

Abstract
The relative stabilities of two fluorous analogs, diisopropyl(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl)silyl and diisopropyl-(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadeca-fluoroundecyl)silyl [C8F17(CH2)nSi(i-Pr)2, where n = 2 or 3], of the standard triisopropylsilyl (TIPS) group are compared in the setting of alcohol protection. The fluorous silyl groups can be installed under standard conditions in comparable yields to the TIPS group, but the derived fluorous silyl ethers are more labile than TIPS ethers towards cleavage by both acids and fluoride.
Keywords: fluorous chemistry, protecting groups, relative reactivity, triisopropylsilyl group
Fluorous protecting groups[1] have been extensively used recently in diversity-oriented synthesis, natural products synthesis and fluorous mixture synthesis. Such groups typically serve a dual role of protecting a given functional group and enabling a fluorous separation. In diversity-oriented synthesis[2] and natural products synthesis,[3] the separation method is usually a fluorous solid-phase extraction,[4] which separates fluorous components from non-fluorous ones. In fluorous mixture synthesis,[5] the method is a fluorous HPLC,[6] which resolves differentially tagged fluorous components of a mixture into individual compounds.
Fluorous protecting groups are usually fashioned after parent (standard) protecting groups, and available protecting groups for alcohols include fluorous analogs of tetrahydropyranyl (THP),[7] p-methoxyben-zyl (PMB),[8] and methoxymethyl (MOM) groups,[9] among others. Assorted fluorous silyl groups are also available,[10] and we have often used a fluorous analog of the parent triisopropylsilyl (TIPS) group that we usually call FTIPS (Figure 1).[11]
Figure 1.

Comparision of fluorous TIPS (FTIPS) and fluorous PMB (FPMB) groups with their organic parents.
An understanding of the relative reactivity and stability of fluorous protecting groups is essential for synthetic planning. Fluorous groups that closely resemble the structure of their parents and that have suitable spacers can be expected to have comparable reactivities to the parent groups.[12] For example, the fluorous PMB (FPMB) group in Figure 1 is structurally very similar to the parent PMB group, and the protected functionality is relatively well insulated from the perfluoroalkyl group (Rf) by a propylene spacer.
In contrast, the parent TIPS group may not be such good model for the FTIPS group for two reasons: 1) the FTIPS group is not an analog of a triisopropylsilyl group but instead a 1°-alkyl(diisopropylsilyl) group, and 2) the ethylene spacer may not be a sufficient insulator. Thus, the FTIPS group may differ in reactivity and stability from TIPS for both steric and electronic reasons. We report here observations showing that two fluorous TIPS groups differ in reactivity from each other and from the parent TIPS group. These observations will help in selection of suitable fluorous protecting groups during synthetic planning.
We first observed differences between TIPS and FTIPS groups during a fluorous mixture synthesis of a natural product called SCH 725674.[13,14] A key step in the pilot (non-fluorous) synthesis of SCH 725674 was selective desilylation of tris-silyl ether 1 bearing one tert-butyldimethylsilyl (TBS) group on the primary alcohol and two TIPS groups on secondary alcohols. Careful treatment of 1 with HCl in MeOH at −5 °C for 1 h[15] removed the TBS group to provide primary alcohol 2 in 87% yield with the benzyl group and the two TIPS groups intact.
A very different result was obtained when an analogous reaction was tried in the fluorous mixture synthesis. Here the precursor was a mixture of four quasi-isomers[16] M-3 with configurations at C3 and C5 coded to the combination of standard and fluorous TIPS groups as indicated. One of the quasi-isomers has the same configurations as 1, so the differing reactivities cannot primarily come from stereochemical differences.
Treatment of M-3 with HCl and MeOH did not give a clean product as above, but instead gave rise to a rather complex mixture. Two spots predominated on TLC analysis and these were isolated by flash chromatography. Both samples lacked the TBS group, but neither contained the target products. The less polar spot, 34% yield, was identified as a mixture of two quasi-isomers M-4 bearing the standard TIPS group on the 2°-alcohol and the fluorous TIPS group on the 1°-alcohol. These products originate from the two starting quasi-isomers with the standard TIPS group on O5, but they are not the target products because the FTIPS group on O2 migrated to the primary alcohol O1. The more polar spot was a mixture of two products M-5, 36% yield, derived from the other two starting quasi-isomers with the FTIPS group on O5. In addition to migration of the one FTIPS group from O2 to Ol, the other FTIPS group was lost entirely. This allowed the partial separation of four products into two fractions since two of the compounds are mono-ols (M-4, less polar) and two are diols (M-5, more polar).
