

Abstract
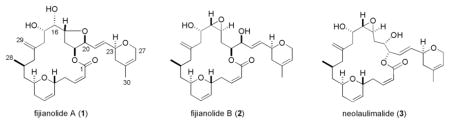
The sponge derived polyketide macrolides fijianolides A (1) and B (2) (a.k.a. isolaulimalide and laulimalide) have taxol-like microtubule-stabilizing activity and the latter exhibits potent cytotoxicity. Insight on the biogeographical and phenotypic variations of Cacospongia mycofijiensis is presented that will enable future study of the biosynthetic pathway that produces the fijianolides. In addition to fijianolides A and B, six new fijianolides, D–I (7–12), were isolated, each with modifications to the C-20 side chain of the macrolide ring. Compounds 7–12 exhibited a range of in vitro activities against HCT-116 and MDA-MB-435 cell lines. Fijianolides 8 and 10 were shown to disrupt interphase and mitotic division but were less potent than 2. An in vivo evaluation of 2 using tumor-bearing SCID mice demonstrated significant inhibition of growth in HCT-116 tumors over 28 days.
Introduction
Attention has been given in recent years to marine-derived polyketides that disrupt tubulin polymerization or promote microtubule assembly. Some consider an important subset of this group to be the 20-membered ring containing compounds characterized simultaneously in 1988 at UCSC 1 as fijianolides A (1) and B (2) from the marine sponge Cacospongia mycofijiensis and at UH as isolaulimalide (1) and laulimalide (2) from a Hyatella sponge.2 The cytotoxicities exhibited by 1 and 2 were recognized early on as being of significance- natural (−) 2 was extremely potent (KB IC50 = 29 nM, 2 MDA-MB-435 IC50 = 5.7 nM3) while natural (−) 1 (HT-29 IC50 = 21 μM 1, KB IC50 > 39 μM,2 MDA-MB-435 IC50 = 2 μM3) was also very active but at reduced potency. Evaluation of synthetic (−) 2 (MDA-MB-435 IC50 = 2 nM) confirmed these bioactivity results.4 Building on this pattern are a host of additional marine-derived polyketides described as potently active against solid tumor cancer cell lines and surveying these structures suggests that the presence of an epoxide functionality may be empowering, as seen above in the relative activities of 2 versus 1. Examples of such epoxide containing polyketides include macrolides of varying ring size with impressive IC50’s in cell-based screens [ring size, cell line tested, and IC50] such as: neolaulimalide (3)5 [21, HT-29 = 20–97 nM], tedanolide C6 [18, HCT-116 = 95 nM], the S-phase cell cycle inhibitor amphidinolide N7 [26, KB = 0.08 nM]. Alternatively, similarly potent marine-derived polyketides lacking an epoxide include: zampanolide8 [20, HT-29 = 2–10 nM], dactylolide9 [20, SK-OV-3 = 3–13 μM], tubulin-binder peloruside A10 [16, HCT-116 GI50 = 14 nM], and taxoid site tubulin binders dictyostatin11 [18, MCF-7 = 2 nM] and the drug candidate discodermolide12 [acyclic, A549 IC50 = 3.5 nM], recently evaluated in Phase I clinical studies.13 (structures SI Figure S1)
The milestone discovery in 1999 revealing that 1 and 2 stabilized microtubules3 was an important additional finding and this conclusion was confirmed independently.14,15 As a related advance, 2 was shown to interact with tubulin at a similar but distinct binding site relative to that of taxol.14–17 These developments, plus the biological data summarized above, motivated eleven total syntheses for 2 from eight different research groups.18 In addition, five different teams seeking to broaden the understanding of cytotoxicity structure activity relationships (CSAR) have prepared 35 synthetic congeners of 2.4,14,19–27 None of the synthetic analogs obtained to date have exhibited greater in vitro cytotoxicty in comparison to 2. The powerful cytotoxin neolaulimalide (3) represents the only additional natural product related to 1 and 2 described, ever. Finally, the chemical instability associated with the epoxide residue of 2 (and possibly 3), which readily rearranges to 1, could be deleterious to further biological exploration of 2, the lead member of this compound class.
Our longstanding belief that the fijianolide class represents an important seed for further therapeutic development stimulated research to amass new information. Described in this report are three complimentary approaches taken to address this possibility. Our campaign consisted of: (a) gathering a biogeographical understanding of the most reliable sponge chemotypes as a source of 2 and new analogues, (b) scaling up the isolation of 2 to launch in vivo trials in tumor bearing mice, and (c) extending the record of CSAR through biological screening of new fijianolides possessing functionality not previously created through synthesis.
Results and Discussion
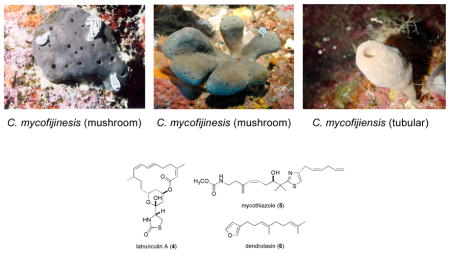
We realized that success in the first project of this study, reisolation of 2 and the discovery of additional new bioactive polyketides from C. mycofijiensis, required obtaining a deeper understanding of the natural history of the sponge. However, completing such a task took nearly a decade and involved an extensive biogeographical study of specimens with unfixed morphology. The sponge populations from Fiji, Vanuatu, and Tonga typically possess a “mushroom” shape and annual study of these specimens, summarized in Table 1, revealed that the major constituents of the Fiji collections were reliably latrunculin A (4)28 and dendrolasin (6),29 whereas taxa from Vanuatu provided 1, 2, 4 and mycothiazole (5),30,31 and, on a single occasion, a small collection from Tonga contained 4 and 5. The other coral reef zones investigated had specimens primarily “tubular” in form. Among these samples, 4 was consistently present and accompanied by 6 in collections from the Solomon Islands and Papua New Guinea, while those from Indonesia also contained 1, 2, and 5. Similar observations have been made by others but the record (see Table S1) is less tidy because of possible confusion in the taxonomic identifications.2,3,5,8,9,32–34
Table 1.
 | ||||
|---|---|---|---|---|
| Entry number | Collection Site | Major Constituents | Morphology | |
| Mushroom | Tubular | |||
| 1 | Fiji c | 4, 6 | X | |
| 2 | Vanuatu d,e | 1, 2, 4, 5 | X | |
| 3 | Tonga | 4, 5 | X | |
| 4 | Solomon Islands | 4, 6 | X | |
| 5 | Papua New Guinea | 4, 6 | X | |
| 6 | Indonesia | 1, 2, 4, 5 | X | |
Previously known as Spongia mycofijiensis.
Sanders, M. L. and Van Soest, R. W. M. Biologie 1996, 88, 117–122.
Kakou, Y.; Crews, P. J. Nat. Prod. 1987, 50, 3, 482–484.
Quinoa, E.; Kakou, Y.; Crews, P. J. Org. Chem. 1988, 53, 3642–4.
Crews, P.; Kakou, Y.; Quinoa, E. J. Am. Chem. Soc. 1988, 110, 4364–68.
