

Abstract
Protein phosphatase Sit4 is required for growth inhibition of Saccharomyces cerevisiae by the antifungals rapamycin and zymocin. Here, we show that the rapamycin effector Tap42, which interacts with Sit4, is dispensable for zymocin action. Although Tap42 binding-deficient sit4 mutants are resistant to zymocin, these mutations also block interaction between Sit4 and the Sit4-associating proteins Sap185 and Sap190, previously shown to mediate zymocin toxicity. Among the four different SAP genes, we found that SAP190 deletions specifically induce rapamycin resistance but that this phenotype is reversed in the additional absence of SAP155. Similarly, the rapamycin resistance of an rrd1Δ mutant lacking the Sit4 interactor Rrd1 specifically requires the Sit4/Sap190 complex. Thus, Sit4/Sap190 and Sit4/Sap155 holophosphatases apparently play opposing roles following rapamycin treatment, although rapamycin inhibition is operational in the absence of all Sap family members or Sit4. We further identified a Sit4-interacting region on Sap185 in sap190Δ cells that mediates Sit4/Sap185 complex formation and is essential for dephosphorylation of Elp1, a subunit of the Elongator complex. This suggests that Sit4/Sap185 and Sit4/Sap190 holophosphatases promote Elongator functions, a notion supported by data showing that their inactivation eliminates Elongator-dependent processes, including tRNA suppression by SUP4 and tRNA cleavage by zymocin.
The antifungals rapamycin and zymocin each inhibit growth of Saccharomyces cerevisiae, leading to accumulation of unbudded yeast cells (8, 58). Although their targets are distinct (11, 23), key events triggered by both antifungals involve Sit4, a multifunctional and growth-relevant type 2A-related protein phosphatase (PP2A) (1, 13, 14, 25, 27, 44, 50). Rapamycin inhibits the target of rapamycin (TOR) kinases, compromising mRNA translation and cell cycle progression (3, 10, 11, 44, 58). During drug-induced TOR inactivation, Tap42-associated phosphatases composed of the PP2A family members Sit4 and Pph21/Phh22 are released from TOR complex 1, enabling dephosphorylation of TOR effectors involved in nutrient availability (Npr1), translation (Gcn2), transcription (Gln3), and TOR regulation itself (Tap42 and Tip41) (6, 10, 29, 32, 45, 55).
When TOR is inhibited by rapamycin, Tap42 is bound by Tip41, promoting Sit4/Tap42 dissociation. Tap42 dissociation may relieve inhibition of Sit4 so as to promote dephosphorylation of downstream targets (13, 29), and in line with this model, Tip41 removal induces rapamycin resistance (29). However, other work has proposed that, following release from TOR complex 1, the Sit4/Tap42 complex is active and that Sit4/Tap42 dissociation is a later event (55). Tap42 binds Sit4 and Pph21/Pph22 independently of their respective interactors, Sap4, Sap155, Sap185, Sap190 or Tpd3, Cdc55, and Rts1 (13, 31, 32, 38). Based on the drug sensitivity of PP2A/Tap42 complexes and data showing that high-copy-number TAP42 enhances the rapamycin resistance of tpd3 and cdc55 mutants (13, 32), TOR signaling appears to involve regulated PP2A/Tap42 association, presumably through changes in the phosphorylation state of Tap42 or Tip41 that are likely to depend on the Sit4 and Pph21/Pph22 phosphatases (29, 32). Intriguingly, SIT4, SAP, PPH21/PPH22, and TAP42 all show genetic interaction with the polymorphic SSD1 (suppressor of SIT4 deletion) locus, whose SSD1-v alleles rescue nonviability or growth defects of sit4Δ, sap4Δ sap155Δ sap185 sap190Δ, pph21Δ pph22Δ, and tap42ts mutants (13, 15, 43, 48, 50).
Though superficially similar to the effect of rapamycin, the G1 arrest triggered by the tRNase toxin zymocin from Kluyveromyces lactis involves cleavage and depletion of tRNAs (23, 28, 37). A key effector role for zymocin toxicity has been assigned to the Elongator complex (18, 24, 40, 53), whose function in the modification of tRNAs (16, 21) is required for anticodon cleavage by zymocin (22, 28, 37). As a result, tRNA modification defects of Elongator mutants protect against the tRNase attack of zymocin (2, 17, 18, 22, 28, 56). Intriguingly, Sit4 inactivation also protects against zymocin and causes tRNA modification defects that are typical of Elongator mutants (21, 22, 25, 27). Moreover, sit4Δ mutants accumulate hyperphosphorylated forms of the Elongator subunit Elp1, demonstrating a function, direct or indirect, of Sit4 in Elp1 dephosphorylation (25, 27). In addition, this Sit4 role requires Sap185 and Sap190, two members of the Sit4-associating protein family (25, 27, 38). Hence, a sap185Δ sap190Δ double mutant displays zymocin resistance, tRNA modification defects, and Elp1 hyperphosphorylation, traits typical of a sit4Δ mutant, while deletions in SAP4 and SAP155 have no such effects (22, 25, 27). However, SAP155 has been shown to be a dosage suppressor of zymocin action and Elp1 dephosphorylation, and these multicopy SAP155 effects are efficiently countered by overexpression of SAP185 or SAP190 to restore Elp1 dephosphorylation and zymocin toxicity (25, 27). This stresses the original proposal that there is competition for Sit4 binding among Sap family members (38) and reinforces the idea that Sap185 and Sap190 specifically mediate Sit4-dependent Elp1 dephosphorylation and zymocin inhibition (25, 27).
Here, we investigate the possibility of Sit4-mediated cross talk between the pathways required for rapamycin and zymocin to inhibit yeast cells. By examining a range of mutations affecting PP2A and Sit4 functions, we found that such mutations mostly have opposite effects on the two antifungals. In particular, and in contrast to the TOR pathway, we found no evidence that the Sit4/Tap42 complex is involved in zymocin action. However, Sit4 complexes involving specific Sap members operate on antifungal sensitivity so that different subsets of Sit4/Sap complexes selectively mediate the response to rapamycin or zymocin. Although sit4 mutants with Tap42 binding defects are zymocin resistant, we show here that they also block the formation of Sit4/Sap complexes required for zymocin inhibition. Thus, the zymocin resistance of such mutants reflects their effects on the binding of multiple Sit4 partners rather than cross talk between the two responses. Finally, we define a Sap185 region required for interaction with Sit4, Sit4/Sap185-dependent dephosphorylation of Elongator subunit Elp1, and nonsense readthrough by an Elongator-dependent tRNA suppressor (SUP4).
MATERIALS AND METHODS
Yeast strains, media, and K. lactis zymocin methods.
The yeast strains and plasmids used for this project are listed in Table 1 and Table 2. Routine yeast growth was in yeast extract, peptone, and dextrose (YPD) or galactose rich or synthetic complete medium (47). For TOR downregulation by poor nutrient supply, we followed a previous protocol (13) using glycerol and ethanol at 2% (vol/vol) each. Testing the effect of rapamycin (Calbiochem) involved the addition (25 to 150 nM) of the antibiotic to YPD plates and growth for 3 days at 23 to 30°C. Assessing zymocin responses of S. cerevisiae strains involved killer eclipse assays as described previously (33) using the K. lactis zymocin producer strain AWJ137 or plate assays with YPD medium containing partially purified zymocin from AWJ137 cell-free filtrates (26). Tenfold serial dilutions of the S. cerevisiae strains were spotted on zymocin-free plates and plates containing 40 to 65% (vol/vol) zymocin (26). Growth was for 3 days at 30°C. To test the effect of TOR downregulation on zymocin action, S. cerevisiae strains were subjected to liquid killer assays as described previously (9). Analysis of gene dosage effects on both antifungals involved transformation (19) with centromeric or multicopy Escherichia coli/yeast shuttle vectors (Table 2). Elongator-dependent tRNA suppression of ade2-1 and can1-100 ocher mutations by SUP4 used plasmid pTC3 (46) (Table 2) and previously described assays (21, 28). Studying the effects of single-, double-, and triple-substitution mutations of the Tap42 binding site of Sit4 on antifungal responses and Elp1 phosphorylation states involved previously described SIT4 alleles (52) carried on single-copy vectors (Table 2) kindly donated by Y. Jiang (University of Pittsburgh, Pittsburgh, PA).
TABLE 1.
