;55:139–146. doi: 10.1016/s0920-9964(01)00276-6. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Neural correlates of tactile prepulse inhibition: a functional MRI study in normal and schizophrenic subjects. Psychiatry Res. 2003a;122:99–113. doi: 10.1016/s0925-4927(02)00123-3. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Effects of acute procyclidine administration on prepulse inhibition of the startle response in schizophrenia: a double-blind, placebo-controlled study. J Psychopharmacol. 2003b;17:89–95. doi: 10.1177/0269881103017001710. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Sex differences in prepulse inhibition deficits in chronic schizophrenia. Schizophr Res. 2004;69:219–235. doi: 10.1016/j.schres.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Association between violent behaviour and impaired prepulse inhibition of the startle response in antisocial personality disorder and schizophrenia. Behav Brain Res. 2005a;158:159–166. doi: 10.1016/j.bbr.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Lack of association between prepulse inhibition and antisaccadic deficits in chronic schizophrenia: implications for identification of schizophrenia endophenotypes. J Psychiatr Res. 2005b;39:227–240. doi: 10.1016/j.jpsychires.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Structural brain correlates of prepulse inhibition of the acoustic startle response in healthy humans. Neuroimage. 2005c;26:1052–1058. doi: 10.1016/j.neuroimage.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Reduced prepulse inhibition in unaffected siblings of schizophrenia patients. Psychophysiology. 2005d;42:588–594. doi: 10.1111/j.1469-8986.2005.00346.x. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. A fMRI investigation of startle gating deficits in schizophrenia patients treated with typical or atypical antipsychotics. Int J Neuropsychopharmacol. 2007a;10:463–477. doi: 10.1017/S1461145706007139. [DOI] [PubMed] [Google Scholar]
- Kumari V, et al. Startle gating in antipsychotic-naive first episode schizophrenia patients: one ear is better than two. Psychiatry Res. 2007b;151:21–28. doi: 10.1016/j.psychres.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Kusljic S, van den Buuse M. Functional dissociation between serotonergic pathways in dorsal and ventral hippocampus in psychotomimetic drug-induced locomotor hyperactivity and prepulse inhibition in rats. Eur J Neurosci. 2004;20:3424–3432. doi: 10.1111/j.1460-9568.2004.03804.x. [DOI] [PubMed] [Google Scholar]
- Kusljic S, van den Buuse M. Differential involvement of 5-HT projections within the amygdala in prepulse inhibition but not in psychotomimetic drug-induced hyperlocomotion. Behav Brain Res. 2006;168:74–82. doi: 10.1016/j.bbr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Kusljic S, et al. Differential role of serotonergic projections arising from the dorsal and median raphe nuclei in locomotor hyperactivity and prepulse inhibition. Neuropsychopharmacology. 2003;28:2138–2147. doi: 10.1038/sj.npp.1300277. [DOI] [PubMed] [Google Scholar]
- Kusljic S, et al. Effects of haloperidol and clozapine on sensorimotor gating deficits induced by 5-hydroxytryptamine depletion in the brain. Br J Pharmacol. 2006;147:800–807. doi: 10.1038/sj.bjp.0706641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacro JP, et al. Prevalence of and risk factors for medication nonadherence in patients with schizophrenia: a comprehensive review of recent literature. J Clin Psychiatry. 2002;63:892–909. doi: 10.4088/jcp.v63n1007. [DOI] [PubMed] [Google Scholar]
- Lahdesmaki J, et al. Alpha2A-adrenoceptors are important modulators of the effects of d-amphetamine on startle reactivity and brain monoamines. Neuropsychopharmacology. 2004;29:1282–1293. doi: 10.1038/sj.npp.1300428. [DOI] [PubMed] [Google Scholar]
- Laplante F, et al. Alterations in behavioral responses to a cholinergic agonist in post-pubertal rats with neonatal ventral hippocampal lesions: relationship to changes in muscarinic receptor levels. Neuropsychopharmacology. 2005;30:1076–1087. doi: 10.1038/sj.npp.1300640. [DOI] [PubMed] [Google Scholar]
- Le Pen G, Moreau JL. Disruption of prepulse inhibition of startle reflex in a neurodevelopmental model of schizophrenia: reversal by clozapine, olanzapine and risperidone but not by haloperidol. Neuropsychopharmacology. 2002;27:1–11. doi: 10.1016/S0893-133X(01)00383-9. [DOI] [PubMed] [Google Scholar]
- Le Pen G, et al. Prepulse inhibition deficits of the startle reflex in neonatal ventral hippocampal-lesioned rats: reversal by glycine and a glycine transporter inhibitor. Biol Psychiatry. 2003;54:1162–1170. doi: 10.1016/s0006-3223(03)00374-3. [DOI] [PubMed] [Google Scholar]
- Le Pen G, et al. Peri-pubertal maturation after developmental disturbance: a model for psychosis onset in the rat. Neuroscience. 2006;143:395–405. doi: 10.1016/j.neuroscience.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Leppert M, et al. Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N Engl J Med. 1990;322:904–908. doi: 10.1056/NEJM199003293221306. [DOI] [PubMed] [Google Scholar]
- Leumann L, et al. Effects of typical and atypical antipsychotics on prepulse inhibition and latent inhibition in chronic schizophrenia. Biol Psychiatry. 2002;52:729–739. doi: 10.1016/s0006-3223(02)01344-6. [DOI] [PubMed] [Google Scholar]
- Libet B, et al. Subjective referral of the timing for a conscious sensory experience. Brain. 1979;102:192–224. doi: 10.1093/brain/102.1.193. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Light GA, Braff DL. Mismatch negativity deficits are associated with poor functioning in schizophrenia patients. Arch Gen Psychiatry. 2005;62:127–136. doi: 10.1001/archpsyc.62.2.127. [DOI] [PubMed] [Google Scholar]
- Light GA, et al. Probing cortical-cortical interactions that underlie the multiple sensory, cognitive, and “real-world” functional deficits in schizophrenia. Behav Brain Sci. 2004;27:799. [Google Scholar]
- Light GA, et al. One year stability of neurophysiological and cognitive endophenotypes of schizophrenia. Proc Am Col Neuropsychopharmacology. 2007a in press. [Google Scholar]
- Light GA, et al. Prepulse inhibition of startle is positively associated with higher order cognition in women. Abstr Soc Neurosciences 806.17 2007b [Google Scholar]
- Light GA, et al. Preattentive sensory processing is associated with cognitive and psychosocial functioning in healthy adults. J Cog Neurosci. 2008;7:120–128. doi: 10.1162/jocn.2007.19.10.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligneau X, et al. Brain histamine and schizophrenia: potential therapeutic applications of H3-receptor inverse agonists studied with BF2.649. Biochem Pharmacol. 2007;73:1215–1224. doi: 10.1016/j.bcp.2007.01.023. [DOI] [PubMed] [Google Scholar]
- Linn GS, et al. Reversal of phencyclidine-induced prepulse inhibition deficits by clozapine in monkeys. Psychopharmacology (Berl) 2003;169:234–239. doi: 10.1007/s00213-003-1533-8. [DOI] [PubMed] [Google Scholar]
- Lipina T, et al. Modulators of the glycine site on NMDA receptors, D-serine and ALX 5407, display similar beneficial effects to clozapine in mouse models of schizophrenia. Psychopharmacology (Berl) 2005;179:54–67. doi: 10.1007/s00213-005-2210-x. [DOI] [PubMed] [Google Scholar]
- Lipina T, et al. The ampakine CX546 restores the prepulse inhibition and latent inhibition deficits in mGluR5-deficient mice. Neuropsychopharmacology. 2007;32:745–756. doi: 10.1038/sj.npp.1301191. [DOI] [PubMed] [Google Scholar]
- Lipska BK, et al. Neonatal excitotoxic hippocampal damage in rats causes post-pubertal changes in prepulse inhibition of startle and its disruption by apomorphine. Psychopharmacology. 1995;122:35–43. doi: 10.1007/BF02246439. [DOI] [PubMed] [Google Scholar]
