

Abstract
We describe the use of singular value decomposition in transforming genome-wide expression data from genes × arrays space to reduced diagonalized “eigengenes” × “eigenarrays” space, where the eigengenes (or eigenarrays) are unique orthonormal superpositions of the genes (or arrays). Normalizing the data by filtering out the eigengenes (and eigenarrays) that are inferred to represent noise or experimental artifacts enables meaningful comparison of the expression of different genes across different arrays in different experiments. Sorting the data according to the eigengenes and eigenarrays gives a global picture of the dynamics of gene expression, in which individual genes and arrays appear to be classified into groups of similar regulation and function, or similar cellular state and biological phenotype, respectively. After normalization and sorting, the significant eigengenes and eigenarrays can be associated with observed genome-wide effects of regulators, or with measured samples, in which these regulators are overactive or underactive, respectively.
DNA microarray technology (1, 2) and genome sequencing have advanced to the point that it is now possible to monitor gene expression levels on a genomic scale (3). These new data promise to enhance fundamental understanding of life on the molecular level, from regulation of gene expression and gene function to cellular mechanisms, and may prove useful in medical diagnosis, treatment, and drug design. Analysis of these new data requires mathematical tools that are adaptable to the large quantities of data, while reducing the complexity of the data to make them comprehensible. Analysis so far has been limited to identification of genes and arrays with similar expression patterns by using clustering methods (4–9).
We describe the use of singular value decomposition (SVD) (10) in analyzing genome-wide expression data. SVD is also known as Karhunen–Loève expansion in pattern recognition (11) and as principal-component analysis in statistics (12). SVD is a linear transformation of the expression data from the genes × arrays space to the reduced “eigengenes” × “eigenarrays” space. In this space the data are diagonalized, such that each eigengene is expressed only in the corresponding eigenarray, with the corresponding “eigenexpression” level indicating their relative significance. The eigengenes and eigenarrays are unique, and therefore also data-driven, orthonormal superpositions of the genes and arrays, respectively.
We show that several significant eigengenes and the corresponding eigenarrays capture most of the expression information. Normalizing the data by filtering out the eigengenes (and the corresponding eigenarrays) that are inferred to represent noise or experimental artifacts enables meaningful comparison of the expression of different genes across different arrays in different experiments. Such normalization may improve any further analysis of the expression data. Sorting the data according to the correlations of the genes (and arrays) with eigengenes (and eigenarrays) gives a global picture of the dynamics of gene expression, in which individual genes and arrays appear to be classified into groups of similar regulation and function, or similar cellular state and biological phenotype, respectively. These groups of genes (or arrays) are not defined by overall similarity in expression, but only by similarity in the expression of any chosen subset of eigengenes (or eigenarrays). Upon comparing two or more similar experiments, with a regulator being overactive or underactive in one but normally expressed in the others, the expression pattern of one of the significant eigengenes may be correlated with the expression patterns of this regulator and its targets. This eigengene, therefore, can be associated with the observed genome-wide effect of the regulator. The expression pattern of the corresponding eigenarray is correlated with the expression patterns observed in samples in which the regulator is overactive or underactive. This eigenarray, therefore, can be associated with these samples.
We conclude that SVD provides a useful mathematical framework for processing and modeling genome-wide expression data, in which both the mathematical variables and operations may be assigned biological meaning.
Mathematical Framework: Singular Value Decomposition
The relative expression levels of N genes of a model organism, which may constitute almost the entire genome of this organism, in a single sample, are probed simultaneously by a single microarray. A series of M arrays, which are almost identical physically, probe the genome-wide expression levels in M different samples—i.e., under M different experimental conditions. Let the matrix ê, of size N-genes × M-arrays, tabulate the full expression data. Each element of ê satisfies 〈n|ê|m〉 ≡ enm for all 1 ≤ n ≤ N and 1 ≤ m ≤ M, where enm is the relative expression level of the nth gene in the mth sample as measured by the mth array.§ The vector in the nth row of the matrix ê, 〈gn| ≡ 〈n|ê, lists the relative expression of the nth gene across the different samples which correspond to the different arrays. The vector in the mth column of the matrix ê, |am〉 ≡ ê|m〉, lists the genome-wide relative expression measured by the mth array.