Yeast strains used in this study
| Strain | Description | Source or reference |
|---|---|---|
| K. lactis | ||
| AWJ137 | α leu2 trp1 [k1+ k2+] (killer and zymocin producer) | 18 |
| S. cerevisiae | ||
| BY4741 | MATahis3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Euroscarf |
| Y06866 | BY4741 but tpd3Δ::kanMX4 | Euroscarf |
| Y01790 | BY4741 but rts1Δ::kanMX4 | Euroscarf |
| rrd1Δ/rrd2Δ | BY4741 but rrd1Δ::HIS3 rrd2Δ::kanMX4 | E. Ogris |
| GAL1-RRD1rrd2Δ | BY4741 but pGAL1-RRD1::HIS3MX6 rrd2Δ::kanMX4 | E. Ogris |
| GAL1-SIT4 | BY4741 but pGAL1-SIT4::HIS3MX6 | E. Ogris |
| KLY101 | BY4741 but ppm1Δ::kanMX4 | S. Clarke |
| KLY102 | BY4741 but ppm2Δ::kanMX4 | S. Clarke |
| YCY1001 | BY4741 but ppm1Δ::kanMX4 ppm2Δ::kanMX4 met15Δ0 | S. Clarke |
| W303-1A | MATaura3-1 leu2-3,112 his3-11,15 trp1-1 ade2-1 can1-100 | Laboratory stock |
| DEY132-1C | W303-1A but pph21::HIS3 MATα | M. Stark |
| DEY132-2C | W303-1A but pph21::URA3 pph22::TRP1 MATα + pPL091 [SSD1-v] | M. Stark |
| JK9-3a | MATaleu2-3,112 ura3-52 trp1 his4 rme1 HMLa | M. Hall |
| TB50a | JK9-3a but his3 instead of his4 | M. Hall |
| JH11-1c | JK9-3a TOR1-1 | M. Hall |
| JH12-17a | JK9-3a TOR2-1 | M. Hall |
| MH284-5a | JK9-3a TOR1-1 TOR2-1 | M. Hall |
| EJ91-1d | TB50a but tip41::kanMX4 | E. Jacinto |
| DJY190 | TB50a but sap190Δ::KlLEU2 | This work |
| DJY41190TB | TB50a but tip41::kanMX4 sap190Δ::KlLEU2 | This work |
| Y3034 | W303-1A but tap42::HIS3 [pRS314(TRP1)-tap42-106] ssd1-d | 15 |
| Y3035 | W303-1A but tap42::HIS3 [pRS314(TRP1)-tap42-109] ssd1-d | 15 |
| TS54-5a | TB50a but tap42::kanMX4 [YCp111(LEU2)-tap42-11] ssd1-d | M. Hall |
| TS54-5b | TS54-5a but SSD1-v | This work |
| LFY3 | W303-1A but elp1Δ::TRP1 | 27 |
| LFY5 | W303-1A but elp3Δ::TRP1 | 25 |
| LFY6 | W303-1A but kti12Δ::TRP1 | 25 |
| 5DWW | W303-1A but elp5Δ::TRP1 | This work |
| 7DWW | W303-1A but elp4Δ::TRP1 | This work |
| CY4029 | W303-1A but SSD1-v1 | 38 |
| CY3938 | CY4029 but sit4Δ::HIS3 | 38 |
| DJY8 | CY4029 but sap4::LEU2 MATα | 25 |
| DJY9 | CY4029 but sap155::HIS3 MATα | 25 |
| CY4917 | CY4029 but sap185::ADE2 | 38 |
| CY4380 | CY4029 but sap190::TRP1 | 38 |
| CY5220 | CY4029 but sap4::LEU2 sap155::HIS3 MATα | 38 |
| DJY10 | CY4029 but sap4::LEU2 sap185::ADE2 MATα | 25 |
| DJY11 | CY4029 but sap4::LEU2 sap190::TRP1 MATα | 25 |
| DJY12 | CY4029 but sap155::HIS3 sap185::ADE2 | 25 |
| CY5224 | CY4029 but sap185::ADE2 sap190::TRP1 | 38 |
| DJY13 | CY4029 but sap155::HIS3 sap190::TRP1 MATα | 25 |
| DJY14 | CY4029 but sap155::HIS3 sap185::ADE2 sap190::TRP1 MATα | 25 |
| DJY15 | CY4029 but sap4::LEU2 sap185::ADE2 sap190::TRP1 MATα | 25 |
| DJY16 | CY4029 but sap4::LEU2 sap155::HIS3 sap190::TRP1 MATα | 25 |
| DJY17 | CY4029 but sap4::LEU2 sap155::HIS3 sap185::ADE2 MATα | 25 |
| CY5236 | CY4029 but sap4::LEU2 sap155::HIS3 sap185::ADE2 sap190::TRP1 | 38 |
| DJY103 | CY4029 but sit4Δ::HIS3 ELP1-HA::KlTRP1 | 27 |
| JETY01 | CY4029 but kan3MX6::pGAL1-HA-SIT4 | This work |
| JETY02 | JETY01 but sap190::SpHIS5 | This work |
| JETY04 | JETY02 but SAP185-c-myc::hphNTI | This work |
| JETY05 | JETY02 but C1 truncation of SAP185-c-myc::hphNTI | This work |
| JETY06 | JETY02 but C2 truncation of SAP185-c-myc::hphNTI | This work |
| JETY07 | JETY02 but C3 truncation of SAP185-c-myc::hphNTI | This work |
| JETY08 | JETY02 but C4 truncation of SAP185-c-myc::hphNTI | This work |
| JETY09 | CY4380 but kan3MX6::pGAL1-HA-SAP185 | This work |
| JETY10 | CY4380 but kan3MX6::pGAL1-HA-SAP185 (truncation N1) | This work |
| JETY11 | CY4380 but kan3MX6::pGAL1-HA-SAP185 (truncation N2) | This work |
| JETY12 | CY4380 but kan3MX6::pGAL1-HA-SAP185 (truncation N3) | This work |
TABLE 2.
Plasmids used in this study
| Plasmid | Description | Source or reference |
|---|---|---|
| pJHW27 | YEplac181 (2μ LEU2) carrying KTI12 | 9 |
| YEpSIT4 | YEplac112 (2μ TRP1) carrying SIT4 | This work |
| psit4Δ | YEpSIT4 carrying sit4::LEU2 disruption | This work |
| pJET2 | YCplac111 (CEN LEU2) carrying ELP1-HA | This work |
| YEpSIT4-HA | YEp (2μ URA3) carrying SIT4-HA | G. Sprague |
| pTC3 | YCp (TRP1) carrying SUP4 | 46 |
| pCB243 | YCp (LEU2) carrying SIT4-HA | 50 |
| pEJ120 | YEplac181 (2μ LEU2) carrying TIP41-HA | E. Jacinto |
| pWAB3 | pGAL-TIP41 (CEN LEU2) carrying GAL1-TIP41 promoter fusion | E. Jacinto |
| p-MycTap42 | YCplac22 (CEN TRP1) carrying c-myc-TAP42 | M. Stark |
| CB2925 | YEp24 (2μ URA3) carrying SAP4 | 38 |
| CB2643 | YEp24 (2μ URA3) carrying SAP155 | 38 |
| CB2819 | YEp24 (2μ URA3) carrying SAP185 | 38 |
| CB2606 | YEp24 (2μ URA3) carrying SAP190 | 38 |
| p42:HA | YCp (CEN LEU2) carrying TAP42-HA | 13 |
| p155:HA | YCp (CEN URA3) carrying SAP155-HA | 38 |
| p185:HA | YCp (CEN URA3) carrying SAP185-HA | 38 |
| p190:HA | YCp (CEN URA3) carrying SAP190-HA | 38 |
| pPL091/092 | pRS315/316 (CEN LEU2/URA3) carrying SSD1-v (JK9-3da allele) | 43 |
| p775 | pRS425 (2μ LEU2) carrying TAP42 | 52 |
| p655 | pRS314 (CEN TRP1) carrying SIT4 | 52 |
| p678 | pRS314 (CEN TRP1) carrying SIT4 mutation E38A | 52 |
| p711 | pRS314 (CEN TRP1) carrying SIT4 mutations E37A E38A | 52 |
| p712 | pRS314 (CEN TRP1) carrying SIT4 mutations L35A E37A E38A | 52 |
Epitope tagging, gene disruptions, and truncations.
Elongator genes were deleted using previously described PCR protocols (18, 24). kti12Δ null mutants were obtained by using the kti12Δ::LEU2 deleter construct from pYF6 (9) or by PCR using knockout primers FW-koKTI12 and RV-koKTI12 (see Table S1 in the supplemental material). SIT4 disruptions involved a sit4::LEU2 cartridge in which SIT4 was disrupted by a 1.8-kb BamHI segment carrying LEU2 from YDpL (5, 27) and cloned into the single BglII site of SIT4. Leu+ transformants carrying the sit4::LEU2 allele were verified by PCR and phenotypic assays indicative of sit4Δ status (25, 27). Elongator subunit Elp1 was tagged at its C terminus with the hemagglutinin (HA) epitope using PCR protocols described previously (18, 34). To detect Sap155, Sap185, Sap190, and Tip41, plasmids with alleles coding for HA-tagged versions of these proteins (Table 2) (13, 29, 38) and originating from K. Arndt (Wyeth-Ayerst Research, NJ), M. Hall (Biozentrum, University of Basel, Basel, Switzerland), and E. Jacinto (UMDNJ-Robert Wood Johnson Medical School, NJ) were used. C-terminal Sap185 truncations were generated in a sap190Δ reporter strain expressing HA-tagged Sit4 under GAL1 promoter control (27, 36). Using PCR-based protocols (30, 34), full-length Sap185 and four derivatives lacking 108 (C1), 208 (C2), 308 (C3), and 408 (C1) amino acid residues were tagged at their C termini with c-Myc using plasmid pYM5 and primer S2-SAP185 in combination with primer S3-SAP185, S3-SAP185-C1, S3-SAP185-C2, S3-SAP185-C3, or S3-SAP185-C4 (see Table S1 in the supplemental material). Similarly, N-terminally HA-tagged Sap185 truncations lacking 200 (N1), 300 (N2), and 400 (N3) residues were generated using PCR protocols with plasmid pFA6-kanMX6-PGAL-3HA (36) and primer F4-SAP185, combined with primer R3-SAP185, R3-SAP185-N1, R3-SAP185-N2, or R3-SAP185-N3 (see Table S1 in the supplemental material). Prior to Western blot analysis, yeast candidate strains expressing the tagged truncations were verified using diagnostic PCR with primers homologous to an internal SAP185 region (SAP185-iFW) or specific to regions upstream (SAP185-FW) and downstream (SAP185-RV2) of the SAP185 gene (see Table S1 in the supplemental material).