- Long LE, et al. Cannabidiol reverses MK-801-induced disruption of prepulse inhibition in mice. Neuropsychopharmacology. 2006;31:795–803. doi: 10.1038/sj.npp.1300838. [DOI] [PubMed] [Google Scholar]
- Lovic V, Fleming AS. Artificially-reared female rats show reduced prepulse inhibition and deficits in the attentional set shifting task-reversal of effects with maternal-like licking stimulation. Behav Brain Res. 2004;148:209–219. doi: 10.1016/s0166-4328(03)00206-7. [DOI] [PubMed] [Google Scholar]
- Ludewig K, Vollenweider FX. Impaired sensorimotor gating in schizophrenia with deficit and with nondeficit syndrome. Swiss Med Wkly. 2002;132:159–165. doi: 10.4414/smw.2002.09873. [DOI] [PubMed] [Google Scholar]
- Ludewig K, et al. Stability of the acoustic startle reflex, prepulse inhibition, and habituation in schizophrenia. Schizophr Res. 2002;55:129–137. doi: 10.1016/s0920-9964(01)00198-0. [DOI] [PubMed] [Google Scholar]
- Ludewig K, et al. Deficits in prepulse inhibition and habituation in never-medicated, first-episode schizophrenia. Biol Psychiatry. 2003;54:121–128. doi: 10.1016/s0006-3223(02)01925-x. [DOI] [PubMed] [Google Scholar]
- Ma J, Leung LS. Schizophrenia-like behavioral changes after partial hippocampal kindling. Brain Res. 2004;997:111–118. doi: 10.1016/j.brainres.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Ma J, Leung LS. The supramammillo-septal-hippocampal pathway mediates sensorimotor gating impairment and hyperlocomotion induced by MK-801 and ketamine in rats. Psychopharmacology (Berl) 2007;191:961–974. doi: 10.1007/s00213-006-0667-x. [DOI] [PubMed] [Google Scholar]
- Ma J, et al. The medial septum mediates impairment of prepulse inhibition of acoustic startle induced by a hippocampal seizure or phencyclidine. Behav Brain Res. 2004;155:153–166. doi: 10.1016/j.bbr.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Mackeprang T, et al. Effects of antipsychotics on prepulse inhibition of the startle response in drug-naive schizophrenic patients. Biol Psychiatry. 2002;52:863–873. doi: 10.1016/s0006-3223(02)01409-9. [DOI] [PubMed] [Google Scholar]
- Malone DT, et al. The effect of SR 141716 and apomorphine on sensorimotor gating in Swiss mice. Pharmacol Biochem Behav. 2004;77:839–845. doi: 10.1016/j.pbb.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology. 1989;2:299–308. doi: 10.1016/0893-133x(89)90035-3. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, et al. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology (Berl) 1988;94:507–514. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- Marquis KL, et al. WAY-163909 [(7bR,10aR)-1,2,3,4,8,9,10,10a-octahydro-7bH-cyclopenta-[b][1,4]diazepino[6,7,1hi]indole]: a novel 5-hydroxytryptamine 2C receptor-selective agonist with preclinical antipsychotic-like activity. J Pharmacol Exp Ther. 2007;320:486–496. doi: 10.1124/jpet.106.106989. [DOI] [PubMed] [Google Scholar]
- McCool MF, et al. Increased auditory startle response and reduced prepulse inhibition of startle in transgenic mice expressing a double mutant form of amyloid precursor protein. Brain Res. 2003;994:99–106. doi: 10.1016/j.brainres.2003.09.025. [DOI] [PubMed] [Google Scholar]
- McDonald MP, et al. Motor deficits in fibroblast growth factor receptor-3 null mutant mice. Behav Pharmacol. 2001;12:477–486. doi: 10.1097/00008877-200111000-00009. [DOI] [PubMed] [Google Scholar]
- McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Brit J Med Psychol. 1961;34:103–116. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- Meincke U, et al. Prepulse inhibition of the acoustically evoked startle reflex in patients with an acute schizophrenic psychosis—a longitudinal study. Eur Arch Psychiatry Clin Neurosci. 2004;254:415–421. doi: 10.1007/s00406-004-0523-0. [DOI] [PubMed] [Google Scholar]
- Metzger KL, et al. Pharmacokinetic and behavioral characterization of a long-term antipsychotic delivery system in rodents and rabbits. Psychopharmacology (Berl) 2007;190:201–211. doi: 10.1007/s00213-006-0616-8. [DOI] [PubMed] [Google Scholar]
- Minassian A, et al. The relationship between sensorimotor gating and clinical improvement in acutely ill schizophrenia patients. Schizophr Res. 2007;89:225–231. doi: 10.1016/j.schres.2006.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa T, et al. Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci USA. 2003;100:8987–8992. doi: 10.1073/pnas.1432926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr D, et al. Accumbal dopamine D2 receptors are important for sensorimotor gating in C3H mice. Neuroreport. 2007;18:1493–1497. doi: 10.1097/WNR.0b013e3282e9a863. [DOI] [PubMed] [Google Scholar]
- Moy SS, et al. Amphetamine-induced disruption of prepulse inhibition in mice with reduced NMDA receptor function. Brain Res. 2006;1089:186–194. doi: 10.1016/j.brainres.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Mukai J, et al. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat Genet. 2004;36:725–731. doi: 10.1038/ng1375. [DOI] [PubMed] [Google Scholar]
- Murphy CA, et al. Latent inhibition, but not prepulse inhibition, is reduced during withdrawal from an escalating dosage schedule of amphetamine. Behav Neurosci. 2001;115:1247–1256. doi: 10.1037//0735-7044.115.6.1247. [DOI] [PubMed] [Google Scholar]
- Myers KM, et al. Partial reversal of phencyclidine-induced impairment of prepulse inhibition by secretin. Biol Psychiatry. 2005;58:67–73. doi: 10.1016/j.biopsych.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Nagai H, et al. Antipsychotics improve Delta9-tetrahydrocannabinol-induced impairment of the prepulse inhibition of the startle reflex in mice. Pharmacol Biochem Behav. 2006;84:330–336. doi: 10.1016/j.pbb.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Nagel J, et al. Effects of an adenosine A2A receptor blockade in the nucleus accumbens on locomotion, feeding, and prepulse inhibition in rats. Synapse. 2003;49:279–286. doi: 10.1002/syn.10240. [DOI] [PubMed] [Google Scholar]
- Nunes Mamede Rosa ML, et al. Isolation-induced changes in ultrasonic vocalization, fear-potentiated startle and prepulse inhibition in rats. Neuropsychobiology. 2005;51:248–255. doi: 10.1159/000085820. [DOI] [PubMed] [Google Scholar]
- Nyffeler M, et al. Maternal immune activation during pregnancy increases limbic GABAA receptor immunoreactivity in the adult offspring: implications for schizophrenia. Neuroscience. 2006;143:51–62. doi: 10.1016/j.neuroscience.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Ojima T, et al. Effects of serotonin-dopamine antagonists on prepulse inhibition and neurotransmitter contents in the rat cortex. Neurosci Lett. 2004;366:130–134. doi: 10.1016/j.neulet.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Ong JC, et al. An investigation of the efficacy of mood stabilizers in rodent models of prepulse inhibition. J Pharmacol Exp Ther. 2005;315:1163–1171. doi: 10.1124/jpet.105.090845. [DOI] [PubMed] [Google Scholar]
- Oranje B, et al. Effects of typical and atypical antipsychotics on the prepulse inhibition of the startle reflex in patients with schizophrenia. J Clin Psychopharmacol. 2002;22:359–365. doi: 10.1097/00004714-200208000-00005. [DOI] [PubMed] [Google Scholar]
- Ouagazzal AM, et al. Drug-induced potentiation of prepulse inhibition of acoustic startle reflex in mice: a model for detecting antipsychotic activity. Psychopharmacology (Berl) 2001a;156:273–283. doi: 10.1007/s002130100763. [DOI] [PubMed] [Google Scholar]
- Ouagazzal AM, et al. Effect of LSD on prepulse inhibition and spontaneous behavior in the rat. a pharmacological analysis and comparison between two rat strains. Neuropsychopharmacology. 2001b;25:565–575. doi: 10.1016/S0893-133X(01)00282-2. [DOI] [PubMed] [Google Scholar]
