

Abstract
Darunavir, a potent antiviral drug, showed an unusual second binding site on the HIV-1 protease dimer surface of the V32I drug resistant mutant and normal binding in the active site cavity. Kinetic analysis for wild type and mutant protease showed mixed-type competitive-uncompetitive inhibition for darunavir and the chemically related amprenavir, while saquinavir showed competitive inhibition. The inhibition model is consistent with the observed second binding site for darunavir and helps to explain its antiviral potency.
HIV-1 protease (PR) has been the target of intensive research for the past two decades that has led to effective drugs for the treatment of HIV/AIDS, i.e. small molecule synthetic protease inhibitors (PIs).1 The PIs have become the paradigm for successful structure-assisted drug design,2 with nine PIs now approved by the FDA for AIDS therapy. The addition of PIs to the antiviral regimens resulted in highly increased survival rates and lower morbidity caused by the disease in the past decade. However, the emergence of drug-resistant HIV has necessitated the recent development of PIs, such as darunavir (DRV) designed to target PR mutants.3,4 DRV has proved highly effective in salvage therapy for patients failing other treatments.5 Success of the design strategy was verified by crystallographic and kinetic analysis of DRV with HIV-1 protease and its mutants.6,7,8,9,10
The clinical PIs have been designed to inhibit the activity of the HIV-1 PR by competitively binding inside the active-site cavity,11,12,13 which is formed by a dimer of two identical 99-residue subunits. Numerous crystal structures of HIV-1 PR complexed with different PIs have shown exclusive binding of the inhibitors in the active-site cavity. Some reversible inhibitor molecules, like beta-lactam compounds14 and Nb-containing polyoxometalates15, were proposed to bind on the surface of the enzyme in uncompetitive or non-competitive manner, respectively, rather than in the usual active site cavity. Also, irreversible inhibitors like haloperidol analogues16 and Cu2+ ions7 can disrupt the HIV-1 PR activity by attaching covalently at surface sites. One peptide inhibitor was observed to bind on the protein surface between PR dimers in the crystal lattice, although this inhibitor showed no specific interactions with PR.17
An unusual second binding site for DRV was observed in the two high-resolution crystal structures of complexes with HIV-1 PR mutants with the single substitutions of V32I (0.84 Å) and M46L (1.22 Å).18 In both structures one inhibitor molecule occupied the active-site cavity, while the other inhibitor molecule was located in a deep groove on the PR surface in the flap region. Thus the PR/DRV2 species was observed in the solid state (Figure 1a). The surface-bound inhibitor molecule formed a number of specific interactions with the mutant PR residues, including hydrogen bonds (Figure 1b), C-H…O and C-H…π contacts, unlike the peptide observed on the flap surface.17 In addition, this second DRV molecule was a different diastereomer relative to the one bound in the active site. Therefore, we concluded that the flap site might serve as an allosteric binding site for DRV, which would explain the unprecedented effectiveness of DRV inhibition against various drug resistant PR variants.19,20
Figure 1.