Immunological techniques.
Detection of tagged proteins by anti-c-Myc and anti-HA antibodies was done as previously described (18). Protein concentrations were determined by the method of Bradford (7), and in Western blots, protein loadings were controlled with a 1:10,000 dilution of an antibody recognizing yeast Pfk1 (phosphofructokinase 1), kindly provided by J. Heinisch (University of Osnabrück, Osnabrück, Germany). Immune precipitation was performed as described previously (18, 57). Analysis of Elp1 phosphorylation states was done essentially as previously described (27) on the basis of mobility shift assays of HA-tagged Elp1 on anti-HA Western blots. In addition, immune detection of Sit4 and Tap42 in whole-cell extracts and immune precipitations used Western blotting and Sit4- and Tap42-specific antibodies (32, 51) kindly donated by J. Broach (Princeton University, Princeton, NJ) and Y. Jiang.
RESULTS
Zymocin inhibition operates independently of Tap42 and the TOR pathway.
Given that Sit4 promotes zymocin toxicity and that PP2A and Sit4 operate in the rapamycin-sensitive TOR pathway, we sought to identify potential phosphatase-mediated overlap between events required for the actions of both antifungals. A comparison between PP2A and Sit4 defects revealed that, in striking contrast to the zymocin resistance of a sit4Δ null mutant, rapamycin-resistant mutants with defects in regulatory genes coding for modifiers (PPM1 and PPM2), activators (RRD1 and RRD2), or subunits (TPD3 and RTS1) of PP2A (14, 31) displayed sensitivity (ppm1Δ ppm2Δ and rts1Δ) or even hypersensitivity (rrd1Δ rrd2Δ and tpd3Δ) to zymocin (see Fig. S1 in the supplemental material). Thus, these PP2A regulators are dispensable for zymocin action, which is consistent with the lack of any evidence implicating PP2A activity itself in zymocin toxicity.
Since the PP2A and Sit4 associator Tap42 has been proposed to mediate all essential functions of these phosphatases, including TOR signaling (13, 15, 52), we next analyzed the role of Tap42 in zymocin toxicity. Studying the original tap42-11ts strain and other rapamycin-resistant tap42ts mutants (13, 15) revealed that at temperatures permissive for growth and drug resistance, the Tap42 defects induced varying degrees of zymocin sensitivity (Fig. 1A). In support of a report on heat-sensitive PP2A/Tap42-11 interaction (52), we found that tap42-11ts cells also suffered from increased Sit4/Tap42-11 dissociation at 37°C (Fig. 1B). Intriguingly, SSD1-v, which genetically interacts with tap42ts alleles (42), countered this Sit4/Tap42-11 dissociation (Fig. 1B) and partially suppressed tap42ts thermosensitivity (Fig. 1A). However, since SSD1-v failed to confer any zymocin protection on the tap42ts mutants (Fig. 1A) and since tap42ts ssd1-d cells were zymocin hypersensitive in the absence of SSD1-v (see Fig. S2B in the supplemental material), we concluded that zymocin acts independently of Tap42 and SSD1 alleles. In line with this notion, we found that deletion of the SSD1 locus in the BY4741 strain background had no effect on zymocin toxicity (data not shown). Consistently, a rapamycin-resistant tip41Δ mutant lacking the Tap42 inhibitor Tip41 (29) was found to be zymocin sensitive, and even increased TIP41 dosage failed to alter this sensitive response (see Fig. S1 in the supplemental material). The latter finding is relevant, since excess levels of Tip41 promote Sit4/Tap42 dissociation (29), so if zymocin action required Sit4/Tap42 interaction, excess Tip41 ought to antagonize zymocin action, a prediction that is not confirmed by our data.
FIG. 1.

Zymocin hypersensitivity of TOR-defective tap42ts mutants. (A) Zymocin profiles. The indicated S. cerevisiae strains (wt, wild type) were serially diluted and replica spotted onto YPD medium lacking (− zymo) or containing (+ zymo) 40% or 50% (vol/vol) zymocin. To assay the impact of SSD1-v on thermosensitivity and zymocin action, the same strains carrying (SSD1-v) or lacking (vector) an extra copy of SSD1-v were grown for 3 days at the temperatures indicated. Thermosensitive (ts), as well as zymocin-resistant (R), -sensitive (S), or -hypersensitive (HS), traits are indicated. (B) SSD1-v suppresses heat-sensitive Sit4/Tap42 dissociation in tap42-11ts cells. Sit4-HA-expressing tap42-11ts cells with (SSD1-v) or without (vector) an extra SSD1-v copy were cultivated at the indicated temperatures and subjected to immune precipitation (IP) using anti-HA antibodies. The presence of Sit4 and Tap42-11 (indicated by arrows) in these precipitates was monitored by immune blotting (IB) using anti-HA and anti-Tap42 antibodies, respectively. In addition, prior to immune precipitation (preIP), the content of Sit4 and Tap42-11 in the input was checked by Western blotting.
To clarify further whether zymocin action requires Sit4/Tap42 interaction, we tested cells grown on glycerol-ethanol, a carbon source known to downregulate the TOR pathway and to disrupt Sit4/Tap42 complexes without affecting Sit4/Sap interactions (13). Under these conditions, Sit4/Tap42 complexes dramatically decreased in comparison to glucose-grown cells with high TOR activity (Fig. 2A) and in contrast to unaltered Sit4/Sap185 interaction in cells grown under both conditions (Fig. 2B). Cells with Sit4/Tap42 interaction defects and grown on glycerol-ethanol showed, if anything, slightly enhanced zymocin sensitivity (Fig. 2C) compared to the sensitive response of glucose-grown cells and the zymocin resistance of a sit4Δ mutant grown under both conditions (Fig. 2C and data not shown). Considering that enhanced Sit4/Tap42 dissociation failed to protect against zymocin, we concluded that Tap42-independent Sit4 complexes are likely to promote zymocin inhibition. In line with this notion, zymocin-specific effector roles have been identified for two Sap (Sit4-associating protein) family members, Sap185 and Sap190 (27). Moreover, our findings that zymocin inhibits rapamycin-resistant mutants with key TOR pathway defects (TOR1-1, TOR2-1, etc.) (see Fig. S2 in the supplemental material and data not shown) and, conversely, that rapamycin is a potent inhibitor of zymocin-resistant mutants with defects in Elongator function (elpΔ and kti12Δ) (see Fig. S2 in the supplemental material) reinforce the ideas that the rapamycin-sensitive TOR pathway is dispensable for zymocin toxicity and that the two antifungal pathways show hardly any phosphatase-mediated cross talk.
FIG. 2.
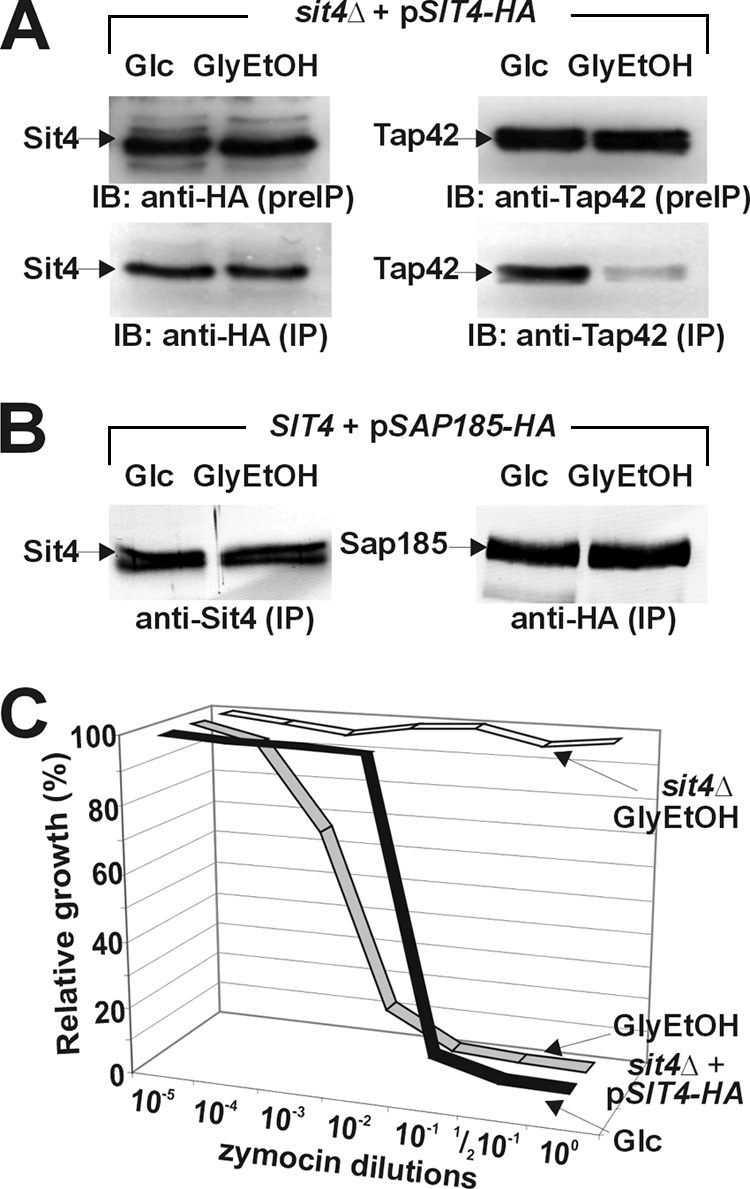
Zymocin inhibition is independent of Sit4/Tap42 interaction. (A) TOR pathway downregulation induces Sit4/Tap42 complex dissociation. Prior to immune precipitation (preIP), protein extracts prepared from Sit4-HA-expressing cells grown under glucose (Glc) or glycerol-ethanol (GlyEtOH), conditions, which promote or downregulate, respectively, TOR pathway signaling, were monitored for expression of Sit4-HA and Tap42 by immune blotting (IB) using anti-HA (top left) and anti-Tap42 (top right) antibodies. Immune precipitates (IP) obtained with the anti-HA antibody were next analyzed in Western blots using anti-HA and anti-Tap42 antibodies to check for the content of Sit4-HA and Tap42, respectively (indicated by arrows). (B) TOR pathway downregulation leaves Sit4/Sap185 interaction unaltered. SAP185-HA-expressing strains were grown as described for panel A and subjected to immune precipitation using anti-HA antibodies. The precipitates were analyzed by anti-Sit4 and anti-HA antibodies in immune blots to monitor the content of Sit4 and Sap185-HA. Note that TOR pathway downregulation did not affect Sit4/Sap185 interaction. (C) TOR pathway downregulation enhances zymocin inhibition. Under TOR-promoting (Glc) or TOR-downregulating (GlyEtOH) conditions, sit4Δ cells carrying SIT4-HA on a single-copy vector were grown in the presence of the indicated zymocin dilutions. Growth is expressed in relation (percentage of optical density at 600 nm) to that of zymocin-minus controls. sit4Δ cells carrying an empty vector and grown under TOR-downregulating conditions were included as a zymocin-resistant control.