- Ozawa K, et al. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Paine TA, et al. Sensitivity of the five-choice serial reaction time task to the effects of various psychotropic drugs in Sprague-Dawley rats. Biol Psychiatry. 2007;62:687–693. doi: 10.1016/j.biopsych.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Palmer AA, et al. Prepulse startle deficit in the Brown Norway rat: a potential genetic model. Behav Neurosci. 2000;114:374–388. doi: 10.1037//0735-7044.114.2.374. [DOI] [PubMed] [Google Scholar]
- Palmer AA, et al. Prenatal protein deprivation in rats induces changes in prepulse inhibition and NMDA receptor binding. Brain Res. 2004;996:193–201. doi: 10.1016/j.brainres.2003.09.077. [DOI] [PubMed] [Google Scholar]
- Palsson E, et al. The amino acid l-lysine blocks the disruptive effect of phencyclidine on prepulse inhibition in mice. Psychopharmacology (Berl) 2007;192:9–15. doi: 10.1007/s00213-006-0683-x. [DOI] [PubMed] [Google Scholar]
- Park C, et al. Deletion in Catna2, encoding alpha N-catenin, causes cerebellar and hippocampal lamination defects and impaired startle modulation. Nat Genet. 2002;31:279–284. doi: 10.1038/ng908. [DOI] [PubMed] [Google Scholar]
- Park WK, et al. KKHA-761, a potent D3 receptor antagonist with high 5-HT1A receptor affinity, exhibits antipsychotic properties in animal models of schizophrenia. Pharmacol Biochem Behav. 2005;82:361–372. doi: 10.1016/j.pbb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Paylor R, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci USA. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peak H. Time order error in successive judgements and in reflexes. I. Inhibition of the judgement and the reflex. J Exper Psychol. 1939;25:535–565. [Google Scholar]
- Peleg-Raibstein D, et al. Differential effects on prepulse inhibition of withdrawal from two different repeated administration schedules of amphetamine. Int J Neuropsychopharmacol. 2006a;9:737–749. doi: 10.1017/S1461145706006493. [DOI] [PubMed] [Google Scholar]
- Peleg-Raibstein D, et al. Withdrawal from repeated amphetamine administration leads to disruption of prepulse inhibition but not to disruption of latent inhibition. J Neural Transm. 2006b;113:1323–1336. doi: 10.1007/s00702-005-0390-5. [DOI] [PubMed] [Google Scholar]
- Perry W, Braff DL. Information-processing deficits and thought disorder in schizophrenia. Am J Psychiatry. 1994;151:363–367. doi: 10.1176/ajp.151.3.363. [DOI] [PubMed] [Google Scholar]
- Perry W, et al. Sensorimotor gating deficits in bipolar disorder patients with acute psychotic mania. Biol Psychiatry. 2001:50418–424. doi: 10.1016/s0006-3223(01)01184-2. [DOI] [PubMed] [Google Scholar]
- Perry W, et al. Information processing deficits in acutely psychotic schizophrenia patients medicated and unmedicated at the time of admission. Am J Psychiatry. 2002;159:1375–1381. doi: 10.1176/appi.ajp.159.8.1375. [DOI] [PubMed] [Google Scholar]
- Perry W, et al. Prepulse inhibition in patients with non-psychotic major depressive disorder. J Affect Disord. 2004;81:179–184. doi: 10.1016/S0165-0327(03)00157-5. [DOI] [PubMed] [Google Scholar]
- Petitto JM, et al. IL-2/15 receptor-beta gene deletion alters neurobehavioral performance. Brain Res. 2002a;929:218–225. doi: 10.1016/s0006-8993(01)03393-5. [DOI] [PubMed] [Google Scholar]
- Petitto JM, et al. Relationship between the development of autoimmunity and sensorimotor gating in MRL-lpr mice with reduced IL-2 production. Neurosci Lett. 2002b;328:304–308. doi: 10.1016/s0304-3940(02)00545-1. [DOI] [PubMed] [Google Scholar]
- Pijlman FT, et al. Behavioural changes after different stress paradigms: prepulse inhibition increased after physical, but not emotional stress. Eur Neuropsychopharmacol. 2003;13:369–380. doi: 10.1016/s0924-977x(03)00040-3. [DOI] [PubMed] [Google Scholar]
- Pillai-Nair N, et al. Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J Neurosci. 2005;25:4659–4671. doi: 10.1523/JNEUROSCI.0565-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnikov MV, et al. Effects of genetic background on neonatal Borna disease virus infection-induced neurodevelopmental damage. I. Brain pathology and behavioral deficits. Brain Res. 2002;944:97–107. doi: 10.1016/s0006-8993(02)02723-3. [DOI] [PubMed] [Google Scholar]
- Podhorna J, Didriksen M. The heterozygous reeler mouse: behavioural phenotype. Behav Brain Res. 2004;153:43–54. doi: 10.1016/j.bbr.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Porras-Garcia E, et al. Purkinje cell loss affects differentially the execution, acquisition and prepulse inhibition of skeletal and facial motor responses in Lurcher mice. Eur J Neurosci. 2005;21:979–988. doi: 10.1111/j.1460-9568.2005.03940.x. [DOI] [PubMed] [Google Scholar]
- Postma P, et al. A behavioural and functional neuroimaging investigation into the effects of nicotine on sensorimotor gating in healthy subjects and persons with schizophrenia. Psychopharmacology. 2006;184:589–599. doi: 10.1007/s00213-006-0307-5. [DOI] [PubMed] [Google Scholar]
- Pothuizen HH, et al. The effects of temporary inactivation of the core and the shell subregions of the nucleus accumbens on prepulse inhibition of the acoustic startle reflex and activity in rats. Neuropsychopharmacology. 2005;30:683–696. doi: 10.1038/sj.npp.1300643. [DOI] [PubMed] [Google Scholar]
- Pothuizen HH, Neumann KR, Feldon J, Yee BK. Selective nucleus accumbens core lesions enhance dizocilpine-induced but not apomorphine-induced disruption of prepulse inhibition in rats. Behav Pharmacol. 2006;17:107–117. doi: 10.1097/01.fbp.0000190683.00232.ec. [DOI] [PubMed] [Google Scholar]
- Pouzet B, et al. Effects of the 5-HT(6) receptor antagonist, SB-271046, in animal models for schizophrenia. Pharmacol Biochem Behav. 2002a;71:635–643. doi: 10.1016/s0091-3057(01)00743-2. [DOI] [PubMed] [Google Scholar]
- Pouzet B, et al. Effects of the 5-HT(7) receptor antagonist SB-258741 in animal models for schizophrenia. Pharmacol Biochem Behav. 2002b;71:655–665. doi: 10.1016/s0091-3057(01)00744-4. [DOI] [PubMed] [Google Scholar]
- Powell KJ, et al. Neonatal ventral hippocampal lesions produce an elevation of DeltaFosB-like protein(s) in the rodent neocortex. Neuropsychopharmacology. 2006;31:700–711. doi: 10.1038/sj.npp.1300883. [DOI] [PubMed] [Google Scholar]
- Powell SB, et al. Isolation rearing-induced deficits in prepulse inhibition and locomotor habituation are not potentiated by water deprivation. Physiol Behav. 2002;77:55–64. doi: 10.1016/s0031-9384(02)00817-x. [DOI] [PubMed] [Google Scholar]
- Powell SB, et al. Dopamine depletion of the nucleus accumbens reverses isolation-induced deficits in prepulse inhibition in rats. Neuroscience. 2003;119:233–240. doi: 10.1016/s0306-4522(03)00122-2. [DOI] [PubMed] [Google Scholar]
- Powell SB, et al. Antipsychotic prophylaxis: initial studies on prepulse inhibition and weight gain with sustained clozapine treatment. Society for Neuroscience Atlanta, GA, Program No 587.12 2006a [Google Scholar]
- Powell SB, et al. Startle gating deficits after isolation rearing: strain differences and effects of sustained clozapine treatment. Biol Psychiatry. 2006b;59:221S. [Google Scholar]
- Quednow BB, et al. Sensorimotor gating and habituation of the startle response in schizophrenic patients randomly treated with amisulpride or olanzapine. Biol Psychiatry. 2006;59:536–545. doi: 10.1016/j.biopsych.2005.07.012. [DOI] [PubMed] [Google Scholar]