Understanding the variations in major constituents vs. collection sites for C. mycofijiensis (Table 1) made it obvious that recollections from Vanuatu35 or Indonesia would be the best starting point to reisolate 1 – 2 plus analogues. A 2002 expedition to Vanuatu provided 3.7 kg (wet weight) of new material (coll. no. 02600) which was extracted using a modified Kupchan-like solvent partition scheme.36 Subsequent LC-MS examination of the four semi-purified fractions (shown in SI, Chart 1) pinpointed the CH2Cl2 sample as rich in a variety of metabolites. Preparative HPLC using a C18 column yielded three known major constituents, latrunculin A (4, 111 mg),28 mycothiazole (5, 112 mg),30 and fijianolide B (2, 55 mg),1 along with one known minor constituent, fijianolide A (1, 11 mg).1 There were an additional six new polyketides isolated with regions of the 1H and 13C NMR spectra remarkably similar to that of 1 or 2. Each of these unique compounds were minor constituents and, after further HPLC purification, designated fijianolides D (7, 6 mg), E (8, 2 mg), F (9, 1 mg), G (10, 1 mg), H (11, 1 mg), and I (12, 1 mg).
The structure elucidation process began by dividing these compounds into two sets, based on comparison to the diagnostic NMR chemical shifts of 1 – 2 at CH-16, CH-17, CH-19, and CH-20. The characteristic patterns for both analogs are as illustrated by the following data, 1: δC/H-16 76/4.11, δC/H-17 78/4.47, δC/H-19 77/5.70, δC/H-20 82/4.45; and 2: δC/H-16 61/2.76, δC/H-17 52/2.95, δC/H-19 73/5.15, δC/H-20 73/4.13. Using these trends it was clear that the analogs of 1 (Table S2, Supporting Information) consisted of 7, 9, 11, and 12 (see Tables S4, S6, S8, S9, Supporting Information), while those of 2 (Table S3, Supporting Information) consisted of 8 and 10 (see Tables S5, S7, Supporting Information). The structural modifications in the new compounds were quickly assessed as being confined to the C-20 side chain, illustrated by the side-by-side comparison of the δC changes for the new analogs relative to the data for 1 and 2. Chemical shift differences of > 2 ppm were observed at only four carbons (C-23, C-25, C-26, and C-27), as shown in Figure 1. The analogs of 1 are covered by entries (a), (c), (e), and (f), while those of 2 are summarized by entries (b) and (d).
Figure 1.

13C NMR difference in δ (>2 ppm) between 1 or 2 vs 7–12: (a) fijianolides A (1) vs D (7) (■); (b) fijianolides B (2) vs E (8) ( ); (c) 1 vs fijianolide F (9) (
); (c) 1 vs fijianolide F (9) (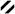 ); (d) 2 vs fijianolide G (10) (
); (d) 2 vs fijianolide G (10) ( ); (e) 1 vs fijianolide H (11) (
); (e) 1 vs fijianolide H (11) (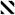 ); (f) 1 vs fijianolide I (12) (
); (f) 1 vs fijianolide I (12) ( ). Carbon δ with Δδ <2 ppm: 1–24, 28–30.
). Carbon δ with Δδ <2 ppm: 1–24, 28–30.
The complete characterization of the four related compounds (7, 9, 11, and 12) began with the analysis of fijianolide D (7) of molecular formula C30H40O8 determined by HRESIMS (m/z = 529.2800 [M+H]+). Entry (a) of Figure 1 illustrates that C-27 of 7 was shifted by more than 95 ppm from that of 1 because of the presence of a carbonyl (δC 165.8), which also accounted for the one additional unit of unsaturation in 7. The presence of an enone was also pinpointed by the HMBC correlation from H-26 (δ 5.80) to C-27. Finally the NMR shifts of 7 shown in Table 2 from C-21 through C-27 were identical to those of similar structural features in oncorhyncolide37 and provided final justification that the C-20 side chain consisted of a substituted 4-methly-6-vinyl-5,6-dihydro-pyran-2-one.
Table 2.
NMR Dataa C-22 to C-27 for 1, 7, 9, 11 and 12
| 1 | 7 | |||||||
|---|---|---|---|---|---|---|---|---|
| position | δC | C Mult. | δH (J in Hz) | δC | C mult. | δH (J in Hz)b | HMBC | |
| 21 | 126.0 | CH | 5.91 ddd (15.6, 6.0, 1.8) | 125.5 | CH | 5.80 ddd (16.0, 5.0, 1.0) | 20, 22 | |
| 22 | 134.0 | CH | 6.06 ddd (15.6, 5.4, 1.8) | 130.0 | CH | 5.95 ddd (15.5, 6.0, 1.5) | 20, 23 | |
| 23 | 73.5 | CH | 3.93 dddd (11.4, 5.4, 4.8, 0.6) | 75.7 | CH | 4.86 ddd (10.5, 5.5, 5.0) | 24, 25 | |
| 24a | 36.1 | CH2 | 2.01 m | 36.2 | CH2 | 2.32-2.28 (H14, H18, H24)* | 25, 26 | |
| 24b | 1.66 dt (16.8, 3.0) | |||||||
| 25 | 131.3 | C | 155.3 | C | ||||
| 26 | 120.5 | CH | 5.13 bs | 117.3 | CH | 5.80 s | 27 | |
| 27a | 65.7 | CH2 | 4.11 brs | 163.7 | C | |||
| 27b | 4.04 brs | |||||||
| 30 | 23.0 | CH3 | 1.50 s | 22.2 | CH3 | 1.97 s | 24, 25, 26 | |
| 9 | 11 | |||||||
| position | δC | C mult. | δH (J in Hz) | HMBCc | δC | C mult. | δH (J in Hz) | COSY |
| 21 | 126.2 | CH | 5.87 dd (13.0, 4.0) | 20. 22 | 125.8 | CH | 5.94 dd (15.0, 4.8) | 20, 22 |
| 22 | 132.5 | CH | 5.90 dd (14.5, 4.5) | 20, 23 | 132.9 | CH | 6.04 dd (15.6, 4.8) | 21 |
| 23 | 64.6 | CH | 4.25 dddd (11.4, 3.6, 2.8, 0.6) | 66.6 | CH | 4.50 dddd (12.0, 4.8, 2.4, 1.0) | 22, 24 | |
| 24a | 36.1 | CH2 | 1.50 m | 25 | 36.1 | CH2 | 1.93 dd (15.0, 10.8) | 23 |
| 24b | 1.39 dd (14.5, 11.5) | 23 | 1.45 m | |||||
| 25 | 59.3 | C | 136.4 | C | ||||
| 26 | 59.5 | CH | 2.68 d (4.2) | 27 | 120.8 | CH | 5.45 brs | 27, 30 |
| 27 | 88.5 | CH | 5.13 dd (12.0, 4.2) | 96.8 | CH | 4.92 brs | 26, 30 | |
| 30 | 21.7 | CH3 | 0.95 s | 24, 25, 26 | 22.5 | CH3 | 1.43 s | 26, 27 |
| OMe | 54.9 | CH3 | 3.34 s | |||||
| 12 | ||||||||
| position | δC | C mult. | δH (J in Hz) | COSY | ||||
| 21 | 126.2 | CH | 5.97 ddd (16.0, 5.5, 1.0) | 20, 22 | ||||
| 22 | 133.4 | CH | 6.07 ddd (15.5, 5.0, 1.0) | 21 | ||||
| 23 | 66.6 | CH | 4.57 dddd (10.7, 4.5, 2.0, 1.0) | 22, 24 | ||||
| 24a | 36.1 | CH2 | 2.15 m | 23 | ||||
| 24b | 1.60 m | 23 | ||||||
| 25 | 136.3 | C | ||||||
| 26 | 121.6 | CH | 5.42 brs | 27, 30 | ||||
| 27 | 89.0 | CH | 5.35 brs | 26, 30 | ||||
| 30 | 22.7 | CH3 | 1.46 s | 26. 27 | ||||
Measured at 500 MHz (1H) and 125 MHz (13C). Recorded in C6D6.
