

Abstract
α-N-acetylgalactosaminidase (α-NAGAL, E.C. 3.2.1.49) is a lysosomal exoglycosidase that cleaves terminal α-N-acetylgalactosamine residues from glycopeptides and glycolipids. In humans, a deficiency of α-NAGAL activity results in the lysosomal storage disorders Schindler and Kanzaki diseases. To better understand the molecular defects in the diseases, we determined the crystal structure of human α-NAGAL after expressing wild type and glycosylation-deficient glycoproteins in recombinant insect cell expression systems. We measured the enzymatic parameters of our purified wild type and mutant enzymes, establishing their enzymatic equivalence. To investigate the binding specificity and catalytic mechanism of the human α-NAGAL enzyme, we determined three crystallographic complexes with different catalytic products bound in the active site of the enzyme. To better understand how individual defects in the α-NAGAL glycoprotein lead to Schindler disease, we have analyzed the effect of disease-causing mutations on the three-dimensional structure.
Keywords: lysosomal storage disease, glycoside hydrolase, Schindler disease, Kanzaki disease, glycoprotein structure, human NAGA gene
Introduction
The human lysosomal enzyme α-NAGAL (E.C. 3.2.1.49) removes terminal α-GalNAc monosaccharides from glycolipids and glycoproteins (primarily O-linked sugars attached to serine and threonine residues) (Fig. 1A). Deficiency in α-NAGAL leads to the lysosomal storage disorder Schindler disease, first identified in 1987.1,2,3 In lysosomal storage disorders, loss of enzyme activity in a patient leads to the accumulation of substrate in the tissues, which ultimately leads to the development of clinical symptoms. In Schindler disease, loss of functional α-NAGAL enzyme activity causes accumulation of glycolipids and glycopeptides, which ultimately results in neurologic and other pathologies.3 Schindler disease phenotypes have been grouped into three classes. The Type I disease is a severe infantile neurodegenerative disorder.4,5 In Type II disease (also known as Kanzaki disease), adult onset of the disease leads to mild cognitive impairments and a characteristic skin lesion, angiokeratoma.6-9 Type III disease displays a spectrum of symptoms including seizures, autistic disorders, and/or cardiomyopathy.8,10,11 There is no treatment for these disorders.
Figure 1.
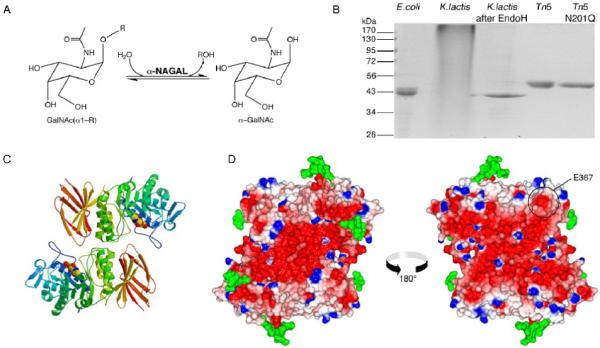
α-NAGAL reaction and overall structure
A. The reaction catalyzed by α-NAGAL. Both substrate and product are in the α anomeric configuration. B. Expression and purification of recombinant human α-NAGAL from different sources. Lane1: E. coli expressed α-NAGAL, Ni-affinity purified from inclusion bodies. Lanes 2 and 3: K. lactis expressed α-NAGAL before and after deglycosylation with Endo H. Lane 4 and 5: Tn5 expression of wild type α-NAGAL and N201Q α-NAGAL (with the 3rd carbohydrate site removed). C. A ribbon diagram of the human α-NAGAL dimer with the enzymatic product α-GalNAc in the active sites. D. An electrostatic map of the dimer showing contoured from -10 kT/e (red) to +10 kT/e (blue). Carbohydrates are shown in green. The left image is in the same orientation as in C, and the right image is rotated 180° about a vertical axis. The surface exposed residue E367 is circled.
In the human genome, the NAGA gene is most closely related to the α-galactosidase A (GLA) gene, having evolved from the same ancestral precursor.12 The corresponding proteins α-NAGAL and α-GAL have 46% amino acid sequence identity, but different substrate specificities. The human α-NAGAL protein, in addition to removing terminal α-GalNAc saccharides, has some reactivity toward substrates with terminal α-galactose saccharides. In fact, the enzyme was originally named α-GAL B and was thought to be an isozyme of α-GAL.13
In contrast, the human α-GAL protein (E.C. 3.2.1.22) removes terminal α-galactose saccharides from substrates, but it shows no enzymatic activity toward substrates with terminal α-GalNAc saccharides. Deficient α-GAL enzyme activity leads to the accumulation of glycoconjugate substrates, primarily globotriaosylceramide (Gb3), which results in Fabry disease.14 Because the GLA gene coding for the human α-GAL protein resides on the X-chromosome, Fabry disease is inherited as an X-linked disorder.
The α-NAGAL and α-GAL proteins also have the ability to convert major blood group antigens.15 The α-NAGAL protein can enzymatically convert the blood group A antigen into blood group O antigen, and the α-GAL protein is able to convert the blood group B antigen into the blood group O antigen. Because type O blood is the universal donor blood type, the α-NAGAL and α-GAL enzymes have been used to seroconvert types A, B, and AB blood into type O blood.16 Individuals with defects in their α-NAGAL or α-GAL proteins abnormally process blood group A or B antigens.17,18
In Fabry disease, human α-GAL deficiency results in the accumulation of its substrates with terminal α-galactose residues, and to a lesser extent, their precursors.14 However, in the Schindler diseases, the substrates that accumulate do not contain terminal α-GalNAc saccharides, but instead contain sialic acid- and galactose-terminal saccharides, similar to those in the lysosomal storage disorders sialidosis and galactosialidosis.19,20 It has been suggested that the α-NAGAL glycoprotein is part of a larger macromolecular assembly (with α-neuraminidase, β-galactosidase, and protective protein), and that loss of functional α-NAGAL leads to malfunction of the complex in the lysosome.3 A second possibility is that in the absence of functional α-NAGAL in the lysosome, other glycosidases such as α-neuraminidase might work in a reverse reaction, acting as glycosyltransferases in the presence of large amounts of enzymatic product.21
Another interesting aspect of α-GAL and α-NAGAL relates to the overlapping specificity of α-NAGAL, which can recognize and hydrolyze substrates with terminal α-GalNAc saccharides and (less efficiently) those with a terminal α-galactose moiety. However, the absence of α-GAL activity in Fabry disease is not compensated by α-NAGAL.
