

In the preceding communication[1] we described our initial attempts to determine the true structures of the vannusals [A (1) and B (2), originally assigned structures, Figure 1]. Herein, we report the total synthesis of the real vannusal B (i.e. structure 4, Figure 1) that served not only to render it available for biological investigations, but also to demystify its true molecular architecture and that of its sibling, vannusal A.
Figure 1.

Originally assigned structures of vannusals A (1) and B (2), final targeted stereoisomer 3 [C10-epi-2], and revised structure 4. BOM = benzyloxymethyl; SEM = trimethylsilylethoxymethyl; TIPS = triisopropylsilyl.
Our initial intelligence gathering efforts led us to the conclusion that (a) the stereochemical details of the “bottom” part of the molecule (i.e. rings A and B, C3, C29, C6, and C7) were most likely correct as reported in the originally assigned structures 1 and 2, and (b) the most likely place for an error was the very “top” part of those structures (i.e. ring E, C25 and C21). Since we had already synthesized three of the four C25/C21 possible diastereomers of the originally assigned structure of vannusal B (2)[2] and found them to be erroneous, we returned to the remaining diastereomer, structure 3 [C25-epi-2, Figure 1], as a possible candidate for the true structure of vannusal B. By virtue of its trans stereochemistry at C25/C21, structure 3 was abandoned earlier on the basis of the large coupling constant observed between H21 and H25 (J = 8.5 Hz) in the trans substituted C21-epi-2 isomer (structure 3, in ref 1). This led us to the chase of several other diastereomers which, as we have seen in the preceding communication,[1] ended by proving that none of them represented the true structure of vannusal B.
With all the evidence in front of us, we were now forced to reconsider the possibility that the C25-epi-2 structure (i.e. 3, Figure 1) may accommodate the observed smaller coupling constant for natural vannusal B (JH25,21 = 1.6 Hz) by virtue of a special conformation. We, therefore, decided to pursue the synthesis of 3 as the possible coveted structure of vannusal B. The selection of structure 3 as our next favorite target defined compounds 5 and 6 (Figure 1) as the required building blocks for its construction. From these two fragments, only 6 needed to be synthesized, since 5 was already available in enantiopure form from our previous studies.[2] Its synthesis begun with the DIBAL-H reduction of racemic 8 [obtained by reaction of the titanium enolate of diketone (±)-7 (TiCl4, Et3N) with acetone][2] which proceeded stereoselectively (ca. 6:1 dr) from the α-face of the molecule and at both carbonyl sites to afford, upon chromatographic purification, pure triol 9 in 64 % overall yield (Scheme 1). The diastereoselective reduction of (±)-8 stands in contrast to the NaBH4 reduction of its TES-protected counterpart, which gave the opposite stereochemistry at C25.[2]
Scheme 1.

Construction of aldehyde (±)-6. Reagents and conditions: (a) TiCl4 (1.3 equiv), Et3N (3.0 equiv), acetone (10 equiv), CH2Cl2, −92 °C, 8 h; (b) DIBAL-H (5.0 equiv), PhMe, −78→0 °C, 30 min, 64 % for two steps; (c) aq. HF:THF (1:4), 25 °C, 18 h, 83 %; (d) iPr2NEt (20 equiv), SEMCl (6.0 equiv), nBu4NI (1.0 equiv), 50 °C, 24 h; KHMDS (3.0 equiv), SEMCl (5.0 equiv), Et3N (10 equiv), THF, −78→25 °C, 1 h, 96 % for the two steps; (e) O3, py (1.0 equiv), CH2Cl2:MeOH (1:1), −78 °C; then Ph3P (5.0 equiv), −78→25 °C, 1 h, 97 %; (f) KH (10 equiv), allyl chloride (30 equiv), HMPA (10 equiv), DME, −10→25 °C, 8 h, 95 %; (g) iPr2NEt (1.0 equiv), 1,2-dichlorobenzene, 200 °C (μ-waves), 20 min; then NaBH4 (10 equiv), MeOH, 1 h, 25 °C, 88 % for two steps; (h) BOMCl (10 equiv), iPr2NEt (30 equiv), nBu4NI (1.0 equiv), CH2Cl2, 50 °C 12 h; (i) O3, py (1.0 equiv), CH2Cl2:MeOH (1:1), −78 °C; then Ph3P (5.0 equiv), −78→25 °C, 1 h, 81 % for two steps; (j) TBSCl (10 equiv), DBU (20 equiv), CH2Cl2, 25 °C, 12 h; (k) O3, py (1.0 equiv), CH2Cl2:MeOH (1:1), −78 °C; then Ph3P (5.0 equiv), −78→25 °C, 1 h, 80 % for two steps. HMPA = hexamethylphosphoramide, DBU = 1,8-diazoicyclo[5.4.0]undec-7-ene.
