

Abstract
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are currently recognized as the most common genetic cause of parkinsonism. Among the large number of LRRK2 mutations identified to date, the G2019S variant is the most common. In Asia, however, another LRRK2 variant, G2385R, appears to occur more frequently. To better understand the contribution of different LRRK2 variants toward disease pathogenesis, we generated transgenic Drosophila over-expressing various human LRRK2 alleles, including wild type, G2019S, Y1699C, and G2385R LRRK2. We found that transgenic flies harboring G2019S, Y1699C, or G2385R LRRK2 variant, but not the wild-type protein, exhibit late-onset loss of dopaminergic (DA) neurons in selected clusters that is accompanied by locomotion deficits. Furthermore, LRRK2 mutant flies also display reduced lifespan and increased sensitivity to rotenone, a mitochondrial complex I inhibitor. Importantly, coexpression of human parkin in LRRK2 G2019S-expressing flies provides significant protection against DA neurodegeneration that occurs with age or in response to rotenone. Together, our results suggest a potential link between LRRK2, parkin, and mitochondria in the pathogenesis of LRRK2-related parkinsonism.
Introduction
Parkinson disease (PD) is a prevalent neurodegenerative movement disorder characterized pathologically by the rather selective loss of dopaminergic (DA) neurons in the substantia nigra pars compacta. Although most cases of PD occur in a sporadic manner, a subset of PD cases is inheritable and attributable to mutations in specific genes, which include α-synuclein, parkin, DJ-1, PINK1, and LRRK2 (Thomas and Beal, 2007; Gupta et al., 2008). Of these, LRRK2 mutations are currently recognized as the most common genetic cause of parkinsonism (Paisán-Ruíz et al., 2004; Zimprich et al., 2004). To date, a large number of LRRK2 mutations have been identified. Among these, the G2019S variant is the most common, with frequencies reaching up to 40% in North African PD populations (Lesage et al., 2006). In Asia, however, another LRRK2 variant, i.e., G2385R, appears to occur more frequently (Tan and Schapira, 2008).
Structurally, LRRK2 is a large protein (2527 aa) comprised of multiple domains, including a GTPase domain and a kinase domain capable of exhibiting a GTP-dependent phosphorylation activity (West et al., 2005). In general, disease-associated mutations of LRRK2 tend to increase its kinase activity and thereby its toxicity (Smith et al., 2006; West et al., 2007). However, significant variations do occur. For example, the I2012T LRRK2 mutant appears to have reduced kinase activity (West et al., 2007), and several LRRK2 mutants also exhibit a propensity to aggregate when expressed in cultured cells (Greggio et al., 2006). Thus, the exact mechanism by which LRRK2 mutations cause disease is not entirely clear.
To understand the contribution of LRRK2 mutations in vivo, a number of groups have recently generated Drosophila models of LRRK2 mutant-induced parkinsonism. Notably, Liu et al. (2008) found that over-expression of human wide-type LRRK2 or the G2019S mutant in flies triggers marked degeneration in their photoreceptor cells and DA neurons in general. Similarly, Imai et al. (2008) observed neurodegeneration in flies expressing Drosophila orthologs of human LRRK2 Y1699C or I2020T mutants, although the degeneration in this case was confined to certain DA neuronal clusters and does not affect the eye. In contrast, Lee et al. (2007) reported that transgenic flies over-expressing orthologous human wild type or R1441C mutant LRRK2 do not exhibit any significant defects in the tissues examined, including DA neurons and muscles.
Here, we report that transgenic Drosophila over-expressing human LRRK2 G2019S, Y1699C, or the G2385R variants, but not the wild-type protein, exhibit late-onset loss of DA neurons in selected clusters that are accompanied by locomotion deficits. Furthermore, LRRK2 mutant flies, especially those expressing the G2019S variant, also display increased sensitivity to rotenone, a mitochondrial complex I inhibitor. Importantly, coexpression of human parkin in LRRK2 G2019S-expressing flies provides significant protection against DA neurodegeneration that occurs with age or in response to rotenone. Together, our results demonstrate that disease-associated LRRK2 variants promote DA neuronal degeneration in vivo and that parkin over-expression mitigates LRRK2-induced neurotoxicity.