Recorded in CDCl3.
Recorded in acetone d6.
Can be interchanged with H-14, H-18. For more complete NMR data of compounds 1, 7, 9, 11, 12 see Supporting Information Tables S2, S4, S6, S8, S9.
A similar analysis provided support for the proposed changes to the pyran ring functionality in the remaining three compounds. The molecular formulas were obtained by HRMS and consisted of: fijianolide F (9) C30H42O9 (m/z = 569.2721 [M+Na]+), fijianolide H (11) C31H44O8 (m/z 513.2846 [M – MeOH + H]+) and fijianolide I (12) C30H42O8 (m/z = 513.2846 [M – H2O + H]+). The presence of the epoxide in 9 was indicated by the large upfield shifts at C-25 (δc 59.3) and C-26 (δc 59.5), illustrated by entry (c) of Figure 1, along with HMBC correlations from H3-30 (δH 0.95) to C-24, C-25, and C-26. The placement of an OH at C-27 (δc 88.5) was substantiated by its chemical shift and an HMBC correlation from H-26 (δH 2.68) to C-27. Fijianolides H (11) and I (12), eluted closely on HPLC and the only difference between the two was the presence in the former of an OCH3 (δ ~ 54.9) at C-27 vs. an OH in the latter. Entry (e) of Figure 1 illustrates that relative to 1, 11 has only one significant change in the 13C chemical shifts, which occurrs at C-27 (11 δc 96.8 vs. 1 δc 65.7). The 1H-1H COSY data of 11 supported the juxtaposition of H3-30, H-26, and H-27 as shown in the structure. The possible placement of the OCH3 group at either C-15 or C-16 was ruled out because the 1H and 13C shifts at these positions are virtually identical between 1, 11 and 12.
The structure elucidation of the remaining compounds, 8 and 10 (Table 3) took advantage of the associations established above. The isomeric relationship observed for 1 vs. 2 was also present for the sets 7 vs. 8 and 9 vs. 10. Also, similar to the situation observed above, using the 13C shift values for 2 as a yardstick for 8 and 10 revealed that C-25 to C-27 was the locus of their structural differences. Entries (b) and (d) of Figure 1 followed the same exact trends as observed above for entries (a) and (c) which supported the final structures of 8 and 10 with the enone and epoxy alcohol group functionalized pyran ring, respectively.
Table 3.
NMR Dataa C-22 to C-27 for 2, 8, 10
| 2 | 8 | 10 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| position | δC | C mult. | δH (J in Hz) | δC | C mult. | δH (J in Hz) | δC | C mult. | δH (J in Hz) | HMBC |
| 21 | 129.0 | CH | 5.74 ddd (16.2, 5.4, 1.8) | 129.2 | CH | 5.68-5.60 m | 129.2 | CH | 5.62 m | 20, 22 |
| 22 | 133.7 | CH | 5.85 ddd (15.6, 4.8, 1.2) | 130.0 | CH | 5.68-5.60 m | 132.5 | CH | 5.62 m | 20, 23 |
| 23 | 73.4 | CH | 3.88 m | 75.7 | CH | 4.33 brdd (14.0, 3.0) | 64.4 | CH | 4.18 m | |
| 24a | 36.1 | CH2 | 1.95 m | 34.4 | CH2 | 1.60 m | 36.1 | CH2 | 1.50 m | |
| 24b | 1.62 m | 1.25 m | 1.27 m | 23 | ||||||
| 25 | 131.3 | C | 155.2 | C | 59.3 | C | ||||
| 26 | 120.5 | CH | 5.13 brs | 117.3 | CH | 5.61 s | 59.5 | CH | 2.68 d (4.2) | 27 |
| 27a | 65.8 | CH2 | 4.11 brs | 163.5 | C | 88.4 | CH | 5.13 d (4.2) | ||
| 27b | 4.04 brs | |||||||||
| 30 | 23.0 | CH3 | 1.51 s | 22.2 | CH3 | 1.22 s | 21.7 | CH3 | 0.93 s | 24, 26 |
Measured at 500 MHz (1H) and 125 MHz (13C). Recorded in C6D6. For more complete NMR data of compounds 2, 8 and 10 see Supporting Information Tables S3, S5 and S7.
The process for determining the stereochemistry of these six new compounds was somewhat involved and only provisional assignments are proposed in the region from C-23 to C-27. First, 2 provided an important anchor point because its configuration of 5R, 9R, 11S, 15S, 16S, 17S, 19S, 20S, 23S has been previously determined by synthesis18 and X-ray analysis.38 Given that 2 rearranges to 1 means that the latter must have 5R, 9R, 11S, 15S, 16S, 17R, 19S, 20S, 23S as well. The next step was to assume that the 23S stereochemistry of 1 and 2 is retained in 7 – 12, owing to the close biosynthetic relationships apparent for these eight compounds. A series of 1D NOE experiments (benzene-d6) were employed to examine the stereochemistry at the remaining three chiral centers in the epoxy pyran rings of 9 and 10. These results appear in Figure 2 and the most important insight was gained from Expt B and D, in which 9 showed dipolar coupling between H-26 and H3-30, requiring both groups to reside on the same face. Given that strong NOE correlations were generally observed for the pyran ring protons in close proximity to each other (such as from H-23 to H-22 and to H-24a, from H3-30 to H-24a and to H-26, and from H-27 to H-26) and the lack of correlations from H-23 to H-27 or to H3-30 suggested that the former is on an opposite molecular face than the latter two. Similar data was observed for 10, which supported assignments of the same stereochemical arrangement. Consistent with these conclusions are the syn-stereochemistry observed between the hydroxyl and epoxy groups in the heterocyclic rings of the withanolide class polyketides from the Solanaceae family and, to our knowledge, there are no examples of compounds with the anti arrangement.39–42 Based on the biogenetic-based assumption of 23S we conditionally assign the additional configurations as 25R, 26R, and 27S for 9 and 10. Finally, the data set obtained did not allow assignment of the stereochemistry at C-27 in 11 and 12.
Figure 2.

1D 1H NMR Difference NOE Enhancementsa Observed for Fijianolide F (9) at H-23,b H-26,c H-27d and H3-30e in C6D6.
