

Abstract
The extent of an ischemic insult is less in brain regions enriched in astrocytes suggesting that astrocytes maintain function and buffer glutamate during ischemia. Astrocytes express a wide variety of potassium channels to support their functions including TREK-2 channels which are regulated by polyunsaturated fatty acids, intracellular acidosis and swelling; conditions that pertain to ischemia. The present study investigated the possible involvement of TREK-2 channels in cultured cortical astrocytes during experimental ischemia (anoxia/hypoglycemia) by examining TREK-2 protein levels, channel activity and ability to clear glutamate. We found that TREK-2 protein levels were increased rapidly within 2 hrs of the onset of simulated ischemia. This increase corresponded to an increase in temperature-sensitive TREK-2-like channel conductance and the ability of astrocytes to buffer extracellular glutamate even during ischemia. Together, these data suggest that up-regulation of TREK-2 channels may help rescue astrocyte function and lower extracellular glutamate during ischemia.
Keywords: glia, TREK-2 channels, ischemia, glutamate uptake
Introduction
Ischemia, glucose deprivation or mild mechanical trauma all result in neuronal death due to glutamate toxicity [1,2]. However, neuronal sensitivity to ischemia is less if neurons are surrounded by astrocytes [3]. One of the major functions of astrocytes is to maintain extracellular homeostasis in the brain by regulating extracellular concentrations of neuroactive substances such as K+, H+, GABA and glutamate. Under normal conditions, astrocytes have a much more hyperpolarized membrane potential (∼30 mV) than neurons, providing a driving force for K+-spatial buffering, glutamate transport, etc. We have previously shown that the highly hyperpolarized membrane potential of some cortical astrocytes is due, in part, to the presence of Kir4.1 inward rectifying K+ channels [4]. Kir4.1 channels are activated by ATP [5,6] and are likely to be inhibited in conditions when ATP is depleted (i.e., during ischemia). This finding suggests that Kir4.1 channels will be metabolically regulated, with activity declining in metabolically compromised conditions [7] perhaps allowing for the activity of other K+ channels to become physiologically important. The role of other K+ channels in astrocytes is supported by the finding that after decrease of Kir4.1 expression either by siRNA or after conditional knock-out of Kir4.1 channels, the membrane potential of the astrocytes is still substantially hyperpolarized [4,8].
In addition to Kir4.1 channels, astrocytes express a wide variety of potassium channels [9] including the TREK-2 tandem-pore domain channels [10,11,12], which are activated by conditions that are likely to pertain during ischemia. TREK-2 channel currents are increased by polyunsaturated fatty acids, intracellular acidosis in the physiological range and by mechanical stretch including swelling [13]. During ischemia, activation of phospholipases promotes liberation and accumulation of arachidonic acid [14], the intracellular pH of astrocytes becomes acidic and astrocytes swell [15,16]. All of these changes can activate TREK-2 channels and therefore, it has been hypothesized that TREK-2 in astrocytes may repolarize astrocytes and help maintain astrocytic function during pathological events such as anoxia, ischemia, hypoxia, hypoglycemia and/or spreading depression [10]. Indeed, Skatchkov et al. [17] and Päsler et al. [18] demonstrated recently roles for tandem pore domain K+ channels in homeostasis of glial cell volume (osmotic regulation) and in glial K+ buffering.
Two comprehensive studies have mapped the distribution of TREK-2 mRNA in rat brain [19,20]. TREK-2c is the splice variant found in abundance in the brain and it has a distinct expression in brain which partially overlaps with that of TREK-1 [20]. Subsequent studies using RT-PCR combined with electrophysiology have also shown expression of TREK-2 in cortical astrocytes [10,11], so it appears that TREK-2 is localized in both neurons and astrocytes.
Recent studies have examined the mRNA levels of TREK-2 after in vivo experimental ischemia in rats [21,22]. In a model of acute cerebral ischemia, TREK-2 mRNA levels were increased in hippocampus and cortex 24 hrs after middle cerebral artery occlusion [22]. In contrast, there were no changes in TREK-2 mRNA levels in rat hippocampus and cortex after 30 days of permanent bilateral carotid artery ligation [21]. These studies did not differentiate between the TREK-2 levels in astrocytes and neurons. Nor did they attempt to determine if increases in TREK-2 mRNA levels corresponded with increases in functional TREK-2 channels.
The purpose of the present study was to determine if TREK-2 channel protein in cultured astrocytes is increased after in vitro experimental ischemia (anoxia/hypoglycemia) and if so, to determine if this increase corresponds with a functional role for TREK-2 channels in ischemic trauma. For comparison, the effects of ischemia on the protein levels of TASK-1, TASK-3 and Kir4.1 potassium channels were examined.
Materials and Methods
Astrocyte Primary Cultures
Primary cultures of astrocytes were prepared from neocortex of 1-2 day old rats as previously described [4] and in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC). Briefly, brains were removed after decapitation and the meninges stripped away to minimize fibroblast contamination. The forebrain cortices were collected and dissociated using the stomacher blender method. The cell suspension was then allowed to filter by gravity through a #60 sieve and then through a #100 sieve. After centrifugation, the cells were suspended in modified Eagle's medium (containing 25 mM glucose, 2 mM glutamine, 1 mM pyruvate and 10% fetal bovine serum, 100 iU/ml penicillin/100 μg/ml streptomycin) and plated in uncoated 75 cm2 flasks at a density of 300,000 cells/cm2. The medium was exchanged with fresh culture medium about every 4 days. At confluence (about 12-14 days), the mixed glial cultures were treated with 50 mM leucine methylester (pH 7.4) for 60 min to effectively kill microglia [23]. Cultures were then allowed to recover for at least one day in growth medium prior to experimentation. Astrocytes were dissociated by trypsinization and reseeded onto the appropriate plates or coverglasses for the experiments. The purity of the astrocyte cultures was greater than 95% as assessed by immunocytochemical staining for glial fibrillary acidic protein (GFAP; data not shown). Astrocytes were used for the experiments between 2 and 6 weeks in vitro.