The combined mass balance of the two products only accounted for about two-thirds of the starting material and there were other unidentified products according to TLC analysis. Nonetheless, the results show clear differences between the stable TIPS groups in 1 and the FTIPS groups in M-3 which either migrated or fell off.
We also encountered untoward liability of the FTIPS group in a recently completed fluorous mixture synthesis of petrocortyne A.[17] In the pilot synthesis of an individual compound, dialkynol (rac)-6a was treated with 2 equiv. of BuLi to generate dianion [(rac)-6b in situ], then iodide (S)-7 was added (Scheme 2). Standard workup and chromatography provided coupled product 8 in 30% yield. Substantial starting material remained, and 8 was the only new product of the reaction.
Scheme 2.

Differences between TIPS and FTIPS groups – petrocortyne setting.
In the fluorous mixture version, the quasi-racemate M-9 with configurations encoded by the FTIPS groups was added to the same dianion (S)-6b. Again, a single new spot appeared on TLC analysis. However, this time the chromographically isolated product (30% yield) was not the pure quasi-racemate M-10. Instead, this was contaminated by a substantial amount (about 50%) of a second component 11 resulting from transfer of the FTIPS groups of 9 to the terminal acetylide of 6. These compounds were not separable, but the structure of 11 was secured by MS and NMR analysis of the mixture. In particular, the 1H NMR spectrum of the mixture exhibited no terminal acetylide proton resonance, so the FTIPS group must be there and not on the alcohol.
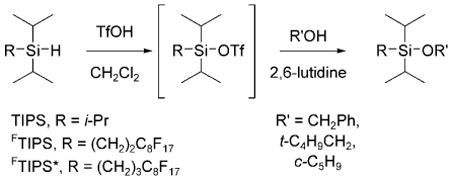
These results prompted us to conduct a short study to more carefully compare the standard TIPS and the first generation FTIPS groups. We also included a new second-generation FTIPS* group with a propylene spacer, Rf(CH2)3Si(i-Pr)2, to help sort out spacer effects from steric effects. The FTIPS* group should be more similar to an n-alkyldiisopropylsilane than the first generation FTIPS group with the ethylene spacer. The synthesis of the corresponding silane precursor, C8F17(CH2)3Si(i-Pr)2H, of the silyl triflate was straight-forward and is detailed below (see Experimental Section).
In a first stage, Table 1, we compared the performance of the three silyl triflates under standard silylation conditions. The goal here was to learn whether they gave comparable yields under comparable conditions and whether the derived products were stable to standard work-up and chromatography. In a standard procedure (see below), the three silyl triflates (TIPS-OTf, FTIPSOTf, FTIPS*OTf) were generated by addition of triflic acid to the corresponding silanes in CH2Cl2 at 0°C. 2,6-Lutidine was added, the solutions were divided into three, and one of the three alcohols (benzyl alcohol, neopentyl alcohol, cyclopentanol) was added to each one. After 15 min at 0 °C, the mixtures were warmed to ambient temperature and stirred for 2 h prior to standard aqueous workup.
Table 1.
Isolated yields of silyl ethers under the standard silylation conditions.
 | |||
|---|---|---|---|
| R-OH | R′OTIPS | R′OFTIPS | R′OFTIPS* |
| C6H5CH2 | 80% | 76% | 76% |
| t-C4H9CH2 | 88% | 79% | 80% |
| c-C5H9 | 76% | 80% | 74% |
Each of the nine crude products was purified by flash chromatography, and their isolated yields are summarized in Table 1. The yields were comparable (74–88%) and there was no evidence of product instability during workup or chromatography. These are not competitive experiments and provide no information of the relative reactivity of the three triflates; however, we do learn that all three triflates can be installed under the same conditions in comparable yields.
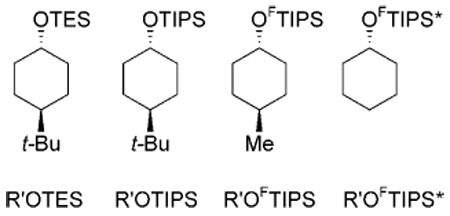
Next, we conducted a series of simple competitive experiments to assess the relative ease of cleavage of the two FTIPS groups compared to either standard TIPS or triethylsilyl (TES). We first prepared the four silyl ethers shown in Table 2 by the same procedure as above and with comparable isolated yields. In the competitive experiments, an equimolar mixture of two or three of the silyl ethers was treated with a standard desilylating reagent. The progress of each reaction was followed by GC as a percentage decrease of the starting amount of silyl ethers, and these values are listed in Table 2 as percent conversion (% conv.) relative to an internal standard (decane). The corresponding alcohols were the only products of both competitive and control experiments, so the difference between the product yield should be [100% — (% conv.)] for each entry. In the case of low conversions (< 10%), the percent conversion was instead estimated from the GC yield of the product. A conversion of 0% means that the alcohol product was not detected by GC.