SVD (10) is then linear transformation of the expression data from the N-genes × M-arrays space to the reduced L-“eigenarrays” × L-“eigengenes” space, where L = min{M, N} (see Fig. 7 in supplemental material at www.pnas.org),
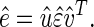 |
1 |
In this space the data are represented by the diagonal nonnegative matrix ê, of size L-eigengenes × L-eigenarrays, which satisfies 〈k|ɛ̂|l〉 ≡ ɛlδkl ≥ 0 for all 1 ≤ k,l ≤ L, such that the lth eigengene is expressed only in the corresponding lth eigenarray, with the corresponding “eigenexpression” level ɛl. Therefore, the expression of each eigengene (or eigenarray) is decoupled from that of all other eigengenes (or eigenarrays). The “fraction of eigenexpression,”
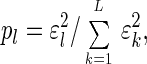 |
2 |
indicates the relative significance of the lth eigengene and eigenarray in terms of the fraction of the overall expression that they capture. Assume also that the eigenexpression levels are arranged in decreasing order of significance, such that ɛ1 ≥ ɛ2 ≥ … ≥ ɛL ≥ 0. “Shannon entropy” of a dataset,
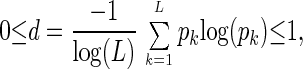 |
3 |
measures the complexity of the data from the distribution of the overall expression between the different eigengenes (and eigenarrays), where d = 0 corresponds to an ordered and redundant dataset in which all expression is captured by a single eigengene (and eigenarray), and d = 1 corresponds to a disordered and random dataset where all eigengenes (and eigenarrays) are equally expressed.
The transformation matrices û and v̂T define the N-genes × L-eigenarrays and the L-eigengenes × M-arrays basis sets, respectively. The vector in the lth row of the matrix v̂T, 〈γl| ≡ 〈l|v̂T, lists the expression of the lth eigengene across the different arrays. The vector in the lth column of the matrix û, |αl〉 ≡ û|l〉, lists the genome-wide expression in the lth eigenarray. The eigengenes and eigenarrays are orthonormal superpositions of the genes and arrays, such that the transformation matrices û and v̂ are both orthogonal
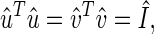 |
4 |
where Î is the identity matrix. Therefore, the expression of each eigengene (or eigenarray) is not only decoupled but also decorrelated from that of all other eigengenes (or eigenarrays). The eigengenes and eigenarrays are unique, except in degenerate subspaces, defined by subsets of equal eigenexpression levels, and except for a phase factor of ±1, such that each eigengene (or eigenarray) captures both parallel and antiparallel gene (or array) expression patterns. Therefore, SVD is data-driven, except in degenerate subspaces.
SVD Calculation.
According to Eqs. 1 and 4, the M-arrays × M-arrays symmetric correlation matrix â = êTê = v̂ɛ̂2v̂T is represented in the L-eigengenes × L-eigengenes space by the diagonal matrix ɛ̂2. The N-genes × N-genes correlation matrix ĝ = êêT = ûɛ̂2ûT is represented in the L-eigenarrays × L-eigenarrays space also by ɛ̂2, where for L = min{M, N} = M, ĝ has a null subspace of at least N − M null eigenvalues. We, therefore, calculate the SVD of a dataset ê, with M ≪ N, by diagonalizing â, and then projecting the resulting v̂ and ɛ̂ onto ê to obtain û = êv̂ɛ̂−1.
Pattern Inference.
The decorrelation of the eigengenes (and eigenarrays) suggests the possibility that some of the eigengenes (and the corresponding eigenarrays) represent independent regulatory programs or processes (and corresponding cellular states). We infer that an eigengene |γl〉 represents a regulatory program or process from its expression pattern across all arrays, when this pattern is biologically interpretable. This inference may be supported by a corresponding coherent biological theme reflected in the functions of the genes, whose expression patterns correlate or anticorrelate with the pattern of this eigengene. With this we assume that the corresponding eigenarray |αl〉 (which lists the amplitude of this eigengene pattern in the expression of each gene |gn〉 relative to all other genes 〈n|αl〉 = 〈gn|γl〉/ɛl) represents the cellular state which corresponds to this process. We infer that the eigenarray |αl〉 represents a cellular state from the arrays whose expression patterns correlate or anticorrelate with the pattern of this eigenarray. Upon sorting of the genes, this inference may be supported by the expression pattern of this eigenarray across all genes, when this pattern is biologically interpretable.