SDS-PAGE and Western Blotting Analysis
Astrocytes were harvested, pelleted and resuspended in homogenization buffer (pH 7.5) containing: (in mM) Tris-HCl 20, NaCl 150, EDTA 1.0, EGTA 1.0, PMSF 1.0, 1% Triton X-100, and an additional mixture of peptide inhibitors (leupeptin, bestatin, pepstatin, and aprotinin). Lysates were mixed with Urea sample buffer (plus dithiothreitol), boiled, spun briefly to pellet debris, and immediately run on 10% SDS-polyacrylamide gels. Protein concentration of cell homogenates was determined with the DC protein assay (Bio-Rad), followed by addition of an appropriate volume of Urea sample buffer (62mM Tris/HCl pH 6.8, 4% SDS, 8M Urea, 20mM EDTA, 5% β-Mercaptoethanol, 0.015% Bromophenol Blue) for a final concentration of 0.5 – 1.5 μg protein/μl, and incubation in a water bath at 100°C for 10 min. Western blotting was performed as previously described [4] using rabbit polyclonal antibodies against TREK-2 (1:1000 dilution; Chemicon or 1:500; Alomone), TASK-1, TASK-3 (both at 1:300, Alomone) or Kir4.1 (1:400, Sigma Chemical Co.). Final detection was performed with enhanced chemiluminescence methodology (SuperSignal® West Dura Extended Duration Substrate; Pierce, Rockford, IL) as described by the manufacturer, and the intensity of the signal measured in a gel documentation system (Versa Doc Model 1000, Bio Rad). In all cases, intensity of the chemiluminescence signal was corrected for minor differences in protein content after densitometry analysis of the India ink stained membrane.
Simulated Ischemia
To simulate ischemia in vitro, cultured astrocytes were subjected to anoxia and hypoglycemia. Astrocytes were plated in 60×15 mm petri dishes for Western blot experiments or 24 well plates for glutamate clearance assays and allowed to grow to confluence. When confluent, anoxia was achieved by removing growth medium from the cultures, rinsing gently by immersion in a bicarbonate-buffered balanced salt solution (BBSS) that contained (in mM): NaCl 127, KCl 3, NaHCO3 19.5, NaH2PO4 1.5, MgSO4 1.5, d-glucose 25, and CaCl2 1, and placing the cultures at 37°C in fresh BBSS previously gassed for 5 min with 5% CO2 and 95% N2. (anoxia buffer; pH 7.4). Hypoxia and hypoglycemia (simulated ischemia) were achieved by decreasing the concentration of d-glucose in these solutions (25% or 10% of normal glucose). The cultures were then placed in a chamber (Billups-Rothenberg, Del Mar, CA) flooded with 5% CO2 and 95% N2 and incubated at 37°C for 24 hrs. Control astrocytes were placed in BBSS (pH 7.4) under normoxic conditions.
Whole Cell Recording
For electrophysiology experiments, astrocytes were grown on coverglasses in 60×15 mm petri dishes and subjected to the conditions described above. Membrane currents were measured using the whole cell voltage-clamp technique as previously described [4]. Electrodes from hard glass (GC-150-10 glass tubing, Clark Electromedical Instruments, England) were pulled in four steps using a Sutter P-97 puller (Novato, CA). After filling with intracellular solution (ICS) containing (in mM): KCl 141, MgCl2 1, CaCl2 1, EGTA 10, HEPES 10, Na2ATP 3, spermine HCl 0.25, pH adjusted to 7.2 with NaOH/HCl, they had resistances of 6-8 MΩ. The extracellular solution (ECS) contained (in mM): NaCl 143, CaCl2 2, KCl 3 mM, MgCl2 2 and HEPES 10. High frequencies (>2 kHz) were cut off, using an Axopatch-200B amplifier and a CV-203BU headstage, and digitized through a DigiData 1200A interface (Axon Instruments, USA). The pClamp 9 (Axon Instrument, USA) software package was used for data acquisition and analysis. For controlling changes in temperature, we used the Dual Heater Controller TC-344A (Warner Instrument Corporation).
Transfection of tsA201 Cells with Rat Kir4.1
Cultures of tsA201 cells (an SV40 transformed variant of the HEK293 human embryonic kidney cell line generously provided by Dr. William Green) were maintained in Dulbecco's modified Eagle's medium (pH 7.4) supplemented with 10 mmol/l glucose, 2 mmol/l L-glutamine, 10% fetal calf serum, and 200 iU/ml penicillin/200 μg/ml streptomycin at 37°C (5% CO2 95% air). Wild-type rat Kir4.1 cDNA was cloned in the pEGFP-C1 expression vector (Clontech), which generates EGFP fusion proteins with EGFP on the N-terminus. The Kir4.1 channel subunit clones were transfected into tsA201 cells using the calcium phosphate transfection technique [24]. Recordings were made from cells 24–72 h after transfection. The chemicals for cultures and for transfections were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Successfully transfected cells were visualized by EGFP-green fluorescence.