Materials and Methods
Fly stocks.
Fly lines for elav-Gal4, dopa decarboxylase gene (ddc)–Gal4, and glass multiple reporter (GMR)--Gal4 were obtained from the Bloomington Stock Center. Human parkin-expressing flies were described previously (Wang et al., 2007). To generate transgenic human LRRK2 flies, human wild type or mutant LRRK2 (Y1699C, G2019S, and G2385R) cDNA containing a myc-tag at the C terminus (West et al., 2007) was inserted into pUAST plasmid and microinjected into Drosophila embryos (BestGene). Sequencing of cloned products was performed before they were microinjected into the embryos.
Immunohistochemistry and scanning electron microscopy.
Immunohistochemical analysis of whole-mount adult fly brains were prepared according to published protocols (Whitworth et al., 2005; Wang et al., 2007) and stained with rabbit anti-tyrosine hydroxylase (anti-TH) (1:200; Pel-Freez Biologicals), rat anti-elav (1:10; Developmental Studies Hybridoma Bank), or rabbit anti-LRRK2 (1:100; Novus Biologicals) as primary antibodies before visualization using confocal microscopy. We analyzed DA neurons in different clusters quantitatively according to the method described by Whitworth et al. (2005). To analyze the morphology of eyes in high definition, fly heads were dissected at day 20 or day 60 after eclosion, mounted, and visualized using the scanning electron microscope Jeol JSM-6300LV.
Climbing assays and rotenone treatment.
Climbing assays were performed according to the method described previously (Wang et al., 2007). Three groups of 20 female adult flies that express the transgenes under the direction of the Ddc–GAL4 driver were anesthetized and placed in a vertical plastic column (length, 25 cm; diameter, 1.5 cm). Age-matched normal flies were used as controls. After a 2 h recovery period from CO2 exposure, flies were gently tapped to the bottom of the column. The number of flies that reached the top of column at 1 min was then counted. Three trials were performed in each experiment at 15 min intervals. The scores represent the mean percentage of flies that reached the top against the total number tested. Results are presented as mean ± SEM of the scores obtained in three independent experiments. In rotenone-treated flies, flies were fed with cornmeal-agar medium containing 50 μm rotenone (Sigma) immediately at after eclosion and during the entire experimental period. This concentration of rotenone is 5–10-fold lower than that originally reported by Coulom and Birman (2004).
Statistical analysis.
Quantitative data are expressed as mean ± SEM, unless otherwise stated. Statistical significance for all the quantitative data obtained was analyzed using the Student's t test (*p < 0.05, **p < 0.001), unless otherwise stated.