The Sit4 partner Sap190 is involved in both rapamycin and zymocin inhibition.
Previous studies of PP2A regulatory (tpd3Δ, cdc55, ppm1Δ ppm2Δ, or rrd1Δ rrd2Δ) and PP2A catalytic (pph21Δ pph22Δ) mutants revealed that deregulation rather than elimination of PP2A activity is associated with rapamycin resistance (13, 32, 54, 59). Similarly, we found that Sit4 misregulation in mutants with defects in members of the Sap family displayed differential rapamycin or zymocin phenotypes (Fig. 3A). Thus, a quadruple sapΔΔΔΔ mutant generated in an SSD1-v background (CY5236) (Table 1) and lacking the SAP4, SAP155, SAP185, and SAP190 genes was found to be rapamycin sensitive but zymocin resistant, both traits shared with a sit4Δ mutant (Fig. 3A). Strains with individual SAP4, SAP155, or SAP185 deletions were susceptible to both antifungals (Fig. 3A). However, removal of Sap190 specifically protected against rapamycin without affecting zymocin inhibition. Furthermore, multiple sap mutants carrying sap190Δ in tandem with sap4Δ, sap185Δ, or sap4Δ sap185Δ null alleles also survived rapamycin (Fig. 3A), stressing that growth inhibition by rapamycin depends on the Sit4/Sap190 complex. Whether this reflects a TOR-embedded role for Sit4/Sap190 in the dephosphorylation of Tip41, a Sit4-dependent phosphoprotein and Tap42 inhibitor (29), is open to question. However, since tip41Δ and sap190Δ single mutants expressed levels of rapamycin resistance similar to that of a sap190Δ tip41Δ double mutant (Fig. 3B), this lack of phenotypic enhancement may be consistent with Tip41 and Sap190 functioning in a shared Sit4-dependent pathway (29).
FIG. 3.

Antifungal profiles of strains carrying single or multiple SAP and TIP41 gene deletions. (A) Effects of SAP gene deletions made in the W303 strain background on rapamycin and zymocin inhibition. The indicated S. cerevisiae strains (all SSD1-v; wt, wild type) were serially diluted and replica spotted onto YPD medium lacking (− rap) or containing (+ rap) 100 nM rapamycin. Growth was scored for 3 to 4 days at 25°C and compared to that of the rapamycin-resistant tap42-11ts control TS54-5a (SSD1-v background) (Table 1). In addition, yeast colony-colony interaction bioassays using the K. lactis zymocin producer AWJ137 (killer) were performed to assess zymocin phenotypes. Here, eclipse formation of S. cerevisiae colonies around the K. lactis killer strain indicated growth inhibition, whereas lack of an eclipse indicated the ability to survive zymocin. (B) Uniform rapamycin survival between sap190Δ and tip41Δ single- and sap190Δ tip41Δ double-deletion strains raised in the JK9-3a background. Tenfold serial dilutions of the indicated mutants were tested against both antifungals essentially as described for panel A. The responses include rapamycin/zymocin resistance (R), sensitivity (S), and hypersensitivity (HS).
Strikingly, in conjunction with a sap155Δ null allele, the rapamycin resistance of sap190Δ, sap185Δ sap190Δ, or sap4Δ sap185Δ sap190Δ mutants was suppressed without altering the respective zymocin phenotypes (Fig. 3A). This indicates that the rapamycin resistance of cells carrying a SAP190 deletion requires Sap155 and stresses that Sit4/Sap190 and Sit4/Sap155 complexes are likely to play opposing roles following rapamycin treatment. In line with this notion, we observed that the rapamycin resistance of an rrd1Δ mutant lacking the Sit4 interactor Rrd1 (59) was suppressed by the quadruple sapΔΔΔΔ mutation and that reintroduction of SAP190 alone rather than SAP155 was sufficient to restore rapamycin resistance (Fig. 4A). Moreover, we found that, in analogy to SAP155 dosage suppression of zymocin (25), SAP155 is also a high-copy-number suppressor of rapamycin (Fig. 4B). This suggests that competition between excess levels of Sap155 and other Sap family members (including Sap190) for Sit4 binding suppresses formation of the Sit4/Sap190 complex, which is particularly important for rapamycin action (Fig. 3A). Consistent with this notion, we observed that the high-copy-number SAP155 effect was efficiently countered by cooverexpression of SAP190 to restore rapamycin sensitivity (not shown) and that the rapamycin-resistant trait of the sap190Δ mutant was insensitive to the SAP155 dosage either alone or in combination with high-copy-number TAP42 (Fig. 4C). In sum, we conclude that while removing or blocking Sit4 phosphatase function in the sit4Δ or sapΔΔΔΔ mutant nullifies zymocin, deregulation of Sit4 activity due to Sap190 suppression (high-copy-number SAP155) or inactivation (sap190Δ) abrogates inhibition by rapamycin. Moreover, while Sap185 and Sap190 share common functions in the zymocin pathway, there is apparently no such redundancy with regard to rapamycin action.
FIG. 4.
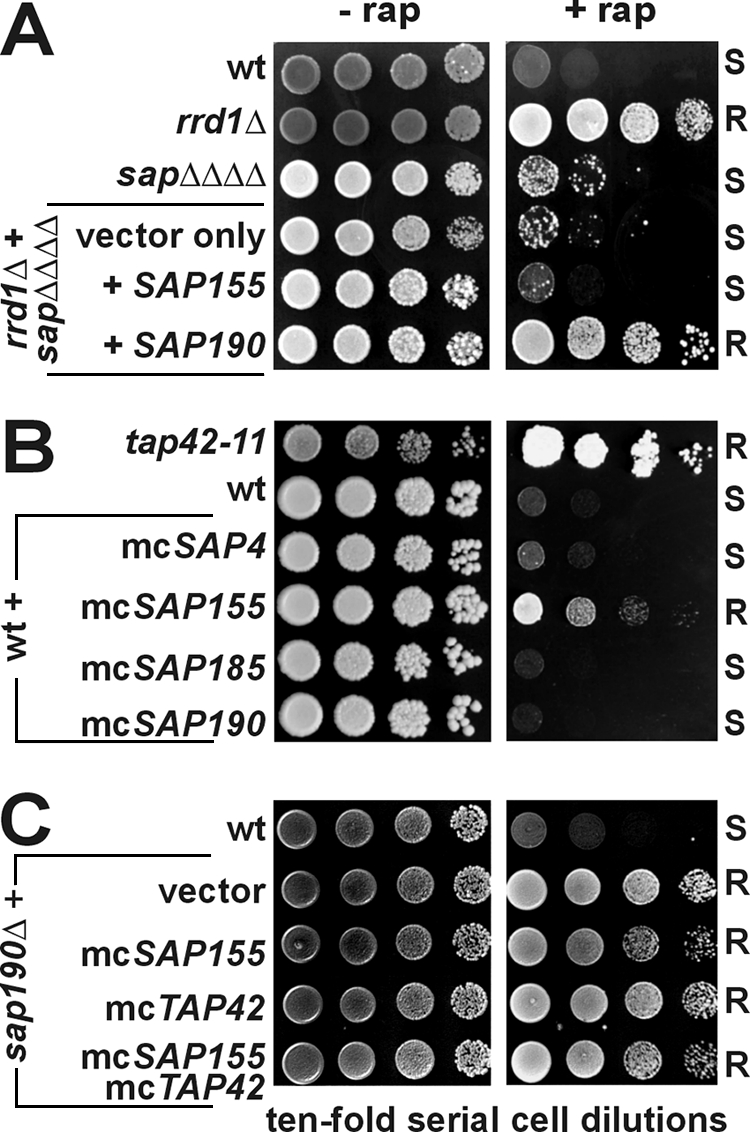
Effects of SAP155 and SAP190 expression on the rapamycin phenotypes of S. cerevisiae wild-type (wt) and deletion strains. The indicated S. cerevisiae strains carrying plasmids with the appropriate SAP and/or TAP42 genes were serially diluted and replica spotted onto YPD medium lacking (− rap) or containing (+ rap) 100 nM rapamycin. Growth was scored for 3 to 4 days at 25°C and compared to that of rapamycin-resistant controls: rrd1Δ (A), tap42-11ts (B), and sap190Δ (C). The responses included rapamycin resistance (R) and sensitivity (S). (A) Sap190 is required for strains lacking the Sit4 interactor Rrd1 to maintain rapamycin resistance. (B) SAP155 overexpression in a wild-type background induces rapamycin resistance. (C) The rapamycin resistance of a sap190Δ mutant is insensitive to the SAP155 and/or TAP42 gene dosage.