Glutamate Clearance Assay
To evaluate the glutamate clearance capacity, primary astrocyte cultures were trypsinized and plated at equal density in 24 well dishes. Glutamate remaining in the medium (BBSS) was determined using the protocol of Abe et al. [25,26]. Briefly, BBSS in each well of the dish was replaced with 300 μl of BBSS containing 200 μM glutamate. After 60 min, the medium was removed and 50 μl of culture supernatant was transferred to 96-well culture plates and the colorometric assay was performed [4,25,26]. The production of MTT formazan was assessed by measurement of absorbance at 550 nm using a microplate reader (Wallac Victor, Perkin Elmer). A standard curve was constructed in each assay using cell-free BBSS containing known concentrations of glutamate. The concentration of extracellular glutamate in the samples was estimated from the standard curve ranging from 6.25 to 600 μM glutamate. As a control for each experiment, BBSS containing 200 μM glutamate was added to empty wells of a 24 well dish (no astrocytes) and processed together with the astrocytes, i.e. 60 min in the incubator until sample collection. For the ischemia experiments, the astrocytes were incubated inside the chamber (Billups-Rothenberg, Del Mar, CA) flooded with 5% CO2 and 95% N2. Statistical significance was considered to be p<0.05 using a two-way ANOVA followed by Tukey's test (Vassar-Stats Statistical Computation Website).
Results
TREK-2 channels are activated by arachidonic acid [27] and increased temperature [28] and have been functionally localized to cortical astrocytes [10,11]. We confirmed these previous findings using whole cell voltage-clamp recording from cultured cortical astrocytes after blocking Kir, KATP, KA, KD and BK channels using a cocktail of inhibitors (Ba2+ 100 μM, TEA 6 mM, 4-AP 4 mM, tolbutamide 500 μM). Fig. (1A) shows that TREK-2-type currents were activated by arachidonic acid (10 μM) and this activation was blocked by quinine (200 μM), a putative blocker of TREK-2 channels. Furthermore, these currents were activated by increasing temperature (Fig. 1B). Similar current responses were observed in 6/10 astrocytes after arachidonic acid (10 μM) and in 7/7 astrocytes after increased temperature (from 23 to 40°C). This response is consistent with that of Gnatenco et al. [10] who reported 50% of astrocytes were activated by arachidonic acid. Taken together, these data confirm that functional TREK-2-type channels are present in our cortical astrocyte cultures.
Fig. (1). TREK-2 currents recorded from cortical astrocytes.
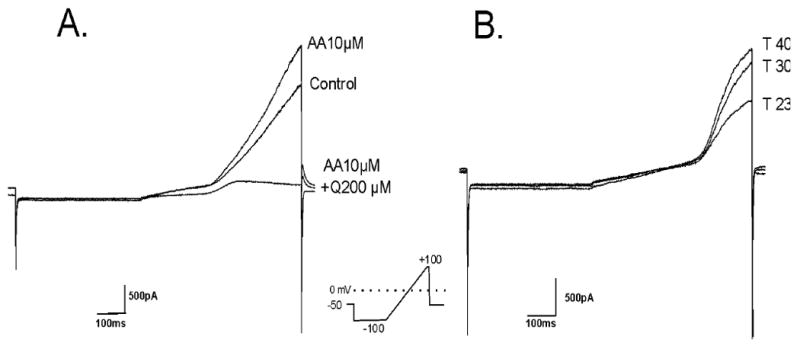
A. Effect of arachidonic acid (10 μM) and quinine (200 μM) on the ramp response of a cortical astrocyte in the presence of other K+ channel blockers (Ba 0.1 mM, TEA 6mM, 4-AP 4mM, tolbutamide 0.5 mM). [K]o = 3 mM.
B. Ramp response of a cortical astrocyte showing activation of current by increasing temperature (23, 30 and 40°C). This effect was obtained in the presence of other K+ channel blockers (as above). [K]o = 3 mM. In both cases, the astrocyte was held at Em.
We next examined the effect of experimental ischemia (anoxia and hypoglycemia) on TREK-2 protein levels in cultured cortical astrocytes. Control astrocytes were incubated at 37°C under normoxic conditions in BBSS, whereas astrocytes subjected to ischemia were incubated at 37°C in BBSS previously gassed with 5% CO2 and 95% N2 with reduced concentrations of d-glucose. Using Western blot, we determined that 24 hours of anoxia/hypoglycemia (either 1/4 or 1/10 of normal glucose) increased TREK-2 protein levels in cortical astrocytes by approximately 3- and 4-fold, respectively (Fig. 2A, B). In subsequent experiments, we determined the onset and recovery of the effect of ischemia on TREK-2 protein levels. We found that TREK-2 protein levels were increased significantly two hours after anoxia in 1/10 normal glucose (Fig. 2C). Furthermore, TREK-2 channel protein levels returned to control within one day of return to normoxic/normoglycemic conditions (Fig. 2D) after prior exposure to 24 hours of experimental ischemia (anoxia in 10% glucose).
Fig. (2). Effect of Hypoxia and Hypoglycemia on TREK-2 Protein Levels in Cultured Cortical Astrocytes.