Interestingly, triol 9 exhibited a small coupling constant between H25 and H21 (JH25,21 = 1.0 Hz), which was rather surprising given the coupling constants between the same protons of its siblings, 9a (JH25,21 = 10.0 Hz),[1] 9b (JH25,21 = 3.5 Hz),[1] and 9c (JH25,21 < 1.0 Hz)[2] (see Figure 2). Particularly striking was the difference between the two trans compounds 9 (JH25,21 = 1.0 Hz) and 9a (JH25,21 = 10.0 Hz). An X-ray crystallographic analysis[3] (see ORTEP drawing, Figure 2) of tetraol 9d (mp 139–141 °C, benzene/MeOH) (obtained from 9 by desilylation with aq. HF as shown in Scheme 1) confirmed the assigned stereochemical relationships within triol 9. These observations underscored the dangers of relying on the coupling constants to assign stereochemistry around 5-membered rings, and provided somewhat of an endorsement of our choice of target 3 as a candidate for the true structure of vannusal B.
Figure 2.

Key 1H NMR coupling constants for tricyclic intermediates 9 (JH25,21 = 1.0 Hz), 9a (JH25,21 = 10.0 Hz), 9b (JH25,21 = 3.5 Hz), and 9c (JH25,21 < 1.0 Hz) (top), and X-ray derived ORTEP drawing of tetraol 9d (bottom).
Returning to the main sequence of Scheme 1, installment of SEM groups on all three hydroxyl groups of intermediate 9 required sequential treatment with SEMCl and iPr2NEt (50 °C, 24 h) first, followed by the use of KHMDS and excess SEMCl to deliver the corresponding tri-SEM derivative 10 (96 % overall yield). The remaining steps to the targeted fragment (±)-6 followed the previously chartered route[1] and proceeded in high overall yield through intermediates 11–13 as summarized in Scheme 1.
The coupling of enantiopure vinyl iodide (−)-5[2] with racemic aldehyde (±)-6 (see Scheme 2) proceeded smoothly through the lithio derivative of 5 (tBuLi) to afford, after removal of the TIPS group (TBAF, 25 °C), a mixture of diastereomeric diols 14a and 14b (84 % for two steps, ca. 1:1 dr), which were chromatographically separated. Based on our previous studies,[4] the undesired diastereomers (such as 14b) had been routinely utilized for reconnaissance studies regarding late stage transformations, particularly the SmI2-cyclization and final global deprotection. Consequently, both coupling products 14a and 14b were separately converted to cyclization precursors 15a ad 15b, respectively, through the same sequence of reactions involving temporary silylation of the primary hydroxyl group (TESCl, imid.), carbonate formation at the secondary position (KHMDS, ClCO2Me), removal of the TES group (HF•py, 89 % yield for 14a, 96 % yield for 14b), and oxidation of the liberated primary hydroxyl group [PhI(OAc)2, AZADO (cat.),[5] 96 % yield for 15a, 97 % yield for 15b] as summarized in Scheme 2.
Scheme 2.