Tap42 binding-deficient sit4 mutants block Sit4/Sap complex formation.
A Tap42 binding site recently identified in Sit4 and Sit4 homologs (52) prompted us to study the zymocin responses of sit4 mutants with Sit4/Tap42 interaction defects (Fig. 5A). A single Tap42 binding site mutation (E38A) reported to leave the Sit4/Tap42 interaction intact (52) had no effect on zymocin action (Fig. 5B). However, multiple mutations ([L35A] E37A E38A) shown to block Sit4/Tap42 interaction (52) copied the zymocin resistance typical of sit4Δ (Fig. 5B) and sap185Δ sap190Δ mutants (27). Given our data that the Sit4/Tap42 complex (Fig. 1 and 2) is dispensable for zymocin action, this was a surprising finding. More strikingly, these Tap42 binding-deficient sit4 mutants also abolished Sit4-dependent dephosphorylation of Elp1, a subunit of the zymocin effector complex Elongator (27), leading to hyperphosphorylated Elp1 forms (Fig. 5B), which again is typical of sit4Δ and sap185Δ sap190Δ mutants (27). Since Tap42 may compete with members of the Sap family for Sit4 binding (13, 29), we examined whether the zymocin resistance of these sit4 mutants might reflect changes in Sit4/Sap interaction in the absence of competition from Tap42. HA-tagged Sap155 and Sap190 were each expressed in SIT4 cells and the Tap42-incompatible sit4 (E37A E38A) mutant (Fig. 5C). Upon immune precipitation using anti-HA antibodies and Western blotting with anti-Sit4 and anti-HA antibodies, it became evident that Sit4/Sap155 and Sit4/Sap190 complexes did not form in the sit4 mutant (Fig. 5C). Consistent with blocked Sit4/Sap interaction, the sit4 mutants ([L35A] E37A E38A) mimicked rapamycin phenotypes typical of sit4Δ mutants, while the single-site mutation (E38A) had no effect compared to SIT4 wild-type cells (Fig. 5D). In contrast to SIT4 wild-type cells with intact Sit4/Tap42 and Sit4/Sap interactions, the Tap42 binding-deficient sit4 mutants no longer allowed SAP155 dosage suppression of rapamycin (Fig. 5D). Again, these data suggest defects in Sit4/Sap complex formation and reinforce our notion that the Tap42 binding defect in the sit4 ([L35A] E37A E38A) mutants prevents the Sit4/Sap interaction required for the multicopy SAP155 effect (Fig. 5D).
FIG. 5.

Properties of Tap42 binding-deficient sit4 mutants. (A) Alignment of the Tap42 binding sites (boxed) of Sit4 and Sit4 homologs (human, PP6; Drosophila, PPV) (52). Substitutions of invariant residues in the different sit4 alleles are shown in boldface. (B) Killer eclipse assays and phosphomodification of Elongator subunit Elp1. ELP1-HA sit4Δ cells carrying wild-type SIT4 or sit4 alleles with the indicated single, double, and triple mutations in the Tap42 binding motif of Sit4 (52) were tested for zymocin inhibition (top) using the killer eclipse assay (33). Zymocin resistance (R) was distinguished from sensitivity (S). Total protein from these ELP1-HA expressers was extracted and immunoblotted using the anti-HA antibody (middle) to distinguish phosphorylated (circled P) Elp1 from unmodified Elp1 (27). Additional Western blots with anti-Pfk1 antibodies (bottom) detected α and β subunits of phosphofructokinase, which served as protein-loading controls. (C) Sit4/Sap155 and Sit4/Sap190 interaction studies. Plasmid-borne alleles of SAP155-HA and SAP190-HA were transformed into the indicated Tap42 binding-deficient sit4 mutants (52) and SIT4 wild-type cells. Following immune precipitation (IP) by anti-HA antibodies, the Sit4 and Sap proteins were detected in these precipitates (upper two blots) using anti-Sit4 and anti-HA antibodies and compared to the protein levels present in the input (preIP) controls (lower two blots). The arrows indicate the positions of Sap155, Sap190, and Sit4. (D) Rapamycin phenotypes. The indicated S. cerevisiae strains (wt, wild type) were assayed for sensitivity to 50 nM rapamycin (+ rap) using the tap42-11 mutant as a rapamycin-resistant control. Growth was for 3 to 4 days at 25°C. Rapamycin responses included resistance (R), sensitivity (S), and hypersensitivity (HS). Note that the Tap42 binding-deficient sit4 mutant (L35A E37A E38A) abolished dosage suppression of rapamycin by SAP155. (E) Heat-inducible zymocin resistance of the sit4-102 mutant, which has a Tap42 binding defect at 37°C. Shown are plate assays of the indicated tester strain dilutions at 30°C (top) and 37°C (bottom) on medium lacking (− zymo) or containing (+ zymo) 45% (vol/vol) zymocin. Sensitivity (S) and resistance (R) to zymocin are indicated. The sit4-102 allele was tested in SSD1-v (sit4-102/v) and ssd1-d (sit4-102/d) backgrounds. Thermosensitivity at 37°C is indicated (ts).
Prompted by these data, we next studied the sit4-102ts allele, which introduces a reverse-of-charge (E38K) substitution (1, 52) within the Tap42 binding site (Fig. 5A) shown to have an impact on Sit/Sap complex formation (Fig. 5C). In an ssd1-d background, the heat-sensitive Tap42 binding defect of the sit4-102ts mutant has been shown to be associated with nonviability at 37°C (Fig. 5E) (1, 38, 50, 52). Consistent with its sit4 suppressor effect, the SSD1-v allele, however, rescues the growth defect of sit4-102 cells at 37°C (Fig. 5E). Intriguingly, while these sit4-102 cells were sensitive to zymocin at 30°C, a shift to 37°C caused a zymocin-resistant trait typical of sit4Δ and sap185Δ sap190Δ mutants (Fig. 5E). Based on our data showing that the Sit4/Tap42 interaction is dispensable for zymocin action and that other mutations in the Tap42 binding site context affect Sit4/Sap interaction, we consider that it is blocked Sit4-102/Sap interaction (rather than defective Sit4-102/Tap42 interaction) that is likely to account for the thermoinducible zymocin resistance of the sit4-102 mutant at 37°C. Taking the data together, we conclude that individual mutations in the Tap42 binding site of Sit4 also compromise Sit4/Sap complex formation and thereby interfere with Sit4 functions, such as Elongator phosphoregulation and zymocin-induced cell death.
Mapping a Sap185 region crucial for Sit4/Sap185 interaction.
To gain further insights into Sit4/Sap holoenzymes, we focused on the Sap family member Sap185 and sought to identify a Sit4-interacting region (SIR185) crucial for Sit4/Sap185 complex formation. Using PCR protocols (30, 34, 36), c-Myc and HA epitope-tagged versions of full-length Sap185 and progressive N- or C-terminal truncations were generated in a sap190Δ strain and subjected to zymocin assays. Here, the rationale was to identify nonfunctional Sap185 variants on the basis of zymocin resistance, a trait associated with Sit4 defects and loss of Elp1 dephosphorylation in a sap185Δ sap190Δ mutant (25, 27). As illustrated by the C-terminal truncation set (Fig. 6A), which removed 108 (C1), 208 (C2), 308 (C3), and 408 (C4) amino acid residues from c-Myc-tagged Sap185, the truncations C1, C2, and C3 and full-length Sap185 were detected at similar levels, together with some low-abundance degradation products in anti-c-Myc Western blots. C4 levels, however, were dramatically decreased, implying instability of this truncation (Fig. 6A). Intriguingly, C1 and C2 conferred zymocin sensitivity typical of full-length Sap185, while C3 and C4 caused resistance (Fig. 6B). Consistent with these readouts, immune precipitation studies revealed Sit4 interaction with C1 and C2 comparable to that with full-length Sap185, while C3 or C4 drastically reduced or abolished Sit4 interaction, respectively (Fig. 6A). Moreover, the ability of sap190Δ cells to mediate Sit4-dependent Elp1 dephosphorylation strictly relied on Sit4 complexed with Sap185 or the truncations C1 and C2 (Fig. 6C). In contrast, the Sit4 binding defects of C3 and C4 caused Elp1 hyperphosphorylation (Fig. 6C) typical of sit4Δ or sap185Δ sap190Δ mutants (27). Based on these Sit4 interaction profiles and zymocin assays, we conclude that in the absence of Sap190, the Sit4/Sap185 complex becomes essential for zymocin inhibition and Elp1 dephosphorylation. The N-terminal Sap185 truncation set (see Fig. S3 in the supplemental material) showed that removal of residues 1 to 200 (N1) had no impact on Sit4 interaction, while larger truncations of 300 (N2) and 400 (N3) residues caused Sit4 binding defects. Although the truncation N1 supported Sit4 binding, it nonetheless failed to confer zymocin sensitivity (see Fig. S3 in the supplemental material). This suggests that the Sap185 N terminus contributes to Sit4 activity independently of promoting Sit4/Sap185 interaction. In sum, genetic SAP185 dissection revealed an SIR185 in the center of Sap185 that promotes Sit4/Sap185 complex formation (Fig. 6D). The SIR185 (residues 200 to 750) overlaps with the SAPS domain conserved in Sap4, Sap155, and Sap190 (38) and their mammalian counterparts PP6R1 to PP6R3 (49) (Fig. 6D and data not shown).