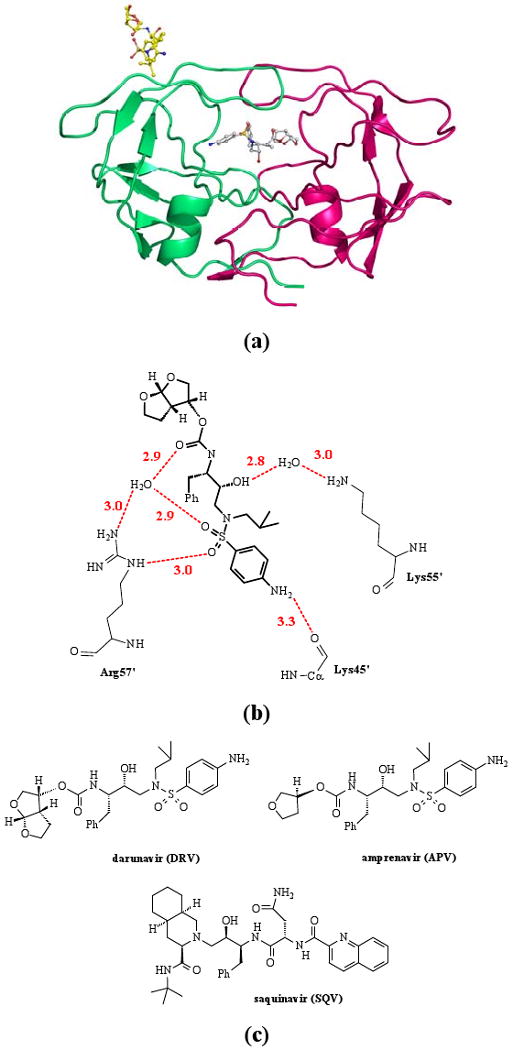
(a) Structure of the PR/DRV2 species observed in the PRV32I complex with DRV;25(b) Hydrogen bond interactions of DRV with PRV32I in the flap site (PDB ID 2HS1). Hydrogen bonds are shown as dashed lines with the distance in Å between donor and acceptor atoms. (c) Chemical structures of the clinical drugs darunavir (DRV), amprenavir (APV) and saquinavir (SQV).
Here, we report on solution measurements of enzyme inhibition kinetics for an optimized wild-type HIV-1 PR denoted PRWT21,22 and the V32I mutant PR (PRV32I) with three PIs: DRV, amprenavir (APV), and saquinavir (SQV) (Figure 1c). The inhibitors DRV and APV share a similar chemical backbone and differ in the presence of one or two THF groups (Figure 1c). In particular DRV and APV share the sulphonamide and aniline group that formed major interactions with the PR in the surface flap site (Figure 1b). The aniline NH2 group formed a hydrogen bond with the carbonyl oxygen of Lys 45, and the sulphonamide oxygens interacted with the side chain of Arg 57. Other water-mediated hydrogen bonds occur between PR and two carbonyl oxygens shared by DRV and APV. Therefore, APV was predicted to bind to the flap site in a manner similar to that observed for DRV. In contrast, SQV was chosen as a negative control since it has a distinctly different geometry, lacking both the sulphonamide and aniline groups, and was not expected to interact similarly in the flap site. The kinetic data for both PRWT and PRV32I demonstrate a mixed type competitive-uncompetitive inhibition for DRV and APV, where two non-mutually exclusive inhibitor sites co-exist in the enzyme, while SQV showed purely competitive inhibition. These data are discussed in relation to other studies suggesting the antiviral effectiveness of DRV arises from an exceptionally slow dissociation rate,23 or inhibition of the formation of PR dimers.24
The kinetic parameters were determined by means of a fluorescence assay using the anthranylyl/p-NO2-Phe containing substrate, which mimics the p2-NC natural cleavage site of the viral Gag polyprotein. The following reactions and kinetic constants were considered in describing the results:

In solution, a ligand-free PR molecule (E) can bind a substrate or inhibitor in its active-site cavity resulting in active ES or inactive EI complexes. Then, the ES may catalyze the hydrolysis of the substrate to generate the products, or it can bind an inhibitor molecule in the surface site to produce the inactive ternary complex ESI. The other species, i.e. inhibited EI form, may bind another inhibitor molecule in the same surface site resulting in a second possible ternary complex EI2. Notably, the EI2 species was observed in the crystal structures of PRV32I and PRM46L complexes with DRV. In this scheme E is the catalytically active dimer of PR as expected in the high concentrations of enzyme used in the kinetic assays. The above scheme therefore describes the mixed-type competitive-uncompetitive inhibition kinetics that is supported by the experimental data. For this type of kinetics the velocity equation is:25
where [S] and [I] are the substrate and inhibitor concentrations, respectively; Vmax is the reaction velocity at the infinitely high [S]; Km is the Michaelis-Menten constant; Ki is the uncompetitive inhibition constant; γKi represents the competitive inhibition constant, while δKi characterizes dissociation of the second inhibitor molecule bound to the surface site from the ternary EI2 complex. The Lineweaver-Burk slope and intercept replots were used to obtain the inhibition constants:
The velocity equation implies that the observed kinetic measurements have following behavior: a) The double-reciprocal plots have no common point of intersection, nor do they intersect at any of the 1/v or 1/[S] axes; b) The Dixon plots (1/v vs. [I]) and the Lineweaver-Burk slope replots are non-linear.