Results
Transgenic LRRK2-expressing flies do not display obvious eye abnormalities
To examine the effects of LRRK2 mutant expression in vivo, we generated transgenic Drosophila expressing myc-tagged human LRRK2 G2019S, Y1699C, or G2385R species under the regulation of the GAL4/upstream activation sequence system, which allows tissue-specific expression of transgenes (Fig. 1A). As a control, we also generated Drosophila producing myc-tagged wild-type human LRRK2. When driven by the pan-neuronal embryonic lethal abnormal vision (elav)–GAL4 driver, the expression of the various transgenic LRRK2 species in the fly's neurons is robust and localized predominantly to the cytoplasm (Fig. 1A; supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Notably, the staining pattern of LRRK2 is rather punctuate, likely a reflection of its localization to membranous and vesicular structures, as reported previously (Biskup et al., 2006). Although LRRK2-expressing cells are devoid of classical Lewy body-like inclusion bodies, mutant LRRK2 species tend to form intracellular aggregates (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Consistent with previous reports (Imai et al., 2008; Liu et al., 2008), we found that neither wild type nor mutant LRRK2 expression appears to compromise the overall anatomical integrity of the fly's brain up to 60 d after eclosion (Fig. 1A) (data not shown for LRRK2 variants), suggesting the LRRK2-induced toxicity, if any, is specific to selected populations of neuronal cells. In view of the conflicting observations regarding the effects on LRRK2 expression on eye maintenance in flies (Imai et al., 2008; Liu et al., 2008), we next expressed the various LRRK2 species in an eye-specific manner via the GMR–GAL4 promoter. Scanning electron microscope analysis reveals no apparent eye abnormalities in these flies at 20 d after eclosion expressing either wild-type or mutant LRRK2 species (Fig. 1B). Similar observations were made when these flies were aged to 60 d; although a very small percentage (<1%) of GMR–GAL4-driven, G2019S-expressing 60-d-old flies do exhibit a rough eye phenotype (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material), which is likely attributable to spontaneous mutational events.
Figure 1.

Expression of LRRK2 transgenes in Drosophila neither affects its overall brain architecture nor triggers obvious eye abnormalities. A, Left, Anti-elav (green) and anti-LRRK2 (red) immunostaining of whole-mount adult brains derived from 2-d-old control or transgenic flies expressing wild-type human LRRK2, as indicated. Right, Enlarged images (and inset) show the localization of elav and LRRK2 signals to the nucleus and cytoplasm, respectively, in the brain of a wild-type LRRK2-expressing fly. An anti-myc immunoblot of brain lysates prepared from 2-d-old control or transgenic flies expressing various LRRK2 species is shown below (genotype: elav-Gal4/+ or elav–Gal4–hLRRK2). B, SEM eye images of 20-d-old GMR-Gal4/+ or GMR–Gal4–hLRRK adult flies. No retinal degeneration was observed in all flies examined.
LRRK2 mutant expression driven by ddc–GAL4 in flies promotes DA neurodegeneration, locomotion deficits, and mortality
To examine the effects of transgenic LRRK2 expression in DA neurons, we used a driver line containing the promoter for the ddc gene (ddc–GAL4) that drives transgene expression specifically in TH-positive neurons, as described previously (Wang et al., 2007). At least six DA neuronal clusters [protocerebral anterior lateral (PAL), PPM1, PPM2, PPM3, PPL1, and PPL2] are recognizable in each of the adult fly brain hemispheres, all of which are amenable to quantitative analysis (Lim and Ng, 2009). At 1 d or 20 d after eclosion, none of the ddc–GAL4-driven LRRK2-expressing flies register appreciable loss of DA neurons in any of these clusters (Fig. 2A; supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). However, in aged flies (60 d old) expressing the pathogenic G2019S and Y1699C LRRK2 mutants, we observed significant degeneration of DA neurons in the PPM clusters 1 and 3 compared with age-matched normal control flies (Fig. 2B,C). This terminal age-linked phenomenon is consistent with the late disease-onset of LRRK2-related parkinsonism. Interestingly, flies expressing the G2385R variant also display similar, age-dependent, pattern of degeneration as the G2019S LRRK2-expressing flies (Fig. 2B,C). In contrast, all the DA neuronal clusters, including PPM 1 and 3, remain unaffected in wild-type LRRK2-expressing flies relative to age-matched control flies (Fig. 2B,C).
Figure 2.

Expression of LRRK2 mutants in flies promotes DA neurodegeneration and concomitant locomotion deficits. A, B, Bar graph showing the number of TH-positive DA neurons in different clusters of various fly species at 20 or 60 d after eclosion, as indicated (n = 10). C, Representative confocal microscopy images showing TH-positive (red) DA neurons in the PPM1 cluster (boxed) of 60-d-old control or LRRK2-expressing flies. Inset, Higher magnification of boxed regions. D, E, Bar graph showing the percentage of various male (D) and female (E) flies at different days after eclosion that reached the top of assay column after 1 min (n = 20) (genotype: ddc–Gal4/+ or ddc–Gal4–hLRRK2).
