

Abstract
DNA-binding proteins are very important constituents of proteomes of all species and play crucial roles in transcription, DNA replication, recombination, repair and other activities associated with DNA. Although a number of DNA-binding proteins have been identified, many proteins involved in gene regulation and DNA repair are likely still unknown because of their dynamic and/or weak interactions with DNA. In this report, we described an approach for the comprehensive identification of DNA-binding proteins with in vivo formaldehyde cross-linking and LC-MS/MS. DNA-binding proteins could be purified via the isolation of DNA-protein complexes and released from the complexes by reversing the cross-linking. By using this method, we were able to identify more than one hundred DNA-binding proteins, such as proteins involved in transcription, gene regulation, DNA replication and repair, and a large number of proteins which are potentially associated with DNA and DNA-binding proteins. This method should be generally applicable to the investigation of other nucleic acid-binding proteins, and hold great potential in the comprehensive study of gene regulation, DNA damage response and repair, as well as many other critical biological processes at proteomic level.
Keywords: Mass spectrometry, Chemical cross-linking, Formaldehyde, DNA-binding protein, Transcription factors, DNA repair
Introduction
Although a number of proteins with specific or general affinity to nucleic acids have been identified, numerous proteins involved in gene regulation, DNA repair and oncogenesis are likely still unknown. Therefore, it has always been of interest to study proteins that interact with nucleic acids, with the motivation to understand fundamental biological processes such as chromatin organization, transcription, DNA replication, recombination and repair, which are often regulated by proteins that bind to nucleic acids.1-3 Given the importance of nucleic acid-binding proteins and their interactions with DNA, RNA, or each other, it is necessary to develop a general analytical technique to identify comprehensively these proteins.
A classical technique used to detect nucleic acid-protein complexes is the electrophoretic mobility shift assay (EMSA), which is based on the principle that the electrophoretic mobility of a protein-nucleic acid complex is typically less than that of the free nucleic acid.4 It is a core technology underlying a wide range of qualitative and quantitative assays for the characterization of protein-nucleic acid interactions.4-6 Although EMSA is commonly used to detect nucleic acid-interacting factors, this assay is usually limited to evaluate in vitro interactions, by incubating a purified protein or a protein mixture with a radioactively labeled DNA probe. Another widely used technique for characterizing DNA-binding proteins and their associated factors is chromatin immunoprecipitation (ChIP), which is employed to study the binding and interaction of post-translationally modified histones or transcription factors with specific DNA sequences.7, 8 ChIP assay allows the detection of in vivo interactions of specific proteins with particular genomic regions in living cells with formaldehyde cross-linking.9, 10 However, both ChIP and EMSA are mainly restricted to known biological targets and have low throughput, making these two methods not suitable for identifying unknown nucleic acid-interacting factors or for studying the dynamics of gene regulation, a complex process requiring the interaction of numerous factors.
Recent advances in mass spectrometry (MS) have greatly facilitated protein identification and quantification.11, 12 In the past several years, hundreds of previously unknown proteins have been identified as nuclear proteins that are potentially involved in the regulation of gene expression, DNA replication and repair.13 The new field of nuclear proteomics has made some promising advances in elucidating the composition and dynamics of protein expression in nucleus and its subcompartments.14-19 For example, nucleolar proteomic studies facilitated the identification of up to approximately 700 proteins from isolated nucleoli in HeLa cells.20-24 However, most of these previous investigations were based on either crude nuclear pellets or purified subcellular compartments. Recently, the nuclear proteome from human Raji lymphoma cells was investigated by using 2D gel and MS.25 In addition, DNA-binding proteins were isolated from the nuclear extracts by using agarose immobilized with calf thymus DNA, where the in vitro interaction between the immobilized DNA and proteins constitutes the principle for the isolation. So far, very few studies have focused on the function-based comprehensive investigation of nucleic acid-binding proteins,26, 27 probably owing to the difficulty in capturing the in vivo DNA-protein interactions at a large scale.
Formaldehyde is a highly reactive reagent, which can freeze the DNA-protein interactions occurring in living cells under physiological conditions in situ, thereby preventing subsequent dissociation and redistribution of proteins while working on sample preparation.10, 27, 28 Formaldehyde cross-linking has been extensively used to study DNA-protein and protein-protein interactions.28-31 Amino and imino groups of amino acids (lysine, arginine and histidine) and DNA (primarily adenine and cytosine) react readily with formaldehyde, leading to the formation of cross-links.10, 32, 33 An attractive feature of formaldehyde cross-linking is that the cross-linking is fully reversible at high temperature (> 67 °C) in aqueous solution.
In this report, we describe an approach for the comprehensive investigation of DNA-binding proteins with in vivo formaldehyde cross-linking. After the cross-linking reaction, cell nuclei were isolated, and the covalently bound DNA-protein complexes were subsequently purified. After purification, the DNA-protein cross-linking was reversed to release the DNA-binding proteins and the liberated DNA was removed by DNase digestion and centrifugal filtration. The purified DNA-binding proteins were resolved by SDS-PAGE, digested in-gel with trypsin, and the digestion mixtures were interrogated by LC-MS/MS. By using this method, we were able to identify more than one hundred DNA-binding proteins according to the Gene Ontology (GO) annotations. In principle, this approach is not limited to the identification of DNA-binding proteins; it is also applicable to the investigation of proteins that bind to RNA.
Materials and Methods
Cell Culture
HL-60 Human acute promyelocytic leukemia cells (ATCC, Manassas, VA) were cultured in Iscove's modified minimal essential medium (IMEM) supplemented with 20% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA), 100 IU/mL of penicillin and 100 μg/mL of streptomycin in 75 cm2 culture flasks. Cells were maintained in a humidified atmosphere with 5% CO2 at 37 °C, with medium renewal of 2-3 times a week depending on cell density.
In Vivo Formaldehyde Cross-linking
HL-60 cells were collected by centrifugation at 300 g for 5 min at 4 °C, and washed with ice-cold PBS to remove culture medium and FBS. In vivo cross-linking was achieved by adding 11% (w/v) formaldehyde to 5 ml of cell suspension in PBS to obtain a final concentration of 1% (w/v). After incubating at room temperature for 10 min, formaldehyde was quenched by the addition of 2.5 M glycine to a final concentration of 125 mM and incubated at room temperature for 5 min. The cross-linked cells were collected by centrifugation (300 g at 4 °C for 5 min) and the cell pellet was washed twice with cold PBS.
Isolation of Nuclei
Nuclei isolation was carried out using a protocol adapted from that reported by Henrich et al.25 The cross-linked HL-60 cell pellet (∼ 4×107 cells) was resuspended in 10 volumes of ice-cold hypotonic lysis buffer A containing 10 mM HEPES (pH 7.4), 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF, and a protease inhibitor cocktail. After incubation on ice for 30 min, NP-40 was added to the lysis buffer until its final concentration reached 0.5% (v/v), and the mixture was incubated on ice for 5-min. Cells were then gently lysed with a Dounce homogenizer with B type pestle (clearance ∼ 0.7 mm) for 10 strokes on ice. The nuclear fraction was collected by centrifugation at 800 g at 4 °C for 5 min, and the resulting crude nuclear pellet was resuspended in buffer B, which contained 250 mM sucrose, 10 mM MgCl2, 20 mM Tris-HCl (pH 7.4) and 1mM DTT. The nuclei suspension was layered over a two-step sucrose gradient cushion [1.3 M sucrose, 6.25 mM MgCl2, 20 mM Tris-HCl (pH 7.4), 0.5 mM DTT above 2.3 M sucrose in 2.5 mM MgCl2 and 20 mM Tris-HCl (pH 7.4)], and centrifuged subsequently at 5000 g at 4 °C for 45 min. The isolated nuclei were washed with buffer A and collected by centrifugation at 1000 g.