Previously, we reported the structures of the human α-GAL22 and chicken α-NAGAL enzymes.23 Those structures allowed us and others to make homology models of the human α-NAGAL enzyme in an effort to understand the molecular defects resulting in disease.23-25 However none of the homology models clarified the above issues. To address these and to establish the molecular basis for Schindler diseases, we performed structural studies of the human α-NAGAL enzyme. Using a recombinant insect cells, we expressed the functional wild type α-NAGAL glycoprotein as well as mutants lacking each of the five N-linked glycosylation sites. We measured the enzymatic activities of our wild type and mutant enzymes and determined the structure of human α-NAGAL to 1.9 Å resolution, revealing the mechanism of the enzyme. To determine the binding specificity and catalytic mechanism of human α-NAGAL, we determined crystallographic complexes with two catalytic products (the α-galactose and α-GalNAc monosaccharides) and a covalent intermediate bound in the enzyme’s active site. To better understand how individual mutations in the NAGA gene lead to Schindler or Kanzaki disease, we analyzed the respective defective enzymes in light of the three-dimensional structure. Overall, these results will lead to better understanding of the molecular defects in Schindler disease and will provide insight into lysosomal storage diseases and protein folding diseases.
Results
Protein expression, purification, and crystallization
In order to generate sufficient glycoprotein for crystallization, we produced human α-NAGAL in several expression systems (Fig 1B). Human α-NAGAL expressed in E. coli remained insoluble, even when expressed as fusion protein with glutathione-S-transferase (data not shown). When expressed in K. lactis yeast, the resulting protein was hyperglycosylated. The K. lactis expressed enzyme could be deglycosylated using the EndoH glycosidase, but the resulting material aggregated, running at the void volume of a size exclusion column. We then expressed the α-NAGAL in Trichoplusia ni Tn5 insect cells using a baculoviral vector, which produced approximately 0.5 mg of purified protein per liter of culture. Subsequently, we generated a stable Tn5 cell line expressing human α-NAGAL, which produced approximately 1-2 mg of pure protein per liter of cell culture; this expression system was used to produce enzyme for structural studies.
Extensive crystallization trials with wild type, fully-glycosylated α-NAGAL resulted in two different crystal forms: needles that diffracted to 8 Å in a synchrotron beam, and stacked plates that diffracted better, but contained significant lattice defects. Since glycosylation can interfere with the formation of well-ordered crystals, we reduced the number of N-linked carbohydrate attachment sites by site-directed mutagenesis, individually changing each N-linked asparagine into a glutamine. We made stable insect cell lines expressing human α-NAGAL with each of the five N-linked glycosylation sites removed. Each of the cell lines yielded expression levels comparable to wild type α-NAGAL, as judged by western blots. One of the proteins, N201Q, with the third N-linked carbohydrate attachment site removed, crystallized as two fused crystals in a single condition after three months. When separated by microdissection, the fragments diffracted to 1.9-2.4 Å resolution, allowing determination of the structures. Details of the cloning, purification, crystallization, and structural analysis are found in the Materials and Methods. Crystallographic statistics are shown in Table 1.
Table 1. Crystallographic statistics.
| Data | ||||
| PDB accession code | 3H53 | 3H54 | 3H55 | 3IGU |
| Ligand soak | None | GalNAc | Gal | F2-α-Gal-TNP |
| X-ray source | Rotating anode | Rotating anode | BNL X6A | BNL X6A |
| Wavelength, Å | 1.54 | 1.54 | 0.979 | 0.980 |
| Space Group | C2 | C2 | C2 | C2 |
| Cell a, b, c in Å | 153.5, 114.3, 68.4 | 153.4, 114.2, 68.3 | 153.9, 114.5, 68.4 | 151.6, 113.6, 68.4 |
| Cell α, β, γ in ° | 90., 96.1, 90. | 90., 95.6, 90. | 90., 96.2, 90. | 90., 96.1, 90. |
| Resolution, Å (last shell) | 50-2.01 (2.08-2.01) | 50-2.2 (2.28-2.2) | 50-1.9 (1.97-1.9) | 50-2.15 (2.23-2.15) |
| No. of Observations (last shell) | 772,346 (60,051) | 356,155 (24,762) | 319,671 (32,173) | 265,173 (21,625) |
| Unique Observations (last shell) | 77,785 (7,283) | 59,205 (5,733) | 87,369 (8,937) | 62,455 (6,056) |
| Completeness, % (last shell) | 99.3 (93.0) | 99.1 (95.9) | 95.8 (98.2) | 99.7 (97.4) |
| Multiplicity (last shell) | 9.9 (8.2) | 6.0 (4.2) | 3.7 (3.6) | 4.2 (3.5) |
| Rsym (last shell) | 0.085 (0.610) | 0.106 (0.674) | 0.117 (0.698) | 0.078 (0.634) |
| <I/σI> (last shell) | 29.0 (3.2) | 15.9 (2.0) | 10.1 (1.7) | 17.6 (1.8) |
| Refinement | ||||
| Rwork / Rfree | 0.161/0.194 | 0.165/0.205 | 0.196/0.235 | 0.164/0.199 |
| No. of Atoms: Protein | 6192 | 6192 | 6192 | 6192 |
| Carbohydrate | 237 | 267 | 285 | 240 |
| Water | 735 | 655 | 898 | 675 |
| Other | 102 | 90 | 26 | 90 |
| Average B factor, Å2: Protein | 26.0 | 33.3 | 20.8 | 26.0 |
| Ligand | 43.8 | 37.0 | 21.7 | 40.1 |
| Ramachandran: Favored | 90.8% | 90.1% | 89.6% | 89.9% |
| Allowed | 8.4% | 9.2% | 9.5% | 9.5% |
| Generous | 0.5% | 0.5% | 0.6% | 0.3% |
| Forbidden | 0.3% | 0.3% | 0.3% | 0.3% |
| RMS Deviations: Bonds, Å | 0.007 | 0.007 | 0.007 | 0.007 |
| Angles, ° | 1.05 | 1.11 | 1.09 | 1.10 |
Rsym=ΣhΣi|Ih,i-<Ih>|/ΣhΣi|Ih,i|, where Ih,i is the ith intensity measurement of reflection h and <Ih> is the average intensity of that reflection.
Rwork, Rfree=Σh|FP-FC|/Σh|FP|, where FC is the calculated and FP is the observed structure factor amplitude of reflection h for the working or free set, respectively.
Ramachandran statistics are calculated in PROCHECK.
Enzyme kinetics
To address the promiscuous substrate specificity in human α-NAGAL, we measured the kinetic parameters KM and kcat of the enzyme using two different synthetic substrates, para-nitrophenyl-α-N-acetylgalactosamine (pNP-α-GalNAc) and para-nitrophenyl-α-galactose (pNP-α-Gal) (Fig. 2). The pNP-α-GalNAc substrate had a markedly lower KM (0.70 mM) compared to pNP-α-Gal (15.2 mM). The N201Q mutant had nearly identical kinetic parameters against the pNP-α-GalNAc substrate, with a KM of 0.89 mM and a kcat of 17.1 sec-1 (compared to 0.70 mM and 16.3 sec-1 for the wild type enzyme). Both the wild type and N201Q mutant enzymes have specificity constants kcat/KM 30-40 times greater for substrates containing α-GalNAc compared with those containing α-galactose. Intriguingly, these data contradict a report that the N201 glycosylation is critical for the enzyme stability and activity.26 The earlier report measured activity from lysates of transiently transfected Chinese hamster ovary (CHO) or COS-1 cells, so the activity differences could result from carbohydrate processing, protein trafficking, or sample preparation. The purified wild type and N201Q mutant proteins expressed in insect cells retained nearly full activity for months at 4°C (N.E.C. and S.C.G., unpublished), but the CHO expressed material lost most of its activity within 72 hours.