Notably, we and others have demonstrated a direct association between DA neurodegeneration and locomotion impairment in flies expressing disease-associated PD-linked proteins (Sang et al., 2007; Wang et al., 2007). To examine whether such a relationship exists in aged LRRK2-expressing flies that exhibit a significant loss of DA neurons, we tested the climbing ability of the various transgenic flies at 60 d after eclosion. Correlating with the significant loss of DA neurons, we found that aged flies expressing either G2019S or Y1699C LRRK2, but not the wild-type protein, recorded dramatically poorer climbing scores (<10% success) compared with control flies, a phenomenon that appears to be independent of gender (Fig. 2D,E). Although G2385R LRRK2-expressing flies at 60 d after eclosion display apparent DA neurodegeneration, unexpectedly their climbing ability is not significantly compromised (Fig. 2D,E). It is possible that G2019S or Y1699C LRRK2 mutant expression may additionally affect the function of surviving DA neurons that otherwise could compensate for the lost neurons. For example, G2019S or Y1699C expression could alter the level of dopamine production. Supporting this, we found that l-DOPA administration markedly improves the climbing ability of aged G2019S- and Y1699C LRRK2-expressing flies (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). Furthermore, HPLC analysis revealed that the level of dopamine in G2019S LRRK2-expressing flies is significantly reduced compared with wild-type LRRK2-expressing flies (supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). Finally, during the course of our examination, we noticed that the viability of the various LRRK2 mutant-expressing flies differs significantly from control flies or those expressing wild-type LRRK2. We found that expression of G2019S; Y1699C and G2385R variants in DA neurons reduced their lifespan, with G2019S LRRK2-expressing flies showing the highest rate of mortality (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material).
Rotenone treatment aggravates LRRK2 mutant-induced DA neurodegeneration
Next, we challenged the various transgenic LRRK2-expressing flies with rotenone to examine the relative susceptibility of their DA neurons to degeneration in response to a PD-linked environmental toxin. We found an accelerated loss of DA neurons in the PPM 2/3 clusters of rotenone-treated flies expressing either G2019S or G2385R LRRK2 alleles that are beyond that occurring in control flies or corresponding untreated flies (Fig. 3A,B). This phenomenon is not observed when LRRK2-expressing flies were treated with vehicle (DMSO) alone (supplemental Fig. S2D, available at www.jneurosci.org as supplemental material). Interestingly, markedly accelerated DA neuronal degeneration in the PAL cluster also occurs in G2385R LRRK2-expressing flies (Fig. 3A,B). In contrast, the rate of DA neurodegeneration in flies expressing wild-type LRRK2 is not enhanced by rotenone treatment relative to control flies (Fig. 3A,B). Surprisingly, rotenone treatment fails to aggravate the phenotype of Y1699C LRRK2-expressing flies at the end of the 15 d experimental period. Our results thus suggest that both G2019S- and G2385R LRRK2-expressing flies are more susceptible to rotenone-induced DA neurodegeneration.
Figure 3.