Isolation of DNA-protein Complexes
DNA-binding proteins were isolated and copurified with genomic DNA as cross-linked DNA-protein complexes. The purification of DNA-protein cross-links was carried out by using a method described by Baker et al.34, 35 with modifications. The isolated nuclei were lysed in 500 DNAzol (Invitrogen) by repeated pipetting with a wide-bore pipette tip. DNA was precipitated by using a half volume of ice-cold 100% ethanol, and incubated at −20 °C for 1 hr. The precipitates were pelletted by centrifugation at 5,000 g at 4 °C for 5 min. The pellet was washed with ice-cold 75% ethanol and resuspended in 50 mM Tris-HCl buffer (pH 7.4). Urea and SDS were added to the suspension until their final concentrations reached 8 M and 2% (w/v), respectively, to denature proteins and to dissociate the non-cross-linked proteins from DNA-protein complexes. The sample was incubated at 37 °C for 30 min with gentle shaking. To the sample solution, an equal volume of 5 M NaCl was added and the resulting mixture was incubated at 37 °C for 30 min. The DNA and its associated proteins were precipitated again by the addition of 0.1 volume of 3 M sodium acetate and 3 volumes of ice-cold ethanol. Precipitated DNA and DNA-protein complexes were collected by centrifugation at 5,000 g at 4 °C for 5 min and washed thrice with ice-cold 75% ethanol to remove salts and detergents.
Cross-linking Reversal and DNA Removal
The purified DNA and DNA-protein complexes were resuspended in 0.5 M sodium acetate, and incubated at 68 °C overnight to reverse the DNA-protein cross-linking. After incubation, DNA was digested with 5 units of DNase I (Worthington Biochemical, Lakewood, NJ) and 5 units of S1 nuclease (Invitrogen) in a solution bearing 0.1 M sodium acetate (pH 5.5), 10 mM MgCl2 and 10 mM ZnCl2 at 37 °C for 1 h. The digested nucleotides were removed by using a Microcon YM-10 centrifugal filter (Millipore, Billerica, MA). The purified DNA-binding proteins were quantified with Bradford Protein Assay kit (Bio-Rad, Hercules, CA). The SDS-PAGE results for the analyses of the purified DNA-protein complexes (i.e., before the reversal of formaldehyde-induced cross-linking) and the purified DNA-binding proteins (i.e., after cross-linking reversal) are shown in Figure S1.
SDS-PAGE Separation and Enzymatic Digestion
The purified DNA-binding proteins were separated by 1D SDS-PAGE using a 12% resolving gel with a 4% stacking gel, and stained with Coomassie blue. The gel was cut into 10 bands, in-gel reduced with dithiothreitol (DTT), alkylated with iodoacetamide (IAA) and digested with trypsin (Promega, Madison, WI). The digested peptides were collected, dried in a Speed-vac, and stored at −20 °C until further analysis.
Western Blotting
For Western blotting analysis, proteins were denatured and reduced by boiling in Laemmli loading buffer containing 80 mM DTT. After SDS-PAGE separation, proteins were transferred to a nitrocellulose membrane under standard conditions. The membrane was blocked with 2% non-fat milk ECL Advance blocking reagent (GE Healthcare, UK) and incubated with primary antibodies at 4 °C overnight. The rabbit polyclonal primary antibodies for MCM2 (Mini chromosome maintenance protein 2, a.k.a. DNA replication licensing factor MCM2) and actin were from Abcam (Cambridge, MA). The membrane was rinsed briefly with two changes of PBS-T washing buffer [PBS with 0.1% (v/v) Tween-20, pH 7.5] and washed with a large amount of washing buffer for 15 min, followed by 3 × 5 min wash with fresh changes of washing buffer at room temperature. After washing, primary antibodies were recognized by incubating with horse radish peroxidase (HRP)-conjugated goat anti-rabbit IgG secondary antibody (Abcam) at room temperature for 1 h. The membrane was washed thoroughly with PBS-T (1× 15 min, then 3 × 5 min). The antibody binding was detected by using ECL Advance Western Blotting Detection Kit (GE Healthcare), and visualized with HyBlot CL Autoradiography Film (Denville Scientific Inc., Metuchen, NJ).
Extraction and Enzymatic Digestion of Nucleic Acids
After the above in-vivo chemical cross-linking, nucleic acids and nucleic acid-protein complexes were isolated from HL-60 cells using a standard phenol extraction protocol36 or the above-described DNAzol method. For phenol extraction, the RNase digestion step was omitted so that both RNA and DNA could be isolated, and this sample was used as a control to estimate the relative amounts of RNA and DNA in the extract. Proteins present in the cross-linked complexes were removed by proteinase K digestion, and the remaining nucleic acids were precipitated by ethanol and digested to mononucleosides by using nuclease P1 (NP1, Sigma-Aldrich, St. Louis, MO) and calf intestinal phosphatase (CIP, Sigma-Aldrich). In this respect, 2 units of NP1 was added to a solution containing 30 μg of DNA, 50 mM sodium acetate and 1.0 mM ZnCl2 (pH 5.5), and the digestion was continued at 37 °C for 12 h. The resulting sample was treated with 20 units of CIP in 50 mM Tris-HCl (pH 8.5) at 37 °C for 3 h. The digestion mixtures were passed through Microcon YM-10 centrifugal filter to remove enzymes and the resulting aliquots were subjected to HPLC analysis.
HPLC Separation
Off-line HPLC separation of nucleoside mixtures was performed on an Agilent 1100 HPLC pump with a 4.6 × 250 mm Grace Apollo C18 column (5 μm in particle size and 300 Å in pore size). A solution of 10 mM ammonium formate (solution A) and a 10 mM ammonium formate/acetonitrile mixture (70/30, v/v, solution B) were used as the mobile phases, and the flow rate was 0.8 mL/min. A gradient (0–5 min, 0–5% B; 5–45 min, 5–30% B, 45–50 min, 30–60% B) was used for the separation of the above nucleoside mixtures. The effluents were monitored by UV detection at 260 nm.
Nanoflow LC-MS/MS Analysis
Online LC-MS/MS analysis was performed on an Agilent 6510 Q-TOF system coupled with an Agilent HPLC-Chip Cube MS interface (Agilent Technologies, Santa Clara, CA). The sample enrichment, desalting, and HPLC separation were carried out automatically on the Agilent HPLC-Chip with an integrated trapping column (40 nL) and a separation column (Zorbax 300SB-C18, 75 μm × 150 mm, 5 μm in particle size). The peptide mixtures for LC-MS/MS analysis were first loaded onto the enrichment column and desalted with a solvent mixture of 0.1% formic acid in CH3CN/H2O (2:98, v/v) at a flow rate of 4 μL/min by using an Agilent 1200 capillary pump. After desalting, the peptide mixture was separated by using an Agilent 1200 nanoflow pump at a flow rate of 300 nL/min with the following gradient: 0-2min, 2% B; 2-10 min, 2-10% B; 10-90 min, 10-30% B; 90-120 min, 30-40% B; 120-130 min, 40-90% B. The gradient was held at 90% B for 5 min, and then changed to 2% B for equilibration for 15 min before the next sample injection. The mobile phases were 0.1% formic acid in H2O (A) and 0.1% formic acid in CH3CN (B).