Figure 2.
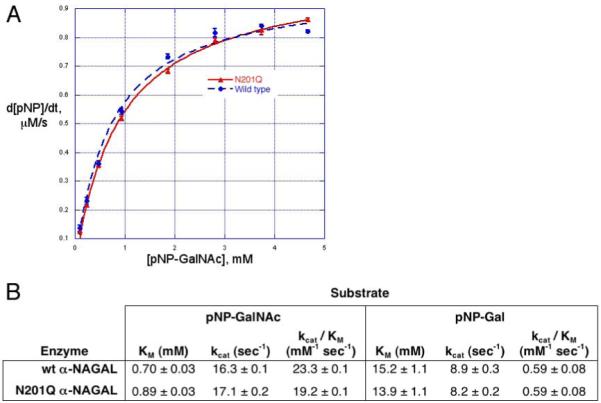
Enzyme kinetics of wild type and N201Q
A. Michaelis-Menten plot of wild type and N201Q human α-NAGAL. Production of pNP from pNP-α-GalNAc was monitored by OD at 400nm. B. Summary of kinetic data on human α-NAGAL. The wild type and N201Q mutant glycoproteins have similar kinetic parameters. The specificity constant kcat/KM for each protein is 30-40 fold greater for the GalNAc substrate compared to the galactose substrate.
Overall description of the structure
Human α-NAGAL is a homodimer with each monomer containing 394 residues (not including the 17 residue signal sequence) divided into two domains (Fig. 1C). Domain 1 forms a (β/α)8 barrel and domain 2 contains eight antiparallel β strands in two β sheets. Consistent with other members of family 27 and clan D glycoside hydrolases27 (and typical for (β/α)8 barrel proteins), the active site is found at the C-terminal end of the β strands in the first domain. The active site is formed by loops C-terminal to six consecutive β strands, strands β1-β6. The residues forming the active site are strictly conserved between human and chicken α-NAGAL, suggesting strong evolutionary pressure on those residues.
Human α-NAGAL is highly negatively charged, with an isoelectric point of 4.85. The structure shows a pronounced negative charge on the surface (Fig. 1D). Since the molecule normally functions in the low pH of the lysosome, the overall charge on the molecule would be approximately neutral at lysosomal pH, but the overall charge on the dimer is predicted to be -28 at neutral pH. Enzymatic studies show that the enzyme functions most efficiently at pH 4.6.3 The molecule is a heavily glycosylated, disulfide rich glycoprotein. The mature wild type protein contains five N-linked glycosylation sites, (N124, N177, N201, N359, and N385), four disulfide bonds (C38-C80, C42-C49, C127-C158, C187-C209) and a free cysteine (C343).
Consistent with their 46% sequence identity, human α-NAGAL and human α-GAL superimpose well, with a root mean square deviation of 1.61 Å for 744 Cα atoms in the dimer. A structure-based sequence alignment (Fig. 3) shows that the secondary structure of the two proteins is well conserved, even in the second domain, where the sequence identity drops to 23%.
Figure 3.
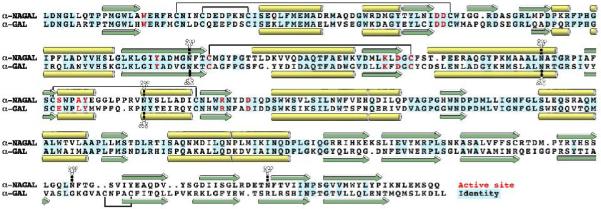
Structural alignment of human α-NAGAL and human α-GAL
The structurally derived sequence alignment shows identities (blue), α helices (yellow), β strands (green), active site residues (red), disulfide bonds (black lines), and N-linked carbohydrates (branched groups).
Active site and ligand binding
The active site of human α-NAGAL is found in the (β/α)8 barrel domain at the C-terminal end of the β strands. Consistent with its exoglycosidase function, the active site forms a small pocket on the surface of the molecule. The residues forming the active site include W33, D78, D79, Y119, C127, K154, D156, C158, S188, A191, Y192, R213, and D217 (Fig. 4A).
Figure 4.

Active site interactions and ligand binding
A. Active site interactions with the GalNAc ligand. Hydrogen bonds are shown in red, van der Waals interactions in blue, with the initial nucleophilic attack shown as a red arrow. B. Active site residues with the GalNAc ligand. Residues are colored as in Figure 2C, and the ligand is shown with σA-weighted 2Fo-Fc electron density contoured at 2σ. C. The galactose ligand with σA-weighted 2Fo-Fc electron density contoured at 1.5σ. D. The glycerol ligand with σA-weighted 2Fo-Fc electron density contoured at 1.2σ. E and F. Two view of the electron density for the 2,2-difluoro-galactose ligand covalently attached to the catalytic nucleophile D156. The map is a σA-weighted 2Fo-Fc electron density contoured at 2.0σ.
To address the overlapping substrate specificity in human α-NAGAL, we determined the structures of complexes of the enzyme with two different catalytic products (N-acetylgalactosamine and galactose) bound to the active site (Figs. 4B and C). Additionally, we determined the structure without a hexose in the active site. In that structure, a cryoprotectant glycerol molecule packs against W33 in the active site, mimicking the position of the 3-, 4-, and 5- carbons and oxygens of the catalytic product (Fig. 4D). The complexes of human α-NAGAL bound to catalytic product reveal how the α-NAGAL enzyme has specificity for substrates with terminal N-acetylgalactosamine or galactose saccharides. The N-acetylated substrate and product make more specific contacts to active site residues (and has a 15-20 fold lower KM) as compared to the galactosylated substrate and product. The N-acetyl group of the substrate makes interactions with S188, A191, and R213 side chains. When the N-acetyl moiety is replaced with a hydroxyl in galactosylated substrates, an additional water molecule fills the space of the carbonyl oxygen in the missing N-acetyl functional group (Fig. 4C). In human α-GAL, space needed for an N-acetyl group on the substrate is blocked by larger E and L residues in place of the smaller S188 and A191 residues in human α-NAGAL; thus human α-GAL substrate specificity is limited to terminal galactose saccharides.22
The active site of human α-NAGAL has anomeric selectivity for its catalytic product, unlike some other members of glycoside hydrolase family 27. The ligands N-acetylgalactosamine and galactose used in the crystallization studies were equilibrium mixtures of α and β anomers, yet the density that appears in the active site reveals only the α anomer. The structures of homologous α-GAL enzymes from rice28 and from Trichoderma reesei29 show anomeric mixtures of their products in the active site. In human α-NAGAL, Y192 (which is not conserved in the rice or fungal structures) and the C127-C158 disulfide select for the α anomer in the product.
In addition to a glycerol molecule in the active site in the unliganded structure, glycerol and other sugar molecules are found in the region surrounding the active site in all three of the structures. Glycerols can mirror the interactions of hexose sugars in their interaction with proteins.30 Since the enzyme cleaves substrates including the blood group A antigen, we used the location of the catalytic product α-GalNAc and the location of the glycerols and sugars to build a model of the blood group A substrate binding to the active site of the enzyme (Fig. 5).