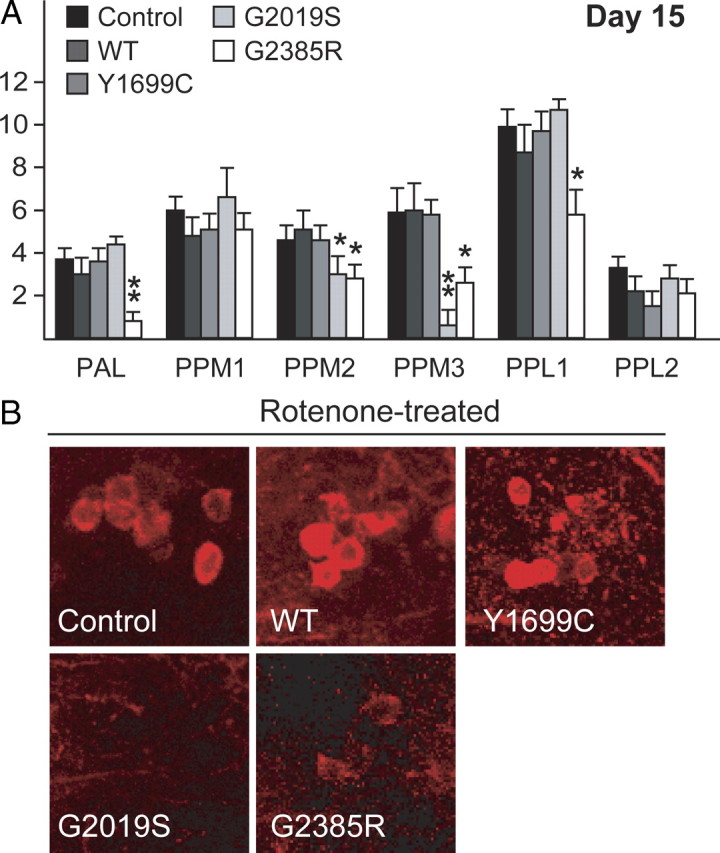
Exposure to rotenone accelerates DA degeneration in LRRK2 G2019 and G2385R mutant flies. A, Bar graph showing the number of TH-positive DA neurons in different clusters of various fly species at 15 d after rotenone treatment (n = 15). B, Representative confocal microscopy images showing TH-positive (red) DA neurons in the PPM3 cluster of various, rotenone-treated fly species, as indicated (genotype: ddc–Gal4/+ or ddc–Gal4–hLRRK2).
Parkin mitigates LRRK2 G2019S mutant-induced DA degeneration
We have previously demonstrated that flies over-expressing wild-type human parkin are significantly protected against rotenone-induced DA neurodegeneration (Wang et al., 2007). To examine whether parkin could mitigate G2019S LRRK2-induced DA neurodegeneration in Drosophila in the presence of rotenone, we generated fly lines that coexpress parkin with either wild-type (WT/PK) or G2019S LRRK2 (G2019S/PK) and repeated our above study with these bigenic flies. Whereas the DA neuron number in wild-type LRRK2-expressing flies remains essentially unchanged in the presence or absence of parkin coexpression, over-expressed parkin expectedly affords significant protection against rotenone-induced acceleration of DA neurodegeneration in LRRK2 G2019S-expressing flies (Fig. 4A,B). Since parkin functions as a broad-spectrum neuroprotectant, we surmised that G2019S/PK flies might also be protected against DA neuronal degeneration promoted by age. Indeed, the PPM 1 and 3 clusters that otherwise registered a marked loss in aged G2019S LRRK2-expressing flies relative to age-matched control flies are now maintained in G2019S/PK flies at day 60 (Fig. 4C). Parkin coexpression thus significantly mitigates DA neurodegeneration induced by LRRK2 G2019S mutant in the presence or absence of externally applied stress.
Figure 4.