FIG. 6.

Use of zymocin as a diagnostic tool to map a SIR on Sap185. (A) sap190Δ deletion strains coexpressing HA-tagged Sit4 and the indicated c-Myc-tagged variants of Sap185 were subjected to immune precipitation (IP) using the anti-c-Myc antibody. The presence of Sit4, as well as full-length and C-terminal truncations of Sap185 (all indicated by arrows), in these precipitates was monitored by immunoblotting (IB) using anti-HA and anti-c-Myc antibodies. In addition, the content of Sit4 (bottom) and the Sap185 variants (not shown) in the inputs (preIP) was checked by IB using anti-HA and anti-c-Myc antibodies, respectively. (B) Sit4 binding deficits of Sap185 truncations C3 and C4 cause zymocin resistance in a sap190Δ deletion mutant. Strains as in panel A were subjected to killer eclipse assays to score zymocin sensitivity (S) or resistance (R). (C) Assaying the Elp1 phosphobalance involved transformation with pELP1-HA, protein extraction, and Western blot analysis using the anti-HA antibody. Elp1 phosphoforms (circled P) were distinguished from unmodified Elp1 by mobility shifts (27). (D) The SIR of Sap185 maps to a central segment conserved in other members of the yeast Sap family. The sketch summarizes SIR mapping data (gray box) on the basis of C- and N-terminal Sap185 truncation sets, C1 to C4 and N1 to N3 (see Fig. S3 in the supplemental material). The regions with the highest similarity between Sap4, Sap155, Sap185, and Sap190 are highlighted as black boxes (38).
Sit4/Sap185 and Sit4/Sap190 promote Elongator-dependent tRNA suppression.
Elongator-dependent tRNA modification is not only required for the action of nonsense and missense tRNA suppressors (21, 28), but is also required for the tRNase activity of the lethal ribotoxin zymocin (16, 22, 28, 37). Since zymocin sensitivity and Elp1 dephosphorylation require Sit4/Sap185 and Sit4/Sap190 phosphatases (25, 27), we sought to strengthen the connection between Sit4 and Elongator function by examining whether the above-mentioned Sap185 truncations also influence Elongator's roles in tRNA modification and nonsense readthrough by the tRNA suppressor SUP4 (21). The tRNATyr encoded by SUP4 carries a uridine in the anticodon wobble position whose modification is Elongator dependent and is required for proper anticodon/codon interaction during suppression of ocher mutations (21, 28). Therefore, Elongator promotes the readthrough of an ade2-1 ocher mutation by SUP4, which yields white colony pigmentation and adenine prototrophy. Conversely, Elongator defects eliminate SUP4 suppression, causing red colony color and adenine auxotrophy (21, 28). We found that full-length Sap185 and the truncations C1 and C2 were able to support ade2-1 readthrough by SUP4, yielding white colonies typical of adenine prototrophs (Fig. 7A). In contrast, red colony pigmentation typical of adenine auxotrophy in the presence of the C3 and C4 truncations indicates lack of SUP4 activity, similar to Elongator (elp1Δ) and sit4Δ mutants (Fig. 7A) (21, 22). Likewise, readthrough of the can1-100 ocher mutation by SUP4, which kills yeast cells in the presence of the toxic amino acid analogue canavanine, was abolished in elp1Δ, elp3Δ, sit4Δ, and sap185Δ sap190Δ mutants, causing canavanine resistance (Fig. 7B). In sum, tRNA modification and ocher suppression by SUP4 not only depend on Elongator function, but also require functional Sit4/Sap185 and Sit4/Sap190 complexes. This is supported by our observation that can1-100 readthrough by SUP4 was intact in a mutant lacking Sap4 and Sap155 (Fig. 7B), which are dispensable for Sit4-dependent Elp1 dephosphorylation (27). In addition, cells that displayed Elp1 hyperphosphorylation as a result of overexpressing the Elongator partner Kti12 (17, 18, 27) also abolished can1-100 readthrough by SUP4, similarly to sit4Δ and sap185Δ sap190Δ mutants (Fig. 7B). Collectively, this shows that Elp1 phosphoregulation via Sit4/Sap185 and Sit4/Sap190 is involved in Elongator-dependent tRNA suppression and potentially in Elongator-dependent tRNA modification.
FIG. 7.

Lack of Sit4/Sap185 and Sit4/Sap190 phosphatase functions abolishes Elongator-dependent tRNA suppression by SUP4. (A) Strains with the indicated backgrounds and carrying plasmid pTC3 (pSUP4), along with the ade2-1 ocher mutation, were grown on YPD medium for 2 days at 30°C. ade2-1 readthrough by SUP4 yielded white colonies and adenine-prototrophy (ade+) typical of the wild-type control, while Elongator inactivation caused antisuppression of SUP4, red colony pigmentation, and adenine auxotrophy (ade−). (B) can1-100 readthrough by SUP4 in the indicated transformants was monitored on the basis of canavanine resistance (R) versus sensitivity (S) using canavanine medium lacking arginine (bottom, + can) or control medium without the toxic amino acid analogue (top, − can). Note that SUP4 readthrough allowed canavanine uptake, leading to can1-100 cell death, while SUP4 antisuppressors survived the toxic amino acid analogue.
DISCUSSION
Sit4 regulatory defects protect against rapamycin.
Unlike the rapamycin-resistant PP2A mutants that we report here to be zymocin sensitive, inactivation of the Sit4 phosphatase catalytic subunit does not protect against rapamycin but causes zymocin resistance. Both traits are copied by a quadruple sap mutant, in which loss of all Sap family members mimics Sit4 inactivation (25, 27, 38, 44). While zymocin inhibition requires either Sap185 or Sap190, rapamycin action depends on Sap190 alone, which implies a TOR-related role for the Sit4/Sap190 complex that is not shared by the Sit4/Sap185 complex. Previously, a sap190Δ deletion in strain Σ1278b was reported to be rapamycin sensitive (44), which is distinct from our drug profiles in the W303 and JK9-3a strains. In support of our view that this difference is likely to be due to genetic variations, Σ1278b displays natural drug tolerance, in contrast to W303 and JK9-3a, and the polysome-to-monosome ratios of Σ1278b are rapamycin insensitive (44). In addition, the rapamycin resistance of a sap190Δ deletion in strain BY4741 (not shown) stresses that the rapamycin inhibition of several yeast strains requires Sap190. As with suppression of zymocin (25), high-copy-number SAP155 also protects against rapamycin, suggesting that excess Sap155 levels may outcompete Sap190 from Sit4 binding, thereby disfavoring Sit4/Sap190 complex formation and mimicking the drug resistance of sap190Δ cells. Work on yeast phosphatase Glc7 (PP1) has shown that overexpression of cytoplasmic regulatory subunits can lead to reduced nuclear PP1 function as a result of cytoplasmic redistribution of Glc7 (41). Thus, competition between different regulatory subunits for a shared phosphatase catalytic subunit is emerging as a common theme, and the correct balance between different holophosphatases is clearly influenced by their relative abundances.
Although Sit4/Sap190 confers rapamycin sensitivity, the quadruple sap mutant is sensitive to the drug, and the rapamycin resistance of sap190Δ cells depends on Sap155 function. Thus, Sit4/Sap155 and Sit4/Sap190 are both involved in the TOR pathway, but they apparently play opposing roles. Clearly, further work is needed to determine how both phosphatase complexes work, but there are several points at which Sit4 may operate in the TOR pathway, including dephosphorylation of the transcription factor Gln3 (4), the protein kinase Npr1 (28, 29, 45), or the Tap42 interactor Tip41 (29). As for the last, the lack of enhancement in rapamycin resistance between sap190Δ or tip41Δ cells alone and a double sap190Δ tip41Δ mutant is consistent with Tip41 and Sap190 functioning in such a shared Sit4 pathway. In sum, misregulation rather than elimination of Sit4 activity protects against rapamycin, suggesting that the drug's effects are particularly sensitive to alterations in the subunit composition and substrate specificity of the Sit4 holophosphatase (32, 44, 52).
The zymocin effector role of Sit4 is TOR independent.
On studying Tap42, which links PP2A and Sit4 to the TOR pathway, we found that rapamycin-resistant tap42ts mutants with Sit4 binding defects are zymocin sensitive (15, 43, 51). Thus, zymocin acts independently of Tap42, a notion supported by our data showing that Sit4/Tap42 dissociation due to TOR pathway downregulation is not protective against zymocin. Hence, rather than requiring Tap42, zymocin is TOR independent and depends on a Sit4 function that is mediated by Sap185 and Sap190 for regulation of Elongator activity (25, 27). In sum, subunit composition is critical for Sit4 phosphatase functions in growth control by zymocin (Sit4/Sap185 and Sit4/Sap190) or rapamycin (Sit4/Tap42, Sit4/Sap190, and Sit4/Sap155) and potentially in other Sit4 functions related to ion homeostasis, oleate toxicity, or telomere integrity (20, 35, 39).