The kinetic data for PRWT and PRV32I with DRV or APV behaved as described above for mixed type inhibition, while the data obtained for SQV agreed with the competitive inhibition model.26 However, in the standard assay for competitive inhibition SQV and DRV show similar values for Ki of 0.4 and 0.5 nM, respectively. Significantly, the Dixon plots were non-linear for PRWT and PRV32I inhibited by DRV and APV, whereas the plots for PRWT and PRV32I inhibited by SQV were linear (Figure 2). The type of inhibition is most clearly evident in the slope replots of the double-reciprocal plots, which showed non-linear dependency (Figure 2d and 2e) for PRWT and PRV32I inhibited by DRV and APV, and were linear for SQV inhibition.
Figure 2.

Dixon plots (1/v vs. [I]): (a) PRWT-DRV, (b) PRWT-APV, (c) PRV32I-SQV; and slope replots for mixed-type competitive-uncompetitive and competitive inhibition: (d) PRWT, (e) PRV32I.
Uncompetitive inhibition by itself is a rare phenomenon in enzyme kinetics,27 although the mixed-type competitive-uncompetitive inhibition is quite common.28,29 On the other hand, the parabolic mixed-type mechanism, which describes the binding of an inhibitor to two non-mutually exclusive sites, is very atypical.30 The latter type of inhibition includes the existence of a ternary EI2 species in which an inhibitor is bound to two different sites in the enzyme. We conclude, based on the observed enzyme inhibition data, that DRV and APV inhibit HIV-1 PR by the parabolic mixed-type competitive-uncompetitive mechanism. The competitive component comes from the usual inhibitor binding in the PR active site cavity. The uncompetitive component characterizes the formation of the unproductive ESI ternary complex signifying that the substrate binding leads to a conformational change allowing an inhibitor molecule to bind to a second site on the PR, which does not overlap with the active site cavity. Finally, the nonlinearity of Dixon plots and Lineweaver-Burk slope replots agrees with the formation of the ternary PR-DRV2 or PR-APV2 species, where both sites are occupied by inhibitor molecules. Hence, we propose that the surface flap site observed in the PRV32I/DRV and PRM46L/DRV crystal structures is responsible for the uncompetitive component of the enzyme inhibition. Conversely, the kinetic data for SQV agree well with the standard competitive inhibition model and binding in the active site cavity, as observed for most clinical inhibitors of HIV-1 PR.
Interestingly, DRV binds with similar affinity to the PR active-site cavity and to the second site as shown by similar values of 18 and 22 nM for uncompetitive and competitive inhibition, respectively (Table 1). However, the competitive inhibition component of APV dominates the uncompetitive one with 4.5 fold smaller γKi compared to Kiuncomp. In contrast, the equilibrium constants for the dissociation of ternary EI2 complexes are comparable for both DRV and APV complexes, with δKi values of 9.0 and 6.4 nM, respectively. Unfortunately, no crystals examined to date have contained the PR-APV2 species, so there is no structural evidence for APV binding at the second flap site.
Table 1.