Figure 5.
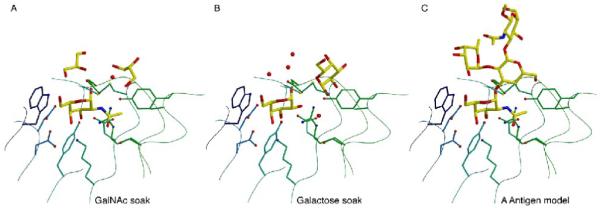
Model of blood group A antigen binding
A. Glycerol molecules cluster around the active site of the GalNAc ligand. B. Galactose and glycerol molecules bound near the active site when galactose is soaked into the active site. C. A docked model of the blood group A antigen bound to the active site, where the locations of the atoms in the model mimic the position of the small molecules bound around the active site of the crystal structures in panels A and B.
Catalytic mechanism
The enzyme operates by a double displacement (or ping-pong) reaction mechanism,31 where the α anomeric substrate is cleaved by two successive nucleophilic attacks on the anomeric carbon, resulting in overall retention of the α anomer in the catalytic product (Fig. 6A). Cleavage of glycosidic linkages using a double displacement reaction mechanism requires two carboxylates, one acting as a nucleophile and one as an acid and then a base during the reaction cycle. Starting with the substrate in the α anomeric configuration, the catalytic carboxylate D156 makes a nucleophilic attack on C1 of the substrate, breaking the glycosidic linkage and releasing the aglycone portion of the product (assisted by the donation of a proton from D217). The resulting covalent intermediate is then hydrolyzed in the second step of the reaction, where a water molecule (deprotonated by D217) makes a nucleophilic attack on C1 of the covalent glycosyl intermediate, regenerating the enzyme and releasing the second half of the cleaved product.
Figure 6.

Reaction mechanism and inactive conformation
A. The double displacement reaction mechanism in human α-NAGAL. D156 acts as the nucleophile and D217 acts as the acid/base. The ligand is bent into a high energy 1S3 skew boat conformation during the reaction. B. Human α-NAGAL without a soaked ligand shows a different conformation from all of the previous structures in the family. The residues D156 and Y192 shift into conformations where the nucleophilic lone pair rotates into a catalytically inactive location.
To examine the structure of the covalent intermediate, trinitrophenyl-2,2-difluoro-α-Gal was soaked into the crystals. The fluorine substituents at the 2 position of the sugar ring slow the second step of the reaction,32 while the excellent trinitrophenol leaving group allows the first step of the reaction to proceed. The covalent intermediate in human α-NAGAL adopts a high energy 1S3 skew boat conformation (Fig. 4E)
The structure of human α-NAGAL reveals a novel active site rearrangement upon hexose ligand binding. When compared to the other structures of family 27 glycoside hydrolases, the unliganded human α-NAGAL shows a unique inactive conformation in the active site (Fig. 6B). When a hexose ligand binds, the active site nucleophile D156 rotates 30° about χ2, leading to a 1.9 Å movement of Oδ1. Additionally, the Y192 side chain rotates, leading to a 1.1 Å shift in the location of the phenolic oxygen Oη. This shift places Oη 2.5 Å from Oδ1 of D156, in an ideal hydrogen bonding distance and geometry. The movement of D156 when a hexose ligand is bound places Oδ2 3.0 Å away from the anomeric carbon of the substrate, with the syn lone pair poised for nucleophilic attack. The Oδ2 of D156 is also 2.8 Å away from the heterocyclic oxygen of the N-acetylgalactosamine ring on the ligand. Thus human α-NAGAL senses the binding of hexose ligand by moving the catalytic aspartic acid into ideal position for nucleophilic attack.
Glycosylation
Despite the existence of highly pure preparations of human α-NAGAL since 1977,13,33 the glycoprotein has resisted structural analysis, and no structure has been reported. The five N-linked glycosylation sites in human α-NAGAL lead to conformational and chemical heterogeneity in the molecule, making it a problematic candidate for structural studies. In order to facilitate structural analysis, we reduced the conformational and chemical heterogeneity of the carbohydrate. First, human α-NAGAL expressed in K. lactis yeast resulted in hyperglycosylated glycoprotein. Enzymatic deglycosylation of that material led to reduced complexity of the carbohydrate, but the material aggregated. Second, we expressed human α-NAGAL in High Five insect cells, which tend to add only paucimannose (GalNAc2Man2-3Fuc0-2) carbohydrate to N-linked attachment sites.34,35 The insect cell-derived human α-NAGAL showed less hyperglycosylation, and it crystallized, but it produced only poorly diffracting crystals. Third, we systematically eliminated each of the five N-linked carbohydrate attachment sites. The mutant N201Q, with the third N-linked carbohydrate site removed, led to well-ordered crystals, allowing us to determine the structure.
The N201Q amino acid substitution is located in a crystal contact in our crystals, explaining why this high-resolution crystal form appears only when the N-linked carbohydrate at N201 is removed. However, removal of all of the carbohydrates from human α-NAGAL leads to insoluble material (N.E.C. and S.C.G., unpublished). Clear electron density appears for three (N124, N177, and N385) of the other four N-linked carbohydrates. Two of them (N124 and N177) are found in carbohydrate-mediated crystal contacts (which might explain the slow growth and very rare appearance of the crystals). Carbohydrate-mediated crystal contacts often produce low-resolution diffraction, but the crystals of human α-NAGAL are very well ordered, with a diffraction limit beyond 2.0 Å resolution. The human α-NAGAL expressed in insect cells contained smaller and less heterogeneous carbohydrates, potentially leading to better ordered crystals. In two of the eight glycosylation sites in the crystallographic asymmetric unit, the entire paucimannose carbohydrate GalNAc2Man3 appears in the electron density (Fig. 7).
Figure 7.
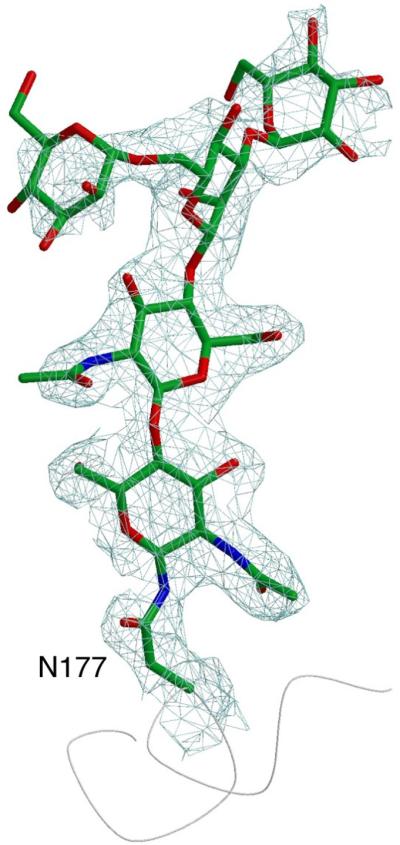
Glycosylation of residue N177
The entire paucimannose carbohydrate is visible in the electron density (a σA-weighted 2Fo-Fc electron density contoured at 1.2σ).