To maintain a stable nanospray during the whole analysis process, the capillary voltage (Vcap) applied to the HPLC-Chip capillary tip was 1900 V. For data collection, the Agilent Q-TOF was operated in an auto (data-dependent) MS/MS mode, where a full MS scan was followed by maximum of eight MS/MS scans (abundance-only precursor selection), with m/z ranges of 350-2000, and 60-2000 for MS and MS/MS scans, respectively. The active (dynamic) exclusion feature was enabled to discriminate against ions previously selected for MS/MS in two sequential scans. The acquisition rates were 6 and 3 spectra/s in MS and MS/MS modes, respectively. For collision-induced dissociation (CID), the collision energy was set at a slope of 3 V/100 Da and an offset of 2.5 V to fragment the selected precursor ions and give MS/MS.
Data Processing
Agilent MassHunter workstation software (Version B.01.03, Agilent Technologies) was used to extract the MS and MS/MS data from the LC-MS/MS results. The extracted LC-MS/MS data were converted to mzData files with MassHunter Qualitative Analysis. Mascot Server 2.2 with Mascot Daemon 2.2.2 (Matrix Science, London, UK) was used for protein and peptide identifications by searching LC-MS/MS data against UniProtKB/Swiss-Prot database (updated weekly). Carbamidomethylation of cysteine residues was used as a fixed modification. Methionine oxidation, serine, threonine and tyrosine phosphorylation were set as variable modifications. Stringent criteria were employed for protein identification. The allowed maximum miscleavages per peptide was one, with a precursor tolerance of 20 ppm and a MS/MS tolerance of 0.6 Da (The average of absolute mass accuracy for all the identified peptides was calculated to be 7.9 ppm). Peptides identified with individual scores at or above the Mascot assigned homology score (p < 0.01 and individual peptide score > 40) were considered as specific peptide sequences. The false discovery rates (FDR, number of random matches divided by the total number of identified peptides) with homology or identity threshold, determined by using decoy database search, were less than 0.95%.
The cellular localization and function of identified proteins were assessed using Gene Ontology database (http://www.geneontology.org), Generic GO Term Mapper (http://go.princeton.edu) and GORetriever (http://www.agbase.msstate.edu).37
Results and Discussion
Strategy for the Identification of DNA-binding Proteins
DNA-binding proteins have a general or specific affinity for single- or double-stranded DNA. Although a number of DNA-binding proteins have been identified, many proteins involved in gene regulation and DNA repair are likely still unknown because of their dynamic and/or weak interactions with DNA. DNA-protein cross-linking induced by ionizing radiation and chemotherapeutic agents such as aldehydes, cisplatin and other metal complexes has been studied as cytotoxic lesions.38 Instead of studying the toxicity of cross-linking between DNA and proteins, we employed the in vivo DNA-protein cross-linking as a strategy to fix the DNA-protein interactions, and used a standard DNA purification method to isolate DNA-binding proteins from complex biological samples. The idea of using immobilized oligodeoxyribonuleotides as probes has been demonstrated for purifying specific DNA-binding proteins, followed by protein identification with MS-based techniques.39, 40 However, the previous methods, such as affinity-DNA probes and EMSA, usually depend on in vitro interactions with specific DNA sequences; therefore, they lack the ability to identify DNA-binding proteins at large scale and with high throughput.
To identify proteins with potential interactions with DNA, in this report, we described an approach for the comprehensive identification of DNA-binding proteins with formaldehyde cross-linking, which can fix DNA-protein interactions in situ and has been widely used in ChIP assays for probing in vivo chromatin structures and dynamics. As depicted in Figure 1, after the cross-linking reaction, cell nuclei were isolated, followed by the purification of cross-linked DNA-protein complexes. After DNA-binding proteins were copurified with genomic DNA, the DNA-protein cross-linking was reversed to release the DNA-binding proteins from DNA. The DNA was removed by using DNase digestion and centrifugation with Microcon centrifugal filters; the released DNA-binding proteins were further fractionated with SDS-PAGE and digested in-gel with trypsin. The extracted peptide mixtures from different gel bands were analyzed using LC-MS/MS, and the LC-MS/MS data were searched against a protein database for protein identification (Figure 1).
Figure 1.
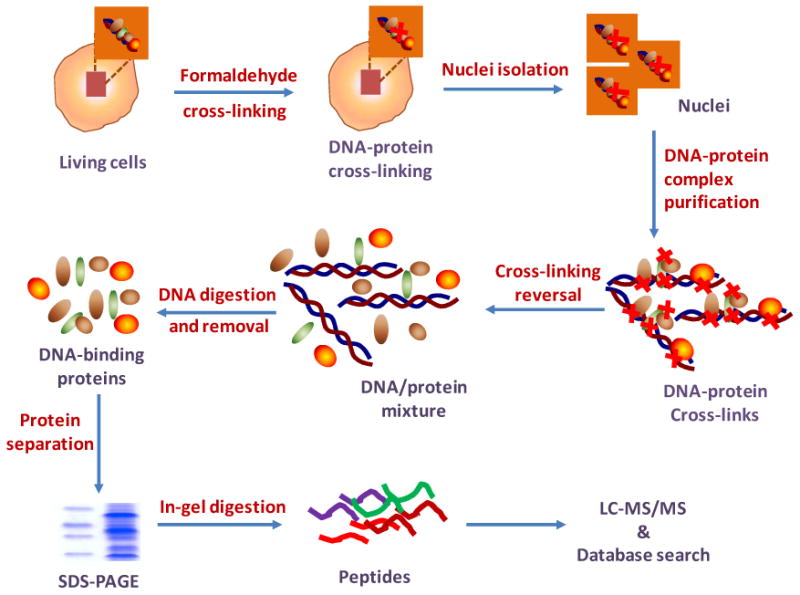
Strategy for the identification of DNA-binding proteins with chemical cross-linking and LC-MS/MS.
In Vivo DNA-protein Cross-linking
Formaldehyde-mediated DNA-protein cross-linking has been used for probing in vivo chromatin structures for more than two decades.33 Formaldehyde is a tight and reactive reagent, which can lead to efficient cross-linking between nucleic acids and proteins within short distance in vivo. The formation of covalent DNA-protein complexes prevents the subsequent dissociation and redistribution of proteins due to changes in physiological condition and/or in the process of sample handling.27 It has been found that formaldehyde is incapable of inducing protein-DNA cross-links in vitro even at extremely high concentrations.33 This suggests that the formaldehyde-mediated in vivo DNA-protein cross-linking is due to physiological DNA-protein interactions.
Aside from nucleic acid-protein cross-linking, protein-protein cross-linking can be generated by formaldehyde in vivo, especially upon long-term incubation and/or with high concentrations of cross-linking reagent. It is well-documented that nucleosomal proteins are normally analyzed following a cross-linking time within 10 min.10 Longer exposure to formaldehyde favors the binding of nucleosome-associated proteins and protein-protein interactions, and most proteins are readily cross-linked following a 20-60 min cross-linking reaction with 1% formaldehyde, based on the study of specific targets of interest.10, 31 However, in our experiment, it is unrealistic to optimize the cross-linking conditions for all DNA-interacting proteins based on specific DNA sequences and protein targets. We tested with different cross-linking reaction time with HL-60 cells and found that longer cross-linking time leads to low yield and poor solubility of DNA-protein complexes. Therefore, we incubated HL-60 cells in 1% formaldehyde for 10 min to generate the DNA-protein cross-links.