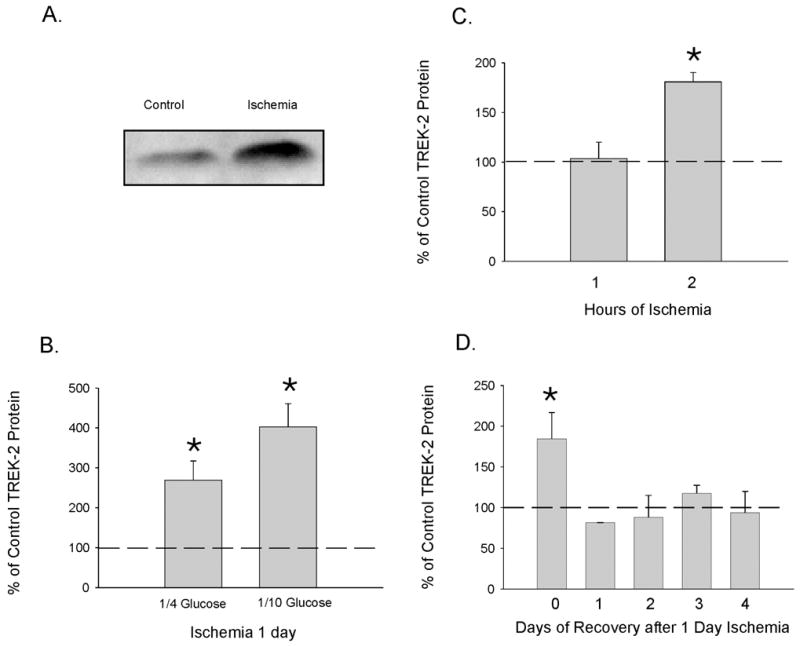
Using Western blot, the levels of TREK-2 protein were determined in control (normoxia and 25 mM glucose) astrocytes and astrocytes exposed to hypoxia and hypoglycemia. A. shows an actual blot from an experiment where astrocytes were exposed to hypoxia and hypoglycemia (2.5 (1/10) mM glucose) for 1 day. B. represents four different experiments (from different astrocyte primary cultures) where the astrocytes were exposed to hypoxia and hypoglycemia (6.25 (1/4) or 2.5 (1/10) mM glucose) for 1 day. The protein levels are presented as % of control, where the control level is indicated by the thick dotted line. Intensity of the chemiluminescence signal was normalized for minor changes in protein content after densitometry analysis of the India ink stained membrane (to measure relative protein concentrations). C. shows the onset of significant elevations in TREK-2 protein after exposure to anoxia and 1/10 glucose. One hour after experimental ischemia there was no difference from control, but by two hours of ischemia TREK-2 protein levels were significantly elevated. The results of 3 separate experiments performed using different astrocyte cultures are shown. The control level is indicated by the thick dotted line. D. shows the recovery of TREK-2 protein levels. After exposure to 1 day of anoxia and 1/10 glucose, astrocytes were placed in normal cell culture medium and returned to the normoxic environment. TREK-2 expression in cultures were determined 0, 1, 2, 3 and 4 days after experimental ischemia by Western blot. The results of 3 separate experiments using different astrocyte cultures are shown. The control level (astrocytes not exposed to ischemia) is indicated by the thick dotted line. For panels B, C and D, the asterisks indicate a significant difference from control (ANOVA followed by Tukey's test; p<0.05).
We also determined the effect of anoxia/hypoglycemia on the protein levels of other potassium channels known to be expressed in cultured astrocytes [4,10]. We found that the expression of TASK-1, TASK-3 and Kir4.1 channels was not altered 24 hours after anoxia/hypoglycemia (1/10 normal glucose) suggesting that experimental ischemia selectively up-regulated TREK-2 channels (Fig. 3).
Fig. (3). Effect of Hypoxia and Hypoglycemia on TASK-1, TASK-3 and Kir4.1 Protein Levels in Cultured Cortical Astrocytes.
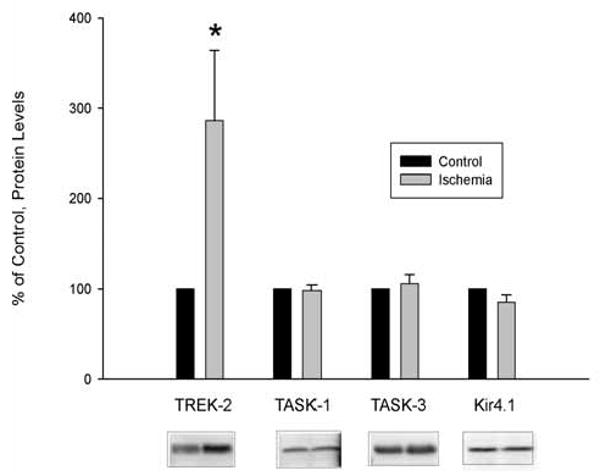
Astrocytes were exposed to hypoxia and hypoglycemia (2.5 (1/10) mM glucose) for 24 hrs and the protein levels of TREK-2, TASK-1, TASK-3 and Kir4.1 were determined by Western blot (representative blots shown below the graph). The data are expressed as % of control (astrocytes not exposed to ischemia). In all, five separate experiments different astrocyte cultures were used. Of these, four experiments were used to determine TREK-2 and TASK-1 protein levels, whereas three were used to determine TASK-3 and Kir4.1 protein levels. In addition, TASK-1 protein levels were determined in a fifth separate experiment. Therefore, the n of the experiments was 4, 5, 3 and 3 for TREK-2, TASK-1, TASK-3 and Kir4.1, respectively.