Table 2.
Percent conversions in competitive experiments with equimolar amounts of two or three TES, TIPS, FTIPS or FTIPS* silyl ethers.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Rgt | t [h] | TES | TIPS | FTIPS | FTIPS* |
| 1 | PPTS[b] | 1 | 91% | NA[a] | 0% | 0% |
| 2 | AcOH[c] | 0.5 | 100% | NA | 0% | 0% |
| 3 | F6SiH2[d] | 0.5 | NA | 0% | 21% | 81% |
| 4 | FeCl3[e] | 1 | NA | NA | 35% | 100% |
| 5 | TBAF[f] | 1 | NA | 0% | 100% | 55% |
| 6 | TBAF[g] | 0.5 | NA | NA | 100% | 3% |
| 7 | TBAF[h] | 1 | 0% | NA | 100% | 27% |
NA is not added.
1 equiv, THF, room temperature.
THF/H2O, 0°C.
0.6 equiv., THF/MeCN/H2O, 0°C.
EtOH, room temperature.
2 equiv., THF, room temperature.
0.7 equiv., THF, 0°C.
1 equiv., THF, room temperature.
Table 2 summarizes the results of several of the more informative competitive experiments. Competitions under acidic conditions are summarized in entries 1–4. The TIPS group and both FTIPS groups are stable for at least 3 h under typical PPTS conditions used to cleave TES groups. The TES group is completely cleaved under these conditions after 3 h (not shown) and is 91% cleaved after 1 h. Likewise, exposure of the TES, FTIPS and FTIPS* silyl ethers to acetic acid in THF/water for 30 min cleaved the TES group but returned the two FTIPS ethers. The TIPS ether is also stable to these conditions (not shown). Treatment of the three component, FTIPS and FTIPS* mixture with hexafluorosilicic acid (SiF6H2) left the TIPS ether unscathed while cleaving most of the FTIPS* ether (81%) and some of the FTIPS ether (21%). Differences in reactivity between the two FTIPS ethers were also observed on treatment with FeCl3 in ethanol. After 1 h at room temperature, the conversion of FTIPS* was 100% while that of FTIPS was only 35%.
Competitive experiments with commercial tetra-butylammonium fluoride (TBAF) in THF are summarized in entries 5–7. Treatment of the TIPS silyl ether and the two FTIPS ethers with 2 equiv. of TBAF returned the TIPS ether unreactive while cleaving all of the FTIPS ether and 55% of FTIPS*. In a more carefully monitored reaction, treatment of the two FTIPS ethers with a limited amount (0.7 equiv.) of TBAF at 0 °C for 30 min gave complete conversion of FTIPS with only 3% conversion of FTIPS*. Finally, in a similar experiment but with the TES ether and 1 equiv. of TBAF, the TES ether was stable, the FTIPS ether was fully cleaved, and the FTIPS* ether was 27% cleaved.
These results provide helpful information about the relative reactivity of the two FTIPS groups compared to each other and to standard silyl groups. Both the FTIPS groups are considerably more labile than TIPS to TBAF. FTIPS is more labile than FTIPS* and is even more labile that TES. It should certainly be possible to cleave FTIPS in the presence of TIPS (as seen in Scheme 1), and it may even be generally possible to cleave FTIPS in the presence of FTIPS*. As implied by the observations of migration or cleavage during the two syntheses (Scheme 1, Scheme 2), FTIPS has to be used with some care under nucleophilic conditions since its reduced steric hindrance (relative to TIPS) and increased electron deficiency on Si (relative to both TIPS and TES) leave it susceptible to nucleophilic attack. That said, we have used this group in many typical synthetic transformations without problems.[11]
Scheme 1.

Differences between TIPS and FTIPS groups – SCH 725674 setting.
Under acidic conditions, TIPS is again the most stable of the four groups, as expected. Both FTIPS and FTIPS* are considerably more stable than TES, presumably for steric reasons. However, the inductive effect of the perfluoroalkyl group of FTIPS with the ethylene spacer now provides stability over the FTIPS* with the propylene spacer. The later is presumably more easily protonated and hence more labile.