Schindler Disease Mutations
Schindler disease is a rare autosomal recessive disorder, with only a score of reported cases, most from the offspring of consanguinous marriages. Most of the mutations causing Schindler disease result in disruption of the hydrophobic core of the protein (Fig. 8), because the affected residues tend to be completely buried (with almost no solvent accessible surface area, Table 2). In one case (D217N), the mutation directly affects the active site of the enzyme, replacing one of the carboxylates required for the double displacement reaction mechanism. A nonsense mutation introduces a stop codon at residue 193, leading to a shortened polypeptide and misfolding of the protein. The E325K substitution leads to the loss of an ion pair between E325 and R316, resulting in the energetically unfavorable clustering of three basic residues (R316, K319, and E325K) within 3 Å of each other. S160 is found buried in a polar pocket, where the serine hydroxyl makes hydrogen bonds to the main chain carbonyl of C158 and to Nη1 of R165. The S160C substitution found in a type III patient weakens those hydrogen bonds and places an unpaired thiol in the vicinity of the C127-C158 active site disulfide bond. The R329 side chain is completely buried in a pocket that is partly hydrophobic and partly polar. Because of the perfectly complementary interactions between R329 and its neighbors, it is likely that the R329Q and R329W mutant proteins are less stable, leading to type II disease. The E367K substitution found in type III patients presents a paradox. The E367 side chain is solvent exposed and the replacement of one surface-exposed charge for another is typically well tolerated by proteins. Insect cell derived E367K protein shows activity comparable to the wild type protein (N.E.C. and S.C.G, unpublished observation). The E367 side chain is surrounded by the I376, S378, Y401, and I403 residues, creating a charge at the center of a predominantly non-polar surface. Intriguingly, this patch makes favorable protein:protein interactions in each of the two independent copies in the asymmetric unit of the crystal.
Figure 8.
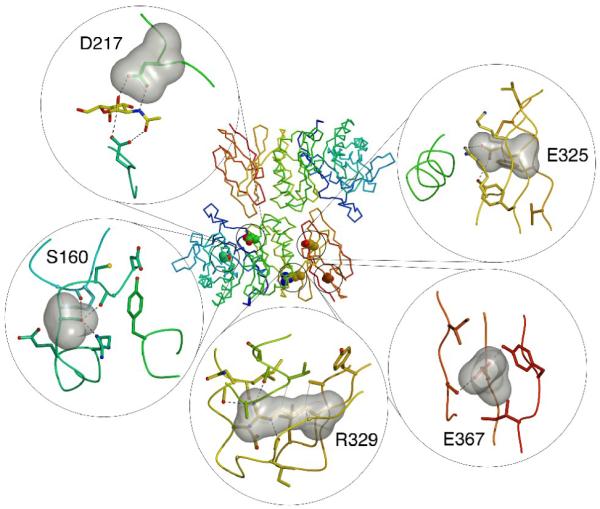
Schindler disease mutations
Five residues are shown in the center panel showing the location of the residue in the polypeptide fold. The panels show a surface representation of the ligand as well as the surrounding residues. Hydrogen bonds are shown as black dashed lines and van der Waals interactions as grey dotted lines.
Table 2. Schindler and Kanzaki disease mutations.
| Mutation | Disease Phenotype | Location | Side Chain Access. Surface Area (Å2) | Proposed Effect |
|---|---|---|---|---|
| S160C | III | Buried | 0 | Disrupts nearby disulfide |
| E193X | II | Surface | 9.8 | Stop codon |
| D217N | III | Active site | 1.9 | Disrupts active site |
| E325K | I, III | Partially Buried | 0 | Disrupts ion pair |
| R329W | II | Buried | 0 | Disrupts hydrophobic core |
| R329Q | II | Buried | 0 | Disrupts hydrophobic core |
| E367K | III | Surface | 37.5 | Disrupts protein protein interaction |
Discussion
The structure of human α-NAGAL reveals a novel conformation of the active site residues in glycoside hydrolase family 27. Human α-NAGAL shifts from a catalytically incompetent to a catalytically competent state upon the binding of substrate. This catalytically inactive form of the protein suggests a new family of mechanism-based inhibitors and pharmacological chaperones in this family of hydrolases. A compound that shifts the catalytic nucleophile into a conformation seen in the unliganded human α-NAGAL would make an effective inhibitor of the enzyme. An inhibitor of human α-NAGAL might make for a useful anti-cancer compound, because serum α-NAGAL can degrade a precursor of macrophage activation, leading to increased progression of, for example, squamous cell carcinomas.36
The structures of human α-NAGAL presented here include one without a hexose ligand, one with a covalent intermediate bound, and two with catalytic product bound. Overall, the structures give views of different stages in the catalytic mechanism as the enzyme binds to low energy conformations of the ligand, and then distorts the ligand into a high energy 1S3 skew boat conformation during the middle of the double displacement reaction. A full analysis of the mechanism of family 27 glycoside hydrolases based upon structures of human α-GAL will appear elsewhere.
Previous work had indicated that the N-linked carbohydrate attached to N201 was critical to the function of the enzyme.26 Our enzymatic and crystallographic studies show that deletion of this site results in a protein with identical activity and stability as wild type. The differences compared to the earlier study raise the possibility that this carbohydrate is necessary not for the stability and activity of the glycoprotein, but for the trafficking of human α-NAGAL to the lysosome.
The structure of human α-NAGAL reveals the molecular basis of Schindler disease. The crystal structure reported here shows that Schindler disease is typically a protein folding disease; thus, it might then be treatable using not only enzyme replacement strategies (such as those used for Fabry and Gaucher patients), but also pharmacological chaperone approaches.37 The collection of small molecule binding sites around the active site opens the door to designing molecules that might act as inhibitors or as chemical chaperones against human α-NAGAL, by taking advantage of the ligand binding features revealed in the human α-NAGAL structure.
The location of the E367 residue on the surface raises the question of how the E367K substitution leads to the loss of enzyme activity in patients. The residue is more than 30 Å from the active site and from the dimer interface, it is surrounded by a patch of mostly non-polar atoms (Fig. 1D), and it is buried by crystal contacts in each of the two independent copies in the crystallographic asymmetric unit. These observations suggest that E367 might be buried in a protein complex in the lysosome. Because the some of the substrates that accumulate in Schindler disease patients have terminal galactose and sialic acid residues, the structure suggests the possibility that α-NAGAL participates in a known complex in the lysosome comprised of α-neuraminidase, β-galactosidase, and the protective protein. The E367K substitution might disrupt this protein complex in the lysosome, leading to the loss of multiple enzymatic activities, and the subsequent accumulation of substrates with terminal α-GalNAc, β-galactose, and sialic acid saccharides.