Several types of biological responses employing cancer tumor cell lines were obtained for the eight polyketide macrolides, 1, 2, and 7 – 12, isolated in this study. One goal was to employ the preclinical paradigm recently developed by the Valeriote group43 that consists of five steps: (a) in vitro evaluation of either solid tumor selectivity or potency against cancer cell lines, (b) IC50 determination, (c) clonogenic assay determination of cell-based cytotoxicity effects at different concentrations and exposure periods, (d) pharmacokinetic assessment at the maximum tolerated dose, and (e) therapeutic assessment in tumor-bearing mice. Another goal was to explore the cytoskeletal effects in screens employed by the Mooberry group.3 A final action was to obtain a broad view of the overall cytotoxicity patterns from the NCI 60 cell line panel.44 The responses for all eight compounds were measured in a disk diffusion soft agar colony formation assay that employed murine tumor cell lines (L1210, C-38), human tumor cell lines (HCT-116, H-125, CEM), and a normal hematopoietic line (CFU-GM). None of the compounds exhibited solid tumor selectivity against cancer cell lines (see data in Table S11 Supporting Information), however, 2 was very potent in the assay. A parallel cytotoxicity evaluation included obtaining IC50 values for all eight compounds against two human cancer cell lines, MDA-MB-435 using the SRB assay45 and HCT-116 using a 72 h cell growth inhibition.46 These data are summarized in Table 4 and show almost equal sensitivity of the two cell lines for the active compounds, with 2 as the most potent against both. In addition, only the two epoxide-containing compounds, 8 and 10, exhibited activity at low μM concentrations against the MDA-MB-435 cell line.
Table 4.
Comparative Sensitivities of Various Cell Lines to the Fijianolides
| Compound | MDA-MB-435 IC50 (μM) | HCT-116 IC50 (μM) |
|---|---|---|
| Fijianolide A (1) | 20.2 | 9.7 |
| Fijianolide B (2) | 0.007 | 0.003 |
| Fijianolide D (7) | 21.4 | 1.9 |
| Fijianolide E (8) | 1.3 | 0.7 |
| Fijianolide F (9) | >50 | >50 |
| Fijianolide G (10) | 9.2 | 14.1 |
| Fijianolide H (11) | >50 | >50 |
| Fijianolide I (12) | >50 | >50 |
All eight compounds were evaluated for disruption of interphase and mitotic microtubules in cell based screens. Our retesting of 1 and 2 verified the different degree of microtubule disruptions previously reported.3 Most importantly, 2 at 2–20 μM showed classic microtubule stabilizing effects including the formation of thick bundles of microtubules, the formation of abnormal circular mitotic spindles, and the capability to initiate apoptosis. Alternatively, 1 at 20 μM promoted an increase in microtubule density but did not stimulate the formation of thick microtubule bundles. Varying degrees of phenotypic microtubule stabilizing effects have been previously noted in the literature for five synthetic analogs of 2 possessing modified 20-membered macrolides but with the C-20 substituent unchanged.21 Subjecting 7 – 12 to similar evaluation at doses up to 50 μM gave the following results. Only fijianolides E (8) and G (10) exhibited a similar action as 2, but at higher concentrations, including increased microtubule density, formation of circular mitotic spindles, and micronucleation. The other four compounds (7, 9, 11 and 12) did not show microtubule or nuclear changes at concentrations up to 50 μM.
We consider 2 to be the best clinical candidate of the fijianolide series and predict that analogs with a modified C-20 side chain will have diminished efficacy due to their significantly lower potency. Given this prediction, 2 was subjected to a clonogenic dose response study,43,47 as shown in Figure 3, prior to its evaluation in tumor-bearing mice. For the clonogenic assay, the hypothesis is that a positive in vivo therapeutic effect will be observed only when the clonogenic cell kill is 90% or greater. Further, the determination of cytotoxicity effects at a number of concentrations and three different exposure durations will allow for the design of the optimal in vivo dose schedule.43 Interesting insights were provided by the data shown in Figure 3, which plots concentration dependent HCT-116 cell survivability as a function of time and concentration. First, there was little effect on cell toxicity for a 2 h exposure at up to 10 μg/mL. Second, for a 24 h exposure, clonogenic cell kill increases to greater than 90% and at 10 ng/mL it remains close to this value for a 1,000-fold increase in concentration. This plateau in the concentration-response curve is indicative of drug cytotoxicity during a short phase of the cell cycle. Finally, for the continuous (168 h) exposure, clonogenic kill at 90% was achieved at 1.2 ng/mL and only 0.01% of cells survived at a concentration of 5 ng/mL. These results predict that a positive in vivo therapeutic effect could be observed against HCT-116 cells through daily dose administration but the exposure of the tumor cells to 2 must be above 1.2 ng/mL for up to 7 days.
Figure 3.

Clonogenic analysis of 2 against HCT-116 cells. Dose-response curve of 2 exposed for 2h ( ), 24h (
), 24h ( ), and 168h (▲) in vitro.
), and 168h (▲) in vitro.
The next step was the pharmacokinetic evaluation of 2 and the results are shown in Figure 4. At 1 min, the plasma level is 160 μg/mL and it decreased to 1 μg/mL at 2 h and then slowly decreased to 210 ng/mL at 24 h. The initial tumor level was 7 μg/mg (primarily a function of the blood content of the tumor). The tumor level in the presence of 2 remained at 4.7 μg/mg from 30 min through 2 h, and finally decreased to 2.8 μg/mg at 24 h. It should be noted that tumor levels were above those of plasma from 1 h onwards and were about 10-fold higher at 24 h. The clonogenic results above allowed for determination of the levels for chronic, daily exposure and the overall pharmacokinetic data suggest that such a schedule should be effective in vivo.
Figure 4.

Pharmacokinetic analysis of 2 in HCT-116 tumor-bearing SCID mice. The observed concentration of 2 in the plasma ( ) and HCT-116 tumor () over a 24h period (1440 minutes). At 24h concentration of 2: plasma = 0.2 ug/mL, tumor = 2.8 ug/mg.
) and HCT-116 tumor () over a 24h period (1440 minutes). At 24h concentration of 2: plasma = 0.2 ug/mL, tumor = 2.8 ug/mg.
It was now appropriate to launch the therapeutic assessment of fijianolide B (2) using the HCT-116 human colon tumor model.48 SCID mice implanted subcutaneously with 106 tumor cells were treated with 2 starting 3 days after tumor inoculation and followed until day 30. The compound was administered as a bolus, intravenous injection daily for 5 days, which corresponds to approximately 12.5 and 25 mg/kg/day, respectively. The average tumor weight as a function of time following tumor inoculation, shown in Figure 5, indicated that the best activity was achieved at 25mg/kg/day. The minimal %T/C values were 80% at day 9 for the lower dose and 11% at day 11 for the higher dose. It should be noted that the body weight of mice receiving all doses increased throughout the 30 days and was identical to untreated controls. The latter response represents sufficient activity to consider 2 as having clinical potential48 and that further therapeutic assessment is warranted. In the near future we expect to add the results from the NCI cell line evaluation, which are now underway.
Figure 5.

In vivo assessment of 2 using HCT-116 tumor-bearing SCID mice. The average tumor weight measured per day after daily inoculation of 2 for 5 days: 25 mg/kg/day (), 12.5 mg/kg/day (▲), and control ().
