

Abstract
Recently, fused in sarcoma/translated in liposarcoma (FUS/TLS) gene, located on chromosome 16p11.2, has been identified as a disease gene in familial amyotrophic lateral sclerosis (FALS). We have analyzed FUS/TLS in a cohort of 52 index cases from seven Italian regions with non-SOD1 and non-TARDBP FALS. We identified a heterozygous c.G1542C missense mutation in a family of northern Italian origin, and a heterozygous c.C1574T missense mutation in a family of Sicilian origin. Both variants are located in exon 15 encoding the RNA-recognition motif, and result in a substitution of an arginine with a serine in position 514 (p.R514S) and substitution of a proline with a leucine at position 525 (p.P525L) respectively. Overall, the two mutations accounted for 3.8% of 52 non-SOD1 and non-TDP43 index cases of FALS. The clinical phenotype was similar within each of the families, with a predominantly upper limb onset in the family carrying the p.R514S mutation and bulbar onset, with very young age and a rapid course in the family carrying the p.P525L mutation.
Keywords: amyotrophic lateral sclerosis, genetics, FUS gene, family pedigrees
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder of cortical, bulbar and spinal motor neurons, clinically characterized by progressive paralysis and ultimately death due to respiratory failure typically within 3 years of symptom onset. Approximately 5% of cases are familial in nature (FALS), whereas the majority of cases occur sporadically in the community (SALS) (Chiò et al, 2008). Among FALS, 15 to 20% are caused by mutations in the superoxide dismutase (SOD1) gene (Rosen et al, 1993), and a further 5 to 10% are due to pathogenic mutations in the TAR DNA binding protein (TARDBP) gene (Sreedharan et al, 2008).
Recently, fused in sarcoma/translated in liposarcoma (FUS/TLS) gene has been identified as a disease gene in FALS (Kwiatkowski et al, 2009; Vance et al 2009). Located on chromosome 16p11.2, this gene encodes for a 526 amino acid protein that binds RNA and is known to be involved in RNA processing. Almost all ALS-linked mutations identified to date are clustered in the RNA-recognition motif domain located at the C terminus of the protein.
In this study, we present genetic analysis data on FUS/TLS in a cohort of 52 index cases from seven Italian regions with non-SOD1 and non-TARDBP FALS to further define the spectrum and frequency of FUS/TLS mutations.
2. Methods
2.1 Subjects
DNA sample from 52 index cases with FALS were collected from 8 Italian ALS referral centres (Torino, Pavia, Modena, Bologna, Siena, Roma, Napoli, and Palermo). All cases were diagnosed as having definite, probable or probable laboratory-supported ALS according to revised El Escorial criteria (Brooks et al, 2001). Mutations in SOD1 and TARDBP genes were excluded before the inclusion in the present study. Control samples were obtained from 280 healthy individuals matched to cases by age, gender, and Italian region of origin. Written informed consent for genetic analysis was obtained from each individual.
2.2 Genetic analysis
Genomic DNA was extracted from peripheral blood with the Biorobot MDX DSP Qiagen. Exons 1–14 of FUS gene were sequenced as previously described.5 Exon 15 of FUS was PCR amplified with the following primers: forward sequence TCGCTGGGTTAGGTAGGAGG, reverse sequence TATTCCAGTTCCTGCTGGGC. PCR products were sequenced using the Big-Dye Terminator v3.1 sequencing kit (Applied Biosystem) and run on an ABIPrism 3100Avant genetic analyzer. Exon 15 was also sequenced in 280 control individuals.
3. Results
We performed mutational screening of the FUS gene in 52 index patients from Italian kindreds with familial ALS. We identified a heterozygous c.G1542C missense mutation in a family of northern Italian (Piedmont) origin (Family A), and a heterozygous c.C1574T missense mutation in a second family of Sicilian origin (Family B). Both variants are located in exon 15 encoding the RNA-recognition motif, and result in a substitution of an arginine with a serine in position 514 (p.R514S) and substitution of a proline with a leucine at position 525 (p.P525L) respectively. DNA was not available from other members of either family, so it was not possible to confirm segregation. However, the mutations were not found in 280 control samples. Apart from a synonymous mutation in exon 15 of another FALS index case (c.G1566A), no other exonic variant was detected.