To determine whether the increased level of TREK-2 channel protein observed by Western blot after ischemic conditions was actually forming functional channels in the astrocytic membrane, we used whole cell voltage-clamp to measure the current of control astrocytes and astrocytes exposed to 24 hours of simulated ischemia (anoxia and 1/10 glucose) in response to a ramp and step protocol and changes in temperature. After simulated ischemia, cortical astrocytes had significantly greater outward current in response to increasing temperature than control cells (Fig. 4) indicating that these TREK-2 channels are functional.
Fig. (4). Effect of Hypoxia and Hypoglycemia on Temperature-Induced Current Responses of Cultured Cortical Astrocytes.
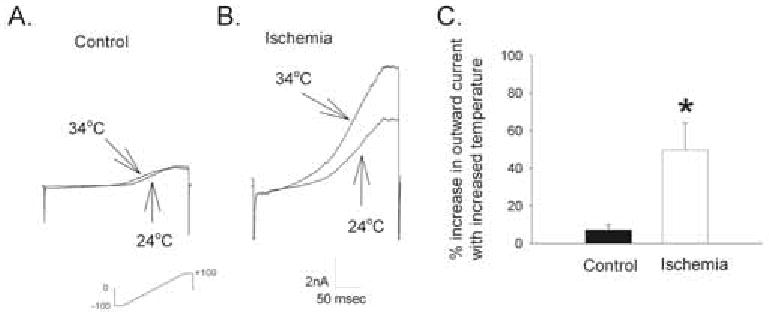
A. Responses of a control astrocyte and B. an astrocyte subjected to simulated ischemia (24 hrs anoxia at reduced glucose (1/10 normal, 2.5 mM)) to a ramp and step protocol at 24°C and 34°C, in the presence of other K+ channel blockers (Ba 0.1 mM, TEA 6mM, 4-AP 4mM, tolbutamide 0.5 mM). [K]o = 3 mM. C: Summary of % increase in outward current with increased temperature, (max current at 34°C over max current at 24°C × 100) of control astrocytes and astrocytes exposed to experimental ischemia. The asterisk indicates a significant difference from control (t-test; p<0.05). (n=5 for each group).
We then measured glutamate clearance capabilities of control and ischemia-treated astrocytes using a colorimetric assay [4,25]. The ability of astrocytes incubated under normoxic or anoxic/hypoglycemic conditions to clear glutamate in the presence and absence of quinine (100 μM) was assessed (Fig. 5). Quinine has been shown to block arachidonic-acid-sensitive K+ channel currents in astrocytes [11], presumably TREK-2 channels. However, since glutamate uptake by cultured cortical astrocytes also depends on Kir4.1 channel expression [4,8], to make sure that using low concentrations of quinine would specifically impair TREK-2-dependent uptake, we first tested the propensity to quinine's blocking of Kir4.1 channels heterologously expressed in tsA201 cells. We found that quinine at 100 μM did not affect currents through Kir4.1 channels, the predominant inward rectifying K+ channel in astrocytes [29,30]. Using a step and ramp protocol as in Figs. (1 and 4), we found that there was no significant difference between the inward currents in Kir4.1 expressing cells in control (-1560 ± 216 pA) and after application of 100 μM quinine to the same cells (-1473 ± 224 pA; n=3) (original data are not shown). This demonstrates a lack of inhibition of Kir4.1 currents by 100 μM quinine. We, therefore, used 100 μM quinine to differentiate between the effects of TREK-2 channels and Kir4.1 channels on glutamate clearance by astrocytes.
Fig. (5). Effect of Ischemia and Quinine on Glutamate Clearance by Rat Cortical Astrocytes.
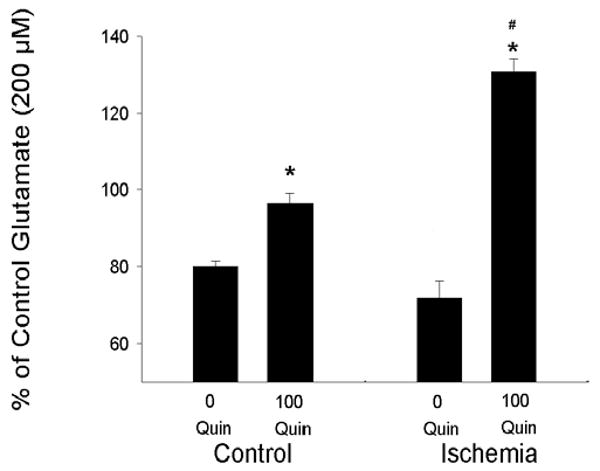
Control astrocytes (incubated for 1 day in BBSS) and astrocytes exposed to ischemic conditions (1 day of anoxia and hypoglycemia 1/10 normal glucose) were incubated for 60 min with 300μl of 200 μM glutamate and 0 or 100 μM quinine. The concentration of glutamate was determined after 60 min and compared with the concentration of glutamate measured in the absence of astrocytes. Data are expressed as % of control glutamate (concentration measured in absence of astrocytes). * indicates significant difference in the presence of quinine from the respective 0 quinine group. # indicates a significant difference in the response of ischemia-treated cells to quinine as compared with quinine-treated control cells (n=6 per group).