Enzyme kinetics and inhibition constants for PRWT and PRV32I with clinical drugs darunavir (DRV), amprenavir (APV), and saquinavir (SQV).
| Mutant | Km (μM) |
kcat (min-1) |
kcat/Km (min-1·μM) |
Relative kcat/Km |
DRV | APV | SQV | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Kiuncomp (nM) |
γKi (nM) (γ) |
δKi (nM) (δ) |
Kiuncomp (nM) |
γKi (nM) (γ) |
δKi (nM) (δ) |
Kicomp (nM) |
|||||
| PRWT | 30 ± 5 | 194 ± 23 | 7.4 ± 1.2 | 1.0 | 18 | 22 (1.2) | 9 (0.5) | 28 | 6.2 (0.22) | 6.4 (0.23) | 0.42 ± 0.07 |
| PRV32I | 80 ± 10 | 105 ± 15 | 1.3 ± 0.2 | 0.3 | 180 | 300 (1.7) | 54 (0.3) | 225 | 101 (0.45) | 63 (0.28) | 9.0 ± 0.9 |
The optimized HIV-1 PR clone with mutations Q7K, L33I, and L63I to diminish the autoproteolysis of the PR, as well as mutations C67A and C95A to prevent cysteine-thiol oxidation was used as wild-type PR enzyme and as a template to introduce the drug resistant mutation V32I. The kinetic parameters and stability of this optimized PR were indistinguishable from those of the unmutated enzyme.21 The PRWT and the mutant were expressed in Escherichia coli BL21 (DE3) and the protein was purified from inclusion bodies as described elsewhere.22 Kinetic parameters were determined by a fluorescence assay with the substrate Abz-Thr-Ile-Nle-pNO2Phe-Gln-Arg-NH2, where Abz is anthranylic acid, Nle is norleucine, based on the p2-NC cleavage site of the natural polyprotein substrate. PR (10 μl, final concentration of ∼ 20-70 nM) was mixed with 98 μl reaction buffer (100 mM MES, pH=5.6, 400 mM NaCl, 1 mM EDTA, 5% glycerol) and 2 μl of DMSO or inhibitor in DMSO, and the mixture was preincubated at 26 °C for 5 min. The reaction was initated by adding 90 μl of substrate (final concentration of 10-70 μM), and the resulting mixture was assayed over 5 min for the increase in fluorescence using 340 nM and 420 nM bandpass filters for the excitation and emission. Data analysis was performed with the program SigmaPlot 8.02. kcat and Km values were obtained by standard data-fitting techniques for a reaction obeying Michaelis-Menten kinetics. In inhibition measurements for DRV or APV, the uncompetitive Kiuncomp values were estimated from intercept replots of the Lineweaver-Burk plots, while the values for competitive γKi and the EI2 dissociation δKi were calculated from the non-linear fit to the Lineweaver-Burk slope replots. The competitive Kicomp values for measurements with SQV were obtained from the IC50 values estimated from an inhibitor dose-response curve with the fluorescent assay using the equation Ki = (IC50 - [E]/2)/(1 + [S]/Km), where [E] and [S] are the PR and substrate concentrations, respectively.26
Introduction of the V32I mutation significantly reduces the PR activity for this substrate based on the p2-NC cleavage site of the viral polyprotein substrate (Table 1). The reduction in PRV32I activity is manifest in an almost three-fold increase of Km and about two-fold decrease in kcat. However, the mutant PR and the wild-type enzyme had similar activity for a different substrate based on the CA-p2 cleavage site, as reported previously.18 The difference in relative activity on the two substrates is likely due to differences in the peptide sequences, which suggests mutation V32I is deleterious for correct polyprotein processing and viral replication. Furthermore, the mutant is resistant to the tested clinical drugs with ∼ 20 fold competitive inhibition for SQV relative to wild type enzyme, and increases in both competitive and uncompetitive inhibition for DRV and APV. Relative to the values for wild type enzyme, the competitive γKi values for the mutant increased by ∼15-fold for both DRV and APV, and uncompetitive Ki values by 8- to 10-fold. Moreover, the equilibrium constants (δKi) representing the dissociation of ternary EI2 complexes increased by 6- and 10-fold for DRV and APV, respectively. Therefore, the V32I mutation is likely to induce resistance to the three tested PIs.