Experimental Section
Preparation of Diisopropyl(4,4,5,5,6,6,7,7,8,8,9,9,10,10, 11,11,11-heptadecafluoroundecyl)silane
A solution of t-BuLi in pentane (1.7M, 100 mL, 170 mmol) was added slowly to a solution of perfluorooctylpropyl iodide (40 g, 68 mmol) in dry Et2O (510 mL) at −78 °C. The internal temperature was maintained below −50 °C throughout. After the addition was completed, the reaction mixture was stirred 30 min at −60 °C, then warmed to −20 °C until the solution became clear. Finally, the mixture was cooled to −78 °C and chlorodiisopropylsilane (12.8 mL, 75 mmol) was added. The reaction mixture was warmed to room temperature and stirred for additional 5 h, then quenched slowly and carefully with water. The aqueous layer was extracted 3 times with Et2O. The combined organic layers were dried over MgSO4, filtered and concentrated under vacuum. The crude product was purified by distillation to afford diisopro-pyl(perfluoroctylpropyl)silane as a colorless oil; yield: 29.4 g (75%), bp 95–100°C/0.1 mbar; 1H NMR (300 MHz, CDCl3): δ = 0.65–0.72 (m, 2H), 1.03 (br, 14H), 1.65–1.76 (m, 2H), 2.02–2.20 (m, 2H), 3.46 (br, 1H); 19F NMR (282.4 MHz, CDCl3): δ = 81.58 (t, J=9.9, 2F), 114.97–115.08 (m, 2F), 122.46 (br, 6F), 123.38 (br, 2F), 124.27 (br, 2F), 126.81 (br, 2F); 13CNMR (75.5 MHz, CDCl3): δ = 8.4, 10.5, 16.4, 18.6, 18.9, 34.5 (t, J=22.1 Hz).
General Procedure for Silylation
To 1 mmol of the corresponding fluorous silane at 0°C was added 1.2 mmol of triflic acid. The mixture was stirred 1 h at 0°C and then warmed to room temperature and stirred 15 h. The resulting triflates were used directly in the protection reaction.
To a 0.1 M solution of the corresponding alcohol (1.1 equiv.) in dichloromethane (DCM) at 0°C were added 2 equiv. of 2,6-lutidine and 1.0 equiv. of the corresponding triflate (0.5 M solution in DCM). The reaction mixture was stirred at 0 °C for 15 min and then warmed to room temperature. After 2 h, the reaction was quenched with NaHCO3 (aqueous solution). The aqueous layer was extracted several times with Et2O, and the organic layers were combined, dried over MgSO4, filtered and concentrated under vacuum. Finally, the protected alcohols were purified by flash column chromatography with hexane.
Supplementary Material
Acknowledgements
We thank the National Institute of General Medical Sciences of the National Institutes of Health for funding this work. A.G.S. thanks the CIPF for a predoctoral fellowship.
Dedicated to Prof. Dr. Armin de Meijere in celebration of his 70th birthday.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.200900061.
References
- 1.Zhang W. In: The Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horvath IT, editors. Weinheim: Wiley-VCH; 2004. pp. 222–235. [Google Scholar]
- 2.a) Dolle RE, Le Bourdonnec B, Goodman AJ, Morales GA, Thomas CJ, Zhang W. J. Comb. Chem. 2008;10:753–802. doi: 10.1021/cc800119z. [DOI] [PubMed] [Google Scholar]; b) Curran DP. Aldrichimica Acta. 2006;39:3–9. [Google Scholar]
- 3.Studer A, Hadida S, Ferritto R, Kim S-Y, Jeger P, Wipf P, Curran DP. Science. 1997;275:823–826. doi: 10.1126/science.275.5301.823. [DOI] [PubMed] [Google Scholar]
- 4.Zhang W, Curran DP. Tetrahedron. 2006;62:11837–11865. doi: 10.1016/j.tet.2006.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Luo ZY, Zhang QS, Oderaotoshi Y, Curran DP. Science. 2001;291:1766–1769. doi: 10.1126/science.1057567. [DOI] [PubMed] [Google Scholar]; b) Curran DP, Zhang QS, Richard C, Lu HJ, Gudipati V, Wilcox CS. J. Am. Chem. Soc. 2006;128:9561–9573. doi: 10.1021/ja061801q. [DOI] [PubMed] [Google Scholar]