Data Normalization.
The decoupling of the eigengenes and eigenarrays allows filtering the data without eliminating genes or arrays from the dataset. We filter any of the eigengenes |γl〉 (and the corresponding eigenarray |αl〉) ê → ê − ɛl|αl〉 〈γl|, by substituting zero for the eigenexpression level ɛl = 0 in the diagonal matrix ɛ̂ and reconstructing the data according to Eq. 1. We normalize the data by filtering out those eigengenes (and eigenarrays) that are inferred to represent noise or experimental artifacts.
Degenerate Subspace Rotation.
The uniqueness of the eigengenes and eigenarrays does not hold in a degenerate subspace, defined by equal eigenexpression levels. We approximate significant similar eigenexpression levels ɛl ≈ ɛl+1 ≈ … ≈ ɛm with ɛl = … = ɛm = 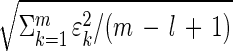 . Therefore, Eqs. 1–4 remain valid upon rotation of the corresponding eigengenes {(|γl〉, … , |γm〉) → R̂(|γl〉, … , |γm〉)}, and eigenarrays {(|αl〉, … , |αm〉) → R̂(|αl〉, … , |αm〉)}, for all orthogonal R̂, R̂TR̂ = Î. We choose a unique rotation R̂ by subjecting the rotated eigengenes to m − l constraints, such that these constrained eigengenes may be advantageous in interpreting and presenting the expression data.
. Therefore, Eqs. 1–4 remain valid upon rotation of the corresponding eigengenes {(|γl〉, … , |γm〉) → R̂(|γl〉, … , |γm〉)}, and eigenarrays {(|αl〉, … , |αm〉) → R̂(|αl〉, … , |αm〉)}, for all orthogonal R̂, R̂TR̂ = Î. We choose a unique rotation R̂ by subjecting the rotated eigengenes to m − l constraints, such that these constrained eigengenes may be advantageous in interpreting and presenting the expression data.
Data Sorting.
Inferring that eigengenes (and eigenarrays) represent independent processes (and cellular states) allows sorting the data by similarity in the expression of any chosen subset of these eigengenes (and eigenarrays), rather than by overall similarity in expression. Given two eigengenes |γk〉 and |γl〉 (or eigenarrays |αk〉 and |αl〉), we plot the correlation of |γk〉 with each gene |gn〉, 〈γk|gn〉/〈gn|gn〉 (or |αk〉 with each array |am〉) along the y-axis, vs. that of |γl〉 (or |αl〉) along the x-axis. In this plot, the distance of each gene (or array) from the origin is its amplitude of expression in the subspace spanned by |γk〉 and |γl〉 (or |αk〉 and |αl〉), relative to its overall expression rn ≡ 〈gn|gn〉−1 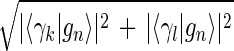 (or rm ≡ 〈am|am〉−1
(or rm ≡ 〈am|am〉−1 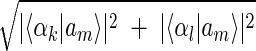 ). The angular distance of each gene (or array) from the x-axis is its phase in the transition from the expression pattern |γl〉 to |γk〉 and back to |γl〉 (or |αl〉 to |αk〉 and back to |αl〉) tan φn ≡ 〈γk|gn〉/〈γl|gn〉, (or tan φm ≡ 〈αk|an〉/〈αl|am〉). We sort the genes (or arrays) according to φn (or φm).
). The angular distance of each gene (or array) from the x-axis is its phase in the transition from the expression pattern |γl〉 to |γk〉 and back to |γl〉 (or |αl〉 to |αk〉 and back to |αl〉) tan φn ≡ 〈γk|gn〉/〈γl|gn〉, (or tan φm ≡ 〈αk|an〉/〈αl|am〉). We sort the genes (or arrays) according to φn (or φm).
Biological Data Analysis: Elutriation-Synchronized Cell Cycle
Spellman et al. (3) monitored genome-wide mRNA levels, for 6,108 ORFs of the budding yeast Saccharomyces cerevisiae simultaneously, over approximately one cell cycle period, T ≈ 390 min, in a yeast culture synchronized by elutriation, relative to a reference mRNA from an asynchronous yeast culture, at 30-min intervals. The elutriation dataset we analyze (see supplemental data and Mathematica notebook at www.pnas.org and at http://genome-www.stanford.edu/SVD/) tabulates the measured ratios of gene expression levels for the N = 5,981 genes, 784 of which were classified by Spellman et al. as cell cycle regulated, with no missing data in the M = 14 arrays.