DRV has several favorable properties for antiviral effectiveness on resistant HIV. DRV is a potent inhibitor due to formation of more extensive hydrogen bond interactions with the PR main chain compared to other PIs, and enthalpically driven binding.9,10,19 DRV showed exceptionally high affinity for HIV-1 PR compared to other PIs in a surface plasmon resonance-based kinetic study, due to a greatly decreased dissociation rate even relative to APV.23 Also, DRV inhibited the formation of PR dimers, unlike other PIs, in cell-based studies with fluorescent-tagged PR monomers.24 It is possible that several of these properties are influenced by DRV binding to a second site, and the current identification of the mixed type competitive-uncompetitive inhibition. The existence of the EI2 species is likely to slow dissociation of the inhibition complex since the DRV bound at the flap site will tend to stabilize the PR dimer with DRV bound in the active site cavity. Clearly, the uncompetitive inhibition component is stronger for DRV than for APV, consistent with the observed difference in dissociation rate of the two PIs.23 Also, the reported inhibition of dimer formation24 may arise from DRV bound to the flap site, since the flaps have important intersubunit interactions. However, this interpretation is more difficult to reconcile with the absence of APV inhibition of PR dimer formation, although our data showed weaker uncompetitive inhibition for APV compared to DRV. Therefore, several diverse assays confirm the unusual properties of DRV binding to PR, which will contribute to its superior antiviral potency.
In conclusion, these kinetic investigations support the existence of a second inhibitor-binding site for DRV and APV on the surface of HIV-1 PR, and the presence of the EI2 species. The enzyme inhibition measurements were described by the parabolic mixed-type competitive-uncompetitive inhibition model. This inhibition model suggests that both DRV and APV can bind to a second site in addition to the active site cavity of the PR. In contrast, the SQV inhibition of PR was purely competitive and consistent with SQV binding only in the active site cavity. Therefore, we proposed that the second flap binding site for DRV reported in the crystal structures with PRV32I and PRM46L is responsible for the observed uncompetitive inhibition component, and predicted that APV will bind to the flap site. These kinetic data confirm the prediction that APV will act similarly to DRV, while SQV cannot due to its different chemical structure. Other small molecules have been shown to bind on the PR surface in the flap region.16 Hence, this second site is a viable target for rational drug design to discover novel uncompetitive and potent inhibitors with exclusive binding to the PR flap region instead of the active site. Such inhibitors may provide new ways of battling the disease and the ubiquitous problem of drug resistance of HIV-1.
Acknowledgments
We thank Jozsef Tozser for valuable comments. This research was supported in part by the Molecular Basis of Disease Program, the Georgia Research Alliance, the Georgia Cancer Coalition, the National Institute of Health grants GM062920 and GM053386.
Abbreviations
- HIV-1
human immunodeficiency virus type 1
- PR
HIV-1 protease
- DRV
darunavir
- APV
amprenavir
- SQV
saquinavir
References
- 1.De Clercq E. New Approaches toward Anti-HIV Chemotherapy. J Med Chem. 2005;48:1–17. doi: 10.1021/jm040158k. [DOI] [PubMed] [Google Scholar]