Isolation of DNA-binding Proteins
The enrichment of DNA-binding proteins was achieved through the isolation of DNA-protein complexes after in vivo chemical cross-linking. To evaluate the enrichment of DNA-binding proteins, we performed Western blotting experiments to assess the amounts of two identified proteins, MCM2 and actin, in whole cell lysates, nuclear fractions, and the enriched fraction of DNA-binding proteins.
DNA replication licensing factor MCM2 belongs to the MCM family proteins, which are DNA-dependent ATPases required for the initiation of eukaryotic DNA replication.41 MCM2 is a component of the prereplicative complex; it is essential for eukaryotic DNA replication and is expressed only in proliferating cells.42 Our Western blotting results revealed that MCM2, a DNA-binding protein, can be enriched by at least 17 fold after the isolation of DNA-protein complexes formed by in vivo chemical cross-linking compared to actin, which was used as an internal standard for this comparison (Figure 2). However, the nuclear fraction showed no significant enrichment of MCM2 with respect to actin. This could be attributed to the fact that a relatively high concentration of actin is present in the nuclear fraction.43 The background signals of MCM2 in high mass ranges in DNA-binding protein fraction could emanate from the undissociated cross-linked nucleotides/or proteins to MCM2. The presence of residual DNA-protein cross-links may lead to the failure in identifying some DNA-binding proteins. In this context, because of the heterogeneity of the cross-linking, i.e., different nucleobases and amino acid residues may participate in the formaldehyde-mediated cross-linking,10, 32, 33 it is difficult to incorporate the protein side-chain modifications, arising from the incomplete cross-linking reversal, into the database search for protein identification.
Figure 2.
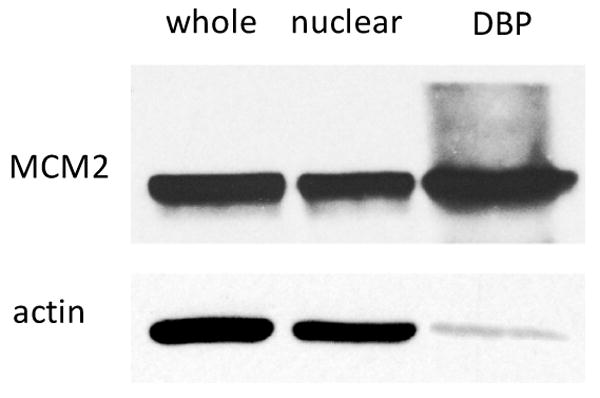
Western blotting of MCM2 was performed in whole cell lysate (whole), nuclear fraction (nuclear), and the DNA-binding protein fraction (DBP) of HL60 cells. Actin was used as the internal standard for quantitative comparison.
It is interesting to observe that actin can also be detected in the isolated DNA-binding protein fraction, which cannot be simply attributed to the contaminations from cytoplasmic or nuclear actin. In fact, it has been reported that actin could be associated with DNA and RNA during transcription.43-45 An alternative explanation is that actin could cross-link to other proteins that can bind to DNA. In a separate experiment, we followed the identical protocol for DNA-protein complex purification and isolated the genomic DNA without chemical cross-linking reaction. The result showed that a relatively small amount of MCM2 can be detected in the DNA fraction (Supporting Information, Figure S2). By contrast, no obvious actin band was visible in the same DNA fraction. This result indicates that the DNA-bound actin might be lost during DNA isolation without cross-linking, or the amount of actin directly associated with DNA, if any, could be very small.
The Selectivity of the Method toward the Isolation of DNA-binding Proteins
Several precautions were exerted to improve the selectivity of the above-described method toward the isolation of DNA-binding proteins. First, we began with isolated nuclei rather than the whole lysate of the formaldehyde-treated cells, which minimizes the contamination of cytosolic proteins. Second, we incubated the isolated protein-DNA complexes in a solution containing high concentrations of urea and SDS to dissociate and remove the non-cross-linked proteins from DNA-protein complexes. Third, we adopted a DNAzol-based protocol for the selective isolation of DNA and DNA-protein cross-links.34 The basis of the DNAzol procedure lies in the use of a novel guanidine-detergent lysis solution that hydrolyzes RNA and allows the selective precipitation of DNA and DNA-protein cross-links from a cell lysate.
While it is difficult to evaluate directly the selectivity of above-described strategy toward the isolation of DNA- over RNA-binding proteins because many proteins can bind both DNA and RNA, we chose to use an indirect method to assess how selective the strategy is toward DNA-binding proteins. In this respect, we isolated DNA and DNA-protein cross-links from HL-60 cells after formaldehyde-induced cross-linking reaction by using two different protocols, i.e., phenol extraction and the DNAzol method. We then removed the proteins by proteinase K treatment, digested the remaining nucleic acids to nucleosides with two enzymes and analyzed the resulting nucleosides by HPLC analysis (See Materials and Methods). As depicted in Figure 3, the amount of ribonucleosides present in the nucleoside mixture emanating from the DNAzol method is much less than that of 2′-deoxynucleosides (<5%, Figure 3B), whereas ribonucleosides are present at a much higher level in the nucleoside mixture arising from the phenol extraction method (Figure 3A). This result, therefore, suggests that the DNAzol method is highly selective toward the isolation of DNA and its associated proteins over RNA and its binding proteins.
Figure 3.
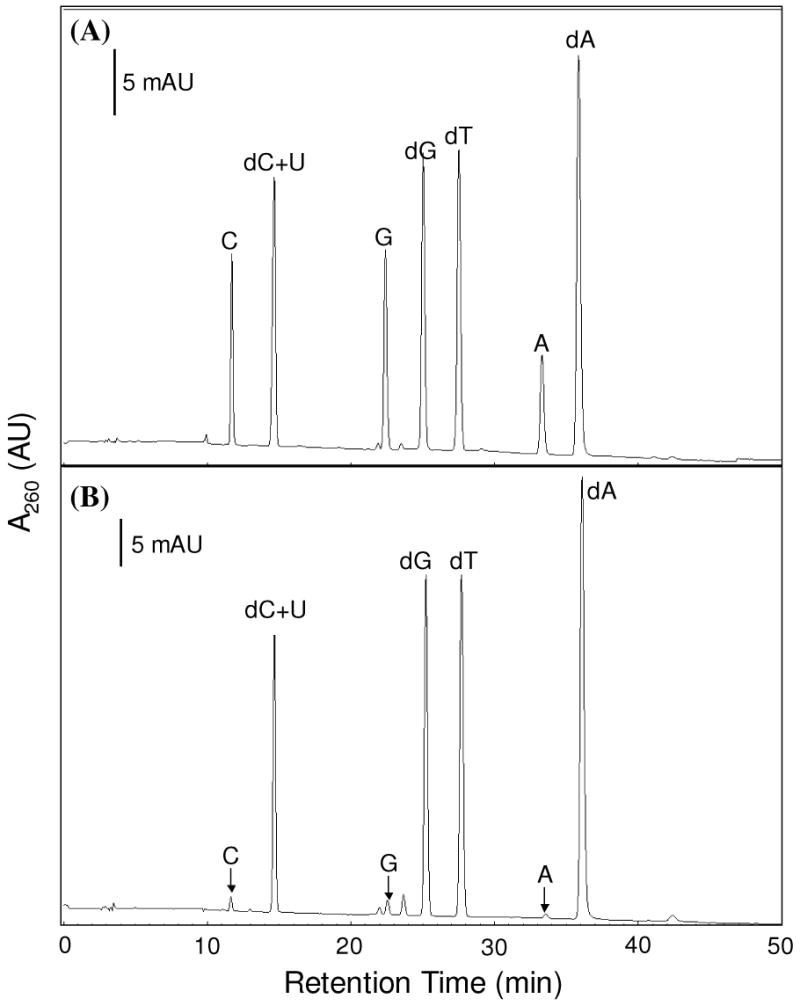
HPLC traces for the separation of nucleoside mixtures arising from the enzymatic digestion of nucleic acids that were isolated from the in-vivo chemically cross-linked DNA-protein complexes by the standard phenol extraction method (A) and the DNAzol method (B). The identities of nucleosides were confirmed by HPLC analysis of authentic compounds. “A”, “C”, “G”, “U” designate the four natural ribonucleosides, and “dA”, “dC”, “dG”, and “dT” represent the four natural 2′-deoxyribonucleosides.