There are a number of noteworthy outcomes from this study. The most significant is that a clear in vivo efficacy of 2 in tumor-bearing mice has been demonstrated and that further therapeutic development of this compound is justified. The side-by-side evaluation of the biological activity properties of 1 and 2 plus those of 7 – 12 provides additional insights in the pharmacophore analysis of this class. Our data indicates the importance of retaining an unmodified 4-methyl-2-vinyl-3,6-dihydro-2H-pyran (MVDP) substituent, but not clear yet is the requirement for 23S vs. 23R stereochemistry. This conclusion is congruent with recent total synthesis of truncated analogs of 2, in which replacement of the MVDP group by H, vinylcyclohexane, or 2-methyl-4-vinylthiazole resulted in diminished in vitro potency.24,26 These insights plus previous CSAR data indicate that the entire structure of 2 is required for the best biological response. A clear picture has now emerged on the sponge chemotypes of C. mycofijiensis that are a reliable source of the most active macrocyclic polyketides in the fijianolide series. It is also evident that other sponges that are a source of 1 – 3 may have been misidentified and should be subject to taxonomic reevaluation. The sponge chemotype information and possible classification updates will be vital to others engaged in biosynthetic discovery efforts to understand the molecular genetics responsible for the biosynthesis of this exciting clinical candidate polyketide class.
Methods
General Experimental Procedures
Optical rotations were obtained on a JASCO DIP-370 digital polarimeter. The NMR spectra were recorded on Varian UNITY INOVA-500 and 600 spectrometers, operating at 500 and 600 MHz for 1H and 125.6 and 150.0 MHz for 13C, respectively. Preparative and semi preparative HPLC were performed using 6 μm and 5 μm C18 ODS columns by means of a single wavelength (λ = 230nm) for compound detection. High resolution mass measurements were obtained from a Mariner ESI-TOF mass spectrometer.
Biological Material, Collection, and Identification
Specimens of Cacospongia mycofijiensis32 (coll. no. 02600) (3.7 kg wet weight) were collected using scuba in 2002 from Mele Bay, Vanuatu, at depths of 15–20 m. Taxonomic identification was based on comparison of the biological features to other samples in our repository. Voucher specimens and underwater photos are available.
Disk Diffusion Soft Agar Colony Formation Assay
An in vitro cell-based assay was employed to identify solid tumor selectivity for pure compounds. The differential cytotoxicity46 is expressed by calculating the zone differential between any solid tumor cell (murine colon C38, human colon HCT-116, human lung H125) and either leukemia cells (murine L1210 or human CEM) or normal cells (CFU-GM). Differential results are designated as (zone units of solid tumor - leukemia tumor, ΔSL), or (zone units of solid tumor – normal cells, ΔSN). If the zone differential is 250 zone units or greater, the activity is considered to demonstrate solid tumor selectivity and such results are bolded. The activity results appear in Table S9.
IC50, Determinations
(a) MDA-MB-435. The SRB assay was used to evaluate the antiproliferative potency of the fijianolides.45 MDA-MB-435 cells were grown in IMEM (Biosource, California) with 10% FBS (Hyclone, Utah) and 25 μg/mL gentamycin. The cells were plated at predetermined densities into 96 well plates and allowed to grow for 24 h before drug addition. Cells were exposed to the fijianolides for 48 h and then fixed and cellular protein measured as described previously.3 Dose response curves were plotted and the IC50 determined for each experiment by linear regression analyses. The numbers represent the mean of 2–4 experiments. (b) HCT-116. The HCT-116 cells are plated at 5×104 cells in T25 tissue culture flasks (Falcon Plastics, New Jersey) with 5 mL media RPMI 1640 (Cellgro, Virginia) supplemented with 15% BCS (Hyclone, Utah), 5% Pen. Strep. and 5% Glutamine (Cellgro). Three days later (cells in logarithmic growth phase; 5×105 cells/flask), test compound is added to the flasks to achieve concentrations ranging from 10 to 10−4 ug/mL. At day 3, the flasks are washed, trypsinized, spun down and the cells counted for both viable and dead cells using (0.08% trypan blue; Gibco, Maryland). Viable cell number as a function of concentration is plotted and the IC50 values are determined by interpolation.
Fijianolide B (2) HPLC Analysis
This analysis was carried out with a Waters 2690 separation module set at 4°C, and a model 2487 UV/Vis detector set at 218 nm. Analytical column of 3.9×150 mm Symmetryshield C18, 5 μm (Waters, Milford, MA) was maintained at 30°C using a Waters temperature control module. The mobile phase was 50% acetonitrile and 50% of 0.1% acetic acid in deionized water with a flow rate of 1mL/min and a run time of 10 min. All the solvents used were HPLC certified solvents from Burdick & Jackson (Honeywell International, Muskegon, MI). The lower limit of quantitation was 100 ng/mL. Under these conditions, the Fijianolide B peak eluted near 7.0 min. Absolute values of Fijianolide B were obtained using a standard curve (5 concentrations) of a stock Fijianolide B solution. Validation of each set of results was accomplished using run standards (stock drug solution dissolved in mobile phase at 1 mg/mL). Fijianolide B (2) was extracted from both plasma and tumor samples by solid phase extraction. Briefly, 0.5 mL of methanol was added to 250 μl of the sample followed by vigorous vortex and centrifuge at 14000 rpm at 4°C. To the supernatant, 1 mL of water was added and diluted to 3 mL with 0.1% acetic acid. Samples were passed through Waters Sep-pak vac 1cc (100mg) C18 cartridges equilibrated with 1 mL of methanol and 1 mL of deionized water. Cartridges were washed with 2 mL of 0.1% acetic acid containing 5% methanol. Fijianolide B was eluted from the cartridges by passing 1.5 mL of 2% formic acid in acetonitrile. Extract was evaporated in a Turbovap LV evaporator (Zymark, Hopkinton, MA) to dryness under a stream of nitrogen at 45°C, reconstituted in 100 μl of mobile phase for HPLC analysis.
Fijianolide B (2) Clonogenic Dose-Response Analysis
Concentration and time-survival studies of fijianolide B were carried out against HCT-116 cells. HCT-116 cells were seeded at 200 to 20,000 cells in 60 mm dishes. Drug was added to the medium (RPMI + 10% FBS) at concentrations of 1 mg/mL and 10-fold dilutions thereof. At either 2 hr or 24 hr, the drug-containing media wass removed and fresh media without drug is added. For continuous exposure to the drug, it remained in contact with the cells for the entire incubation period. The dishes were incubated for 7 days, media removed and the colonies stained with methylene blue. Colonies containing 50 cells or more were counted. The results were normalized to an untreated control. Plating efficiency for the untreated cells was about 90%. Repeat experiments were carried out to define the cell survival range between 100% and 0.1% survival. The results of this analysis are shown in Figure 3.