3.1 Family A (Piedmont, northern Italy)
The index case IV-1 (Figure 1, Supplementary Table1) developed lower limb weakness at 36 years of age. His symptoms progressed rapidly to involve neck and respiratory muscles with generalized hyperreflexia. EMG at the time of presentation confirmed acute and chronic alterations in all limbs and the thoracic muscles. CSF examination and MRI of the brain and the spinal cord were normal. The patient died of respiratory failure 22 months after symptom onset.
Figure 1.
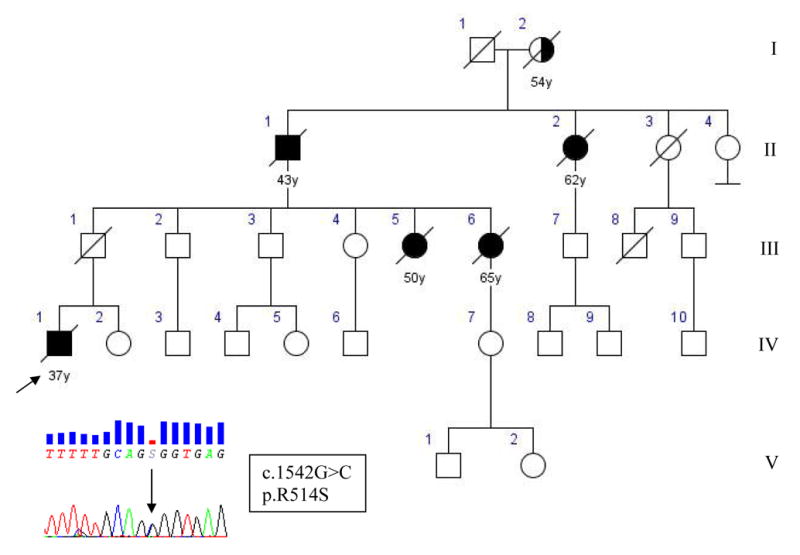
Pedigree of family A with chromatogram of part of exon 15 of FUS showing single base change c.1542G>C in the index patient, substituting arginine for serine in position 514. Square indicates male; circle, female; slash, deceased; solid symbol, affected; and arrow, index patient.
His father (III-1), an obligate gene carrier, died at 69 years of age from lung cancer without signs of ALS or neuromuscular disorders. Two paternal sisters of the index case developed hands and shoulder girdle weakness at age 48 (III-5) and 64 (III-6), respectively. Symptoms were rapidly progressive, were associated with generalized hyperreflexia, and they died from respiratory failure 25 and 15 months after onset. The paternal grandfather (II-1) of the index case developed asymmetrical shoulder girdle weakness at age 41, and died from respiratory failure 22 months later. His sister (II-2) developed right hand wasting at 60 years of age. Her symptoms spread rapidly to involve her bulbar musculature and she died from respiratory failure 19 months after symptom onset. The mother of II-1 and II-2 (I-1) presented with right shoulder atrophy at 52 and died from respiratory failure 2 years later. Autopsy material was not available from any of the deceased family members.
3.2 Family B (Sicily, southern Italy)
The index case IV-3 (Figure 2, Supplementary Table2) presented at 21 years of age with a three-month history of rapidly progressive bulbar dysfunction. Her symptoms spread quickly to involve her upper limbs with generalized hyperreflexia. EMG at the time of presentation confirmed active and chronic denervation in upper limbs. CSF examination, biochemical profile, and MRI brain and spinal cord were normal. The patient died of respiratory failure less than one year after the onset of ALS.
Figure 2.
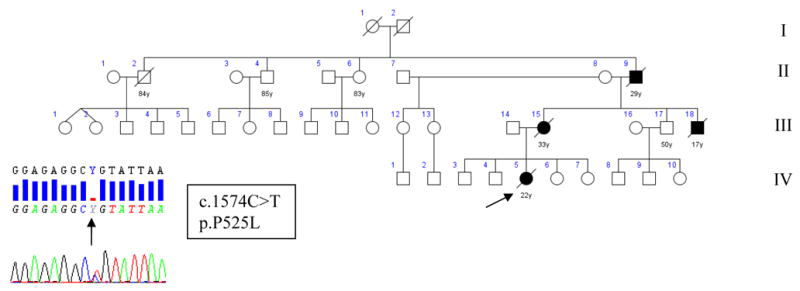
Pedigree of family B. Pedigree of family B with chromatogram of part of exon 15 of FUS showing single base change c.1574C>T in the index patient, substituting proline for leucine in position 525. Square indicates male; circle, female; slash, deceased; solid symbol, affected; and arrow, index patient.