Identification and Characterization of Proteins with In Vivo DNA-Protein Cross-linking
The current protocol with in vivo DNA-protein chemical cross-linking enabled us to enrich the DNA-binding proteins through the isolation of DNA-protein complexes. The cross-linking generated by formaldehyde stabilizes DNA-protein complexes and allows the capture of transient interactions between DNA and proteins. Based on this protocol, we were able to identify 780 proteins with Mascot database search (Supporting Information, Table S1).
To further understand the distribution and function of the identified proteins, we investigated the GO annotations of the identified proteins using Gene Ontology database (http://www.geneontology.org), Generic GO Term Mapper (http://go.princeton.edu) and GORetriever (http://www.agbase.msstate.edu).37 The comprehensive GO annotations for the identified proteins were summarized in the Supporting Information (Table S2). Among the proteins with GO annotations, 305 unique proteins were classified as nuclear proteins (GO: 0005634), which represented 39.1% of all proteins identified (Figure 4A). The presence of a relatively high percentage of nuclear proteins in the purified fraction could be attributed to the isolation of the DNA-protein complexes. In addition to the nuclear localization, 40.8% of the identified proteins were annotated as cytoplasmic proteins (GO: 0005737), followed by membrane (GO: 0016020, 15.4%), mitochondrial (GO: 0005739, 7.4%), ribosomal (GO: 0005840, 7.2%) proteins, etc. Because of the lack of annotation information, more than 10% of the identified proteins cannot be classified based on GO slim terms. On the other hand, many proteins can be grouped into multiple GO categories, which can cause overlaps. Therefore, the sum of percentages of GO categories is over 100%. It is also worth noting that many proteins were only annotated with their major cellular localizations and functions. For instance, actin is also localized in the nucleus and is associated with gene regulation (Figure 2)43-45; however, the current GO databases classify actin only as a cytoplasmic protein.
Figure 4.
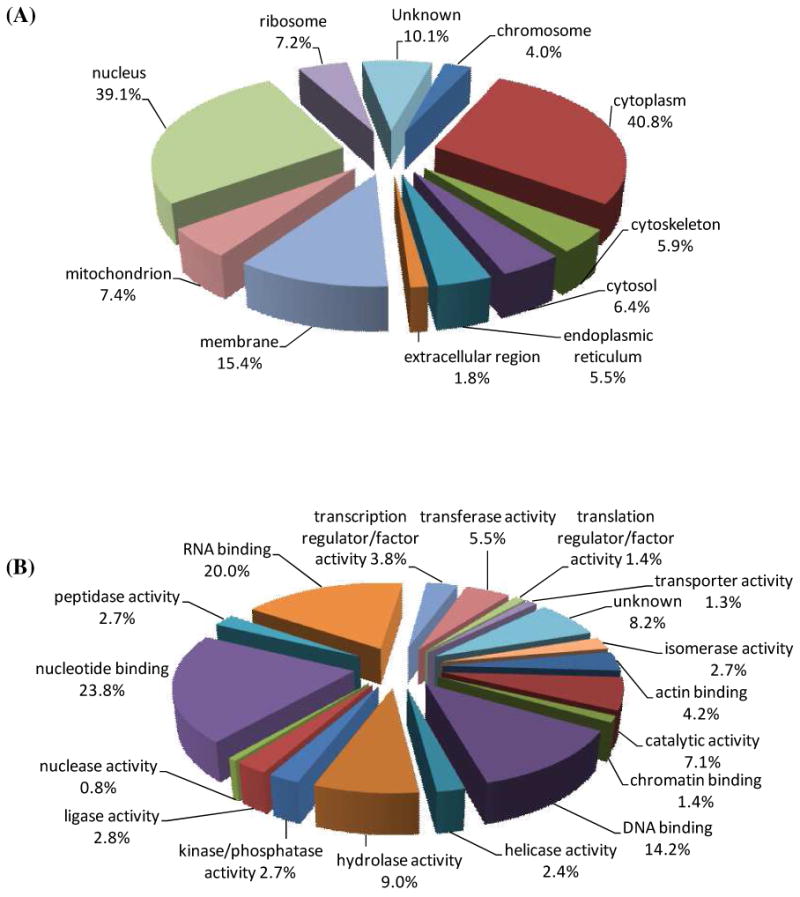
The identified proteins were classified using GO annotations. (A) Distribution of proteins in major cellular localizations; (B) Classification of proteins according to their function. Percentages indicate the identified proteins in each category relative to the total number of identified proteins.
Apart from the classification of the cellular distribution of the identified proteins, we also organized the identified proteins, according to their biological functions (Figure 4B). Among the identified proteins with GO molecular function annotation, 111 proteins (14.2% of the identified proteins) were clearly annotated as DNA-binding proteins (GO: 0003677), including double-stranded/single-stranded DNA binding, damaged DNA binding, sequence-specific/structure-specific DNA binding, transcription regulation, DNA helicase/ligase activity, etc. More functional categories were shown as nucleotide-binding (GO: 0000166, 23.8%), RNA-binding (GO: 0003723, 20.0%), hydrolase activity (GO: 0016787, 9.0%), etc. The identification of proteins without known DNA-binding activity could arise from the cross-linking of these proteins with DNA-binding proteins through protein-protein interactions. Additionally, some of these proteins may bear DNA binding capabilities that have yet been characterized. Furthermore, formaldehyde may also give rise to non-specific protein-protein cross-links, thereby resulting in the isolation and identification of proteins that are not capable of binding to DNA or its associated proteins. Guisan et al.46 showed that the addition of dextran could improve the specificity of aldehyde-induced cross-linking of purified proteins. In addition, it was found that weakly bound protein-protein complexes could be selectively adsorbed on lowly activated anionic exchange support, thereby preserving the weak protein-protein interactions.47 These methods are, however, not applicable for the formaldehyde-induced in-vivo cross-linking discussed in the current paper. In the future, it is important to develop strategies which can further minimize or avoid the formation of non-specific protein-protein complexes during the formaldehyde-induced in-vivo cross-linking.