There was no significant difference in glutamate clearance by astrocytes subjected to normoxic conditions and astrocytes subjected to ischemic conditions (Fig. 5). Sixty min after incubation with 300 μl of 200 μM glutamate in serum-free medium, astrocytes cultured under normoxic conditions cleared 20% of the glutamate from the medium and this effect was inhibited by quinine (Fig. 5). Similarly, astrocytes exposed to ischemic conditions were also able to effectively clear glutamate from the medium (a 28% reduction in 60 min). Strikingly, the inhibition of clearance by quinine (100 μM) in ischemic cells was significantly greater than inhibition in control cells (Fig. 5) indicating a major contribution of TREK-2 channels in glutamate clearance by astrocytes exposed to experimental ischemia. All data are expressed as % of control glutamate where 100% is the measured concentration of glutamate after serum free medium with 200 μM glutamate was incubated for 60 min without astrocytes. In the experiments described in Fig. (5), the concentration of glutamate in dishes with no astrocytes remained at approximately 200 μM.
Discussion
The ability of astrocytes to regulate the extracellular concentrations of neuroactive substances such as K+ and glutamate depends upon the presence of K+ channels in their membranes and the hyperpolarized membrane potential of these cells [4,8,32]. We and others have previously shown that the highly hyperpolarized membrane potential of glial cells is largely due to the presence of Kir4.1 inward rectifying K+ channels [4,8,31], although Kir4.1 channels are not the only channels contributing to the overall negative membrane potential of glia [4,17, 8]. Particularly in astrocytes, after knock-down [4] or pharmacological blockade (Fig. 1) of Kir4.1-containing channels, inward currents are dramatically reduced whereas robust outward currents remain under physiological K+ conditions. This current profile is consistent with the presence of tandem-pore domain K+ channels [33] in astrocytes.
In the present study, we used Western blot, electrophysiology, pharmacology and a glutamate clearance assay to determine the properties and the putative functional role of TREK-2 channels in astrocytes during experimental ischemia. We confirm that TREK-2 channels are present in cultured astrocytes under control conditions (as determined by I/V curve and pharmacology and temperature-sensitivity) and that these channels are functionally up-regulated during ischemic conditions. The ability of ischemia to increase TREK-2 levels is relatively selective as expression of several other potassium channels in astrocytes (TASK-1, TASK-3 and Kir4.1) remains unchanged.
After 2 hours of experimental ischemia, expression of TREK-2 protein was increased to 175% of control, suggesting that regulation of TREK-2 protein changes may occur at the translational and not transcriptonal level. There are a number of post-transcriptional mechanisms that could be involved: 1) regulation of translation by factors binding to the UTR of mRNA [34], 2) liberation of TREK-2 mRNA from production bodies or p-bodies [35] and/or 3) decreased degradation of protein [36]. All of these processes would result in increased TREK-2 protein within the astrocyte. Alternatively, rapid changes in mRNA expression could account for these differences, although Xu et al. [21] reported no changes in TREK-2 mRNA levels in rat hippocampus and cortex after 30 days of permanent bilateral carotid artery ligation.
We determined if this increased TREK-2 protein corresponded with an increase in functional TREK-2 channels in the astrocytic membrane. Using whole-cell voltage-clamp recording and increases in temperature to activate TREK-2 channels [28], we found much larger outward currents in astrocytes exposed to ischemic conditions than in control astrocytes (Fig. 4). Furthermore, there was a much greater activation of outward currents recorded from “ischemic” astrocytes in response to increased temperature known to increase TREK-2 channel activity [28]. This suggests that not only are TREK-2 protein levels increased in these astrocytes, but also that they are forming functional channels in the astrocytic membrane.
To determine if functional up-regulation of TREK-2 channels could help to protect neurons during ischemic events, we evaluated the ability of astrocytes to clear glutamate. Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system [37,38]. Rapid removal of glutamate from the extracellular space is required for the survival and normal function of neurons. High-affinity Na+-dependent electrogenic transporters maintain low extracellular glutamate concentration. Although the glutamate transporters are expressed in both astrocytes and neurons, astrocytes are the cell type primarily responsible for glutamate uptake [39,40]. Extracellular glutamate levels are increased following ischemia, hypoglycemia and trauma [41] and if in vitro extracellular glutamate levels increase >100 μM for longer than 5 min, neuronal death can occur [42]. It has recently been shown that reactive astrocytes are neuroprotective during brain ischemia in vivo and this neuroprotection is mediated, in part, by glutamate transport [43].
Glutamate clearance depends upon glial cells having a hyperpolarized membrane potential [4,8,44]. During ischemia when ATP is reduced, Kir4.1 channels should not be functioning optimally and unless compensatory changes in other K+ channels occur, the membrane potential of astrocytes will be depolarized resulting in decreased glutamate clearance.
Using a colorimetric assay to assess glutamate clearance, we found no significant difference in the ability of control astrocytes and astrocytes subjected to ischemic conditions to clear glutamate. The finding that glutamate clearance is comparable in both normal and ischemic astrocytes suggests that TREK-2 channel up-regulation may compensate during ischemic conditions. To test this, we determined the contribution of glutamate clearance due to TREK-2 channels in both the control and ischemic conditions. The most notable difference observed was the amount of clearance inhibited by 100 μM quinine; a concentration of quinine that blocks arachidonic acid sensitive K+ channels (some of 2P-domain channels including TREK-2) in astrocytes [11], but not Kir4.1 channels (see results). This suggests a much greater contribution of TREK-2 channels to glutamate clearance by astrocytes exposed to ischemic conditions. Moreover, after blockade of TREK-2 channels in astrocytes subjected to ischemia, there is an apparent release of glutamate from the cells. Although the mechanism of this glutamate release is not known, it could perhaps be due to reverse transport by the glutamate transporter [45] or release from hemichannels [46] that are known to be opened during metabolic inhibition [47]. Disregarding the mechanism, these data do suggest that TREK-2 channels help to rescue astrocytic buffering of glutamate.