Pattern Inference.
Consider the 14 eigengenes of the elutriation dataset. The first and most significant eigengene |γ1〉, which describes time invariant relative expression during the cell cycle (Fig. 8a at www.pnas.org), captures more than 90% of the overall relative expression in this experiment (Fig. 8b). The entropy of the dataset, therefore, is low d = 0.14 ≪ 1. This suggests that the underlying processes are manifested by weak perturbations of a steady state of expression. This also suggests that time-invariant additive constants due to uncontrolled experimental variables may be superimposed on the data. We infer that |γ1〉 represents experimental additive constants superimposed on a steady gene expression state, and assume that |α1〉 represents the corresponding steady cellular state. The second, third, and fourth eigengenes, which show oscillations during the cell cycle (Fig. 8c), capture about 3%, 1%, and 0.5% of the overall relative expression, respectively. The time variation of |γ3〉 fits a normalized sine function of period T, 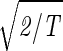 sin(2πt/T). We infer that |γ3〉 represents expression oscillation, which is consistent with gene expression oscillations during a cell cycle. The time variations of the second and fourth eigengenes fit a cosine function of period T with
sin(2πt/T). We infer that |γ3〉 represents expression oscillation, which is consistent with gene expression oscillations during a cell cycle. The time variations of the second and fourth eigengenes fit a cosine function of period T with 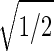 the amplitude of a normalized cosine with this period,
the amplitude of a normalized cosine with this period, 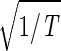 cos 2πt/T. However, while |γ2〉 shows decreasing expression on transition from t = 0 to 30 min, |γ4〉 shows increasing expression. We infer that |γ2〉 and |γ4〉 represent initial transient increase and decrease in expression in response to the elutriation, respectively, superimposed on expression oscillation during the cell cycle.
cos 2πt/T. However, while |γ2〉 shows decreasing expression on transition from t = 0 to 30 min, |γ4〉 shows increasing expression. We infer that |γ2〉 and |γ4〉 represent initial transient increase and decrease in expression in response to the elutriation, respectively, superimposed on expression oscillation during the cell cycle.
Data Normalization.
We filter out the first eigengene and eigenarray of the elutriation dataset, ê → êC = ê − ɛ1|α1〉 〈γ1|, removing the steady state of expression. Each of the elements of the dataset êC, 〈n|êC|m〉 ≡ eC,nm, is the difference of the measured expression of the nth gene in the mth array from the steady-state levels of expression for these gene and array as calculated by SVD. Therefore, eC,nm2 is the variance in the measured expression of the nth gene in the mth array. Let êLV tabulate the natural logarithm of the variances in elutriation expression, such that each element of êLV satisfies 〈n|êLV|m〉 ≡ log(eC,nm2) for all 1 ≤ n ≤ N and 1 ≤ m ≤ M, and consider the eigengenes of êLV (Fig. 9a in supplemental material at www.pnas.org). The first eigengene |γ1〉LV, which captures more than 80% of the overall information in this dataset (Fig. 9b), describes a weak initial transient increase superimposed on a time-invariant scale of expression variance. The initial transient increase in the scale of expression variance may be a response to the elutriation. The time-invariant scale of expression variance suggests that a steady scale of experimental as well as biological uncertainty is associated with the expression data. This also suggests that time-invariant multiplicative constants due to uncontrolled experimental variables may be superimposed on the data. We filter out |γ1〉LV, removing the steady scale of expression variance, êLV → êCLV = êLV − ɛ1,LV|α1〉LV LV〈γ1|.