To gain further knowledge about the identified DNA-binding proteins, we extracted the comprehensive GO annotations including the molecular function and biological process, which are summarized in the Supporting Information (Table S2). According to the GO annotations, at least 67 identified proteins are involved in gene transcription, and 20 identified proteins are involved in DNA replication. For instance, 23 transcription factors were identified in our experiment (Table 1). In addition, almost a complete family of DNA replication licensing factors was identified in our experiments, which include all six subunits of the MCM protein complex, i.e., MCM2 through MCM7. These six subunits form a ring-shaped heterohexameric ATPase involved in DNA replication.42, 48
Table 1.
A list of transcription factors identified in the present study.
| Uniprot ID | Protein Name | Gene Name | MW (Da) | Function |
|---|---|---|---|---|
| ENOA_HUMAN | Alpha-enolase | ENO1 | 47169 | transcription factor activity, transcription repressor activity, transcription corepressor activity |
| ARNT_HUMAN | Aryl hydrocarbon receptor nuclear translocator | ARNT | 86636 | transcription factor activity, transcription coactivator activity, signal transduction |
| CREB1_HUMAN | cAMP response element-binding protein | CREB1 | 36688 | transcription factor activity, sequence-specific DNA binding, signal transduction |
| CNBP_HUMAN | Cellular nucleic acid-binding protein | CNBP | 19463 | single-stranded DNA binding, single-stranded RNA binding, transcription activity |
| DBPA_HUMAN | DNA-binding protein A | CSDA | 40090 | double-stranded DNA binding, transcription factor activity, transcription corepressor activity |
| EDF1_HUMAN | Endothelial differentiation-related factor 1 | EDF1 | 16369 | regulation of transcription, transcription factor activity, transcription coactivator activity |
| ELF1_HUMAN | ETS-related transcription factor Elf-1 | ELF1 | 67456 | sequence-specific DNA binding, transcription activator activity, transcription factor activity, transcription repressor activity |
| FUBP1_HUMAN | Far upstream element-binding protein 1 | FUBP1 | 67560 | single-stranded DNA binding, transcription factor activity |
| HCLS1_HUMAN | Hematopoietic lineage cell-specific protein | HCLS1 | 53998 | transcription factor activity, regulation of transcription |
| ROAA_HUMAN | Heterogeneous nuclear ribonucleoprotein A/B | HNRPAB | 36225 | transcription factor activity, positive regulation of gene-specific transcription |
| HMGB2_HUMAN | High mobility group protein B2 | HMGB2 | 24034 | DNA bending activity, base-excision repair, DNA replication, transcription factor activity |
| HMGA1_HUMAN | High mobility group protein HMG-I/HMG-Y | HMGA1 | 11676 | AT DNA binding, transcription factor binding, transcription factor activity |
| HCFC1_HUMAN | Host cell factor | HCFC1 | 208732 | transcription factor activity, transcription coactivator activity, regulation of transcription |
| MEF2D_HUMAN | Myocyte-specific enhancer factor 2D | MEF2D | 55938 | transcription factor activity, regulation of transcription |
| YBOX1_HUMAN | Nuclease-sensitive element-binding protein 1 | YBX1 | 35924 | transcription factor activity, regulation of transcription, transcription repressor activity |
| NDKB_HUMAN | Nucleoside diphosphate kinase B | NME2 | 17298 | transcription factor activity, regulation of transcription |
| PA2G4_HUMAN | Proliferation-associated protein 2G4 | PA2G4 | 43787 | transcription factor activity, regulation of transcription |
| MAX_HUMAN | Protein max | MAX | 18275 | transcription regulator activity, transcription factor activity, transcription cofactor activity |
| STAT3_HUMAN | Signal transducer and activator of transcription 3 | STAT3 | 88068 | transcription factor activity, regulation of transcription, transcription factor binding |
| TADBP_HUMAN | TAR DNA-binding protein 43 | TARDBP | 44740 | transcription factor activity, regulation of transcription |
| SPT6H_HUMAN | Transcription elongation factor SPT6 | SUPT6H | 199073 | transcription factor activity, regulation of transcription |
| TIF1B_HUMAN | Transcription intermediary factor 1-beta | TRIM28 | 88550 | transcription factor activity, regulation of transcription, transcription corepressor activity |
| ZN207_HUMAN | Zinc finger protein 207 | ZNF207 | 50751 | transcription factor activity, regulation of transcription |
Besides the proteins involved in transcription and replication, we were able to identify at least 26 proteins involved in DNA damage and repair processes, such as base-excision repair, nucleotide-excision repair and double-strand break repair, as shown in Table 2. It is worth noting that some of identified proteins involved in DNA damage response and repair, however, are not annotated as DNA-binding proteins in the current GO databases. These proteins include MMS19 nucleotide excision repair protein homolog (MMS19), FACT complex subunit SPT16 (SUPT16H), transitional endoplasmic reticulum ATPase (VCP), etc. Because of the lack of proper GO annotations, these proteins were not counted as DNA-binding proteins depicted in Figure 4B.
Table 2.
A list of identified proteins involved in DNA repair.
| Uniprot ID | Protein Name | Gene Name | MW (Da) | Function |
|---|---|---|---|---|
| KU70_HUMAN | ATP-dependent DNA helicase 2 subunit 1 | XRCC6 | 69843 | DNA ligation, double-strand break repair, DNA recombination |
| KU86_HUMAN | ATP-dependent DNA helicase 2 subunit 2 | XRCC5 | 82705 | DNA recombination, double-strand break repair |
| DDB1_HUMAN | DNA damage-binding protein 1 | DDB1 | 126968 | damaged DNA binding, nucleotide-excision repair, DNA damage removal |
| DNL1_HUMAN | DNA ligase 1 | LIG1 | 101736 | ligase activity, DNA replication, DNA repair |
| MCM7_HUMAN | DNA replication licensing factor MCM7 | MCM7 | 81308 | DNA replication, transcription, response to DNA damage stimulus |
| TOP2A_HUMAN | DNA topoisomerase 2-alpha | TOP2A | 174385 | DNA topoisomerase activity, DNA-dependent ATPase activity, DNA repair |
| APEX1_HUMAN | DNA-(apurinic or apyrimidinic site) lyase | APEX1 | 35555 | lyase activity, base-excision repair, transcription coactivator activity |
| PRKDC_HUMAN | DNA-dependent protein kinase catalytic subunit | PRKDC | 469089 | DNA-dependent protein kinase activity, DNA recombination, DNA repair |
| SP16H_HUMAN | FACT complex subunit SPT16 | SUPT16H | 119914 | DNA replication, DNA repair, nucleosome disassembly |
| HMG1X_HUMAN | High mobility group protein 1-like 10 | HMG1L10 | 24218 | protein binding, base-excision repair, regulation of transcription |
| HMGB1_HUMAN | High mobility group protein B1 | HMGB1 | 24894 | base-excision repair, signal transduction, transcription factor binding |
| HMGB2_HUMAN | High mobility group protein B2 | HMGB2 | 24034 | DNA bending activity, base-excision repair, DNA replication, transcription factor activity |