- Radant AD, et al. Successful multi-site measurement of antisaccade performance deficits in schizophrenia. Schizophr Res. 2007;89:320–329. doi: 10.1016/j.schres.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Rajakumar N, et al. Altered neurotrophin receptor function in the developing prefrontal cortex leads to adult-onset dopaminergic hyperresponsivity and impaired prepulse inhibition of acoustic startle. Biol Psychiatry. 2004;55:797–803. doi: 10.1016/j.biopsych.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Caine SB. Dopamine D1 and D2 agonist effects on prepulse inhibition and locomotion: comparison of Sprague-Dawley rats to Swiss-Webster, 129X1/SvJ, C57BL/6J, and DBA/2J mice. J Pharmacol Exp Ther. 2005;312:733–741. doi: 10.1124/jpet.104.074468. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Caine SB. Effects of selective dopamine D1-like and D2-like agonists on prepulse inhibition of startle in inbred C3H/HeJ, SPRET/EiJ, and CAST/EiJ mice. Psychopharmacology (Berl) 2007;191:731–739. doi: 10.1007/s00213-006-0511-3. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, et al. The dopamine D2, but not D3 or D4, receptor subtype is essential for the disruption of prepulse inhibition produced by amphetamine in mice. J Neurosci. 1999;19:4627–4633. doi: 10.1523/JNEUROSCI.19-11-04627.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph-Williams RJ, et al. Differential effects of direct and indirect dopamine agonists on prepulse inhibition: a study in D1 and D2 receptor knock-out mice. J Neurosci. 2002;22:9604–9611. doi: 10.1523/JNEUROSCI.22-21-09604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph-Williams RJ, et al. Dopamine D1 rather than D2 receptor agonists disrupt prepulse inhibition of startle in mice. Neuropsychopharmacology. 2003a;28:108–118. doi: 10.1038/sj.npp.1300017. [DOI] [PubMed] [Google Scholar]
- Ralph-Williams RJ, et al. Valproate attenuates hyperactive and perseverative behaviors in mutant mice with a dysregulated dopamine system. Biol Psychiatry. 2003b;53:352–359. doi: 10.1016/s0006-3223(02)01489-0. [DOI] [PubMed] [Google Scholar]
- Rasmussen BA, et al. Long-term effects of developmental PCP administration on sensorimotor gating in male and female rats. Psychopharmacology (Berl) 2007;190:43–49. doi: 10.1007/s00213-006-0584-z. [DOI] [PubMed] [Google Scholar]
- Razoux F, et al. Ketamine, at a dose that disrupts motor behavior and latent inhibition, enhances prefrontal cortex synaptic efficacy and glutamate release in the nucleus accumbens. Neuropsychopharmacology. 2007;32:719–727. doi: 10.1038/sj.npp.1301057. [DOI] [PubMed] [Google Scholar]
- Rehn AE, et al. An animal model of chronic placental insufficiency: relevance to neurodevelopmental disorders including schizophrenia. Neuroscience. 2004;129:381–391. doi: 10.1016/j.neuroscience.2004.07.047. [DOI] [PubMed] [Google Scholar]
- Rich BA, et al. An investigation of prepulse inhibition in pediatric bipolar disorder. Bipolar Disord. 2005;7:198–203. doi: 10.1111/j.1399-5618.2005.00183.x. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, et al. Corticotropin-releasing factor receptors CRF1 and CRF2 exert both additive and opposing influences on defensive startle behavior. J Neurosci. 2004;24:6545–6552. doi: 10.1523/JNEUROSCI.5760-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risterrucci C, et al. Functional magnetic resonance imaging reveals similar brain activity changes in two different animal models of schizophrenia. Psychopharmacology. 2005;180:724–734. doi: 10.1007/s00213-005-2204-8. [DOI] [PubMed] [Google Scholar]
- Roegge CS, et al. Histamine H1 receptor involvement in prepulse inhibition and memory function: relevance for the antipsychotic actions of clozapine. Pharmacol Biochem Behav. 2007;86:686–692. doi: 10.1016/j.pbb.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E, et al. Neurobehavioral and immunological consequences of prenatal immune activation in rats. Influence of antipsychotics. Neuropsychopharmacology. 2007;32:1791–1804. doi: 10.1038/sj.npp.1301292. [DOI] [PubMed] [Google Scholar]
- Rosa ML, et al. Routine post-weaning handling of rats prevents isolation rearing-induced deficit in prepulse inhibition. Braz J Med Biol Res. 2005;38:1691–1696. doi: 10.1590/s0100-879x2005001100018. [DOI] [PubMed] [Google Scholar]
- Roy MA, et al. Selecting a control group in studies of the familial coaggregation of two disorders: a quantitative genetics perspective. Am J Med Genet. 1997;74:296–303. doi: 10.1002/(sici)1096-8628(19970531)74:3<296::aid-ajmg11>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Rueter LE, et al. Chronic low dose risperidone and clozapine alleviate positive but not negative symptoms in the rat neonatal ventral hippocampal lesion model of schizophrenia. Psychopharmacology (Berl) 2004;176:312–319. doi: 10.1007/s00213-004-1897-4. [DOI] [PubMed] [Google Scholar]
- Russig H, et al. Apomorphine-induced disruption of prepulse inhibition that can be normalised by systemic haloperidol is insensitive to clozapine pretreatment. Psychopharmacology (Berl) 2004;175:143–147. doi: 10.1007/s00213-004-1810-1. [DOI] [PubMed] [Google Scholar]
- Sakaue M, et al. The 5-HT1A receptor agonist MKC-242 reverses isolation rearing-induced deficits of prepulse inhibition in mice. Psychopharmacology (Berl) 2003;170:73–79. doi: 10.1007/s00213-003-1515-x. [DOI] [PubMed] [Google Scholar]
- Sallinen J, et al. Pharmacological characterization and CNS effects of a novel highly selective alpha2C-adrenoceptor antagonist JP-1302. Br J Pharmacol. 2007;150:391–402. doi: 10.1038/sj.bjp.0707005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salum C, et al. Dopamine and nitric oxide interaction on the modulation of prepulse inhibition of the acoustic startle response in the Wistar rat. Psychopharmacology (Berl) 2006;185:133–141. doi: 10.1007/s00213-005-0277-z. [DOI] [PubMed] [Google Scholar]
- Sandager-Nielsen K, et al. Effects of postnatal anoxia on striatal dopamine metabolism and prepulse inhibition in rats. Pharmacol Biochem Behav. 2004;77:767–774. doi: 10.1016/j.pbb.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Sandner G, et al. Effects of ketamine and apomorphine on inferior colliculus and caudal pontine reticular nucleus evoked potentials during prepulse inhibition of the startle reflex in rats. Behav Brain Res. 2002;128:161–168. doi: 10.1016/s0166-4328(01)00273-x. [DOI] [PubMed] [Google Scholar]
- Schmitt A, et al. Altered NMDA receptor expression and behavior following postnatal hypoxia: potential relevance to schizophrenia. J Neural Transm. 2007;114:239–248. doi: 10.1007/s00702-006-0440-7. [DOI] [PubMed] [Google Scholar]
- Schneider M, Koch M. Chronic pubertal, but not adult chronic cannabinoid treatment impairs sensorimotor gating, recognition memory, and the performance in a progressive ratio task in adult rats. Neuropsychopharmacology. 2003;28:1760–1769. doi: 10.1038/sj.npp.1300225. [DOI] [PubMed] [Google Scholar]
- Schneider M, Koch M. Behavioral and morphological alterations following neonatal excitotoxic lesions of the medial prefrontal cortex in rats. Exp Neurology. 2005;195:185–198. doi: 10.1016/j.expneurol.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Schneider M, et al. Behavioral effects in adult rats of chronic prepubertal treatment with the cannabinoid receptor agonist WIN55,212-2. Behav Pharmacol. 2005;16:447–454. doi: 10.1097/00008877-200509000-00018. [DOI] [PubMed] [Google Scholar]
- Schneider T, et al. Environmental enrichment reverses behavioral alterations in rats prenatally exposed to valproic acid: issues for a therapeutic approach in autism. Neuropsychopharmacology. 2006;31:36–46. doi: 10.1038/sj.npp.1300767. [DOI] [PubMed] [Google Scholar]
- Schwabe K, Koch M. Role of the medial prefrontal cortex in N-methyl-d-aspartate receptor antagonist induced sensorimotor gating deficit in rats. Neurosci Lett. 2004;355:5–8. doi: 10.1016/j.neulet.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Schwabe K, et al. Effects of neonatal lesions of the medial prefrontal cortex on adult rat behaviour. Behav Brain Res. 2004;153:21–34. doi: 10.1016/j.bbr.2003.10.030. [DOI] [PubMed] [Google Scholar]