Fijianolide B (2) Pharmacokinetic Analysis
SCID mice bearing HCT-116 subcutaneous tumors (approximately 100 mg each) received 0.5 mg/mouse (25 mg/kg) of fijinanolide B in 0.25 mL by intravenous administration. Fijinanolide B (2) was first prepared at 10 mg/mL in DMSO, mixed 1:1 with a Cremophor/Propylene glycol (60:40, v:v) solution and then diluted 20-fold with saline. Plasma and tumor samples were obtained at 1′, 15′, 30′, 1h, 2h, 6h and 24h from individual mice. Upon collection, tumor samples were immediately placed in ice-cold saline (3 mL per g of tissue) and homogenized on ice. The homogenate was centrifuged and the supernatant assayed for 2 as described above. The overall results are shown in Figure 4.
Fijianolide B (2) Therapeutic Assessment
The in vivo therapeutic assessment trial was carried out using the HCT-116 human colon tumor model as previously described.48 Individual mouse body weights for each experiment were within 5 g and all mice are over 20 g at the start of therapy. The mice were supplied food and water ad libidum. SCID mice were pooled, implanted subcutaneously with 106 tumor cells, and pooled again before distribution to treatment and control groups (5 mice per group). Treatment with 2 was started 3 days after tumor inoculation; mice were sacrificed 30 days later. Tumor weights were estimated using 2-dimensional caliper measurements done three times per week using the formula: Tumor Weight (mg) = (a × b2)/2, where a and b are the tumor length and width in mm, respectively, and the median calculated as an indication of antitumor effectiveness. The parameter, %T/C, was determined after each measurement and the minimum value reported as therapeutic efficacy. All mice were weighed at the time of tumor measurement. Fijianolide B was prepared identically to that described above for the pharmacokinetic studies at both 1 and 2 mg/mL for intravenous administration as 0.25 mL volumes via the tail vein. The drug was administered as a bolus injection daily for 5 days, which corresponds to approximately 12.5 and 25 mg/kg/day, respectively. The overall results are shown in Figure 5.
Extraction and Isolation
Samples were preserved in the field according to our standard laboratory procedures36 and stored in a cold room until extraction was performed. The sponge was extracted 3× with methanol and then the resultant oil was partitioned using a modified Kupchen-type solvent partition scheme as described previously.36 Pure compounds were obtained as follows: (Chart 1 SI) a portion of the 02600 CH2Cl2 (DCM) extract was fractionated using preparatory reversed-phase gradient HPLC (50:50 CH3CN/H2O up to 55:45 over 50 minutes) to give 11 fractions. Preparatory fraction 5 (P5, 70.1 mg) and 7 (P7, 38.1 mg) were then separated using semi preparative reversed-phase gradient HPLC (30:70 CH3CN/H2O up to 80:20 over 50 minutes) to give 3 fractions (P5, F1-3) and 4 fractions (P7, F1-4). Fractions P5F1 and 2 were then further purified to yield 10 (1.3 mg) and 8 (1.7 mg). Fraction P5F3 gave pure 2 (55.2 mg). Fractions P7F2 and F4 yielded pure 7 (6.3 mg) and 1 (10.7 mg). P7 F1 was further separated to give pure 9 (1.4 mg) and purification of P7F3 resulted in 11 (1.0 mg) and 12 (1.2 mg).
Fijianolide A (1): amorphous glass-like solid; [α]23D - 44.4 (c 0.9, CHCl3) LRESITOFMS m/z 515 [M + H]+; 1H and 13C NMR data (C6D6) in Figure S2, S3 and Table S2 were in agreement with literature values.1
Fijianolide B (2): amorphous glass-like solid; [α]23D - 143.7 (c 0.8, CHCl3) LRESITOFMS m/z 515 [M + H]+; 1H and 13C NMR data (C6D6) in Figure S4, S5 and Table S3 were in agreement with literature values.1
Latrunculin A (4): white oil; LRESITOFMS m/z 404 [M - H2O + H]+; 1H and 13C NMR (CDCl3) data were in agreement with literature values.28
Mycothiazole (5): whitish powder; LRESITOFMS m/z 405 [M + H]+; 1H and 13C NMR (CDCl3) data were in agreement with literature values.30,31
Fijianolide D (7): amorphous glass-like solid; [α]27D - 9.0 (c 1.0, C6H6); 1H and 13C NMR data in Figure S6, S7 and Table S4. HRESITOFMS m/z 529.2800 [M + H]+ (calcd for C30H41O8, 529.2801).
Fijianolide E (8): amorphous glass-like solid; [α]27D - 3.8 (c 0.2, C6H6); 1H and 13C NMR data in Figure S8, S9 and Table S5. HRESITOFMS m/z 529.2801 [M + H]+ (calcd for C30H41O8, 529.2801).
Fijianolide F (9): amorphous glass-like solid; [α]23D - 17.0 (c 0.35, CH3OH); 1H and 13C NMR data in Figure S10, S11 and Table S6. HRESITOFMS m/z 569.2721 [M + Na]+ (calcd for C30H42O9Na, 569.2727).
Fijianolide G (10): amorphous glass-like solid; [α]23D - 153.3 (c 0.3, CH3OH); 1H and 13C NMR data in Figure S12, S13 and Table S7. HRESITOFMS m/z 569.2721 [M + Na]+ (calcd for C30H42O9Na, 569.2727).
Fijianolide H (11): amorphous glass-like solid; [α]23D - 16.0 (c 0.25, CH3OH); 1H and 13C NMR data in Figure S14, S15 and Table S8. HRESITOFMS m/z 513.2846 [M – MeOH + H]+ (calcd for C30H41O7, 513.2854).
Fijianolide I (12): amorphous glass-like solid; [α]23D - 46.6 (c 0.6, CH3OH); 1H and 13C NMR data in Figure S16, S17 and Table S9. HRESITOFMS m/z 513.2846 [M – H2O + H]+ (calcd for C30H41O7, 513.2854).
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health RO1 CA 47135 and funding from the William Randolph Hearst Foundation (SLM). We wish to give special thanks to M. Amos and the Vanuatu Ministry of Fisheries. We are also grateful to C. Boot, P. Ralifo, P. Wenzel and the members of the Hideaway Island diving staff for their generous assistance in the repeated collection of C. mycofijiensis. The technical assistance of P. Hills is gratefully acknowledged.
Abbreviations
- SCID
severe combined immuno-deficiency
- MVDP
4-methyl-2-vinyl-3,6-dihydro-2H-pyran
- CSAR
cyotoxicity structure activity relationship
- SRB
sulforhodamine B. Cell lines (panel name, type)
- HCT-116
human colon
- HT29
human colon
- SK-OV-3
human ovarian
- KB
human carcinoma
- MDA-MB-435
human breast
- MCF-7
human breast
- A549
human non-small cell lung
- CCRF
human leukemia
- CEM
human leukemia
- CFU-GM
human normal
- L1210
murine leukemia
- C38
murine colon.