The normalized elutriation dataset êN, where each of its elements satisfies 〈n|êN|m〉 ≡ sign(eC,nm)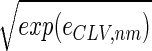 , tabulates for each gene and array expression patterns that are approximately centered at the steady-state expression level (i.e., of approximately zero arithmetic means), with variances which are approximately normalized by the steady scale of expression variance (i.e., of approximately unit geometric means). The first and second eigengenes, |γ1〉N and |γ2〉N, of êN (Fig. 1a), which are of similar significance, capture together more than 40% of the overall normalized expression (Fig. 1b). The time variations of |γ1〉N and |γ2〉N fit normalized sine and cosine functions of period T and initial phase θ ≈ 2π/13,
, tabulates for each gene and array expression patterns that are approximately centered at the steady-state expression level (i.e., of approximately zero arithmetic means), with variances which are approximately normalized by the steady scale of expression variance (i.e., of approximately unit geometric means). The first and second eigengenes, |γ1〉N and |γ2〉N, of êN (Fig. 1a), which are of similar significance, capture together more than 40% of the overall normalized expression (Fig. 1b). The time variations of |γ1〉N and |γ2〉N fit normalized sine and cosine functions of period T and initial phase θ ≈ 2π/13, 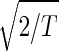 sin(2πt/T − θ) and
sin(2πt/T − θ) and 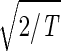 cos(2πt/T − θ), respectively (Fig. 1c). We infer that |γ1〉N and |γ2〉N represent cell cycle expression oscillations, and assume that the corresponding eigenarrays |α1〉N and |α2〉N represent the corresponding cell cycle cellular states. Upon sorting of the genes (and arrays) according to |γ1〉N and |γ2〉N (and |α1〉N and |α2〉N), the initial phase θ ≈ 2π/13 can be interpreted as a delay of 30 min between the start of the experiment and that of the cell cycle stage G1. The decay to zero in the time variation of |γ2〉N at t = 360 and 390 min can be interpreted as dephasing in time of the initially synchronized yeast culture.
cos(2πt/T − θ), respectively (Fig. 1c). We infer that |γ1〉N and |γ2〉N represent cell cycle expression oscillations, and assume that the corresponding eigenarrays |α1〉N and |α2〉N represent the corresponding cell cycle cellular states. Upon sorting of the genes (and arrays) according to |γ1〉N and |γ2〉N (and |α1〉N and |α2〉N), the initial phase θ ≈ 2π/13 can be interpreted as a delay of 30 min between the start of the experiment and that of the cell cycle stage G1. The decay to zero in the time variation of |γ2〉N at t = 360 and 390 min can be interpreted as dephasing in time of the initially synchronized yeast culture.
Figure 1.

Normalized elutriation eigengenes. (a) Raster display of v̂NT, the expression of 14 eigengenes in 14 arrays. (b) Bar chart of the fractions of eigenexpression, showing that |γ1〉N and |γ2〉N capture about 20% of the overall normalized expression each, and a high entropy d = 0.88. (c) Line-joined graphs of the expression levels of |γ1〉N (red) and |γ2〉N (blue) in the 14 arrays fit dashed graphs of normalized sine (red) and cosine (blue) of period T = 390 min and phase θ = 2π/13, respectively.
Data Sorting.
Consider the normalized expression of the 14 elutriation arrays {|am〉} in the subspace spanned by |α1〉N and |α2〉N, which is assumed to approximately represent all cell cycle cellular states (Fig. 2a). All arrays have at least 25% of their normalized expression in this subspace, with their distances from the origin satisfying 0.5 ≤ rm < 1, except for the eleventh array |a11〉. This suggests that |α1〉N and |α2〉N are sufficient to approximate the elutriation array expression. The sorting of the arrays according to their phases {φm}, which describes the transition from the expression pattern |α2〉N to |α1〉N and back to |α2〉N, gives an array order which is similar to that of the cell cycle time points measured by the arrays, an order that describes the progress of the cell cycle expression from the M/G1 stage through G1, S, S/G2, and G2/M and back to M/G1.
Figure 2.

Normalized elutriation expression in the subspace associated with the cell cycle. (a) Array correlation with |α1〉N along the y-axis vs. that with |α2〉N along the x-axis, color-coded according to the classification of the arrays into the five cell cycle stages, M/G1 (yellow), G1 (green), S (blue), S/G2 (red), and G2/M (orange). The dashed unit and half-unit circles outline 100% and 25% of overall normalized array expression in the |α1〉N and |α2〉N subspace. (b) Correlation of each gene with |γ1〉N vs. that with |γ2〉N, for 784 cell cycle regulated genes, color-coded according to the classification by Spellman et al. (3).