- 2.Wlodawer A, Vondrasek J. Inhibitors of HIV-1 Protease: A Major Success of Structure-Assisted Drug Design. Annu Rev Biophys Biomol Struct. 1998;27:249–284. doi: 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh AK, Chapsla BD, Weber IT, Mitsuya H. Design of protease inhibitors targeting protein backbone: an effective strategy for combating drug-resistance. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 4.Weber IT, Kovalevsky AY, Harrison RW. Structures of HIV Protease Guide Inhibitor Design to Overcome Drug Resistance. In: Caldwell GW, Atta-ur-Rahman, Player MR, Choudhary MI, editors. Frontiers in Drug Design & Discovery. Vol. 3. Bentham Science Publishers; 2007. pp. 45–62. [Google Scholar]
- 5.Katlama C, Esposito R, Gatell JM, Goffard JC, Grinsztejn B, Pozniak A, Rockstroh J, Stoehr A, Vetter N, Yeni P, Parys W, Vangeneugden T. Efficacy and safety of TMC114/ritonavir in treatment-experienced HIV patients: 24-week results of POWER 1. AIDS. 2007;21:395–402. doi: 10.1097/QAD.0b013e328013d9d7. [DOI] [PubMed] [Google Scholar]
- 6.Miller M, Schneider J, Sathyanarayana BK, Toth M, Marshall GR, Clawson L, Selk L, Kent SBH, Wlodawer A. Structure of Complex of Synthetic HIV-1 Protease with a Substrate-Based Inhibitor at 2.3 Å Resolution. Science. 1989;246:1149–1152. doi: 10.1126/science.2686029. [DOI] [PubMed] [Google Scholar]
- 7.Danielson H, Lindgren MT, Markgren PO, Nillroth U. Investigation of an Allosteric Site of HIV-1 Proteinase Involved in Inhibition by Cu2+ Adv Experim Med Biol. 1998;436:99–103. doi: 10.1007/978-1-4615-5373-1_13. [DOI] [PubMed] [Google Scholar]
- 8.Kovalevsky AY, Tie Y, Liu F, Boross PI, Wang YW, Leshchenko S, Ghosh AK, Harrison RW, Weber IT. Effectiveness of Nonpeptide Clinical Inhibitor TMC114 on HIV-1 Protease with Highly Drug Resistant Mutations D30N, I50V and L90M. J Med Chem. 2006;49:1379–1387. doi: 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King NM, Prabu-Jeyabalan M, Nalivaika EA, Wigerinck P, de Béthune MP, Schiffer CA. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol. 2004;78:12012–12021. doi: 10.1128/JVI.78.21.12012-12021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High Resolution Crystal Structures of HIV-1 Protease with a Potent Non-peptide Inhibitor (UIC-94017) Active against Multi-Drug Resistant Clinical Strains. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 11.Lam PYS, Jadhav PK, Eyermann CJ, Hodge CN, Ru Y, Bacheler LT, Meek JL, Otto MJ, Rayner MM, Wong YN, Chang CH, Weber PC, Jackson DA, Sharpe TR, Erickson-Viitanen S. Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors. Science. 1994;263:380–384. doi: 10.1126/science.8278812. [DOI] [PubMed] [Google Scholar]
- 12.Kim EE, Baker CT, Dwyer MD, Murcko MA, Rao BG, Tung RD, Navia MA. Crystal Structure of HIV-1 Protease in Complex with VX-478, a Potent and Orally Bioavailable Inhibitor of the Enzyme. J Am Chem Soc. 1995;117:1181–1182. [Google Scholar]
- 13.Kaldor SW, Kalish VJ, Davies JF, II, Shetty BV, Fritz JE, Appelt K, Burgess JA, Campanale KM, Chirgadze NY, Clawson DK, Dressman BA, Hatch SD, Khalil DA, Kosa MB, Lubbehusen PP, Muesing MA, Patick AK, Reich SH, Su KS, Tatlock JH. Viracept (Nelfinavir Mesylate, AG1343): A Potent, Orally Bioavailable Inhibitor of HIV-1 Protease. J Med Chem. 1997;40:3979–3985. doi: 10.1021/jm9704098. [DOI] [PubMed] [Google Scholar]
- 14.Sperka T, Pitlik J, Bagossi P, Tozser J. Beta-lactam Compounds as Apparently Uncompetitive Inhibitors of HIV-1 Protease. Bioorg Med Chem Lett. 2005;15:3086–3090. doi: 10.1016/j.bmcl.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Judd DA, Nettles JH, Nevis N, Snyder JP, Liotta DC, Tang J, Ermolieff J, Schinazi RF, Hill CL. Polyoxometalate HIV-1 Protease Inhibitors. A New Mode of Protease Inhibition. J Am Chem Soc. 2001;123:886–897. doi: 10.1021/ja001809e. [DOI] [PubMed] [Google Scholar]