- 6.a) Zhang W. J. Fluorine Chem. 2008;129:910–919. [Google Scholar]; b) Curran DP. In: The Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horvath IT, editors. Weinheim: Wiley-VCH; 2004. pp. 101–127. [Google Scholar]; c) reverse fluorous FSE. Matsugi M, Curran DP. Org. Lett. 2004;6:2717–2720. doi: 10.1021/ol049040o. [DOI] [PubMed] [Google Scholar]
- 7.a) Wipf P, Reeves JT. Tetrahedron Lett. 1999;40:4649–4652. [Google Scholar]; b) Wipf P, Reeves JT. Tetrahedron Lett. 1999;40:5139–5142. [Google Scholar]
- 8.Curran DP, Furukawa T. Org. Lett. 2002;4:2233–2235. doi: 10.1021/ol026084t. [DOI] [PubMed] [Google Scholar]
- 9.Curran DP, Ogoe C. QSAR Comb. Sci. 2006;25:732–735. [Google Scholar]
- 10.a) Studer A, Curran DP. Tetrahedron. 1997;53:6681–6696. [Google Scholar]; Studer A, Jeger P, Wipf P, Curran DP. J. Org. Chem. 1997;62:2917–2924. doi: 10.1021/jo970095w. [DOI] [PubMed] [Google Scholar]; b) Rover S, Wipf P. Tetrahedron Lett. 1999;40:5667–5670. [Google Scholar]; Manzoni L. Chem. Commun. 2003:2930–2931. doi: 10.1039/b311448a. [DOI] [PubMed] [Google Scholar]; c) Manzoni L, Castelli R. Org. Lett. 2004;6:4195–4198. doi: 10.1021/ol048474g. [DOI] [PubMed] [Google Scholar]; d) Tripathi S, Misra K, Sanghvi YS. Org. Prep. Proced. Int. 2005;37:257–263. [Google Scholar]; e) Fustero S, Sancho AG, Chiva G, Sanz-Cervera JF, del Pozo C, Acena JL. J. Org. Chem. 2006;71:3299–3302. doi: 10.1021/jo052569u. [DOI] [PubMed] [Google Scholar]
- 11.a) Lu Y, Zhang W. e-EROS (Encycl. Reagents Org. Synth.) reagent number rn00424; fluorous TIPS triflate. [Google Scholar]; b) Zhang W, Luo Z, Chen CHT, Curran DP. J. Am. Chem. Soc. 2002;124:10443–10450. doi: 10.1021/ja026947d. [DOI] [PubMed] [Google Scholar]; c) Zhang QS, Rivkin A, Curran DP. J. Am. Chem. Soc. 2002;124:5774–5781. doi: 10.1021/ja025606x. [DOI] [PubMed] [Google Scholar]; d) Yang F, Newsome JJ, Curran DP. J. Am. Chem. Soc. 2006;128:14200–14205. doi: 10.1021/ja064812s. [DOI] [PubMed] [Google Scholar]; e) Jung W-H, Guyenne S, Riesco-Fagundo C, Mancuso J, Nakamura S, Curran DP. Angew. Chem. 2008;120:1146–1149. doi: 10.1002/anie.200704893. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:1130–1133. doi: 10.1002/anie.200704893. [DOI] [PubMed] [Google Scholar]
- 12.a) Gladysz JA. In: The Handbook of Fluorous Chemistry. Gladysz JA, Curran DP, Horvath IT, editors. Weinheim: Wiley-VCH; 2004. pp. 41–55. [Google Scholar]; b) Curran DP, Wang X, Zhang QS. J. Org. Chem. 2005;70:3716–3719. doi: 10.1021/jo050116j. [DOI] [PubMed] [Google Scholar]
- 13.a) Yang S-W, Chan T-Z, Terracciano J, Loebenberg D, Patel M, Chu M. J. Antibiot. 2005;58:535–538. doi: 10.1038/ja.2005.74. [DOI] [PubMed] [Google Scholar]; b) Wang X. PhD Thesis. University of Pittsburgh: 2009. [Google Scholar]
- 14.a) The fluorous silane precursors to make these compounds are commercially available from Fluorous Technologies, Inc. www.fluorous.com; b) DPC owns an equity interest in this company.
- 15.Khan AT, Mondal E. Synlett. 2003:694–696. [Google Scholar]
- 16.Zhang QS, Curran DP. Chem. Eur. J. 2005;11:4866–4880. doi: 10.1002/chem.200500076. [DOI] [PubMed] [Google Scholar]
- 17.a) Kim JS, Lim YJ, Im KS, Jung JH, Shim CJ, Lee CO, Hong J, Lee H. J. Nat. Prod. 1999;62:554–559. doi: 10.1021/np9803427. [DOI] [PubMed] [Google Scholar]; b) Seo Y, Cho KW, Rho J-R, Shin J, Sim CJ. Tetrahedron. 1998;54:447–462. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.