Paradoxically, we show here that Tap42 binding-deficient sit4 mutants (52) survive zymocin and display Elp1 hyperphosphorylation, both traits typical of sit4Δ and sap185Δ sap190Δ mutants. However, additional prevention of Sit4/Sap complex formation, which is crucial for zymocin action, fully explains the traits of these Tap42 binding-deficient sit4 mutants. Hence although Tap42 interaction has been proposed to account for the vital function(s) of Sit4 on the basis of these Tap42 binding-deficient sit4 alleles (52), our data showing that they also block Sit4/Sap interactions reinforce the original model (38), in which the Sap family members have been proposed to mediate the essential nature of Sit4 in ssd1-d cells. Importantly, our data demonstrating that the heat-sensitive Tap42 binding defect of the sit4-102ts mutant, which is lethal at 37°C in an ssd1-d background, coincides with zymocin resistance at 37°C in the SSD1-v background strongly suggests that the sit4-102 allele also affects the formation of Sit4-102/Sap complexes required for zymocin toxicity. In line with this, sit4-102 is a reverse-of-charge (E38K) substitution in the Tap42 binding site, independent mutations ([L35A] E37A E38A) of which also compromise Sit4/Sap interaction, as shown here. Such an additional Sap binding deficit of the Sit4-102 variant may provide an explanation as to why elevated SAP gene dosage is able to suppress the thermosensitivity of sit4-102ts cells in the ssd1-d background at 37°C, as previously reported (38), since higher than normal levels of Sap proteins may compensate for compromised Sit4-102/Sap interaction and thus rescue cell growth under otherwise nonpermissive conditions (13, 38, 52). In support of this scenario, which implies that the thermosensitivity of sit4-102ts cells is not necessarily linked to the Tap42 binding defect alone, loss of interaction between Sit4 and the Sap members can indeed be lethal in a quadruple sapΔΔΔΔ mutant lacking a functional Sap complement (38).
Since we have shown here that Tap42 is dispensable for zymocin action and that sit4-102 cells survive zymocin under conditions of Sit4-102/Tap42 dissociation, we conclude that defective Sit4-102/Sap interaction is likely to induce protection against zymocin. In further support of Tap42-independent Sit4 complexes, Sit4/Tap42 and Sit4/Sap holoenzymes apparently form in mutually exclusive manners involving ∼5 and 50% of cellular Sit4 protein, respectively (13, 32). Whether Tap42 is a chaperone that assists the formation of drug-responsive phosphatases, including the Sit4/Sap190 complex, or targets PP2A enzymes to distinct subcellular locations, similar to the Drosophila Tap42 homolog (12), is not known. However, we consider it unlikely that Sit4/Sap phosphatases generally require Tap42 binding to Sit4 and favor a working model in which the formation of Sit4/Tap42 and Sit4/Sap complexes involves overlapping binding motifs on Sit4 so that binding site mutations can provoke interaction defects with both Tap42 and Sap family members. In line with this model, the Tap42 binding defects studied here concomitantly blocked the formation of Sit4/Tap42 and Sit4/Sap complexes, and in mammals, the formation of mutually exclusive PP2A complexes has been shown to involve overlapping binding sites for the Tap42 homolog α4 and regulatory subunit A (42).
Zymocin as a tool to study Sit4/Sap complex formation and function.
As reported here, SAP185 gene dissection has identified an SIR185 that is crucial for the formation and activity of the Sit4/Sap185 phosphatase. The SIR185 spans a central segment of ∼550 residues within the SAPS domain (38), which is conserved in Sap4, Sap155, and Sap190, as well as PP6R1 to PP6R3, which partner with PP6, the human homolog of Sit4 (49). Whether the SIR185 mediates direct or indirect interaction with Sit4 is not known. However, on the basis of Elp1 dephosphorylation assays, our analysis shows that, in addition to the SIR185, flanking Sap185 segments are required for full Sit4 phosphatase activity. These may contribute to further Sit4 regulation and/or Elp1 specificity of the Sit4/Sap185 phosphatase complex. Elongator phosphoregulation by direct or indirect actions of the Sit4/Sap185 and Sit4/Sap190 complexes clearly promotes the tRNA modification function of the Elongator complex. This is evidenced by the capacity of Sit4/Sap185 and Sit4/Sap190 to mediate ade2-1 and can1-100 nonsense readthrough by the Elongator-dependent suppressor tRNA SUP4. Obviously, this role for Sit4 in Elongator function is also required for cytotoxicity of zymocin, whose tRNase γ-toxin subunit cleaves tRNA anticodons that are modified in an Elongator-dependent manner (21, 28, 37). Apparently, Sap185 and Sap190 (rather than Sap4, Sap155, or Tap42) effect or activate this Sit4 role in Elongator function, reinforcing that it is the subunit composition of Sit4 complexes that confers specificity on the phosphatases (38, 49). This is entirely consistent with a recent report that sap185Δ sap190Δ mutants display tRNA modification defects that are typical of sit4Δ and Elongator-related mutants and are instrumental in abrogating the tRNase attack of zymocin (2, 21, 22, 28, 37, 56). Summing up, we conclude that in spite of a shared requirement for Sit4, the antifungals zymocin and rapamycin operate through independent pathways that do not involve Sit4 phosphatase-mediated cross talk.
Supplementary Material
Acknowledgments
We thank K. Arndt, J. Broach, S. Clarke, K. Düvel, E. Jacinto, Y. Jiang, M. Hall, J. Heinisch, E. Ogris, T. Powers, and G. Sprague for antibodies, plasmid DNAs, and yeast strains and T. Hahn for technical assistance.
R.S. appreciates project support by Fonds der Chemischen Industrie, Deutsche Forschungsgemeinschaft (DFG Scha750/2), and the Biotechnology and Biological Sciences Research Council (BBSRC) (BB/F019106/1). M.J.R.S. also acknowledges support through a BBSRC grant (BB/F0191629/1).
Footnotes
Published ahead of print on 1 September 2009.
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Arndt, K. T., C. A. Styles, and G. R. Fink. 1989. A suppressor of a HIS4 transcriptional defect encodes a protein with homology to the catalytic subunit of protein phosphatases. Cell 56:527-537. [DOI] [PubMed] [Google Scholar]
- 2.Bär, C., R. Zabel, S. Liu, M. J. R. Stark, and R. Schaffrath. 2008. A versatile partner of eukaryotic protein complexes that is involved in multiple biological processes: Kti11/Dph3. Mol. Microbiol. 69:1221-1233. [DOI] [PubMed] [Google Scholar]
- 3.Barbet, N. C., U. Schneider, S. B. Helliwell, I. Stansfield, M. F. Tuite, and M. N. Hall. 1996. TOR controls translation initiation and early G1 progression in yeast. Mol. Biol. Cell 7:25-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck, T., and M. N. Hall. 1999. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402:689-692. [DOI] [PubMed] [Google Scholar]
- 5.Berben, G., J. Dumont, V. Gilliquet, P. A. Bolle, and F. Hilger. 1991. The YDp plasmids: a uniform set of vectors bearing versatile gene disruption cassettes for Saccharomyces cerevisiae. Yeast 7:475-477. [DOI] [PubMed] [Google Scholar]
- 6.Bertram, P. G., J. H. Choi, J. Carvalho, T. F. Chan, W. Ai, and X. F. Zheng. 2002. Convergence of TOR-nitrogen and Snf1-glucose signaling pathways onto Gln3. Mol. Cell. Biol. 22:1246-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 8.Butler, A. R., J. H. White, and M. J. Stark. 1991. Analysis of the response of Saccharomyces cerevisiae cells to Kluyveromyces lactis toxin. J. Gen. Microbiol. 137:1749-1757. [DOI] [PubMed] [Google Scholar]
- 9.Butler, A. R., J. H. White, Y. Folawiyo, A. Edlin, D. Gardiner, and M. J. Stark. 1994. Two Saccharomyces cerevisiae genes which control sensitivity to G1 arrest induced by Kluyveromyces lactis toxin. Mol. Cell. Biol. 14:6306-6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherkasova, V. A., and A. G. Hinnebusch. 2003. Translational control by TOR and TAP42 through dephosphorylation of eIF2α kinase GCN2. Genes Dev. 17:859-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crespo, J. L., and M. N. Hall. 2002. Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 66:579-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cygnar, K. D., X. Gao, D. Pan, and T. P. Neufeld. 2005. The phosphatase subunit tap42 functions independently of target of rapamycin to regulate cell division and survival in Drosophila. Genetics 170:733-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Como, C. J., and K. T. Arndt. 1996. Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev. 10:1904-1916. [DOI] [PubMed] [Google Scholar]
- 14.Düvel, K., and J. R. Broach. 2004. The role of phosphatases in TOR signaling in yeast. Curr. Top. Microbiol. Immunol. 279:19-38. [DOI] [PubMed] [Google Scholar]
- 15.Düvel, K., A. Santhanam, S. Garrett, L. Schneper, and J. R. Broach. 2003. Multiple roles of Tap42 in mediating rapamycin-induced transcriptional changes in yeast. Mol. Cell 11:1467-1478. [DOI] [PubMed] [Google Scholar]
- 16.Esberg, A., B. Huang, M. J. Johansson, and A. S. Byström. 2006. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell 24:139-148. [DOI] [PubMed] [Google Scholar]
- 17.Fichtner, L., F. Frohloff, K. Bürkner, M. Larsen, K. D. Breunig, and R. Schaffrath. 2002. Molecular analysis of KTI12/TOT4, a Saccharomyces cerevisiae gene required for Kluyveromyces lactis zymocin action. Mol. Microbiol. 43:783-791. [DOI] [PubMed] [Google Scholar]
- 18.Frohloff, F., L. Fichtner, D. Jablonowski, K. D. Breunig, and R. Schaffrath. 2001. Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J. 20:1993-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gietz, D., A. St. Jean, R. A. Woods, and R. H. Schiestl. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 20:1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayashi, N., T. Nomura, N. Sakumoto, Y. Mukai, Y. Kaneko, S. Harashima, and S. Murakami. 2005. The SIT4 gene, which encodes protein phosphatase 2A, is required for telomere function in Saccharomyces cerevisiae. Curr. Genet. 47:359-367. [DOI] [PubMed] [Google Scholar]
- 21.Huang, B., M. J. O. Johanson, and A. S. Byström. 2005. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 11:424-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang, B., J. Lu, and A. S. Byström. 2008. A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA 14:2183-2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jablonowski, D., and R. Schaffrath. 2007. Zymocin, a composite chitinase and tRNase killer toxin from yeast. Biochem. Soc. Trans. 35:1533-1537. [DOI] [PubMed] [Google Scholar]
- 24.Jablonowski, D., F. Frohloff, L. Fichtner, M. J. Stark, and R. Schaffrath. 2001. Kluyveromyces lactis zymocin mode of action is linked to RNA polymerase II function via Elongator. Mol. Microbiol. 42:1095-1106. [DOI] [PubMed] [Google Scholar]
- 25.Jablonowski, D., A. R. Butler, L. Fichtner, D. Gardiner, R. Schaffrath, and M. J. Stark. 2001. Sit4p protein phosphatase is required for sensitivity of Saccharomyces cerevisiae to Kluyveromyces lactis zymocin. Genetics 159:1479-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jablonowski, D., L. Fichtner, F. Frohloff, and R. Schaffrath. 2003. Chitin binding capability of the zymocin complex from Kluyveromyces lactis, p. 191-194. In K. Wolf, K. D. Breunig, and G. Barth (ed.), Non-conventional yeasts in genetics, biochemistry and biotechnology. Springer-Verlag, New York, NY.