Footnotes
Supporting Information Available: Eleven tables and 32 figures are provided which include the isolation scheme, 1D and 2D NMR data along with the cytotoxicity in zone units from the disk diffusion soft agar colony formation assay46 and LCMS analysis for compounds 1 and 2 and 7–12. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Quinoa E, Kakou Y, Crews P. Fijianolides, polyketide heterocycles from a marine sponge. J Org Chem. 1988;53:3642–3644. [Google Scholar]
- 2.Corley DG, Herb R, Moore RE, Scheuer PJ, Paul VJ. Laulimalides - new potent cyto-toxic macrolides from a marine sponge and a nudibranch predator. J Org Chem. 1988;53:3644–3646. [Google Scholar]
- 3.Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Laulimalide and isolaulimalide, new paclitaxel-like microtubule stabilizing agents. Cancer Res. 1999;59:653–660. [PubMed] [Google Scholar]
- 4.Gallagher BM, Fang FG, Johannes CW, Pesant M, Tremblay MR, Zhao HJ, Akasaka K, Li XY, Liu JK, Littlefield BA. Synthesis and biological evaluation of (−)-laulimalide analogues. Bioorg Med Chem Lett. 2004;14:575–579. doi: 10.1016/j.bmcl.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka J, Higa T, Bernardinelli G, Jefford CW. New cytotoxic macrolides from the sponge Fasciospongia rimosa. Chem Lett. 1996:255–256. [Google Scholar]
- 6.Chevallier C, Bugni TS, Feng XD, Harper MK, Orendt AM, Ireland CM. Tedanolide C: a potent new 18-membered-ring cytotoxic macrolide isolated from the Papua New Guinea marine sponge Ircinia sp. J Org Chem. 2006;71:2510–2513. doi: 10.1021/jo052285+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishibashi M, Yamaguchi N, Sasaki T, Kobayashi J. Amphidinolide-N, a novel 26-membered macrolide with remarkably potent cytotoxicity from the cultured marine dinoflagellate Amphidinium sp. J Chem Soc Chem Commun. 1994:1455–1456. [Google Scholar]
- 8.Tanaka J, Higa T. Zampanolide, a new cytotoxic macrolide from a marine sponge. Tetrahedron Lett. 1996;37:5535–5538. [Google Scholar]
- 9.Cutignano A, Bruno I, Bifulco G, Casapullo A, Debitus C, Gomez-Paloma L, Riccio R. Dactylolide, a new cytotoxic macrolide from the Vanuatu sponge Dactylospongia sp. Eur J Org Chem. 2001:775–778. [Google Scholar]
- 10.Hood KA, West LM, Rouwe B, Northcote PT, Berridge MV, Wakefield SJ, Miller JH. Peloruside A, a novel antimitotic agent with paclitaxel-like microtubule-stabilizing activity. Cancer Res. 2002;62:3356–3360. [PubMed] [Google Scholar]
- 11.Isbrucker RA, Cummins J, Pomponi SA, Longley RE, Wright AE. Tubulin polymerizing activity of dictyostatin-1, a polyketide of marine sponge origin. Biochem Pharmacol. 2003;66:75–82. doi: 10.1016/s0006-2952(03)00192-8. [DOI] [PubMed] [Google Scholar]
- 12.Isbrucker RA, Gunasekera SP, Longley RE. Structure-activity relationship studies of discodermolide and its semisynthetic acetylated analogs on microtubule function and cytotoxicity. Cancer Chemother Pharmacol. 2001;48:29–36. doi: 10.1007/s002800100287. [DOI] [PubMed] [Google Scholar]
- 13.Mita A, Lockhart AC, Chen TL, Bochinski K, Curtright J, Cooper W, Hammond L, Rothenberg M, Rowinsky E, Sharma S. A phase I pharmacokinetic (PK) trial of XAA296A (Discodermolide) administered every 3 wks to adult patients with advanced solid malignancies. J Clin Oncol. 2004;22:133S–133S. [Google Scholar]
- 14.Pryor DE, O’Brate A, Bilcer G, Diaz JF, Wang YF, Wang Y, Kabaki M, Jung MK, Andreu JM, Ghosh AK, Giannakakou P, Hamel E. The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry. 2002;41:9109–9115. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- 15.Gaitanos TN, Buey RM, Diaz JF, Northcote PT, Teesdale-Spittle P, Andreu JM, Miller JH. Peloruside A does not bind to the taxoid site on β-tubulin and retains its activity in multidrug-resistant cell lines. Cancer Res. 2004;64:5063–5067. doi: 10.1158/0008-5472.CAN-04-0771. [DOI] [PubMed] [Google Scholar]
- 16.Peloruside A (S7) was concluded to have identical tubulin site binding properties in comparison to 2. Also, 2 and S7 each were synergistic with seven other taxol-site binding agents but not with each other.
- 17.Hamel E, Day BW, Miller JH, Jung MK, Northcote PT, Ghosh AK, Curran DP, Cushman M, Nicolaou KC, Paterson I, Sorensen EJ. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol Pharmacol. 2006;70:1555–1564. doi: 10.1124/mol.106.027847. [DOI] [PubMed] [Google Scholar]
- 18.Mulzer J, Ohler E. Microtubule-stabilizing marine metabolite laulimalide and its derivatives: Synthetic approaches and antitumor activity. Chem Rev. 2003;103:3753–3786. doi: 10.1021/cr940368c. [DOI] [PubMed] [Google Scholar]
- 19.Ahmed A, Hoegenauer EK, Enev VAS, Hanbauer M, Kaehlig H, Ohler E, Mulzer J. Total synthesis of the microtubule stabilizing antitumor agent laulimalide and some nonnatural analogues: The power of Sharpless’ asymmetric epoxidation. J Org Chem. 2003;68:3026–3042. doi: 10.1021/jo026743f. [DOI] [PubMed] [Google Scholar]
- 20.Wender PA, Hegde SG, Hubbard RD, Zhang L, Mooberry SL. Synthesis and biological evaluation of (−)-laulimalide analogues. Org Lett. 2003;5:3507–3509. doi: 10.1021/ol035339f. [DOI] [PubMed] [Google Scholar]
- 21.Mooberry SL, Randall-Hlubek DA, Leal RM, Hegde SG, Hubbard RD, Zhang L, Wender PA. Microtubule-stabilizing agents based on designed laulimalide analogues. Proc Natl Acad Sci U S A. 2004;101:8803–8808. doi: 10.1073/pnas.0402759101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paterson I, Bergmann H, Menche D, Berkessel A. Synthesis of novel 11-desmethyl analogues of laulimalide by Nozaki-Kishi coupling. Org Lett. 2004;6:1293–1295. doi: 10.1021/ol049791q. [DOI] [PubMed] [Google Scholar]
- 23.Gallagher BM, Zhao HJ, Pesant M, Fang FG. Synthesis of 8-(S)-methoxy-11-desmethyl laulimalide: a novel laulimalide analogue. Tetrahedron Lett. 2005;46:923–926. [Google Scholar]
- 24.Paterson I, Menche D, Hakansson AE, Longstaff A, Wong D, Barasoain I, Buey RM, Diaz JF. Design, synthesis and biological evaluation of novel, simplified analogues of laulimalide: modification of the side chain. Bioorg Med Chem Lett. 2005;15:2243–2247. doi: 10.1016/j.bmcl.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 25.Wender PA, Hilinski MK, Soldermann N, Mooberry SL. Total synthesis and biological evaluation of 11-desmethyllaulimalide, a highly potent simplified laulimalide analogue. Org Lett. 2006;8:1507–1510. doi: 10.1021/ol060233g. [DOI] [PubMed] [Google Scholar]