Because |α1〉N is correlated with the arrays |a4〉, |a5〉, |a6〉, and |a7〉 and is anticorrelated with |a13〉 and |a14〉, we associate |α1〉N with the cell cycle cellular state of transition from G1 to S, and −|α1〉N with the transition from G2/M to M/G1. Similarly, |α2〉N is correlated with |a2〉 and |a3〉, and therefore we associate |α2〉N with the transition from M/G1 to G1. Also, |α2〉N is anticorrelated with |a8〉 and |a10〉, and therefore we associate −|α2〉N with the transition from S to S/G2. With these associations the phase of |a1〉, φ1 = −θ ≈ −2π/13, corresponds to the 30-min delay between the start of the experiment and that of the cell cycle stage G1, which is also present in the inferred cell cycle expression oscillations |γ1〉N and |γ2〉N.
Consider also the expression of the 5,981 genes {|gn〉} in the subspace spanned by |γ1〉N and |γ2〉N, which is inferred to approximately represent all cell cycle expression oscillations (Fig. 10 in supplemental material at www.pnas.org). One may expect that genes that have almost all of their normalized expression in this subspace with rn ≈ 1 are cell cycle regulated, and that genes that have almost no expression in this subspace with rn ≈ 0, are not regulated by the cell cycle at all. Indeed, of the 784 genes that were classified by Spellman et al. (3) as cell cycle regulated, 641 have more than 25% of their normalized expression in this subspace (Fig. 2b). We sort all 5,981 genes according to their phases {φn}, to describe the transition from the expression pattern |γ2〉N to that of |γ1〉N and back to |γ2〉N, starting at φ1 ≈ −2π/13. One may expect this to order the genes according to the stages in the cell cycle in which their expression patterns peak. However, for the 784 cell cycle regulated genes this sorting gives a classification of the genes into the five cell cycle stages, which is somewhat different than the classification by Spellman et al. This may be due to the poor quality of the elutriation expression data, as synchronization by elutriation was not very effective in this experiment. For the α factor-synchronized cell cycle expression there is much better agreement between the two classifications (Fig. 5b).
Figure 5.

Rotated normalized α factor, CLB2, and CLN3 expression in the subspace associated with the cell cycle. (a) Array correlation with |α1〉RN along the y-axis vs. that with |α2〉RN along the x-axis, color-coded according to the classification of the arrays into the five cell cycle stages, M/G1 (yellow), G1 (green), S (blue), S/G2 (red), and G2/M (orange). The dashed unit and half-unit circles outline 100% and 25% of overall normalized array expression in the |α1〉RN and |α2〉RN subspace. (b) Correlation of each gene with |γ1〉RN vs. that with |γ2〉RN, for 638 cell cycle regulated genes, color-coded according to the classification by Spellman et al. (3).
With all 5,981 genes sorted, the gene variations of |α1〉N and |α2〉N fit normalized sine and cosine functions of period Z ≡ N − 1 = 5,980 and initial phase θ ≈ 2π/13, −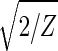 sin(2πz/Z − θ) and
sin(2πz/Z − θ) and 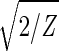 cos(2πz/Z − θ), respectively, where z ≡ n − 1 (Fig. 3 b and c). The sorted and normalized elutriation expression fit approximately a traveling wave of expression, varying sinusoidally across both genes and arrays, such that the expression of the nth gene in the mth array satisfies 〈n|êN|m〉 ∝ −2 cos[2π(t/T − z/Z)]/
cos(2πz/Z − θ), respectively, where z ≡ n − 1 (Fig. 3 b and c). The sorted and normalized elutriation expression fit approximately a traveling wave of expression, varying sinusoidally across both genes and arrays, such that the expression of the nth gene in the mth array satisfies 〈n|êN|m〉 ∝ −2 cos[2π(t/T − z/Z)]/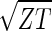 (Fig. 3a).
(Fig. 3a).
Figure 3.

Genes sorted by relative correlation with |γ1〉N and |γ2〉N of normalized elutriation. (a) Normalized elutriation expression of the sorted 5,981 genes in the 14 arrays, showing traveling wave of expression. (b) Eigenarrays expression; the expression of |α1〉N and |α2〉N, the eigenarrays corresponding to |γ1〉N and |γ2〉N, displays the sorting. (c) Expression levels of |α1〉N (red) and |α2〉N (green) fit normalized sine and cosine functions of period Z ≡ N − 1 = 5,980 and phase θ ≈ 2π/13 (blue), respectively.