- 27.Jablonowski, D., L. Fichtner, M. J. R. Stark, and R. Schaffrath. 2004. The yeast Elongator histone acetylase requires Sit4-dependent dephosphorylation for toxin-target capacity. Mol. Biol. Cell 15:1459-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jablonowski, D., S. Zink, C. Mehlgarten, G. Daum, and R. Schaffrath. 2006. tRNAGlu wobble uridine methylation by Trm9 identifies Elongator's key role for zymocin-induced cell death in yeast. Mol. Microbiol. 59:677-688. [DOI] [PubMed] [Google Scholar]
- 29.Jacinto, E., B. Guo, K. T. Arndt, T. Schmelzle, and M. N. Hall. 2001. TIP41 interacts with TAP42 and negatively regulates the TOR signaling pathway. Mol. Cell 8:1017-1026. [DOI] [PubMed] [Google Scholar]
- 30.Janke, C., M. M. Magiera, N. Rathfelder, C. Taxis, S. Reber, H. Maekawa, A. Moreno-Borchart, G. Doenges, E. Schwob, E. Schiebel, and M. Knop. 2004. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21:947-962. [DOI] [PubMed] [Google Scholar]
- 31.Jiang, Y. 2006. Regulation of the the cell cycle by protein phosphatase 2A in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 70:440-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang, Y., and J. R. Broach. 1999. Tor proteins and protein phosphatase 2A reciprocally regulate Tap42 in controlling cell growth in yeast. EMBO J. 18:2782-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kishida, M., M. Tokunaga, Y. Katayose, H. Yajima, A. Kawamura-Watabe, and F. Hishinuma. 1996. Isolation and genetic characterization of pGKL killer-insensitive mutants (iki) from Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 60:798-801. [DOI] [PubMed] [Google Scholar]
- 34.Knop, M., K. Siegers, G. Pereira, W. Zachariae, B. Winsor, K. Nasmyth, and E. Schiebel. 1999. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15:963-972. [DOI] [PubMed] [Google Scholar]
- 35.Lockshon, D., L. E. Surface, E. O. Kerr, M. Kaeberlein, and B. K. Kennedy. 2007. The sensitivity of yeast mutants to oleic acid implicates the peroxisome and other processes in membrane function. Genetics 175:77-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953-961. [DOI] [PubMed] [Google Scholar]
- 37.Lu, J., B. Huang, A. Esberg, M. J. Johansson, and A. S. Byström. 2005. The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. RNA 11:1648-1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luke, M. M., F. Della Seta, C. J. Di Como, H. Sugimoto, R. Kobayashi, and K. T. Arndt. 1996. The SAP, a new family of proteins, associate and function positively with the SIT4 phosphatase. Mol. Cell. Biol. 16:2744-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manlandro, C. M., D. H. Haydon, and A. G. Rosenwald. 2005. Ability of Sit4p to promote K+ efflux via Nha1p is modulated by Sap155p and Sap185p. Eukaryot. Cell 4:1041-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otero, G., J. Fellows, Y. Li, T. de Bizemont, A. M. Dirac, C. M. Gustafsson, H. Erdjument-Bromage, P. Tempst, and J. Q. Svejstrup. 1999. Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol. Cell 3:109-118. [DOI] [PubMed] [Google Scholar]
- 41.Pinsky, B. A., C. V. Kotwaliwale, S. Y. Tatsutani, C. A. Breed, and S. Biggins. 2006. Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing Glc7. Mol. Cell. Biol. 26:2648-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prickett, T. D., and D. L. Brautigan. 2004. Overlapping binding sites in protein phosphatase 2A for association with regulatory A and α-4 (mTap42) subunits. J. Biol. Chem. 279:38912-38920. [DOI] [PubMed] [Google Scholar]
- 43.Reinke, A., S. Anderson, J. M. McCaffery, J. Yates III, S. Aronova, S. Chu, S. Fairclough, C. Iverson, K. P. Wedaman, and T. Powers. 2004. TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J. Biol. Chem. 279:14752-14762. [DOI] [PubMed] [Google Scholar]
- 44.Rohde, J. R., S. Campbell, S. A. Zurita-Martinez, N. S. Cutler, M. Ashe, and M. E. Cardenas. 2004. TOR controls transcriptional and translational programs via Sap-Sit4 protein phosphatase signaling effectors. Mol. Cell. Biol. 24:8332-8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmidt, A., T. Beck, A. Koller, J. Kunz, and M. N. Hall. 1998. The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J. 17:6924-6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw, K. J., and M. V. Olson. 1984. Effects of altered 5′-flanking sequences on the in vivo expression of a Saccharomyces cerevisiae tRNATyr gene. Mol. Cell. Biol. 4:657-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherman, F. 2002. Getting started with yeast. Methods Enzymol. 305:3-41. [DOI] [PubMed] [Google Scholar]
- 48.Stark, M. J. 1996. Yeast protein serine/threonine phosphatases: multiple roles and diverse regulation. Yeast 12:1647-1675. [DOI] [PubMed] [Google Scholar]
- 49.Stefansson, B., and D. L. Brautigan. 2006. Protein phosphatase 6 subunit with conserved Sit4-associated protein domain targets IκBɛ. J. Biol. Chem. 281:22624-22634. [DOI] [PubMed] [Google Scholar]
- 50.Sutton, A., D. Immanuel, and K. T. Arndt. 1991. The SIT4 protein phosphatase functions in late G1 for progression into S phase. Mol. Cell. Biol. 11:2133-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang, H., and Y. Jiang. 2003. The Tap42-protein phosphatase type 2A catalytic subunit complex is required for cell cycle-dependent distribution of actin in yeast. Mol. Cell. Biol. 23:3116-3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang, H., X. Wang, and Y. Jiang. 2003. Interaction with Tap42 is required for the essential function of Sit4 and type 2A phosphatases. Mol. Biol. Cell 14:4342-4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winkler, G. S., T. G. Petrakis, S. Ethelberg, M. Tokunaga, H. Erdjument-Bromage, P. Tempst, and J. Q. Svejstrup. 2001. RNA polymerase II elongator holoenzyme is composed of two discrete subcomplexes. J. Biol. Chem. 276:32743-32749. [DOI] [PubMed] [Google Scholar]
- 54.Wu, J., T. Tolstykh, J. Lee, K. Boyd, J. B. Stock, and J. R. Broach. 2000. Carboxyl methylation of the phosphoprotein phosphatase 2A catalytic subunit promotes its functional association with regulatory subunits in vivo. EMBO J. 19:5672-5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan, G., X. Shen, and Y. Jiang. 2006. Rapamycin activates Tap42-associated phosphatases by abrogating their association with Tor complex 1. EMBO J. 25:3546-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zabel, R., C. Bär, C. Mehlgarten, and R. Schaffrath. 2008. Yeast alpha-tubulin suppressor Ats1/Kti13 relates to the Elongator complex and interacts with Elongator partner protein Kti11. Mol. Microbiol. 69:175-187. [DOI] [PubMed] [Google Scholar]
- 57.Zachariae, W., T. H. Shin, M. Galova, B. Obermaier, and K. Nasmyth. 1996. Identification of subunits of the anaphase-promoting complex of Saccharomyces cerevisiae. Science 274:1201-1204. [DOI] [PubMed] [Google Scholar]
- 58.Zaragoza, D., A. Ghavidel, J. Heitman, and M. C. Schultz. 1998. Rapamycin induces the G0 program of transcriptional repression in yeast by interfering with the TOR signaling pathway. Mol. Cell. Biol. 18:4463-4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng, Y., and Y. Jiang. 2005. The yeast phosphotyrosyl phosphatase activator is part of the Tap42-phosphatase complexes. Mol. Biol. Cell 16:2119-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.