| MMS19_HUMAN | MMS19 nucleotide excision repair protein homolog | MMS19 | 113290 | nucleotide-excision repair, transcription |
| NONO_HUMAN | Non-POU domain-containing octamer-binding protein | NONO | 54232 | RNA splicing, DNA replication, DNA repair |
| PARP1_HUMAN | Poly [ADP-ribose] polymerase 1 | PARP1 | 113084 | transferase activity, DNA repair, response to DNA damage stimulus |
| PCNA_HUMAN | Proliferating cell nuclear antigen | PCNA | 28769 | DNA binding, nucleotide-excision repair, DNA replication |
| RENT1_HUMAN | Regulator of nonsense transcripts 1 | UPF1 | 124345 | helicase activity, hydrolase activity, DNA replication, DNA repair |
| RBM14_HUMAN | RNA-binding protein 14 | RBM14 | 69492 | RNA binding, DNA recombination, DNA repair, transcription |
| RUVB2_HUMAN | RuvB-like 2 | RUVBL2 | 51157 | damaged DNA binding, DNA recombination, DNA repair |
| SFPQ_HUMAN | Splicing factor, proline- and glutamine-rich | SFPQ | 76149 | RNA splicing, DNA recombination, DNA repair, response to DNA damage stimulus |
| SMC1A_HUMAN | Structural maintenance of chromosomes protein 1A | SMC1A | 143233 | DNA repair, DNA damage response, signal transduction |
| SMC3_HUMAN | Structural maintenance of chromosomes protein 3 | SMC3 | 141542 | protein binding, ATPase activity, DNA repair, signal transduction |
| SODC_HUMAN | Superoxide dismutase [Cu-Zn] | SOD1 | 15936 | double-strand break repair, DNA fragmentation during apoptosis |
| TERA_HUMAN | Transitional endoplasmic reticulum ATPase | VCP | 89322 | ATPase activity, double-strand break repair, response to DNA damage stimulus |
| TP53B_HUMAN | Tumor suppressor p53-binding protein 1 | TP53BP1 | 213574 | transcription activator activity, damaged DNA binding, DNA repair |
| RD23B_HUMAN | UV excision repair protein RAD23 homolog B | RAD23B | 43171 | damaged DNA binding, single-stranded DNA binding, nucleotide-excision repair |
Conclusions
We described an approach for the comprehensive investigation of DNA-binding proteins with in vivo formaldehyde cross-linking. DNA-binding proteins can be purified via the isolation of DNA-protein complexes and released from the complexes by reversing the DNA-protein cross-linking. By using this method, we were able to identify more than one hundred DNA-binding proteins, including those involved in transcription, gene regulation, DNA replication and repair, and a large number of proteins which are potentially associated with DNA and DNA-binding proteins. This method should be generally applicable to the investigation of other nucleic acid-binding proteins, and hold great potential in the comprehensive study of gene regulation, DNA damage response and repair, as well as many other important biological processes at proteomic level. We believe that this function-oriented protein purification strategy may serve as a valuable tool for studying in vivo DNA-protein interaction and dynamic cellular responses to perturbations and stress, not only by identifying potential protein targets, transcription factors or DNA repair enzymes, but also by determining protein modifications and differential expression by incorporating other techniques, e.g., stable isotope labeling.
Supplementary Material
The list of proteins identified, comprehensive GO annotations, SDS-PAGE and Western blotting results. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank the National Institutes of Health for supporting this research (Grant No. R01 CA116522).
References
- 1.Carey M, Smale ST. Transcriptional regulation in eukaryotes: concepts, strategies, and techniques. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.: 2000. [Google Scholar]
- 2.Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 3.Naar AM, Lemon BD, Tjian R. Transcriptional coactivator complexes. Annu Rev Biochem. 2001;70:475–501. doi: 10.1146/annurev.biochem.70.1.475. [DOI] [PubMed] [Google Scholar]
- 4.Garner MM, Revzin A. The use of gel-electrophoresis to detect and study nucleic-acid protein interactions. Trends Biochem Sci. 1986;11:395–396. [Google Scholar]
- 5.Fried MG. Measurement of protein-DNA interaction parameters by electrophoresis mobility shift assay. Electrophoresis. 1989;10:366–376. doi: 10.1002/elps.1150100515. [DOI] [PubMed] [Google Scholar]
- 6.Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2:1849–61. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen HW, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 8.Spencer VA, Sun JM, Li L, Davie JR. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods. 2003;31:67–75. doi: 10.1016/s1046-2023(03)00089-6. [DOI] [PubMed] [Google Scholar]
- 9.Kuo MH, Allis CD. In vivo cross-linking and immunoprecipitation for studying dynamic Protein:DNA associations in a chromatin environment. Methods. 1999;19:425–433. doi: 10.1006/meth.1999.0879. [DOI] [PubMed] [Google Scholar]
- 10.Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem Sci. 2000;25:99–104. doi: 10.1016/s0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- 11.Ahn NG, Shabb JB, Old WM, Resing KA. Achieving in-depth proteomics profiling by mass spectrometry. ACS Chem Biol. 2007;2:39–52. doi: 10.1021/cb600357d. [DOI] [PubMed] [Google Scholar]
- 12.Cravatt BF, Simon GM, Yates JR. The biological impact of mass-spectrometry-based proteomics. Nature. 2007;450:991–1000. doi: 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- 13.Nordhoff E, Lehrach H. Identification and characterization of DNA-binding proteins by mass spectrometry. Adv Biochem Eng Biotechnol. 2007;104:111–195. doi: 10.1007/10_2006_037. [DOI] [PubMed] [Google Scholar]
- 14.Barthelery M, Salli U, Vrana KE. Nuclear proteomics and directed differentiation of embryonic stem cells. Stem Cells Dev. 2007;16:905–919. doi: 10.1089/scd.2007.0071. [DOI] [PubMed] [Google Scholar]
- 15.Barthelery M, Salli U, Vrana KE. Enhanced nuclear proteomics. Proteomics. 2008;8:1832–1838. doi: 10.1002/pmic.200700841. [DOI] [PubMed] [Google Scholar]
- 16.Escobar MA, Hoelz DJ, Sandoval JA, Hickey RJ, Grosfeld JL, Malkas LH. Profiling of nuclear extract proteins from human neuroblastoma cell lines: the search for fingerprints. J Pediatr Surg. 2005;40:349–358. doi: 10.1016/j.jpedsurg.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 17.Jung E, Hoogland C, Chiappe D, Sanchez JC, Hochstrasser DF. The establishment of a human liver nuclei two-dimensional electrophoresis reference map. Electrophoresis. 2000;21:3483–3487. doi: 10.1002/1522-2683(20001001)21:16<3483::AID-ELPS3483>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 18.Malmstrom J, Larsen K, Malmstrom L, Tufvesson E, Parker K, Marchese J, Williamson B, Patterson D, Martin S, Juhasz P, Westergren-Thorsson G, Marko-Varga G. Nanocapillary liquid chromatography interfaced to tandem matrix-assisted laser desorption/ionization and electrospray ionization-mass spectrometry: Mapping the nuclear proteome of human fibroblasts. Electrophoresis. 2003;24:3806–3814. doi: 10.1002/elps.200305619. [DOI] [PubMed] [Google Scholar]