Coupling of fragments (−)-5 and (±)-6. Reagents and conditions: (a) (−)-5 (1.3 equiv), tBuLi (2.6 equiv), THF, −78→−40 °C, 40 min; then (±)-6 (1.0 equiv), −40→0 °C, 20 min; (b) TBAF (1.0 M in THF, 2.0 equiv), THF, 25 °C, 8 h, 84 % (ca. 1:1 dr) for the two steps, 14a and 14b chromatographically separated; c) TESCl (2.0 equiv), imid (10 equiv), CH2Cl2, 25 °C, 30 min; (d) KHMDS (10 equiv), ClCO2Me (20 equiv), Et3N (20 equiv), THF, −78→25 °C, 30 min; (e) HF•py / py (1:4), 0→25 °C, 12 h, 89 % for sequence 14a→15a, 96 % for sequence 14b→15b (for three steps); (f) PhI(OAc)2 (2.0 equiv), AZADO (0.1 equiv), CH2Cl2, 25 °C, 24 h, (96 % for 15a, 97 % for 15b); (g) aq. HF:THF (1:4), 25 °C, 1.5 h, 81 %.
First to be advanced from this point was intermediate 15a possessing our favorite diastereomeric configuration. Much to our disappointment, precursor 15a with its three SEM groups in place failed to undergo the desired SmI2-induced ring closure as several of its previous siblings,[1, 2] thus prompting us to modify its structure as a means to adjust its reactivity. To this end, 15a was treated with aq. HF to afford, cleanly, dihydroxy substrate 16 (81 % yield) through selective cleavage of the C22 and C26 SEM groups (Scheme 2). Pleasantly, substrate 16 underwent the desired SmI2-induced cyclization to afford polycyclic structure 17 as the major product (67 % yield), possessing, however, the undesired C10/C28 stereochemistry as shown in Scheme 3. The obligatory inversion of the stereochemistry at C10 and C28 of product 17 required a sequence that initially involved selective temporary acetylation at C28 (Ac2O, Et3N, 4-DMAP, 100 % yield), installation of a SEM group at C22 (SEMCl, iPr2NEt), and deacetylation (DIBAL-H) to furnish diol 19, in 83 % overall yield. Selective xanthate formation at C28 followed by syn-elimination (NaH, CS2, MeI, 79 % yield; then μ-waves; 185 °C, 88 % yield) to afford, after SEM protection of the C26-OH group, conjugate diene 20 (78 % yield). The latter compound was converted to the desired C10/C28 diastereomer 21 through the previously developed sequence involving hydroboration/oxidation (ThexBH2; BH3; H2O2, 71% yield) and phenylselenenylation/syn-elimination (oNO2C6H4 SeCN, nBu3P; H2O2, 86 % yield). Intermediate 21 was then transformed to diol 22 through sequential silylation (KHMDS, TESCl, 93 % yield) and cleavage of the BOM groups (LiDBB, 85 % yield). The final three steps to structure 3 involving sequential oxidation of the primary hydroxyl group within 22 [PhI(OAc)2, TEMPO, 88 % yield], acetylation of the secondary hydroxyl group (Ac2O, Et3N, 4-DMAP, 100 % yield), and global desilylation (aq. HF, 90 % yield) proceeded smoothly to afford vannusal B structure 3, but not before the completion of the synthesis of vannusal B structure 4 (vide infra), which we will describe next because of its special significance.
Scheme 3.