Biological Data Analysis: α Factor-Synchronized Cell Cycle and CLB2 and CLN3 Overactivations
Spellman et al. (3) also monitored genome-wide mRNA levels, for 6,108 yeast ORFs simultaneously, over approximately two cell cycle periods, in a yeast culture synchronized by α factor, relative to a reference mRNA from an asynchronous yeast culture, at 7-min intervals for 119 min. They also measured, in two independent experiments, mRNA levels of yeast strain cultures with overactivated CLB2, which encodes a G2/M cyclin, both at t = 40 min relative to their levels at the start of overactivation at t = 0. Two additional independent experiments measured mRNA levels of strain cultures with overactivated CLN3, which encodes a G1/S cyclin, at t = 30 and 40 min relative to their levels at the start of overactivation at t = 0. The dataset for the α factor, CLB2, and CLN3 experiments we analyze (see supplemental data and Mathematica notebook at www.pnas.org) tabulates the ratios of gene expression levels for the N = 4,579 genes, 638 of which were classified by Spellman et al. as cell cycle regulated, with no missing data in the M = 22 arrays.
After data normalization and degenerate subspace rotation (see Appendix in supplemental material at www.pnas.org), the time variations of |γ1〉RN and |γ2〉RN fit normalized sine and cosine functions of two 66-min periods during the cell cycle, from t = 7 to 119 min, and initial phase θ ≈ π/4, respectively (Fig. 4c). While |γ2〉RN describes steady-state expression in the CLB2- and CLN3-overactive arrays, |γ1〉RN describes underexpression in the CLB2-overactive arrays and overexpression in the CLN3-overactive arrays.
Figure 4.

Rotated normalized α factor, CLB2, and CLN3 eigengenes. (a) Raster display of v̂RNT, where |γ1〉RN = R̂2R̂1|γ1〉N, |γ2〉RN = R̂1|γ2〉N, and |γ3〉RN = R̂2|γ3〉N. (b) |γ1〉RN, |γ2〉RN and |γ3〉RN capture 20% of the overall normalized expression each. (c) Expression levels of |γ1〉RN (red) and |γ2〉RN (blue) fit dashed graphs of normalized sine (red) and cosine (blue) of period T/2 = 66 min and phase π/4, respectively, and |γ3〉RN (green) fits dashed graph of normalized sine of period T = 112 min and phase −π/8, from t = 7 to t = 119 min during the cell cycle.
Upon sorting the 4,579 genes in the subspace spanned by |γ1〉RN and |γ2〉RN (Fig. 5b), |γ1〉RN is correlated with genes that peak late in the cell cycle stage G1 and early in S, among them CLN3, and we associate |γ1〉RN with the cell cycle expression oscillations that start at the transition from G1 to S and are dependent on CLN3, which encodes a G1/S cyclin. Also, |γ1〉RN is anticorrelated with genes that peak late in G2/M and early in M/G1, among them CLB2, and therefore we associate −|γ1〉RN with the oscillations that start at the transition from G2/M to M/G1 and are dependent on CLB2, which encodes a G2/M cyclin. Similarly, |γ2〉RN is correlated with genes that peak late in M/G1 and early in G1, anticorrelated with genes that peak late in S and early in S/G2, and uncorrelated with CLB2 and CLN3. We, therefore, associate |γ2〉RN with the oscillations that start at the transition from M/G1 to G1 (and appear to be CLB2- and CLN3-independent), and −|γ2〉RN with the oscillations that start at the transition from S to S/G2 (and appear to be CLB2- and CLN3-independent).
Upon sorting the 22 arrays in the subspace spanned by |α1〉RN and |α2〉RN (Fig. 5a), |α1〉RN is correlated with the arrays |a13〉 and |a14〉, as well as with |a21〉 and |a22〉, which measure the CLN3-overactive samples. We therefore associate |α1〉RN with the cell cycle cellular state of transition from G1 to S, which is simulated by CLN3 overactivation. Also, |α1〉RN is anticorrelated with the arrays |a9〉 and |a10〉, as well as with |a19〉 and |a20〉, which measure the CLB2-overactive samples. We associate −|α1〉RN with the cellular transition from G2/M to M/G1, which is simulated by CLB2 overactivation. Similarly, |α2〉RN appears to be correlated with |a2〉, |a3〉, |a11〉, and |a12〉, anticorrelated with |a6〉, |a7〉, |a16〉, and |a17〉, and uncorrelated with |a19〉, |a20〉, |a21〉, or |a22〉. We therefore associate |α2〉RN with the cellular transition from M/G1 to G1 (which appears to be CLB2- and CLN3-independent), and −|α2〉RN with the cellular transition from S to S/G2 (which also appears to be CLB2- and CLN3-independent).