- Schwienbacher I, et al. Dopamine D1 receptors and adenosine A1 receptors in the rat nucleus accumbens regulate motor activity but not prepulse inhibition. Eur J Pharmacol. 2002;444:161–169. doi: 10.1016/s0014-2999(02)01622-9. [DOI] [PubMed] [Google Scholar]
- Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova S, et al. Inactivation of the 5-HT(7) receptor partially blocks phencyclidine-induced disruption of prepulse inhibition. Biol Psychiatry. 2008;63:98–105. doi: 10.1016/j.biopsych.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, et al. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilling PD, Feifel D. SR146131, a cholecystokinin-A receptor agonist, antagonizes prepulse inhibition deficits produced by dizocilpine and DOI. Psychopharmacology (Berl) 2002;164:285–293. doi: 10.1007/s00213-002-1214-z. [DOI] [PubMed] [Google Scholar]
- Shilling PD, et al. The effects of systemic NT69L, a neurotensin agonist, on baseline and drug-disrupted prepulse inhibition. Behav Brain Res. 2003;143:7–14. doi: 10.1016/s0166-4328(03)00037-8. [DOI] [PubMed] [Google Scholar]
- Shilling PD, et al. Neurotensin agonists block the prepulse inhibition deficits produced by a 5-HT2A and an alpha1 agonist. Psychopharmacology (Berl) 2004;175:353–359. doi: 10.1007/s00213-004-1835-5. [DOI] [PubMed] [Google Scholar]
- Shilling PD, et al. Strain differences in gating-disruptive effects of apomorphine: relationship to accumbens gene expression. Abstr Soc Biol Psychiatry. 2008 doi: 10.1016/j.biopsych.2007.10.015. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishkina GT, et al. Influence of neonatal short-term reduction in brainstem alpha2A-adrenergic receptors on receptor ontogenesis, acoustic startle reflex, and prepulse inhibition in rats. Behav Neurosci. 2004;118:1285–1292. doi: 10.1037/0735-7044.118.6.1285. [DOI] [PubMed] [Google Scholar]
- Shoemaker JM, et al. Quetiapine produces a prolonged reversal of the sensorimotor gating-disruptive effects of basolateral amygdala lesions in rats. Behav Neurosci. 2003;117:136–143. doi: 10.1037//0735-7044.117.1.136. [DOI] [PubMed] [Google Scholar]
- Shoemaker JM, et al. Prefrontal D1 and ventral hippocampal N-methyl-d-aspartate regulation of startle gating in rats. Neuroscience. 2005;135:385–394. doi: 10.1016/j.neuroscience.2005.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum FW, et al. Genetic alteration of anxiety and stress-like behavior in mice lacking CaMKIV. Mol Pain. 2005;1:22. doi: 10.1186/1744-8069-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sills TL. Amphetamine dose dependently disrupts prepulse inhibition of the acoustic startle response in rats within a narrow time window. Brain Res Bull. 1999;48:445–448. doi: 10.1016/s0361-9230(99)00036-2. [DOI] [PubMed] [Google Scholar]
- Silva RC, et al. Unilateral electrical stimulation of the inferior colliculus of rats modifies the prepulse modulation of the startle response (PPI): effects of ketamine and diazepam. Behav Brain Res. 2005;160:323–330. doi: 10.1016/j.bbr.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Siuciak JA, et al. CP-809,101, a selective 5-HT2C agonist, shows activity in animal models of antipsychotic activity. Neuropharmacology. 2007;52:279–290. doi: 10.1016/j.neuropharm.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Ehlers CL. Enhanced prepulse inhibition following adolescent ethanol exposure in Sprague–Dawley rats. Alcohol Clin Exp Res. 2005;29:829–836. doi: 10.1097/01.alc.0000183024.47167.27. [DOI] [PubMed] [Google Scholar]
- Smith SJ, Lees AJ. Abnormalities of the blink reflex in Gilles de la Tourette syndrome. J Neurol Neurosurg Psychiatry. 1989;52:895–898. doi: 10.1136/jnnp.52.7.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobin C, et al. Associations between prepulse inhibition and executive visual attention in children with the 22q11 deletion syndrome. Mol Psychiatry. 2005a;10:553–562. doi: 10.1038/sj.mp.4001609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobin C, et al. Lower prepulse inhibition in children with the 22q11 deletion syndrome. Am J Psychiatry. 2005b;162:1090–1099. doi: 10.1176/appi.ajp.162.6.1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag TA, et al. Removal of short-term isolation stress differentially influences prepulse inhibition in APO-SUS and APO-UNSUS rats. Behav Brain Res. 2003;141:171–175. doi: 10.1016/s0166-4328(02)00364-9. [DOI] [PubMed] [Google Scholar]
- Sorenson CA, Swerdlow NR. The effect of tail pinch on the acoustic startle response in rats. Brain Res. 1982;247:105–113. doi: 10.1016/0006-8993(82)91032-0. [DOI] [PubMed] [Google Scholar]
- Sotoyama H, et al. Neonatal exposure to epidermal growth factor induces dopamine D(2)-like receptor supersensitivity in adult sensorimotor gating. Psychopharmacology. 2007;191:783–792. doi: 10.1007/s00213-006-0595-9. [DOI] [PubMed] [Google Scholar]
- Spencer CM, et al. Exaggerated behavioral phenotypes in Fmr1/ Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X-related proteins. Hum Mol Genet. 2006;15:1984–1994. doi: 10.1093/hmg/ddl121. [DOI] [PubMed] [Google Scholar]
- Spielewoy C, Markou A. Strain-specificity in nicotine attenuation of phencyclidine-induced disruption of prepulse inhibition in mice: relevance to smoking in schizophrenia patients. Behav Genet. 2004;34:343–354. doi: 10.1023/B:BEGE.0000017878.75206.fd. [DOI] [PubMed] [Google Scholar]
- Spooren W, et al. Pharmacological and genetic evidence indicates that combined inhibition of NR2A and NR2B subunit containing NMDA receptors is required to disrupt prepulse inhibition. Psychopharmacology (Berl) 2004;175:99–105. doi: 10.1007/s00213-004-1785-y. [DOI] [PubMed] [Google Scholar]
- Stanhope KJ, et al. The muscarinic receptor agonist xanomeline has an antipsychotic-like profile in the rat. J Pharmacol Exp Ther. 2001;299:782–792. [PubMed] [Google Scholar]
- Stevenson CW, Gratton A. Role of basolateral amygdala dopamine in modulating prepulse inhibition and latent inhibition in the rat. Psychopharmacology. 2004;176:139–145. doi: 10.1007/s00213-004-1879-6. [DOI] [PubMed] [Google Scholar]
- Suemaru K, et al. Nicotine blocks apomorphine-induced disruption of prepulse inhibition of the acoustic startle in rats: possible involvement of central nicotinic alpha7 receptors. Br J Pharmacol. 2004;142:843–850. doi: 10.1038/sj.bjp.0705855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugar C, et al. Subtyping the group of schizophrenias using neurophysiological endophenotypes. Abstr Soc Neuroscience 226.6 2007 [Google Scholar]
- Svenningsson P, et al. Diverse psychotomimetics act through a common signaling pathway. Science. 2003;302:1412–1415. doi: 10.1126/science.1089681. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR. Cortico–striatal substrates of cognitive, motor and sensory gating: Speculations and implications for psychological function and dysfunction. In: Panksepp J, editor. Advances in biological psychiatry. Vol. 2. JAI Press Inc; Greenwich CT: 1996. pp. 179–208. [Google Scholar]
- Swerdlow NR. Much ado about (almost) nothing: response to ‘prepulse lost and regained’. Psychopharmacology. 2005;179:893–894. doi: 10.1007/s00213-004-2113-2. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA. Prepulse inhibition of acoustic startle in rats after lesions of the pedunculopontine nucleus. Behav Neurosci. 1993a;107:104–117. doi: 10.1037//0735-7044.107.1.104. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA. Clozapine and haloperidol in an animal model of sensorimotor gating deficits in schizophrenia. Pharmacol Biochem Behav. 1993b;44:741–744. doi: 10.1016/0091-3057(93)90193-w. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA. Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizphr Bull. 1998;24:285–301. doi: 10.1093/oxfordjournals.schbul.a033326. [DOI] [PubMed] [Google Scholar]