In summary, TREK-2 channels are functionally up-regulated in astrocytes after ischemia. Taken together, these data suggest that up-regulation of TREK-2 channels may help maintain the membrane potential of astrocytes and lower extracellular glutamate and K+ concentrations during ischemia.
Acknowledgments
The authors thank Paola López Pieraldi and Natalia Skachkova for their superior technical assistance. This publication was made possible by NIH grant numbers S11-NS48201 (MJE), U54 NS039408 (SNS), S06 GM050695 (MJE) and G12 RR03035 (supports core facilities used in this study) from NINDS, NIGMS and NCRR, respectively. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Footnotes
This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
References
- 1.Liu B, Liao M, Mielke JG, et al. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci. 2006;26:5309–19. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bell JD, Ai J, Chen Y, Baker A. Mild in vitro trauma induces rapid Glur-2 endocytosis, robustly augments calcium permeability and enhances susceptibility to secondary excitotoxic insult in cultured Purkinje cells. Brain. 2007;130:2528–42. doi: 10.1093/brain/awm164. [DOI] [PubMed] [Google Scholar]
- 3.Cervós-Navarro J, Diemer NH. Selective vulnerability in brain hypoxia. Crit Rev Neurobiol. 1991;6:149–82. [PubMed] [Google Scholar]
- 4.Kucheryavykh YV, Kucheryavykh LY, Nichols CG, et al. Down-regulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–81. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- 5.Bredt DS, Wang TL, Cohen NA, Guggino WB, Snyder SH. Cloning and expression of two brain-specific inwardly rectifying potassium channels. Proc Natl Acad Sci, USA. 1995;92:6753–7. doi: 10.1073/pnas.92.15.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takumi T, Ishii T, Horio Y, et al. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem. 1995;270:16339–46. doi: 10.1074/jbc.270.27.16339. [DOI] [PubMed] [Google Scholar]
- 7.Loussouarn G, Pike LJ, Ashcroft FM, Makhina EN, Nichols CG. Dynamic sensitivity of ATP-sensitive K(+) channels to ATP. J Biol Chem. 2001;276:29098–103. doi: 10.1074/jbc.M102365200. [DOI] [PubMed] [Google Scholar]
- 8.Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–65. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verkhratsky A, Steinhäuser C. Ion channels in glial cells. Brain Res Rev. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- 10.Gnatenco C, Han J, Snyder AK, Kim D. Functional expression of TREK-2 K+ channel in cultured rat brain astrocytes. Brain Res. 2002;931:56–67. doi: 10.1016/s0006-8993(02)02261-8. [DOI] [PubMed] [Google Scholar]
- 11.Ferroni S, Valente P, Caprini M, Nobile M, Schubert P, Rapisarda C. Arachidonic acid activates an open rectifier potassium channel in cultured rat cortical astrocytes. J Neurosci Res. 2003;72:363–72. doi: 10.1002/jnr.10580. [DOI] [PubMed] [Google Scholar]
- 12.Eaton MJ, Makarov F, Veh RW, et al. Functional expression of tandem pore potassium channels in glial cells. Washington, DC: Society for Neuroscience; 2003. Program No. 53.16. 2003 Abstract Viewer/Itinerary Planner. Online. [Google Scholar]
- 13.Kim D. Fatty acid-sensitive two-pore domain K+ channels. TIPS. 2003;24:648–54. doi: 10.1016/j.tips.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 14.Rehncrona S, Westerberg E, Akesson B, Siesjö B. Brain cortical fatty acids and phospholipids during and following complete and severe incomplete ischemia. J Neurochem. 1982;38:84–93. doi: 10.1111/j.1471-4159.1982.tb10857.x. [DOI] [PubMed] [Google Scholar]
- 15.Kempski O, von Rosen S, Weigt H, Staub F, Peters J, Baethmann A. Glial ion transport and volume control. Ann NY Acad Sci. 1991;633:306–17. doi: 10.1111/j.1749-6632.1991.tb15622.x. [DOI] [PubMed] [Google Scholar]
- 16.Plesnila N, Haberstok J, Peters J, Kolbl I, Baethmann A, Staub F. Effect of lactacidosis on cell volume and intracellular pH of astrocytes. J Neurotrauma. 1999;16:831–41. doi: 10.1089/neu.1999.16.831. [DOI] [PubMed] [Google Scholar]
- 17.Skatchkov SN, Eaton MJ, Shuba YM, et al. Tandem-pore domain potassium channels are functionally expressed in retinal (Müller) glial cells. Glia. 2006;53:266–76. doi: 10.1002/glia.20280. [DOI] [PubMed] [Google Scholar]
- 18.Päsler D, Gabriel S, Heinemann U. Two-pore-domain potassium channels contribute to neuronal potassium release and glial potassium buffering in the rat hippocampus. Brain Res. 2007;1173:14–26. doi: 10.1016/j.brainres.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 19.Talley EM, Solórzano G, Lei Q, Kim D, Bayliss DA. CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci. 2001;21:7491–505. doi: 10.1523/JNEUROSCI.21-19-07491.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu W, Schlichthörl G, Hirsch JR, et al. Expression pattern and functional characteristics of two novel splice variants of the two-pore-domain potassium channel TREK-2. J Physiol. 2002;539:657–68. doi: 10.1113/jphysiol.2001.013432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X, Pan Y, Wang X. Alterations in the expression of lipid and mechano-gated two-pore domain potassium channel genes in rat brain following chronic cerebral ischemia. Brain Res Mol Brain Res. 2004;120:205–9. doi: 10.1016/j.molbrainres.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Li ZB, Zhang HX, Li LL, Wang XL. Enhanced expressions of arachidonic acid-sensitive tandem-pore domain potassium channels in rat experimental acute cerebral ischemia. Biochem Biophys Res Commun. 2005;327:1163–9. doi: 10.1016/j.bbrc.2004.12.124. [DOI] [PubMed] [Google Scholar]