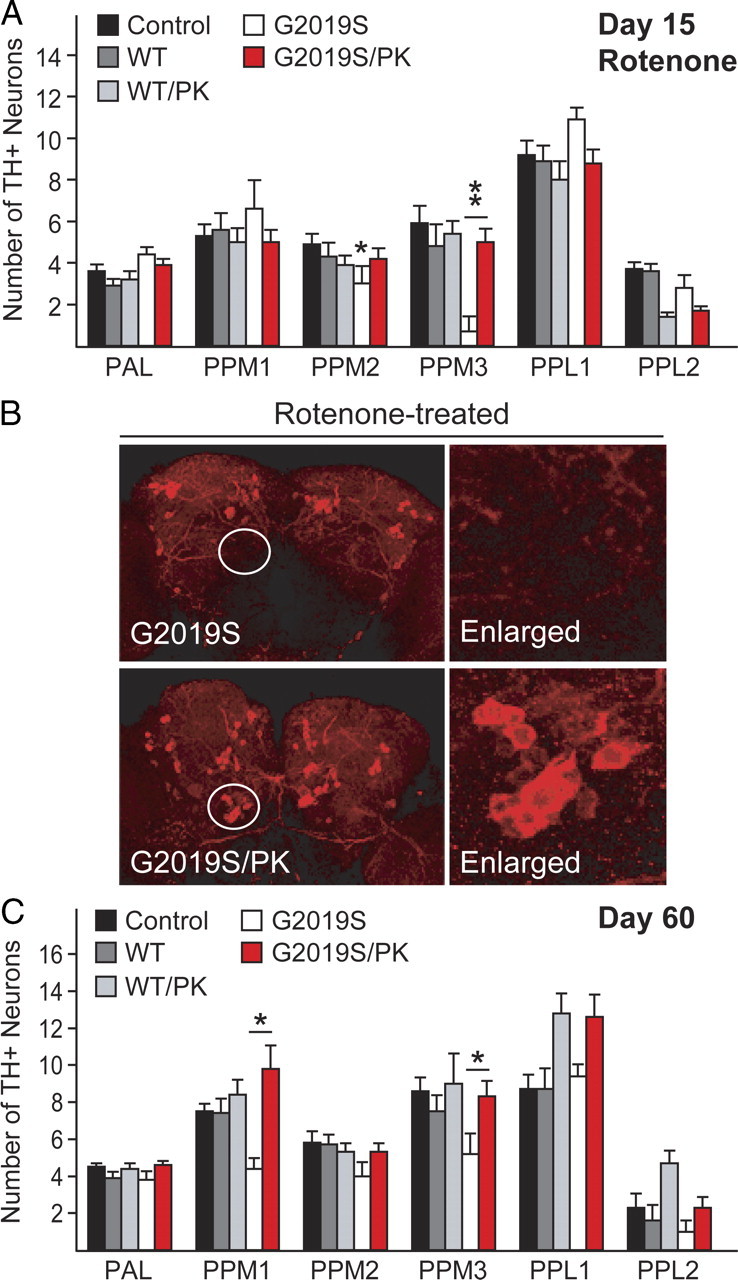
Parkin coexpression mitigates DA degeneration in LRRK2 G2019S-expressing flies in the presence or absence of rotenone. A, Bar graph showing the number of TH-positive DA neurons in different clusters of various fly species at 15 d after rotenone treatment (n = 15). B, Representative confocal microscopy images showing TH-positive (red) DA neurons in whole-mount adult brains derived from rotenone-treated flies expressing either LRRK2 G2019S alone or in the presence of parkin coexpression. Right, PPM3 cluster (circled) are shown at higher magnification. C, Bar graph showing the number of TH-positive DA neurons in different clusters of the various fly species at 60 d after eclosion, as indicated (n = 8) (genotype: ddc–Gal4/+ or ddc–Gal4–hLRRK2 or ddci-Gal4–hLRRK2;ddc–Gal4–hparkin).
Discussion
The major finding of our present study conducted in the Drosophila is that rotenone aggravates, whereas parkin mitigates, the otherwise late-onset loss of DA neurons triggered by the expression of LRRK2 G2019S mutant. Furthermore, we also demonstrate that two other LRRK2 variants, including G2385R, which is considered to be a risk allele among Asian population, produce similar phenotypes as LRRK2 G2019S mutant when expressed as transgenes in flies. Our study suggests that although a significant length of time is normally required for disease-associated LRRK2 mutants to manifest their toxicity, the pathogenic process can be hastened by an exogenous inhibitor of mitochondrial complex I, which implies a gene–environment interaction. Moreover, parkin rescues both mutant LRRK2 and rotenone-enhanced DA neuronal cell loss. This putative link between LRRK2, parkin, and mitochondria has direct mechanistic and therapeutic implications for LRRK2-induced parkinsonism.