Materials and Methods
Molecular biology
Human NAGA cDNA was purchased from Open Biosystems. The ORF, including the native human signal sequence, was PCR amplified using Phusion polymerase (NEB) with primers that added a hexahistidine tag and stop codon to the C terminus. For the stably transfected insect cells, the PCR product was gel purified, incubated with Taq polymerase to add 3′ A-overhangs, and cloned into the pIB/V5-His-TOPO TA vector as per the manufacturer’s instruction (Invitrogen). For baculoviral expression, the cDNA was also cloned into the vector pAcSec1 (Orbigen), with the insect gp64 signal sequence replacing the native human signal sequence. Recombinant virus was generated using Sapphire baculoviral DNA according to the manufacturer’s protocol (Orbigen), and plaque purified. Clones were analyzed by restriction enzyme digest and sequenced. Site-directed mutagenesis (Phusion, NEB) was performed to make five additional constructs (N124Q, N177Q, N201Q, N359Q, and N385Q), each lacking one N-linked glycosylation site. Residue numbering begins with the first amino acid of the 17 residue signal sequence, and the mature polypeptide begins at L18.
Cell culture
Sequence-verified recombinant plasmids were transfected into Tn5 (High Five) insect cells (Invitrogen) using Cellfectin II (Invitrogen). Polyclonal cell lines were established by using selective media containing 100 ug/ml blastocidin, as per the manufacturer’s protocols (Invitrogen). After approximately 10 days of selection, the cell lines were expanded from T-flasks to 30 ml shaker flasks. SFX-Insect serum free media (HyClone) was used at all times. For large-scale expression, either 1L cultures in 3L baffled polycarbonate Fernbach flasks (Corning) were grown, or 5 L batch cultures in a Wave Bioreactor 2/10EH disposable unit (GE Healthcare). In either case, cultures were inoculated at 5 × 105 cells/ml in fresh serum-free media, and grown for 3-5 days until viability dropped below 60-70%.
We expressed α-NAGAL both in baculovirally infected and in stably transfected insect cell expression systems; the latter produced more reproducible and slightly higher expression levels. Typically, three 5L batches of the polyclonal α-NAGAL High-Five cell lines were grown in a Wave bioreactor, and the resulting culture media pooled and used for purification. The recombinant protein was secreted into the culture supernatant.
Protein purification
E. coli expressed α-NAGAL was Ni-affinity purified under denaturing conditions as per manufacturer’s protocols (Qiagen). K. lactis expressed α-NAGAL was Ni-affinity purified from spent culture media after addition of phosphate buffer and sodium chloride. Insect cells were pelleted by centrifugation at 800g for 10 min, and the supernatant clarified by centrifuging for 45 min at 10,000g. After concentrating and buffer exchange of the supernatant by tangential flow filtration, the protein was purified using Ni affinity and anion exchange chromatography. Supernatant was exchanged into 50 mM sodium phosphate pH 7.0, 250 mM sodium chloride, passaged over a 10 ml Ni-Sepharose-FF column (GE Healthcare), and then eluted with an imidazole gradient (0-400 mM) over 50 column volumes on an FPLC at 4° C. α-NAGAL eluted at approximately 150 mM imidazole. Fractions with purified α-NAGAL were pooled, and dialyzed against an excess of 20 mM Bis-Tris pH 6.0. The dialyzed fractions were then passed over a 3 ml SOURCE15Q anion-exchange column (GE Healthcare). Proteins were separated with a salt gradient using buffer A (20 mM Bis-Tris pH 6.0) and buffer B (20 mM Bis-Tris pH 6.0, 1 M sodium chloride), going from 0% to 30% B over 100 column volumes at 4° C. Pure α-NAGAL eluted between 70 and 130 mM NaCl. The fractions were pooled, washed several times with 20 mM Bis-Tris pH 6.0, 0.01% sodium azide, and concentrated to 20 mg/ml.
Kinetic assays
A kinetic assay measuring hydrolysis of pNP-α-GalNAc and pNP-α-Gal (each from Toronto Research Chemicals) was adapted from a Sigma assay. Stocks of wild type and N201Q α-NAGAL proteins were made at 0.9 μM, based on UV-absorption at 280 nm and an extinction coefficient of 2.1 mg/ml protein/OD280. Enzyme and substrate solutions were equilibrated to 37°C. 10 μl of the enzyme stock was added to 140 μl of 100 mM citrate/phosphate buffer pH 4.5, with eight substrate concentrations (0.01 to 5 mM for pNP-α-GalNAc and 1 to 50 mM for pNP-α-Gal). Every 30 seconds for four minutes, 10 μl aliquots were removed and added to 290 μl of 200 mM borate buffer, pH 9.8. Absorbance at 400 nm was measured in a microplate reader. Blank values were subtracted, and absorbance of pNP at 400 nm was converted to [pNP] using an extinction coefficient of 18.1 mM-1 cm-1. Triplicate or quadruplicate measurements were averaged and used to calculate standard deviations. The slopes of the plots of [pNP] vs. time were calculated using the LINEST function of Excel. The change in [pNP] over time was plotted (with errors bars from the LINEST fit) as a function of substrate concentration, and a weighted fit to a Michaelis-Menten hyperbolic curve was used to obtain KM, Vmax, and kcat in KaleidaGraph.
Crystallography
Crystals of α-NAGAL mutant N201Q (with the third glycosylation site removed) were grown in 8-16% PEG3350, 60 mM citric acid, 40 mM Bis-Tris propane (pH 4.1) by hanging-drop vapor diffusion at 20° C. Despite extensive crystallization efforts, only two crystals, each 100-200um, grew fused together in a single condition after three months. The two crystals were separated into four fragments using MicroTools (MiTeGen). The crystals were harvested into 20% PEG3350, 60 mM citric acid, 40 mM Bis-Tris propane, transferred to cryoprotectant solution (20% PEG3350, 60 mM citric acid, 40 mM Bis-Tris propane, and 20% [v:v] glycerol) for two minutes, and then rapidly cooled into liquid nitrogen. Data collection was performed on a RU-H3R rotating copper-anode generator (Rigaku) and at Brookhaven National Laboratory beamline X6A. For N-acetylgalactosamine soaks, crystals stored in cryoprotectant were thawed into a cryoprotectant solution containing 200 mM N-acetylgalactosamine and rapidly cooled again into liquid nitrogen. For the N-acetylgalactosamine soak, an unexpected power outage resulted in the loss of a crystal after 60° of data collection at 1.9 Å resolution, necessitating collection from a second crystal fragment (of lesser quality at 2.4 Å resolution) and merging the data from the two (at 2.2 Å resolution). For the galactose soak, 20% D-galactose was added to the harvest buffer prior to data collection. Ice rings in the diffraction images from the galactose-soaked crystal limited the quality and completeness of the x-ray data. For the covalent intermediate structure, TNP-2-deoxy-2,2-difluoro-α-Gal was prepared as described,38 and 50mM of compound was added to the crystal for 10 minutes prior to cryocooling and x-ray data collection. Diffraction data were processed with HKL2000.39 Chicken α-NAGAL 23 (PDB ID: 1KTB) was used as a molecular replacement model in the CCP4 program AMoRe.40 Atomic models were built into electron density using the program O,41 and refinement and water placement were done using REFMAC5.40 The test set, comprised a random 5% of the reflections, was matched for the three structures reported here. Ramachandran plots were computed using PROCHECK.42 Sequence alignments were calculated with ClustalW,43 coordinates were superimposed using LSQMAN,44 and accessible surface areas were calculated in AREAIMOL.40 Molecular docking studies were performed in O41 and in Pymol.45 Figures were made in MolScript,46 POVScript+,47 Pymol,45 and ccp4mg.40
Acknowledgements
We thank Matt Metcalf and Yurie Kim for contributions to the project. We thank the Wave Research Initiative for use of the Wave equipment. We thank John Burand and Woojin Kim for assistance with insect cell culture. We thank Harry Brumer, Dina Ivanen, and Anna Kulminskaya for the gift of TNP-2-deoxy-2,2-difluoro-α-Gal. We gratefully acknowledge Jean Jankonic, Marc Allaire, and Vivian Stojanoff at the X6A beam line, funded by the National Institute of General Medical Sciences, National Institute of Health under agreement GM-0080. This work was funded by NIH grant R01 DK76877 to S.C.G. and by NSF Integrative Graduate Education and Research Traineeship 0654128 to N.E.C.