Synthesis of vannusal B structure 3. Reagents and conditions: (a) SmI2 (0.1 M in THF, 10 equiv), HMPA (30 equiv), THF, −20→25 °C, 20 min, 67 %; (b) Ac2O (20 equiv), Et3N (20 equiv), 4-DMAP (0.2 equiv), CH2Cl2, 25 °C, 30 min, 100 %; (c) SEMCl (10 equiv), iPr2NEt (30 equiv), CH2Cl2, 50 °C, 18 h; (d) DIBAL-H (5.0 equiv), CH2Cl2, −78 °C, 30 min, 83 % for two steps; (e) NaH (10 equiv), CS2 (3.0 equiv), THF, 0→25 °C, 30 min; then CH3I (6.0 equiv), 0→25 °C, 1 h, 79 %; then 185 °C (μ–waves), 1,2-dichlorobenzene, 15 min, 88 %; (f) KHMDS (4.0 equiv), SEMCl (4.0 equiv), Et3N (8.0 equiv), THF, −50→25 °C, 20 min, 78 %; (g) ThexBH2 (5.0 equiv), THF, −10→25 °C, 30 min; then BH3•THF (15 equiv), 25 °C, 1 h; then 30 % H2O2/3 N NaOH (1:1 dr), 25→45 °C, 30 min; 71 % (1:1.3 mix); (h) oNO2C6H4SeCN (3.0 equiv), nBu3P (9.0 equiv), py (12.0 equiv), THF, 25 °C, 10 min; then 30 % H2O2, 25→45 °C, 30 min, 86 %; (i) KHMDS (6.0 equiv), TESCl (4.0 equiv), Et3N (8.0 equiv), THF, −50→25 °C, 20 min, 93 %; (j) LiDBB (excess), THF, −78→−50 °C, 30 min, 85 %; (k) PhI(OAc)2 (4.0 equiv), TEMPO (2.0 equiv), CH2Cl2, 25 °C, 15 h, 88 %; (l) Ac2O (30 equiv), Et3N (60 equiv), 4–DMAP (2.0 equiv), CH2Cl2, 25 °C, 24 h, 100 %; (m) aq. HF:THF (1:3), 25 °C, 6 h, 90 %. KHMDS = potassium hexamethyldisilyazide, TEMPO = 2,2,6,6-teramethyl-1-piperidinyloxy free radical, Thexyl = thexylborane, LiDBB = Lithium di-tert-butylbiphenyl.
Scheme 4 summarizes the total synthesis of vannusal B structure 4. As it turned out, this synthesis was shorter and more efficient than that of vannusal B structure 3. Thus, remarkably and in stark contrast to the attempted cyclization of its diastereomeric counterpart (15a), the SmI2-mediated ring closure of precursor 15b proceeded smoothly and in high yield (82 %) to afford polycyclic compound 24 as a single diastereomer. Furthermore and much to our delight, the two newly formed stereogenic centers C10 and C28 possessed the correct stereochemistry relative to the adjacent quaternary centers, needing no configurational adjustment as previously required. Placement of a TES group on the C28 hydroxyl group of 24 (KHMDS, TESCl), followed by removal of the two BOM groups (LiDBB), led to diol 25, in 78 % overall yield for the two steps. Selective oxidation of the primary alcohol within the latter compound was best achieved through the use of PhI(OAc)2 in the presence of 1-Me-AZADO[5] as a catalyst, affording aldehyde 26 whose remaining hydroxyl group was acetylated (Ac2O, 4-DMAP) to give protected vannusal B structure 26 (87% yield for the two steps). Finally, the coveted structure 4 was generated from 26, in 85% yield, by exposure to aq. HF at ambient temperature.
Scheme 4.

Completion of the revised structure of vannusal B (4). Reagents and conditions: (a) SmI2 (0.1 M in THF, 10 equiv), HMPA (30 equiv), THF, −20→25 °C, 30 min, 82 %; (b) KHMDS (5.0 equiv), TESCl (10 equiv), Et3N (10 equiv), THF, −78→25 °C, 20 min, 94 %; (c) LiDBB (excess), THF, −78→−50°C, 30 min, 83 %; (d) PhI(OAc)2 (2.0 equiv), 1-Me-AZADO (0.2 equiv), CH2Cl2, 25 °C, 18 h; (e) Ac2O (10 equiv), Et3N (20 equiv), 4-DMAP (1.0 equiv), CH2Cl2, 25 °C, 18 h, 87 % for two steps; (f) aq. HF:THF (1:4→1:3), 25 °C, 3 h, 85 %; (g) NaBH4 (20 equiv), MeOH, 20 min, 90 %.