- 26.Wender PA, Hilinski MK, Skaanderup PR, Soldermann NG, Mooberry SL. Pharmacophore mapping in the laulimalide series: Total synthesis of a vinylogue for a late-stage metathesis diversification strategy. Org Lett. 2006;8:4105–4108. doi: 10.1021/ol061619u. [DOI] [PubMed] [Google Scholar]
- 27.Faveau C, Mondon M, Gesson JP, Mahnke T, Gebhardt S, Koert U. Synthetic studies on a phenyl-laulimalide analogue. Tetrahedron Lett. 2006;47:8305–8308. [Google Scholar]
- 28.Kashman Y, Groweiss A, Shmueli J. Latrunculin, a new 2-thiazolidinone macrolide from the marine sponge Latrunculia magnifica. Tetrahedron Lett. 1980;21:3629–3632. [Google Scholar]
- 29.Kakou Y, Crews P, Bakus GJ. Dendrolasin and latrunculin A from the Fijian sponge Spongia mycofijiensis and an associated nudibranch Chromodoris lochi. J Nat Prod. 1987;50:482–484. [Google Scholar]
- 30.Crews P, Kakou Y, Quinoa E. Mycothiazole, a polyketide heterocycle from a marine sponge. J Am Chem Soc. 1988;110:4365–4368. [Google Scholar]
- 31.Sonnenschein RN, Johnson TA, Tenney K, Valeriote FA, Crews P. A reassignment of (−)-mycothiazole and the isolation of a related diol. J Nat Prod. 2006;69:145–147. doi: 10.1021/np0503597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanders ML, van Soest RWM. A revised classification of Spongia mycofijiensis. Biologie. 1996;88:117–122. [Google Scholar]
- 33.Gulavita NK, Gunasekera SP, Pomponi SA. Isolation of latrunculin-A, 6,7-epoxylatrunculin-A, fijianolide-A, and euryfuran from a new genus of the family Thorectidae. J Nat Prod. 1992;55:506–508. [Google Scholar]
- 34.In a correction to the literature it was noted that the following three sponges are actually identical including: an unidentifiable specimen from American Samoa (providing 1, 4, s3), Cacospongia mycofijiensis (a.k.a. Spongia mycofijiensis), from Fiji (providing 4, 6), and Hyatella sp. from Indonesia (providing 1, 2, and 4). Related observations (Table S1) are that a Vanuatu sponge identified as Dactylospongia sp. was reported as a source of 1, 2, s2, 4 and 5, while a sponge designated as C. mycofijiensis from the Marshall Islands contained 1, 2, and 4. Further examination of the literature shows 4 was isolated from one other sponge genera, Fasciospongia rimosa (Japan), that also afforded polyketide macrolides 1, 2, s1, or 3.
- 35.An additional analog, fijianolide C, was previously isolated from a Vanuatu sponge (coll. no. 00600) but not fully characterized. Details of the isolation are included in a 2001 Ph.D. dissertation: Carroll, J. Sponge Derived Marine Natural Products as Pharmaceutical Leads, University of California, Santa Cruz. pp.142–194.
- 36.Thale Z, Johnson T, Tenney K, Wenzel PJ, Lobkovsky E, Clardy J, Media J, Pietraszkiewicz H, Valeriote FA, Crews P. Structures and cytotoxic properties of sponge-derived bisannulated acridines. J Org Chem. 2002;67:9384–9391. doi: 10.1021/jo026459o. [DOI] [PubMed] [Google Scholar]
- 37.Needham J, Andersen RJ, Kelly MT. Oncorhyncolide, a novel metabolite of a bacterium isolated from seawater. Tetrahedron Lett. 1991;32:315–318. [Google Scholar]
- 38.Jefford CW, Bernardinelli G, Tanaka J, Higa T. Structures and absolute configurations of the marine toxins, latrunculin A and laulimalide. Tetrahedron Lett. 1996;37:159–162. [Google Scholar]
- 39.Bado S, Mareggiani G, Amiano N, Burton G, Veleiro AS. Lethal and sublethal effects of withanolides from Salpichroa origanifolia and analogues on Ceratitis capitata. J Agric Food Chem. 2004;52:2875–2878. doi: 10.1021/jf035508a. [DOI] [PubMed] [Google Scholar]
- 40.Nicotra VE, Gil RR, Vaccarini C, Oberti JC, Burton G. 15,21-Cyclowithanolides from Jaborosa bergii. J Nat Prod. 2003;66:1471–1475. doi: 10.1021/np030248c. [DOI] [PubMed] [Google Scholar]
- 41.Tettamanzi MC, Veleiro AS, de la Fuente JR, Burton G. Withanolides from Salpichroa origanifolia. J Nat Prod. 2001;64:783–786. doi: 10.1021/np010010t. [DOI] [PubMed] [Google Scholar]
- 42.Zhu XH, Takagi M, Ikeda S, Midzuki K, Nohara T. Withanolide-type steroids from Solanum cilistum. Phytochemistry. 2001;56:741–745. doi: 10.1016/s0031-9422(00)00487-8. [DOI] [PubMed] [Google Scholar]
- 43.Subramanian B, Nakeff A, Tenney K, Crews P, Gunatilaka L, Valeriote FA. A new paradigm for the development of anticancer agents from natural products. J Exp Ther Oncol. 2006;5:195–204. [PMC free article] [PubMed] [Google Scholar]
- 44.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigrowolff A, Graygoodrich M, Campbell H, Mayo J, Boyd M. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor-cell lines. J Natl Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 45.Scudiero DA, Monks A, Sausville EA. Cell line designation change: Multidrug-resistant cell line in the NCI anticancer screen. J Natl Cancer Inst. 1998;90:862–862. doi: 10.1093/jnci/90.11.862. [DOI] [PubMed] [Google Scholar]
- 46.Valeriote FA, Grieshaber CK, Media J, Pietraszkewicz H, Hoffman J, Pan M, McLaughlin S. Discovery and development of anticancer agents from plants. J Exp Ther Oncol. 2002;2:228–236. doi: 10.1046/j.1359-4117.2002.01038.x. [DOI] [PubMed] [Google Scholar]
- 47.Corbett T, Valeriote F, Lorusso P, Polin L, Panchapor C, Pugh S, White K, Knight J, Demchik L, Jones J, Jones L, Lowichik N, Biernat L, Foster B, Wozniak A, Lisow L, Valdivieso M, Baker L, Leopold W, Sebolt J, Bissery MC, Mattes K, Dzubow J, Rake J, Perni R, Wentland M, Coughlin S, Shaw JM, Liversidge G, Liversidge E, Bruno J, Sarpotdar P, Moore R, Patterson G. Tumor-models and the discovery and secondary evaluation of solid tumor active agents. Int J Pharma. 1995;33:102–122. [Google Scholar]
- 48.Bruce WR, Meeker BE, Powers WE, Valeriote FA. Comparison of dose- and time-survival curves for normal hematopoietic and lymphoma colony-forming cells exposed to vinblastine, vincristine, arabinocytosine, and amethopterin. J Natl Cancer Inst. 1969;42:1015–1025. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.