- Swerdlow, et al. Central dopamine hyperactivity in rats mimics abnormal acoustic startle response in schizophrenics. Biol Psychiatry. 1986;21:23–33. doi: 10.1016/0006-3223(86)90005-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. The neural substrates of sensorimotor gating of the startle reflex: a review of recent findings and their implications. J Psychopharmacol. 1992;6:176–190. doi: 10.1177/026988119200600210. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. A preliminary assessment of sensorimotor gating in patients with Obsessive Compulsive Disorder (OCD) Biol Psychiatry. 1993a;33:298–301. doi: 10.1016/0006-3223(93)90300-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Men are more inhibited than women by weak prepulses. Biol Psychiatry. 1993b;34:253–261. doi: 10.1016/0006-3223(93)90079-s. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Assessing the validity of an animal model of sensorimotor gating deficits in schizophrenic patients. Arch Gen Psychiatry. 1994;51:139–154. doi: 10.1001/archpsyc.1994.03950020063007. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Impaired prepulse inhibition of acoustic and tactile startle in patients with Huntington’s Disease. J Neurol Neurosurg Psychiat. 1995a;58:192–200. doi: 10.1136/jnnp.58.2.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, et al. “Normal” personality correlates of sensorimotor, cognitive, and visuospatial gating. Biol Psychiatry. 1995b;37:286–299. doi: 10.1016/0006-3223(94)00138-S. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Changes in sensorimotor inhibition across the menstrual cycle: Implications for neuropsychiatric disorders. Biol Psychiatry. 1997;41:452–460. doi: 10.1016/S0006-3223(96)00065-0. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Discrepant findings of clozapine effects on prepulse inhibition of startle: is it the route or the rat. Neuropsychopharmacology. 1998;18:50–56. doi: 10.1016/S0893-133X(97)00110-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Animal models of deficient sensorimotor gating: What we know, what we think we know, and what we hope to know soon. Behav Pharmacol. 2000a;111:185–204. doi: 10.1097/00008877-200006000-00002. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Towards understanding the biology of a complex phenotype: rat strain and substrain differences in the sensorimotor gating-disruptive effects of dopamine agonists. J Neurosci. 2000b;20:4325–4336. doi: 10.1523/JNEUROSCI.20-11-04325.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) 2001a;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Tactile prepuff inhibition of startle in children with Tourette’s syndrome: in search of an “fMRI-friendly” startle paradigm. Biol Psychiatry. 2001b;50:578–585. doi: 10.1016/s0006-3223(01)01164-7. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Matching strategies for drug studies of prepulse inhibition in humans. Behav Pharmacol. 2001c;12:45–52. doi: 10.1097/00008877-200102000-00005. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Prestimulus effects on startle magnitude: sensory or motor. Behavioral Neuroscience. 2002a;116:672–681. doi: 10.1037//0735-7044.116.4.672. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Prestimulus modification of the startle reflex: Relationship to personality and physiological markers of dopamine function. Biol Psychology. 2002b;62:17–26. doi: 10.1016/s0301-0511(02)00090-x. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Startle gating in rats is disrupted by chemical inactivation but not D2 stimulation of the dorsomedial thalamus. Brain Research. 2002c;953:246–254. doi: 10.1016/s0006-8993(02)03298-5. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Sensitivity to sensorimotor gating-disruptive effects of apomorphine in two outbred parental rat strains and their F1 and N2 progeny. Neuropsychopharmacology. 2003a;28:226–234. doi: 10.1038/sj.npp.1300035. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Amphetamine effects on prepulse inhibition across species: Replication and parametric extension. Neuropsychopharmacology. 2003b;28:640–650. doi: 10.1038/sj.npp.1300086. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Memantine effects on startle gating in rats: pretreatments with quetiapine and haloperidol. Society for Neuroscience, Program No 858.17, Washington, DC 2003c [Google Scholar]
- Swerdlow NR, et al. Prestimulus modification of the startle reflex: relationship to personality and physiological markers of dopamine function. Biol Psychology. 2003d;62:17–26. doi: 10.1016/s0301-0511(02)00090-x. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Sensitivity to drug effects on prepulse inhibition in inbred and outbred rat strains. Pharmacol Biochem Behav. 2004a;77:291–302. doi: 10.1016/j.pbb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. The ventral hippocampal regulation of prepulse inhibition and its disruption by apomorphine in rats are not mediated via the fornix. Neuroscience. 2004b;123:675–85. doi: 10.1016/j.neuroscience.2003.08.028. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Weak prepulses inhibit but do not elicit startle in rats and humans. Biol Psychiatry. 2004c;55:1195–1198. doi: 10.1016/j.biopsych.2004.02.030. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Heritable differences in the dopaminergic regulation of sensorimotor gating: II. Temporal, pharmacologic and generational analyses of apomorphine effects on prepulse inhibition. Psychopharmacology. 2004d;174:452–462. doi: 10.1007/s00213-003-1480-4. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Effects of memantine on startle gating in normal adult men. Proc Am Coll Neuropsychopharm; Waikoloa, HI. 2005a. [Google Scholar]
- Swerdlow NR, et al. Prepulse inhibition of perceived stimulus intensity: paradigm assessment. Biol Psychology. 2005b;69:133–147. doi: 10.1016/j.biopsycho.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Reduced startle gating after D1 blockade: effects of concomitant D2 blockade. Pharm Biochem Behav. 2005c;82:293–299. doi: 10.1016/j.pbb.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Antipsychotic effects on prepulse inhibition in normal ’low gating’ humans and rats. Neuropsychopharmacology. 2006a;31:2011–2021. doi: 10.1038/sj.npp.1301043. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Convergence and divergence in the neurochemical regulation of prepulse inhibition of startle and N40 suppression in rats. Neuropsychopharmacology. 2006b;31:506–515. doi: 10.1038/sj.npp.1300841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Forebrain D1 function and sensorimotor gating in rats: effects of D1 blockade, frontal lesions and dopamine denervation. Neurosci Lett. 2006c;402:40–45. doi: 10.1016/j.neulet.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Heritable differences in the dopaminergic regulation of behavior in rats: relationship to D2-like receptor G-protein function. Neuropsychopharmacology. 2006d;31:721–729. doi: 10.1038/sj.npp.1300877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Separable noradrenergic and dopaminer-gic regulation of prepulse inhibition in rats: implications for predictive validity and Tourette Syndrome. Psychopharmacology (Berl) 2006e;186:246–254. doi: 10.1007/s00213-006-0374-7. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. Startle gating deficits in a large cohort of patients with schizophrenia: relationship to medications, symptoms, neurocognition, and level of function. Arch Gen Psychiatry. 2006f;63:1325–1335. doi: 10.1001/archpsyc.63.12.1325. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, et al. The “low gating” phenotype in rats: antipsychotic sensitivity, heritability and sex differences. Biol Psychiatry. 2006g;59:220S. [Google Scholar]