- 19.Tan F, Li GS, Chitteti BR, Peng ZH. Proteome and phosphoproteome analysis of chromatin associated proteins in rice (Oryza sativa) Proteomics. 2007;7:4511–4527. doi: 10.1002/pmic.200700580. [DOI] [PubMed] [Google Scholar]
- 20.Andersen JS, Lam YW, Leung AKL, Ong SE, Lyon CE, Lamond AI, Mann M. Nucleolar proteome dynamics. Nature. 2005;433:77–83. doi: 10.1038/nature03207. [DOI] [PubMed] [Google Scholar]
- 21.Andersen JS, Lyon CE, Fox AH, Leung AKL, Lam YW, Steen H, Mann M, Lamond AI. Directed proteomic analysis of the human nucleolus. Curr Biol. 2002;12:1–11. doi: 10.1016/s0960-9822(01)00650-9. [DOI] [PubMed] [Google Scholar]
- 22.Coute Y, Burgess JA, Diaz JJ, Chichester C, Lisacek F, Greco A, Sanchez JC. Deciphering the human nucleolar proteome. Mass Spectrom Rev. 2006;25:215–234. doi: 10.1002/mas.20067. [DOI] [PubMed] [Google Scholar]
- 23.Hinsby AM, Kiemer L, Karlberg EO, Lage K, Fausboll A, Juncker AS, Andersen JS, Mann M, Brunak S. A wiring of the human nucleolus. Mol Cell. 2006;22:285–295. doi: 10.1016/j.molcel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Scherl A, Coute Y, Deon C, Calle A, Kindbeiter K, Sanchez JC, Greco A, Hochstrasser D, Diaz JJ. Functional proteomic analysis of human nucleolus. Mol Biol Cell. 2002;13:4100–4109. doi: 10.1091/mbc.E02-05-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henrich S, Cordwell SJ, Crossett B, Baker MS, Christopherson RI. The nuclear proteome and DNA-binding fraction of human Raji lymphoma cells. Biochim Biophys Acta, Proteins Proteomics. 2007;1774:413–432. doi: 10.1016/j.bbapap.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 26.Afjehi-Sadat L, Engidawork E, Slavc I, Lubec G. Comparative proteomic analysis of nucleic acid-binding proteins in ten human tumor cell lines. Int J Oncol. 2006;28:173–190. [PubMed] [Google Scholar]
- 27.Vigneault F, Guerin SL. Regulation of gene expression: probing DNA-protein interactions in vivo and in vitro. Expert Rev Proteomics. 2005;2:705–718. doi: 10.1586/14789450.2.5.705. [DOI] [PubMed] [Google Scholar]
- 28.Jackson V. Formaldehyde cross-linking for studying nucleosomal dynamics. Methods. 1999;17:125–139. doi: 10.1006/meth.1998.0724. [DOI] [PubMed] [Google Scholar]
- 29.Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- 30.Guerrero C, Tagwerker C, Kaiser P, Huang L. An integrated mass spectrometry-based proteomic approach - Quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol Cell Proteomics. 2006;5:366–378. doi: 10.1074/mcp.M500303-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Vasilescu J, Guo XC, Kast J. Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics. 2004;4:3845–3854. doi: 10.1002/pmic.200400856. [DOI] [PubMed] [Google Scholar]
- 32.Metz B, Kersten GFA, Hoogerhout P, Brugghe HF, Timmermans HAM, de Jong A, Meiring H, ten Hove J, Hennink WE, Crommelin DJA, Jiskoot W. Identification of formaldehyde-induced modifications in proteins - Reactions with model peptides. J Biol Chem. 2004;279:6235–6243. doi: 10.1074/jbc.M310752200. [DOI] [PubMed] [Google Scholar]
- 33.Solomon MJ, Varshavsky A. Formaldehyde-mediated DNA protein crosslinking - a probe for in vivo chromatin structures. Proc Natl Acad Sci USA. 1985;82:6470–6474. doi: 10.1073/pnas.82.19.6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barker S, Murray D, Zheng J, Li L, Weinfeld M. A method for the isolation of covalent DNA-protein crosslinks suitable for proteomics analysis. Anal Biochem. 2005;344:204–215. doi: 10.1016/j.ab.2005.06.039. [DOI] [PubMed] [Google Scholar]
- 35.Barker S, Weinfeld M, Zheng J, Li L, Murray D. Identification of mammalian proteins cross-linked to DNA by ionizing radiation. J Biol Chem. 2005;280:33826–33838. doi: 10.1074/jbc.M502477200. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.: 2001. [Google Scholar]
- 37.McCarthy FM, Bridges SM, Wang N, Magee GB, Williams WP, Luthe DS, Burgess SC. AgBase: a unified resource for functional analysis in agriculture. Nucleic Acids Res. 2007;35:D599–D603. doi: 10.1093/nar/gkl936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reardon JT, Cheng Y, Sancar A. Repair of DNA-protein cross-links in mammalian cells. Cell Cycle. 2006;5:1366–1370. doi: 10.4161/cc.5.13.2892. [DOI] [PubMed] [Google Scholar]
- 39.Nordhoff E, Krogsdam AM, Jorgensen HF, Kallipolitis BH, Clark BFC, Roepstorff P, Kristiansen K. Rapid identification of DNA-binding proteins by mass spectrometry. Nat Biotechnol. 1999;17:884–888. doi: 10.1038/12873. [DOI] [PubMed] [Google Scholar]
- 40.Yaneva M, Tempst P. Affinity capture of specific DNA-binding proteins for mass spectrometric identification. Anal Chem. 2003;75:6437–6448. doi: 10.1021/ac034698l. [DOI] [PubMed] [Google Scholar]
- 41.Yan H, Gibson S, Tye BK. Mcm2 and Mcm3, 2 proteins important for Ars activity, are related in structure and function. Genes Dev. 1991;5:944–957. doi: 10.1101/gad.5.6.944. [DOI] [PubMed] [Google Scholar]
- 42.Ramnath N, Hernandez FJ, Tan DF, Huberman JA, Natarajan N, Beck AF, Hyland A, Todorov IT, Brooks JSJ, Bepler G. MCM2 is an independent predictor of survival in patients with non-small-cell lung cancer. J Clin Oncol. 2001;19:4259–4266. doi: 10.1200/JCO.2001.19.22.4259. [DOI] [PubMed] [Google Scholar]
- 43.Valkov NI, Ivanova MI, Uscheva AA, Krachmarov CP. Association of actin with DNA and nuclear matrix from guerin ascites tumor cells. Mol Cell Biochem. 1989;87:47–56. doi: 10.1007/BF00421082. [DOI] [PubMed] [Google Scholar]
- 44.Nakayasu H, Yoshimura Y, Ueda K. Association of actin-filaments with the nuclear matrix from bovine lymphocytes. Cell Struct Funct. 1981;6:424–424. [Google Scholar]
- 45.Verheijen R, Vanvenrooij W, Ramaekers F. The nuclear matrix - structure and composition. J Cell Sci. 1988;90:11–36. doi: 10.1242/jcs.90.1.11. [DOI] [PubMed] [Google Scholar]
- 46.Fuentes M, Segura RL, Abian O, Betancor L, Hidalgo A, Mateo C, Fernandez-Lafuente R, Guisan JM. Determination of protein-protein interactions through aldehyde-dextran intermolecular cross-linking. Proteomics. 2004;4:2602–2607. doi: 10.1002/pmic.200300766. [DOI] [PubMed] [Google Scholar]
- 47.Fuentes M, Mateo C, Pessela BCC, Guisan JM, Fernandez-Lafuente R. Purification, stabilization, and concentration of very weak protein-protein complexes: Shifting the association equilibrium via complex selective adsorption on lowly activated supports. Proteomics. 2005;5:4062–4069. doi: 10.1002/pmic.200401270. [DOI] [PubMed] [Google Scholar]
- 48.Chong JPJ, Mahbubani HM, Khoo CY, Blow JJ. Purification of an Mcm-containing complexes a component of the DNA-replication licensing system. Nature. 1995;375:418–421. doi: 10.1038/375418a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The list of proteins identified, comprehensive GO annotations, SDS-PAGE and Western blotting results. This material is available free of charge via the Internet at http://pubs.acs.org.