Accession numbers: Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers 3H53, 3H54, 3H55, and 3IGU.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Diggelen OP, Schindler D, Kleijer WJ, Huijmans JM, Galjaard H, Linden HU, Peter-Katalinic J, Egge H, Dabrowski U, Cantz M. Lysosomal α-N-acetylgalactosaminidase deficiency: a new inherited metabolic disease. Lancet. 1987;2:804. doi: 10.1016/s0140-6736(87)92542-6. [DOI] [PubMed] [Google Scholar]
- 2.Schindler D, Bishop DF, Wolfe DE, Wang AM, Egge H, Lemieux RU, Desnick RJ. Neuroaxonal dystrophy due to lysosomal α-N-acetylgalactosaminidase deficiency. N. Engl. J. Med. 1989;320:1735–40. doi: 10.1056/NEJM198906293202606. [DOI] [PubMed] [Google Scholar]
- 3.Desnick RJ, Schindler D. α-N-Acetylgalactosaminidase Deficiency: Schindler Disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edit. McGraw-Hill; New York: 2001. pp. 3483–3505. [Google Scholar]
- 4.van Diggelen OP, Schindler D, Willemsen R, Boer M, Kleijer WJ, Huijmans JG, Blom W, Galjaard H. α-N-acetylgalactosaminidase deficiency, a new lysosomal storage disorder. J. Inherit. Metab. Dis. 1988;11:349–57. doi: 10.1007/BF01800424. [DOI] [PubMed] [Google Scholar]
- 5.Wang AM, Schindler D, Desnick R. Schindler disease: the molecular lesion in the α-N-acetylgalactosaminidase gene that causes an infantile neuroaxonal dystrophy. J. Clin. Invest. 1990;86:1752–6. doi: 10.1172/JCI114901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang AM, Kanzaki T, Desnick RJ. The molecular lesion in the α-N-acetylgalactosaminidase gene that causes angiokeratoma corporis diffusum with glycopeptiduria. J. Clin. Invest. 1994;94:839–45. doi: 10.1172/JCI117404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chabas A, Coll MJ, Aparicio M, Rodriguez Diaz E. Mild phenotypic expression of α-N-acetylgalactosaminidase deficiency in two adult siblings. J Inherit Metab Dis. 1994;17:724–31. doi: 10.1007/BF00712015. [DOI] [PubMed] [Google Scholar]
- 8.Keulemans JL, Reuser AJ, Kroos MA, Willemsen R, Hermans MM, van den Ouweland AM, de Jong JG, Wevers RA, Renier WO, Schindler D, Coll MJ, Chabas A, Sakuraba H, Suzuki Y, van Diggelen OP. Human α-N-acetylgalactosaminidase (α-NAGA) deficiency: new mutations and the paradox between genotype and phenotype. J. Med. Genet. 1996;33:458–64. doi: 10.1136/jmg.33.6.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kodama K, Kobayashi H, Abe R, Ohkawara A, Yoshii N, Yotsumoto S, Fukushige T, Nagatsuka Y, Hirabayashi Y, Kanzaki T. A new case of α-N-acetylgalactosaminidase deficiency with angiokeratoma corporis diffusum, with Meniere’s syndrome and without mental retardation. Br. J. Dermatol. 2001;144:363–8. doi: 10.1046/j.1365-2133.2001.04028.x. [DOI] [PubMed] [Google Scholar]
- 10.Bakker HD, de Sonnaville ML, Vreken P, Abeling NG, Groener JE, Keulemans JL, van Diggelen OP. Human α-N-acetylgalactosaminidase (α-NAGA) deficiency: no association with neuroaxonal dystrophy? Eur. J. Hum. Genet. 2001;9:91–6. doi: 10.1038/sj.ejhg.5200598. [DOI] [PubMed] [Google Scholar]
- 11.Chabas A, Duque J, Gort L. A new infantile case of alpha-N-acetylgalactosaminidase deficiency. Cardiomyopathy as a presenting symptom. J Inherit Metab Dis. 2007;30:108. doi: 10.1007/s10545-006-0470-1. [DOI] [PubMed] [Google Scholar]
- 12.Wang AM, Bishop DF, Desnick RJ. Human α-N-acetylgalactosaminidase-molecular cloning, nucleotide sequence, and expression of a full-length cDNA. Homology with human α-galactosidase A suggests evolution from a common ancestral gene. J. Biol. Chem. 1990;265:21859–66. [PubMed] [Google Scholar]
- 13.Dean KJ, Sung SS, Sweeley CC. The identification of α-galactosidase B from human liver as an α-N-acetylgalactosaminidase. Biochem. Biophys. Res. Commun. 1977;77:1411–7. doi: 10.1016/s0006-291x(77)80136-8. [DOI] [PubMed] [Google Scholar]
- 14.Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A Deficiency: Fabry Disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edit. McGraw-Hill; New York: 2001. pp. 3733–3774. [Google Scholar]
- 15.Olsson ML, Clausen H. Modifying the red cell surface: towards an ABO-universal blood supply. Br J Haematol. 2008;140:3–12. doi: 10.1111/j.1365-2141.2007.06839.x. [DOI] [PubMed] [Google Scholar]
- 16.Liu QP, Sulzenbacher G, Yuan H, Bennett EP, Pietz G, Saunders K, Spence J, Nudelman E, Levery SB, White T, Neveu JM, Lane WS, Bourne Y, Olsson ML, Henrissat B, Clausen H. Bacterial glycosidases for the production of universal red blood cells. Nat Biotechnol. 2007;25:454–64. doi: 10.1038/nbt1298. [DOI] [PubMed] [Google Scholar]
- 17.Kimura A, Kanekura T, Saito Y, Sagawa K, Nosaka M, Kanzaki T, Tsuji T. Blood group A glycosphingolipid accumulation in the hair of patients with alpha-N-acetylgalactosaminidase deficiency. Life Sci. 2005;76:1817–24. doi: 10.1016/j.lfs.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 18.Asfaw B, Ledvinova J, Dobrovolny R, Bakker HD, Desnick RJ, van Diggelen OP, de Jong JG, Kanzaki T, Chabas A, Maire I, Conzelmann E, Schindler D. Defects in degradation of blood group A and B glycosphingolipids in Schindler and Fabry diseases. J Lipid Res. 2002;43:1096–104. doi: 10.1194/jlr.m100423-jlr200. [DOI] [PubMed] [Google Scholar]
- 19.d’Azzo A, Andria G, Strisciuglio P, Galjaard H. Galactosialidosis. In: Scriver CS, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edit. McGraw-Hill; New York: 2001. pp. 3811–3826. [Google Scholar]
- 20.Thomas GH. Disorders of Glycoprotein Degradation: α-Mannosidosis, β-Mannosidosis, Fucosidosis, and Sialidosis. In: Scriver CS, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edit. McGraw-Hill; New York: 2001. pp. 3507–3533. [Google Scholar]