- Swerdlow NR, et al. Multi-site studies of acoustic startle and prepulse inhibition in humans: Initial experience and methodological considerations based on studies by the Consortium on the Genetics of Schizophrenia. Schizophrenia Research. 2007;92:237–251. doi: 10.1016/j.schres.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, et al. A novel rat strain with enhanced sensitivity to the effects of dopamine agonists on startle gating. Pharmacol Biochem Behav. 2008;88:280–290. doi: 10.1016/j.pbb.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szumlinski KK, et al. Behavioral and neurochemical phenotyping of Homer1 mutant mice: possible relevance to schizophrenia. Genes Brain Behav. 2005;4:273–288. doi: 10.1111/j.1601-183X.2005.00120.x. [DOI] [PubMed] [Google Scholar]
- Tadros MG, et al. Neuroprotective effect of taurine in 3-nitropropionic acid-induced experimental animal model of Huntington’s disease phenotype. Pharmacol Biochem Behav. 2005;82:574–582. doi: 10.1016/j.pbb.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Takahashi M, et al. Sustained brain-derived neurotrophic factor up-regulation and sensorimotor gating abnormality induced by postnatal exposure to phencyclidine: comparison with adult treatment. J Neurochem. 2006;99:770–780. doi: 10.1111/j.1471-4159.2006.04106.x. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. Neural circuits containing pallidotegmental GABAergic neurons are involved in the prepulse inhibition of the startle reflex in mice. Biol Psychiatry. 2007;62:148–157. doi: 10.1016/j.biopsych.2006.06.035. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, et al. Roles of the glutamate receptor epsilon2 and delta2 subunits in the potentiation and prepulse inhibition of the acoustic startle reflex. Eur J Neurosci. 2001;14:153–160. doi: 10.1046/j.0953-816x.2001.01620.x. [DOI] [PubMed] [Google Scholar]
- Tan SE. Prenatal amphetamine exposure alters behavioral reactivity to amphetamine in rats. Neurotoxicol Teratol. 2003;25:579–585. doi: 10.1016/s0892-0362(03)00047-3. [DOI] [PubMed] [Google Scholar]
- Tanaka K, et al. Psychostimulant-induced attenuation of hyperactivity and prepulse inhibition deficits in Adcyap1-deficient mice. J Neurosci. 2006;26:5091–5097. doi: 10.1523/JNEUROSCI.4376-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, et al. Transgenic mice expressing mutant (N279K) human tau show mutation dependent cognitive deficits without neurofibrillary tangle formation. FEBS Lett. 2005;579:5704–5712. doi: 10.1016/j.febslet.2005.09.047. [DOI] [PubMed] [Google Scholar]
- Tejkalova H, et al. Does neonatal brain ischemia induces schizophrenia-like behaviour in young adult rats? Physiol Res. 2007;56:815–823. doi: 10.33549/physiolres.931056. [DOI] [PubMed] [Google Scholar]
- Tenn CC, et al. Amphetamine-sensitized animals show a sensorimotor gating and neurochemical abnormality similar to that of schizophrenia. Schizophr Res. 2003;64:103–114. doi: 10.1016/s0920-9964(03)00009-4. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, et al. Chronic, intermittent exposure to chlorpyrifos in rats: protracted effects on axonal transport, neurotrophin receptors, cholinergic markers, and information processing. J Pharmacol Exp Ther. 2007;322:1117–1128. doi: 10.1124/jpet.107.125625. [DOI] [PubMed] [Google Scholar]
- Thomsen M, et al. Decreased prepulse inhibition and increased sensitivity to muscarinic, but not dopaminergic drugs in M5 muscarinic acetylcholine receptor knockout mice. Psychopharmacology (Berl) 2007;192:97–110. doi: 10.1007/s00213-006-0682-y. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, et al. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Tsai G, et al. Gene knockout of glycine transporter 1: characterization of the behavioral phenotype. Proc Natl Acad Sci USA. 2004;101:8485–8490. doi: 10.1073/pnas.0402662101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, et al. Effect of prefrontal cortex inactivation on behavioral and neurochemical abnormalities in rats with excitotoxic lesions of the entorhinal cortex. Synapse. 2007;61:391–400. doi: 10.1002/syn.20383. [DOI] [PubMed] [Google Scholar]
- Ukai M, Okuda A. Endomorphin-1, an endogenous mu-opioid receptor agonist, improves apomorphine-induced impairment of prepulse inhibition in mice. Peptides. 2003;24:741–744. doi: 10.1016/s0196-9781(03)00123-2. [DOI] [PubMed] [Google Scholar]
- Umeda K, et al. Effects of mood stabilizers on the disruption of prepulse inhibition induced by apomorphine or dizocilpine in mice. Eur J Pharmacol. 2006;553:157–162. doi: 10.1016/j.ejphar.2006.09.050. [DOI] [PubMed] [Google Scholar]
- Valls-Sole J, et al. Abnormalities of prepulse inhibition do not depend on blink reflex excitability: a study in Parkinson’s disease and Huntington’s disease. Clin Neurophysiol. 2004;115:1527–1536. doi: 10.1016/j.clinph.2004.02.014. [DOI] [PubMed] [Google Scholar]
- van den Buuse M. Prepulse inhibition of acoustic startle in spontaneously hypertensive rats. Behav Brain Res. 2004;154:331–337. doi: 10.1016/j.bbr.2004.02.021. [DOI] [PubMed] [Google Scholar]
- van den Buuse M, Gogos A. Differential effects of antipsychotic drugs on serotonin-1A receptor-mediated disruption of prepulse inhibition. J Pharmacol Exp Ther. 2007;320:1224–1236. doi: 10.1124/jpet.106.113084. [DOI] [PubMed] [Google Scholar]
- van den Buuse M, et al. Prepulse inhibition of acoustic startle in aromatase knock-out mice: effects of age and gender. Genes Brain Behav. 2003;2:93–102. doi: 10.1034/j.1601-183x.2003.00014.x. [DOI] [PubMed] [Google Scholar]
- van den Buuse M, et al. Effect of adrenalectomy and corticosterone replacement on prepulse inhibition and locomotor activity in mice. Br J Pharmacol. 2004;142:543–550. doi: 10.1038/sj.bjp.0705511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Buuse M, et al. Enhanced effect of dopaminergic stimulation on prepulse inhibition in mice deficient in the alpha subunit of G(z) Psychopharmacology. 2005a;183:358–367. doi: 10.1007/s00213-005-0181-6. [DOI] [PubMed] [Google Scholar]
- van den Buuse M, et al. Reduced effects of amphetamine on prepulse inhibition of startle in gastrin-deficient mice. Neurosci Lett. 2005b;373:237–242. doi: 10.1016/j.neulet.2004.10.013. [DOI] [PubMed] [Google Scholar]
- van der Elst MC, et al. Cocaine strongly reduces prepulse inhibition in apomorphine-susceptible rats, but not in apomorphine-unsusceptible rats: regulation by dopamine D2 receptors. Behav Brain Res. 2006;175:392–398. doi: 10.1016/j.bbr.2006.09.014. [DOI] [PubMed] [Google Scholar]
- van der Elst MC, et al. Differences in the cellular mechanism underlying the effects of amphetamine on prepulse inhibition in apomorphine-susceptible and apomorphine-unsusceptible rats. Psychopharmacology (Berl) 2007;190:93–102. doi: 10.1007/s00213-006-0587-9. [DOI] [PubMed] [Google Scholar]
- van der Linden D, et al. Disrupted sensorimotor gating due to mental fatigue: preliminary evidence. Int J Psychophysiol. 2006;62:168–174. doi: 10.1016/j.ijpsycho.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, et al. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J Neurosci. 2005;25:4169–4180. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanover KE, et al. Pharmacological and behavioral profile of N-(4-fluorophenylmethyl)-N-(1-methylpiperidin-4-yl)-N′-(4-(2-methylpropylo xy)phenylmethyl) carbamide (2R,3R)-dihy-droxybutanedioate (2:1) (ACP-103), a novel 5-hydroxytryptamine (2A) receptor inverse agonist. J Pharmacol Exp Ther. 2006;317:910–918. doi: 10.1124/jpet.105.097006. [DOI] [PubMed] [Google Scholar]