Consistent with the report by Imai et al. (2008), we found that mutant LRRK2-mediated degeneration occurs only toward the terminal age of the fly and is restricted to selected clusters of DA neurons, whereas transgenic flies expressing wild-type LRRK2 are spared from this age-associated phenotype. This is unlike parkin null flies, where significant DA degeneration could be observed as early as 1 d after eclosion (Wang et al., 2007), a phenotype consistent with the earlier onset of parkin-related disease. Notably, our and other investigator's (Lee et al., 2007; Imai et al., 2008) findings are in stark contrast to the report by Liu et al. (2008) where degeneration in Drosophila expressing either wild type or G2019S LRRK2 was observed to occur nonselectively across all the DA neuronal clusters. Furthermore, Liu et al. (2008) also detected significant retinal degeneration in their transgenic LRRK2 flies at as early as 3 weeks of age after eclosion. This phenomenon is neither observed in our current study with flies at 20 or 60 d after eclosion nor in studies reported by other investigators (Lee et al., 2007; Imai et al., 2008). The significant phenotypic variations between our LRRK2 and the other reported models (Lee et al., 2007; Imai et al., 2008) and the one generated by Liu et al. (2008) may be attributable the different fly strains used or the use of artificial constructs optimized for protein expression (Smith et al., 2006) in Liu et al. (2008).
To date, the exact mechanism by which LRRK2 mutations cause disease remains elusive, but increased kinase activity of the protein is thought to be an important contributor to its neurotoxicity (West et al., 2005; Greggio et al., 2006; Smith et al., 2006). That being the case, elevating the expression of wild-type LRRK2 expression should in theory result in a mathematical increase in LRRK2-mediated catalysis and thereby neurotoxicity in susceptible host cells. However, our results, as well as those from two related studies (Lee et al., 2007; Imai et al., 2008), would suggest that a mere enhancement of wild-type LRRK2 expression alone, either in the presence or absence of rotenone, is not sufficient to phenocopy the effects produced by LRRK2 mutations in vivo. A tempting speculation is that wild-type LRRK2 normally exists in an inactivated state in vivo independent of its expression levels. However, this does not adequately explain the phenotype of flies expressing the G2385R polymorphic variant, which according to in vitro kinase activity assessments, behaves like the wild-type protein (West et al., 2007). Perhaps the G2385R mutation, which resides at the C-terminal end outside the catalytic core domain of the protein, promotes LRRK2 deregulation in vivo in an activity-independent manner. Remarkably, G2385R-expressing flies exposed to rotenone display marked degeneration in several DA neuronal clusters, including PAL, PPM2, and PPL1, that are otherwise unaffected by normal aging. On the other hand, Y1699C-expressing flies are relatively resistant to the effects of rotenone treatment. Although this could reflect the utilization of different disease mechanisms by various LRRK2 variants, our previous studies revealed that the extent of cell death triggered by LRRK2 G2385R expression in response to stress is comparable with that brought about by the G2019S or Y1699C mutant (West et al., 2007). Thus, the mechanism accounting for the relative resistance of Y1699C LRRK2-expression toward rotenone-induced neurodegeneration remains elusive. The differential sensitivity between G2019S and Y1699C LRRK2 flies to rotenone does suggest different mechanisms of neuronal degeneration between the LRRK2 variants. Notably, a number of studies examining the effects of Y1699C LRRK2 expression relative to other disease-associated LRRK2 mutants in other models have also recorded dissimilar outcomes. For example, MacLeod et al. (2006) found that, whereas G2019S LRRK2 expression in primary cortical neurons results in significant shortening of neurite length and branching, the phenomenon is not appreciably observed in cortical neurons transfected with Y1699C LRRK2. Notwithstanding this, it appears that LRRK2 G2385R variant could exert marked neurotoxicity when over-expressed in Drosophila. Our study thus provides the first in vivo evidence supporting the association of LRRK2 G2385R variant, otherwise known as the “Asian Allele,” with an increased risk for PD.