- 21.Shaikh FA, Withers SG. Teaching old enzymes new tricks: engineering and evolution of glycosidases and glycosyl transferases for improved glycoside synthesis. Biochem Cell Biol. 2008;86:169–77. doi: 10.1139/O07-149. [DOI] [PubMed] [Google Scholar]
- 22.Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human α-galactosidase. J Mol Biol. 2004;337:319–35. doi: 10.1016/j.jmb.2004.01.035. [DOI] [PubMed] [Google Scholar]
- 23.Garman SC, Hannick L, Zhu A, Garboczi DN. The 1.9 Å structure of α-N-acetylgalactosaminidase: molecular basis of glycosidase deficiency diseases. Structure. 2002;10:425–434. doi: 10.1016/s0969-2126(02)00726-8. [DOI] [PubMed] [Google Scholar]
- 24.Sakuraba H, Matsuzawa F, Aikawa S, Doi H, Kotani M, Nakada H, Fukushige T, Kanzaki T. Structural and immunocytochemical studies on α-N-acetylgalactosaminidase deficiency (Schindler/Kanzaki disease) J Hum Genet. 2004;49:1–8. doi: 10.1007/s10038-003-0098-z. [DOI] [PubMed] [Google Scholar]
- 25.Kanekura T, Sakuraba H, Matsuzawa F, Aikawa S, Doi H, Hirabayashi Y, Yoshii N, Fukushige T, Kanzaki T. Three dimensional structural studies of α-N-acetylgalactosaminidase (α-NAGA) in α-NAGA deficiency (Kanzaki disease): different gene mutations cause peculiar structural changes in α-NAGAs resulting in different substrate specificities and clinical phenotypes. J Dermatol Sci. 2005;37:15–20. doi: 10.1016/j.jdermsci.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 26.Ohta M, Ohnishi T, Ioannou YA, Hodgson ME, Matsuura F, Desnick RJ. Human α-N-acetylgalactosaminidase: site occupancy and structure of N-linked oligosaccharides. Glycobiology. 2000;10:251–61. doi: 10.1093/glycob/10.3.251. [DOI] [PubMed] [Google Scholar]
- 27.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fujimoto Z, Kaneko S, Momma M, Kobayashi H, Mizuno H. Crystal structure of rice α-galactosidase complexed with D-galactose. J Biol Chem. 2003;278:20313–8. doi: 10.1074/jbc.M302292200. [DOI] [PubMed] [Google Scholar]
- 29.Golubev AM, Nagem RA, Brandao Neto JR, Neustroev KN, Eneyskaya EV, Kulminskaya AA, Shabalin KA, Savel’ev AN, Polikarpov I. Crystal structure of α-galactosidase from Trichoderma reesei and its complex with galactose: implications for catalytic mechanism. J Mol Biol. 2004;339:413–22. doi: 10.1016/j.jmb.2004.03.062. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Jaramillo FJ, Gonzalez-Ramirez LA, Albert A, Santoyo-Gonzalez F, Vargas-Berenguel A, Otalora F. Structure of concanavalin A at pH 8: bound solvent and crystal contacts. Acta Crystallogr D Biol Crystallogr. 2004;60:1048–56. doi: 10.1107/S0907444904007000. [DOI] [PubMed] [Google Scholar]
- 31.Koshland DE. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. Cambridge Philos. Soc. 1953;28:416–436. [Google Scholar]
- 32.Ly HD, Howard S, Shum K, He S, Zhu A, Withers SG. The synthesis, testing and use of 5-fluoro-α-D-galactosyl fluoride to trap an intermediate on green coffee bean α-galactosidase and identify the catalytic nucleophile. Carbohydr. Res. 2000;329:539–47. doi: 10.1016/s0008-6215(00)00214-7. [DOI] [PubMed] [Google Scholar]
- 33.Schram AW, Hamers MN, Tager JM. The identity of α-galactosidase B from human liver. Biochim. Biophys. Acta. 1977;482:138–44. doi: 10.1016/0005-2744(77)90361-8. [DOI] [PubMed] [Google Scholar]
- 34.Mehndiratta P, Walton WJ, Hare JT, Pulido S, Parthasarathy G, Emmett MR, Marshall AG, Logan TM. Expression, purification, and characterization of avian Thy-1 from Lec1 mammalian and Tn5 insect cells. Protein Expr Purif. 2004;33:274–87. doi: 10.1016/j.pep.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Kost TA, Condreay JP, Jarvis DL. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat Biotechnol. 2005;23:567–75. doi: 10.1038/nbt1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddi AL, Sankaranarayanan K, Arulraj HS, Devaraj N, Devaraj H. Serum α-N-acetylgalactosaminidase is associated with diagnosis/prognosis of patients with squamous cell carcinoma of the uterine cervix. Cancer Lett. 2000;158:61–4. doi: 10.1016/s0304-3835(00)00502-4. [DOI] [PubMed] [Google Scholar]
- 37.Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nat Rev Genet. 2002;3:954–66. doi: 10.1038/nrg963. [DOI] [PubMed] [Google Scholar]
- 38.Brumer H, 3rd, Sims PF, Sinnott ML. Lignocellulose degradation by Phanerochaete chrysosporium: purification and characterization of the main α-galactosidase. Biochem J. 1999;339(Pt 1):43–53. [PMC free article] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW, editor. Methods in Enzymology: Macromolecular Crystallography, part A. Vol. 276. Academic Press; 1997. pp. 307–326. J. R. M. S. [DOI] [PubMed] [Google Scholar]
- 40.Collaborative Computational Project, N The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 41.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A. 1991;47:110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 42.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallog. 1993;26:283–291. [Google Scholar]
- 43.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleywegt GJ, Read RJ. Not your average density. Structure. 1997;5:1557–1569. doi: 10.1016/s0969-2126(97)00305-5. [DOI] [PubMed] [Google Scholar]
- 45.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; Palo Alto, CA, USA: 2008. [Google Scholar]
- 46.Kraulis PJ. MOLSCRIPT: A Program to Produce Both Detailed and Schematic Plots of Protein Structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
- 47.Fenn TD, Ringe D, Petsko GA. POVScript+: a program for model and data visualization using persistence of vision ray-tracing. J. Appl. Cryst. 2003;36:944–947. [Google Scholar]