- Varty GB, et al. Isolation rearing of mice induces deficits in prepulse inhibition of the startle response. Behav Brain Res. 2006;169:162–167. doi: 10.1016/j.bbr.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Venables P. Input dysfunction in schizophrenia. Prog Exp Pers Res. 1964;1:1–47. [PubMed] [Google Scholar]
- Vollenweider FX, et al. Opposite effects of 3,4-methylenediox-ymethamphetamine (MDMA) on sensorimotor gating in rats versus healthy humans. Psychopharmacology (Berl) 1999;143:365–372. doi: 10.1007/s002130050960. [DOI] [PubMed] [Google Scholar]
- Vollenweider FX, et al. Clozapine enhances prepulse inhibition in healthy humans with low but not with high prepulse inhibition levels. Biol Psychiatry. 2006;60:597–603. doi: 10.1016/j.biopsych.2006.03.058. [DOI] [PubMed] [Google Scholar]
- Volz M, et al. Temporal course of emotional startle modulation in schizophrenia patients. Int J Psychophysiol. 2003;49:123–137. doi: 10.1016/s0167-8760(03)00100-4. [DOI] [PubMed] [Google Scholar]
- von Goethe JW. The sorcerer’s apprentice 1779 [Google Scholar]
- Wang C, et al. Blockade of phencyclidine-induced cortical apoptosis and deficits in prepulse inhibition by M40403, a superoxide dismutase mimetic. J Pharmacol Exp Ther. 2003a;304:266–271. doi: 10.1124/jpet.102.041798. [DOI] [PubMed] [Google Scholar]
- Wang JH, et al. Reduced startle habituation and prepulse inhibition in mice lacking the adenosine A2A receptor. Behav Brain Res. 2003b;143:201–207. doi: 10.1016/s0166-4328(03)00036-6. [DOI] [PubMed] [Google Scholar]
- Wardas J, et al. Influence of CGS 21680, a selective adenosine A(2A) agonist, on the phencyclidine-induced sensorimotor gating deficit and motor behaviour in rats. Psychopharmacology (Berl) 2003;168:299–306. doi: 10.1007/s00213-003-1439-5. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, et al. Neonatal impact of leukemia inhibitory factor on neurobehavioral development in rats. Neurosci Res. 2004;48:345–353. doi: 10.1016/j.neures.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Weber M, Swerdlow NR. Rat strain differences in startle gating-disruptive effects of apomorphine occur with both acoustic and visual prepulses. Pharmacol Biochem Behav. 2008;88:306–311. doi: 10.1016/j.pbb.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weike AI, et al. Effective neuroleptic medication removes prepulse inhibition deficits in schizophrenia patients. Biol Psychiatry. 2000;47:61–70. doi: 10.1016/s0006-3223(99)00229-2. [DOI] [PubMed] [Google Scholar]
- Weike AI, et al. Sensorimotor gating and attitudes related to schizotypal proneness. Psychol Rep. 2001;88:1035–1045. doi: 10.2466/pr0.2001.88.3c.1035. [DOI] [PubMed] [Google Scholar]
- Weil ZM, et al. Melatonin receptor (MT1) knockout mice display depression-like behaviors and deficits in sensorimotor gating. Brain Res Bull. 2006;68:425–429. doi: 10.1016/j.brainresbull.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Weiss IC, et al. Isolation rearing-induced disruption of prepulse inhibition: further evidence for fragility of the response. Behav Pharmacol. 1999;10:139–149. doi: 10.1097/00008877-199903000-00003. [DOI] [PubMed] [Google Scholar]
- Weiss IC, et al. Strain differences in the isolation-induced effects on prepulse inhibition of the acoustic startle response and on locomotor activity. Behav Neurosci. 2000;114:364–373. [PubMed] [Google Scholar]
- Wiedholz LM, et al. Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ’schizophrenia-related’ behaviors. Mol Psychiatry. 2008 doi: 10.1038/sj.mp.4002056. in press. [DOI] [PubMed] [Google Scholar]
- Wilmouth CE, Spear LP. Withdrawal from chronic nicotine in adolescent and adult rats. Pharmacol Biochem Behav. 2006;85:648–657. doi: 10.1016/j.pbb.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf R, et al. Acute or subchronic clozapine treatment does not ameliorate prepulse inhibition (PPI) deficits in CPB-K mice with low levels of hippocampal NMDA receptor density. Psychopharmacology (Berl) 2007;194:93–102. doi: 10.1007/s00213-007-0824-x. [DOI] [PubMed] [Google Scholar]
- Wolinsky TD, et al. The trace amine 1 receptor knockout mouse: an animal model with relevance to schizophrenia. Genes Brain Behav. 2007;6:628–639. doi: 10.1111/j.1601-183X.2006.00292.x. [DOI] [PubMed] [Google Scholar]
- Wynn JK, et al. Prepulse facilitation and prepulse inhibition in schizophrenia patients and their unaffected siblings. Biol Psychiatry. 2004;55:518–523. doi: 10.1016/j.biopsych.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Wynn JK, et al. Sensorimotor gating, orienting and social perception in schizophrenia. Schizophr Res. 2005;73:319–325. doi: 10.1016/j.schres.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Wynn JK, et al. Effects of olanzapine, risperidone and haloperidol on prepulse inhibition in schizophrenia patients: A double-blind, randomized controlled trial. Schizophr Res. 2007;95:134–142. doi: 10.1016/j.schres.2007.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, et al. Norepinephrine transporter blockade can normalize the prepulse inhibition deficits found in dopamine transporter knockout mice. Neuropsychopharmacology. 2006;31:2132–2139. doi: 10.1038/sj.npp.1301009. [DOI] [PubMed] [Google Scholar]
- Yee BK, et al. Apomorphine-induced prepulse inhibition disruption is associated with a paradoxical enhancement of prepulse stimulus reactivity. Neuropsychopharmacology. 2004;29:240–248. doi: 10.1038/sj.npp.1300323. [DOI] [PubMed] [Google Scholar]
- Yee BK, et al. A schizophrenia-related sensorimotor deficit links alpha 3-containing GABAA receptors to a dopamine hyperfunction. Proc Natl Acad Sci USA. 2005;102:17154–17159. doi: 10.1073/pnas.0508752102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeomans JS, et al. Midbrain pathways for prepulse inhibition and startle activation in rat. Neuroscience. 2006;142:921–929. doi: 10.1016/j.neuroscience.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Yukawa K, et al. Reduced prepulse inhibition of startle in STAT6-deficient mice. Int J Mol Med. 2005;16:673–675. [PubMed] [Google Scholar]
- Zajaczkowski W, et al. A competitive antagonist of NMDA receptors CGP 40116 attenuates experimental symptoms of schizophrenia evoked by MK-801. Pol J Pharmacol. 2003;55:703–711. [PubMed] [Google Scholar]
- Zhang WN, et al. Effects of hippocampal N-methyl-d-aspartate infusion on locomotor activity and prepulse inhibition: differences between the dorsal and ventral hippocampus. Behav Neurosci. 2002a;116:72–84. doi: 10.1037//0735-7044.116.1.72. [DOI] [PubMed] [Google Scholar]
- Zhang WN, et al. Prepulse inhibition in rats with temporary inhibition/inactivation of ventral or dorsal hippocampus. Pharmacol Biochem Behav. 2002b;73:929–940. doi: 10.1016/s0091-3057(02)00936-x. [DOI] [PubMed] [Google Scholar]
- Zhang M, et al. Effect of dopamine D3 antagonists on PPI in DBA/2J mice or PPI deficit induced by neonatal ventral hippocampal lesions in rats. Neuropsychopharmacology. 2006;31:1382–1392. doi: 10.1038/sj.npp.1300985. [DOI] [PubMed] [Google Scholar]
- Zhang L, et al. Minocycline attenuates hyperlocomotion and prepulse inhibition deficits in mice after administration of the NMDA receptor antagonist dizocilpine. Neuropsychopharmacology. 2007a;32:2004–2010. doi: 10.1038/sj.npp.1301313. [DOI] [PubMed] [Google Scholar]
- Zhang M, et al. Effects of antipsychotics and selective D(3) antagonists on PPI deficits induced by PD 128907 and apomorphine. Behav Brain Res. 2007b;182:1–11. doi: 10.1016/j.bbr.2007.04.021. [DOI] [PubMed] [Google Scholar]