- 23.Simmons ML, Murphy S. Induction of nitric oxide synthase in glial cells. J Neurochem. 1992;59:897–905. doi: 10.1111/j.1471-4159.1992.tb08328.x. [DOI] [PubMed] [Google Scholar]
- 24.Eertmoed AL, Vallejo YF, Green WN. Transient expression of heteromeric ion channels. Methods Enzymol. 1998;293:564–85. doi: 10.1016/s0076-6879(98)93034-8. [DOI] [PubMed] [Google Scholar]
- 25.Abe K, Abe Y, Saito H. Evaluation of L-glutamate clearance capacity of cultured rat cortical astrocytes. Biol Pharm Bull. 2000;23:204–7. doi: 10.1248/bpb.23.204. [DOI] [PubMed] [Google Scholar]
- 26.Abe K, Misawa M. Amyloid Beta protein enhances the clearance of extracellular L-glutamate by cultured rat cortical astrocytes. Neurosci Res. 2003;45:25–31. doi: 10.1016/s0168-0102(02)00190-6. [DOI] [PubMed] [Google Scholar]
- 27.Bang H, Kim Y, Kim D. TREK-2, a new member of the mechanosensitive tandem-pore K(+) channel family. J Biol Chem. 2000;275:17412–9. doi: 10.1074/jbc.M000445200. [DOI] [PubMed] [Google Scholar]
- 28.Kang D, Choe C, Kim D. Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J Physiol. 2005;564:103–16. doi: 10.1113/jphysiol.2004.081059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poopalasundaram S, Knott C, Shamotienko OG, et al. Glial heterogeneity in expression of the inwardly rectifying K(+) channel, Kir4.1, in adult rat CNS. Glia. 2000;30:362–72. doi: 10.1002/(sici)1098-1136(200006)30:4<362::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 30.Higashi K, Fujita A, Inanobe A, et al. An inwardly rectifying K+ channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–31. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- 31.Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–40. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neusch C, Papdopoulos N, Mϋller M, et al. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: Impact on extracellular K+ regulation. J Neurophysiol. 2006;95:1843–52. doi: 10.1152/jn.00996.2005. [DOI] [PubMed] [Google Scholar]
- 33.Goldstein SAN, Bockenhauer D, O'Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nature Reviews. 2001;2:175–84. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- 34.Wilkie GS, Dickson KS, Gray NK. Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. TIBS. 2003;28:182–8. doi: 10.1016/S0968-0004(03)00051-3. [DOI] [PubMed] [Google Scholar]
- 35.Marx J. P-bodies mark the spot for controlling protein production. Science. 2005;310:764–5. doi: 10.1126/science.310.5749.764. [DOI] [PubMed] [Google Scholar]
- 36.Chua B, Kao RL, Rannels DE, Morgan HE. Inhibition of protein degradation by anoxia and ischemia in perfused rat hearts. J Biol Chem. 1979;254:6617–23. [PubMed] [Google Scholar]
- 37.Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- 38.Erecinska M, Silver IA. Metabolism and role of glutamate in mammalian brain. Prog Neurobiol. 1990;35:245–96. doi: 10.1016/0301-0082(90)90013-7. [DOI] [PubMed] [Google Scholar]
- 39.Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knock-out of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–86. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka K, Watase K, Manabe T, et al. Epilepsy and exacerbation of brain injurty in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 41.Schousboe A, Frandsen A. In: CNS Neurotransmitters and Neuromodulators: Glutamate. Stone TW, editor. Boca Raton, FL: CRC Press; 1995. pp. 239–51. [Google Scholar]
- 42.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxity in cortical cell culture. J Neurosci. 1987;7:357–68. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Lundkvist A, Andersson D, et al. Protective role of reactive astrocytes in brain ischemia. J Cerebral Blood Flow Metab. 2007;28:468–81. doi: 10.1038/sj.jcbfm.9600546. [DOI] [PubMed] [Google Scholar]
- 44.Brew H, Attwell D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature. 1987;327:707–9. doi: 10.1038/327707a0. [DOI] [PubMed] [Google Scholar]
- 45.Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–6. doi: 10.1038/348443a0. [DOI] [PubMed] [Google Scholar]
- 46.Ye ZC, Wyeth MS, Baltan-Tekkok S, Ransom BR. Functional hemichannels in astrocytes: a novel mechanism of glutamate release. J Neurosci. 2003;23:3588–96. doi: 10.1523/JNEUROSCI.23-09-03588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Contreras JE, Sánchez HA, Eugenin EA, et al. Metabolic inhibition induces opening of upapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc Natl Acad Sci, USA. 2002;99:495–500. doi: 10.1073/pnas.012589799. [DOI] [PMC free article] [PubMed] [Google Scholar]