- 16.De Voss JJ, Sui Z, DeCamp DL, Salto R, Babe LM, Craik CS, Ortiz de Montellano PR. Haloperidol-based Irreversible Inhibitors of the HIV-1 and HIV-2 Proteases. J Med Chem. 1994;37:665–673. doi: 10.1021/jm00031a017. [DOI] [PubMed] [Google Scholar]
- 17.Brynda J, Rezacova P, Fabry M, Horejsi M, Stouracova R, Soucek M, Hradilek M, Konvalinka J, Sedlacek J. Inhibitor Binding at the Protein Interface in Crystals of a HIV-1 Protease Complex. Acta Crystallogr. 2004;D60:1943–1948. doi: 10.1107/S0907444904021572. [DOI] [PubMed] [Google Scholar]
- 18.Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Loius JM, Harrison RW, Weber IT. Ultra-high resolution Crystal Structure of HIV-1 Protease Mutant Reveals Two Binding Sites for Clinical Inhibitor TMC114. J Mol Biol. 2006;363:161–173. doi: 10.1016/j.jmb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-Tetrahydrofuranylurethane-Containing Nonpeptidic Protease Inhibitor (PI) TMC-114 (UIC-94017) with Potent Activity against Multi-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune MP. TMC114, a Novel Human Immunodeficiency Virus Type 1 Protease Inhibitor Active against Protease Inhibitor-Resistant Viruses, Including a Broad Range of Clinical Isolates. Antimicrob Agents Chemother. 2005;49:2314–2321. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 22.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug resistant mutants of HIV-1 protease: High resolution crystal structures of the mutant protease/substrate analog complexes. Prot Struct Funct Genet. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 23.Dierynck I, De Wit M, Gustin E, Keuleers I, Vandersmissen J, Hallenberger S, Hertogs K. Binding kinetics of darunavir to human immunodeficiency virus type 1 protease explain the potent antiviral activity and high genetic barrier. J Virol. 2007;81:13845–51. doi: 10.1128/JVI.01184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh Y, Matsumi S, Das D, Amano M, Davis DA, Li J, Leschenko S, Baldridge A, Shioda T, Yarchoan R, Ghosh AK, Mitsuya H. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J Biol Chem. 2007;282:28709–20. doi: 10.1074/jbc.M703938200. [DOI] [PubMed] [Google Scholar]
- 25.Segel IH. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems. John Wiley & Sons, Inc.; 1993. [Google Scholar]
- 26.Maibaum J, Rich DH. Inhibition of porcine pepsin by two substrate analogues containing statine: the effect of histidine at the P2 subsite on the inhibition of aspartic proteinases. J Med Chem. 1988;31:625–629. doi: 10.1021/jm00398a022. [DOI] [PubMed] [Google Scholar]
- 27.Cornish-Bowden A. Why is Uncompetitive Inhibition So Rare? FEBS. 1986;203:3–6. doi: 10.1016/0014-5793(86)81424-7. [DOI] [PubMed] [Google Scholar]
- 28.Porcelli F, Delfini M, Del Guidice MR. The Kinetic Inhibition of Acetylcholinesterase from Human Erythrocyte by Tacrine and Some Tacrine Derivatives. Bioorg Chem. 1999;27:197–205. [Google Scholar]
- 29.Jin P, Bhattacharya SK. Anaerobic removal of pentachlorophenol in presence of zinc. J Envir Eng. 1996;122:590–598. [Google Scholar]
- 30.Lorey S, Stockel-Maschek A, Faust J, Brandt W, Stiebitz B, Gorrell MD, Kahne T, Mrestani-Klaus C, Wrenger S, Reinhold D, Ansorge S, Neubert K. Different Modes of Dipeptidyl Peptidase IV (CD26) Inhibition by Oligopeptides Derived from the N-terminus of HIV-1 Tat Indicate at Least Two Inhibitor Binding Sites. Eur J Biochem. 2003;270:2147–2156. doi: 10.1046/j.1432-1033.2003.03568.x. [DOI] [PubMed] [Google Scholar]