With all 4,579 genes sorted the gene variations of |α1〉RN and |α2〉RN fit normalized sine and cosine functions of period Z ≡ N − 1 = 4,578 and initial phase π/8, respectively (Fig. 6 b and c). The normalized and sorted cell cycle expression approximately fits a traveling wave, varying sinusoidally across both genes and arrays. The normalized and sorted expression in the CLB2- and CLN3-overactive arrays approximately fits standing waves, constant across the arrays and varying sinusoidally across genes only, which appear similar to −|α1〉RN and |α1〉RN, respectively (Fig. 6a).
Figure 6.

Genes sorted by relative correlation with |γ1〉RN and |γ2〉RN of rotated normalized α factor, CLB2, and CLN3. (a) Normalized expression of the sorted 4,579 genes in the 22 arrays, showing traveling wave of expression from t = 0 to 119 min during the cell cycle and standing waves of expression in the CLB2- and CLN3-overactive arrays. (b) Eigenarrays expression; the expression of |α1〉RN and |α2〉RN, the eigenarrays corresponding to |γ1〉RN and |γ2〉RN, displays the sorting. (c) Expression levels of |α1〉RN (red) and |α2〉RN (green) fit normalized sine and cosine functions of period Z ≡ N − 1 = 4,578 and phase π/8 (blue), respectively.
Conclusions
We have shown that SVD provides a useful mathematical framework for processing and modeling genome-wide expression data, in which both the mathematical variables and operations may be assigned biological meaning.
Supplementary Material
Acknowledgments
We thank S. Kim for insightful discussions, G. Sherlock for technical assistance and careful reading, and J. Doyle and P. Green for thoughtful reviews of this manuscript. This work was supported by a grant from the National Cancer Institute (National Institutes of Health, CA77097). O.A. is an Alfred P. Sloan and U.S. Department of Energy Postdoctoral Fellow in Computational Molecular Biology, and a National Human Genome Research Institute Individual Mentored Research Scientist Development Awardee in Genomic Research and Analysis (National Institutes of Health, 1 K01 HG00038-01). P.O.B. is an Associate Investigator of the Howard Hughes Medical Institute.
Abbreviation
- SVD
singular value decomposition
Footnotes
In this report, m̂ denotes a matrix, |v〉 denotes a column vector, and 〈u| denotes a row vector, such that m̂|v〉, 〈u|m̂, and 〈u|v〉 all denote inner products and |v〉〈u| denotes an outer product.
References
- 1.Fodor S P, Rava R P, Huang X C, Pease A C, Holmes C P, Adams C L. Nature (London) 1993;364:555–556. doi: 10.1038/364555a0. [DOI] [PubMed] [Google Scholar]
- 2.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 3.Spellman P T, Sherlock G, Zhang M Q, Iyer V R, Anders K, Eisen M B, Brown P O, Botstein D, Futcher B. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roth F P, Hughes J D, Estep P W, Church G M. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- 5.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alon U, Barkai N, Notterman D A, Gish K, Ybarra S, Mack D, Levine A J. Proc Natl Acad Sci USA. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T R. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavazoie S, Hughes J D, Campbell M J, Cho R J, Church G M. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 9.Brown M P S, Grundy W N, Lin D, Cristianini N, Sugnet C W, Furey T S, Ares M, Jr, Haussler D. Proc Natl Acad Sci USA. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Golub G H, Van Loan C F. Matrix Computation. 3rd Ed. Baltimore: Johns Hopkins Univ. Press; 1996. [Google Scholar]
- 11.Mallat S G. A Wavelet Tour of Signal Processing. 2nd Ed. San Diego: Academic; 1999. [Google Scholar]
- 12.Anderson T W. Introduction to Multivariate Statistical Analysis. 2nd Ed. New York: Wiley; 1984. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.