The 500 MHz 1H NMR spectrum of synthetic vannusal B structure 4 appeared excitingly close to the 600 MHz 1H NMR spectrum of natural vannusal B that we had in our possession,[6] a fact that piqued our enthusiasm for the soon to be completed structure 3 (see Scheme 3), which still commanded our main attention. Our arrival at vannusal B structure 3 (Scheme 3), however, was met with grief, for the 600 MHz 1H NMR spectrum of this compound did not match that (also 600 MHz) of the natural product. Thankfully, this time our disappointment was short lived, for upon obtaining a 600 MHz 1H NMR spectrum of synthetic structure 4 (Scheme 4), to which we immediately returned, we realized that it was identical in every detail to that (600 MHz) of natural vannusal B! Indeed, structure 4 represents the true structure, including absolute configuration, of vannusal B as proven by comparison of its 1H and 13C NMR and CD spectra with those of the natural product. Furthermore, the 1H and 13C NMR spectral data of the NaBH4 reduction product of synthetic vannusal B (27, Scheme 4) also matched those of its counterpart obtained from natural vannusals A and B,[6, 7] providing further support of structure 4 as the true structure of vannsual B. The structure of crystalline synthetic vannusal B (mp >200 °C decomposition, EtOAc/THF) was ultimately confirmed by X-ray crystallographic analysis[8] (see ORTEP drawing, Figure 3). The structural differences between the originally assigned and revised structures of vannusal B are located mainly in the “right” domain of the molecule (C10, C28, C13, C14, C26, C17, C18 and C21 stereocenters inverted), a circumstance that apparently arose from the difficulty in relating the C7 and C10 stereocenters in the original structural studies. This challenge could only be solved either by X-ray crystallographic analysis, or chemical synthesis. In the end it was done by both, the latter preceding the former, demonstrating the facilitating nature of total synthesis in structural elucidation even in this modern era.
Figure 3.
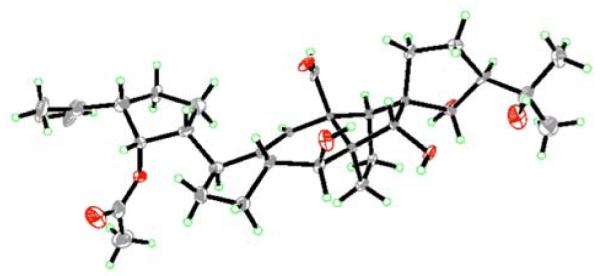
X-ray derived ORTEP drawing of vannusal B (4) (Thermal ellipsoids are shown at 30 % probability).
The described chemistry underscores once again the indispensable roles of total synthesis in the structural elucidation of scarce natural products and in rendering them in sufficient quantities for further investigations in those cases where their scarcity stymie such studies.
Supplementary Material
Footnotes
We thank Professor Graziano Guella for samples and NMR spectra of vannusals A and B, and for helpful discussions. We thank Drs. D. H. Huang, G. Siuzdak and R. Chadha for NMR spectroscopic, mass spectroscopic and X-ray crystallographic assistance, respectively. Financial support for this work was provided by the NSF (fellowship to A. Ortiz), the Skaggs Institute for Research, and a grant from the National Institutes of Health (USA).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from authors.
References
- [1].Nicolaou KC, Zhang H, Ortiz A. Angew. Chem. 2009 doi: 10.1002/anie.200902028. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009. submitted, preceding communication in this issue.
- [2].Nicolaou KC, Zhang H, Ortiz A, Dagneau P. Angew. Chem. 2008;120:8733–8738. doi: 10.1002/anie.200804228. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:8605–8610. doi: 10.1002/anie.200804228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]. CCDC-726955 contains the supplementary crystallographic data for 9d and is available free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- [4].Details of these studies will be reported in the full account of this work.
- [5].Shibuya M, Tomizawa M, Suzuki I, Iwabuchi Y. J. Am. Chem. Soc. 2006;128:8412–8413. doi: 10.1021/ja0620336. [DOI] [PubMed] [Google Scholar]
- [6].Guella G, Dini F, Pietra F. Angew. Chem. 1999;111:1217–1220. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1134::AID-ANIE1134>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 1999;38:1134–1136. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1134::AID-ANIE1134>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- [7].We thank Professor Graziano Guella for samples of vannusals A and B and 1H and 13C NMR spectra of vannusals A and B and their reduction derivative 27.
- [8]. CCDC-726954 contains the supplementary crystallographic data for vannusal B (4) and is available from The Cambridge Crystallographic Data Centre free of charge via www.ccdc.cam.ac.uk/data_request/cif.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.