Both her mother (III-13) and a maternal uncle (III-16) died at 33 and 17, from aggressive bulbar-onset motor neuron disease leading to respiratory failure in less than one year. Her maternal grandfather (II-7) developed rapidly progressive upper limb atrophy and weakness and respiratory failure and died less than 2 years later due to respiratory failure.
4. Discussion
In the present paper, we screened a cohort of 52 Italian patients diagnosed with FALS and identified two multi-generational families carrying mutations in the FUS gene. The two mutations accounted for 3.8% of 52 non-SOD1 and non-TDP43 index cases of FALS, a frequency similar to that found in series of 197 index cases of British and Australian FALS kindreds (4.1%) (Vance et al, 2009). Although it was not possible to confirm segregation of the mutations with disease within the families, these variants are likely to be pathogenic, as they were not found in 280 normal Italian control subjects. Furthermore, although the c.G1542C variant has not been previously reported, a G to T transversion at the same base pair leading to the same p.R514S amino acid change has previously been identified in a FALS case (Kwiatkowski et al, 2009). Finally, the c.C1574T mutation was also previously identified in a familial case (Kwiatkowski et al, 2009).
The clinical phenotype was similar within each of the families. Family A was characterized by a relatively young mean age at onset (50 years, SD 11.1), predominantly upper limb onset, clear UMN involvement, and rapid progression to respiratory failure and death. Mean disease duration from onset was 21.2 months (SD 3.7). One obligate carrier (III-1) deceased at 69 without any sign of ALS, indicating a possible reduced penetrance.
In contrast to Family A, the clinical phenotype of Family B is dominated by bulbar dysfunction (presenting symptom in 3 out of four cases), a young age at onset (mean, 23.7; SD 7.1) and rapid progression (death in less than 12 months in 3 out of four of the cases). The clinical pattern observed in our family is similar to that already reported for this mutation (mean age of onset = 22 years, mean disease duration = 6 months) (Kwiatkowski et al, 2009). confirming that this particular FUS mutation results in a highly aggressive form of ALS.
In summary, the identification of two Italian kindreds of different origin with FALS due to FUS/TLS missense mutations indicates the widespread diffusion of this mutation. Together with the figures reported in other studies (Vance et al, 2009), it seems that the occurrence of FUS/TLS mutations is about 4% of non-SOD1 and non-TDP43 FALS.
Supplementary Material
Acknowledgments
The work was supported by Ministero della Salute, Ricerca Sanitaria Finalizzata 2007 (to AC, GR and GM); Fondazione Vialli e Mauro for ALS, Torino (to AC and GM); and Regione Piemonte, Progetti Finalizzati (to GR). This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (Z01-AG000949-02).
Footnotes
Disclosure statement
The authors have no conflicts of interests.
Data contained in the manuscript being submitted have not been previously published, have not been submitted elsewhere and will not be submitted elsewhere while under consideration at Neurobiology of Aging.
Other members of the ITALSGEN Consortium: Fabio Giannini (Siena), Claudia Ricci (Siena), Cristina Moglia (Turin), Federica Lombardo (Turin), Luca Sbaiz (Turin), Stefania Cammarosano (Turin), Gioacchino Tedeschi (Naples), Patrizia Sola (Modena), Ilaria Bartolomei (Bologna), Kalliopi Marinou (Milan), Laura Papetti (Milan), Amelia Conte (Rome), Marco Luigetti (Rome), Piera Paladino (Palermo), Claudia Caponnetto (Genua), Gabriele Siciliano (Pisa)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brooks BR, Miller RG, Swash M, Munsat TL World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- Chiò A, Traynor BJ, Lombardo F, Fimognari M, Calvo A, Ghiglione P, Mutani R, Restagno G. Prevalence of SOD1 mutations in the Italian ALS population. Neurology. 2008;70:533–7. doi: 10.1212/01.wnl.0000299187.90432.3f. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak–Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH., Jr Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.