Finally, we demonstrated that parkin coexpression with LRRK2 G2019S mutant in flies affords considerable protection against DA neurodegeneration during normal aging or in response to rotenone. This is consistent with the widely accepted neuroprotective role of parkin and also with our previous demonstration that parkin could mitigate rotenone-mediated toxicity both in vitro and in vivo (Wang et al., 2005, 2007). Notably, parkin appears to interact with LRRK2 physically (Smith et al., 2005). Whether parkin exerts its protective effects via its direct association with mutant LRRK2 species and/or indirectly either by reducing the level of oxidative stress associated with aging or rotenone exposure, or by promoting mitochondrial homeostasis, remains to be clarified. Nonetheless, the potential involvement of mitochondrial dysfunction in LRRK2-induced degeneration (as implicated by rotenone-treatment studies) is mechanistically exciting. Already, robust evidence now supports a functional interaction between parkin and PINK1 in maintaining mitochondrial homeostasis. Furthermore, DJ-1 also appears to participate directly or indirectly in ensuring proper mitochondrial function (Andres-Mateos et al., 2007; Thomas and Beal, 2007). A link between parkin and LRRK2 via mitochondria could potentially help elucidate a common pathway underlying PD pathogenesis that would be of immense therapeutic relevance.
Footnotes
This work was supported by grants from the Singhealth Foundation, Duke–NUS Graduate Medical School and Singapore Millennium Foundation. C.-H.N. is supported by the Singapore National Medical Research Council and Singapore Millennium Foundation. F.Y. is supported by Temasek Life Sciences Laboratory. T.M.D. and V.L.D. are supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grant P50 NS38377. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. We thank Henry Zhu Liqun and Dr. Eva Andres-Mateos for their assistance with HPLC measurement of dopamine.
References
- Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807–14812. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Coulom H, Birman S. Chronic exposure to rotenone models sporadic Parkinson's disease in Drosophila melanogaster. J Neurosci. 2004;24:10993–10998. doi: 10.1523/JNEUROSCI.2993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Gupta A, Dawson VL, Dawson TM. What causes cell death in Parkinson's disease? Ann Neurol. 2008;64:S3–S15. doi: 10.1002/ana.21573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun. 2007;358:534–539. doi: 10.1016/j.bbrc.2007.04.156. [DOI] [PubMed] [Google Scholar]
- Lesage S, Dürr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A, Brice A. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N Engl J Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- Lim KL, Ng CH. Genetic models of Parkinson disease. Biochim Biophys Acta. 2009;1792:604–615. doi: 10.1016/j.bbadis.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, Ross CA, Montell C, Smith WW. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A. 2008;105:2693–2698. doi: 10.1073/pnas.0708452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Sang TK, Chang HY, Lawless GM, Ratnaparkhi A, Mee L, Ackerson LC, Maidment NT, Krantz DE, Jackson GR. A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J Neurosci. 2007;27:981–992. doi: 10.1523/JNEUROSCI.4810-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Tan EK, Schapira AH. Uniting Chinese across Asia: the LRRK2 Gly2385Arg risk variant. Eur J Neurol. 2008;15:203–204. doi: 10.1111/j.1468-1331.2007.02053.x. [DOI] [PubMed] [Google Scholar]
- Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16:R183–R194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. 2005;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- Wang C, Lu R, Ouyang X, Ho MW, Chia W, Yu F, Lim KL. Drosophila overexpressing parkin R275W mutant exhibits dopaminergic neuron degeneration and mitochondrial abnormalities. J Neurosci. 2007;27:8563–8570. doi: 10.1523/JNEUROSCI.0218-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci U S A. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]