

Abstract
Porphyromonas gingivalis, a periodontal pathogen, secretes outer membrane vesicles (MVs) that contain major virulence factors, including proteases termed gingipains (Arg-gingipain [Rgp] and Lys-gingipain [Kgp]). We recently showed that P. gingivalis MVs swiftly enter host epithelial cells via an endocytosis pathway and are finally sorted to lytic compartments. However, it remains unknown whether MV entry impairs cellular function. Herein, we analyzed cellular functional impairment following entry of P. gingivalis into epithelial cells, including HeLa and immortalized human gingival epithelial (IHGE) cells. After being taken up by endocytic vacuoles, MVs degraded the cellular transferrin receptor (TfR) and integrin-related signaling molecules, such as paxillin and focal adhesion kinase (FAK), which resulted in depletion of intracellular transferrin and inhibition of cellular migration. Few Rgp-null MVs entered the cells, and these negligibly degraded TfR, whereas paxillin and FAK degradation was significant. In contrast, Kgp-null MVs clearly entered the cells and degraded TfR, while they scarcely degraded paxillin and FAK. In addition, both wild-type and Kgp-null MVs significantly impaired cellular migration, whereas the effect of Rgp-null MVs was limited. Our findings suggest that, following entry of P. gingivalis MVs into host cells, MV-associated gingipains degrade cellular functional molecules such as TfR and paxillin/FAK, resulting in cellular impairment, indicating that P. gingivalis MVs are potent vehicles for transmission of virulence factors into host cells and are involved in the etiology of periodontitis.
Bacteria have evolved mechanisms for the secretion of virulence factors into host cells; these virulence factors alter host cell biology and enable bacterial colonization (11). Bacterial outer membrane vesicles (MVs), ubiquitously shed from gram-negative bacteria by a mechanism involving cell wall turnover, consist of a subset of outer membrane and soluble periplasmic components (54). This extracellular secretion system likely plays a part in the strategy utilized by bacterial pathogens to modulate host defense and response and impair host cell function. For example, Pseudomonas aeruginosa (3), Helicobacter pylori (19), and Actinobacillus actinomycetemcomitans (8), as well as pathogenic and nonpathogenic Escherichia coli (7, 51), secrete MVs that contain toxins, proteases, adhesins, and lipopolysaccharide. Therefore, it has been proposed that MVs are bacterial “bombs” (30). However, the molecular mechanism of MV entry into host cells is unclear, while it also remains unknown whether MV-associated virulence factors have cytotoxic effects within the invaded cells. A recent study showed that enterotoxigenic E. coli MVs containing heat-labile enterotoxin and other bacterial envelope components were taken up by a human epithelial cell line via cellular lipid rafts, after which intracellular MVs accumulated in nonacidified compartments inaccessible to the extracellular milieu (28). In addition, a very recent study found that Pseudomonas aeruginosa MVs deliver multiple virulence factors, including β-lactamase, alkaline phosphatase, hemolytic phospholipase C, and Cif (cystic fibrosis transmembrane conductance regulator inhibitory factor), directly into the host cytoplasm via fusion of MVs with lipid rafts in the host plasma membrane, which has an effect on host cell biology (5). Thus, it is likely MVs function as specifically targeted transport vehicles to mediate entry of bacterial virulence factors into host cells. However, no other related results have been reported.
Periodontitis, one of the most common infectious diseases seen in humans (52), is characterized by gingival inflammation, as well as loss of connective tissue and bone from around the roots of the teeth, which leads to eventual tooth exfoliation. Porphyromonas gingivalis is considered to be a bona fide pathogen that causes several forms of severe periodontal disease. The bacterium releases MVs in an extracellular manner; these MVs retain the full components of outer membrane constituents, including lipopolysaccharide, muramic acid, a capsule, fimbriae, and proteases termed gingipains (13, 35). Fimbriae reportedly mediate bacterial adherence to and entry into periodontal cells (2), while gingipains, which consist of arginine (Arg-gingipain [Rgp])- and lysine (Lys-gingipain [Kgp])-specific cysteine proteinases, contribute to the destruction of periodontal tissues (24). Gingipains degrade collagen and fibronectin and inhibit interactions between host cells and the extracellular matrix. In addition, they degrade various cytokines, resulting in a disturbance of the host cytokine network. Therefore, fimbriae and gingipains are responsible for the adhesive and proteolytic abilities of MVs, which, together with the small size of MVs (20 to 500 nm), are suspected of enabling MVs to penetrate an intact mucosa and enter underlying host tissues (34).
Very recently, we showed that P. gingivalis MVs swiftly enter HeLa and immortalized human gingival epithelial (IHGE) cells in a fimbria-dependent manner (10). At 15 min after addition of MVs to cell cultures, most were observed to be associated with the cellular plasma membrane, whereas they were scarcely found within the cells. Nevertheless, the number of intracellular MVs increased with incubation time, and nearly all had entered the cells at 90 min. These intracellular MVs were subsequently routed to early endosomal compartments, and then they were sorted to lysosomal compartments within 90 min, suggesting that intracellular MVs are ultimately degraded by the cellular digestive machinery. However, P. gingivalis MVs remained for over 24 h and significantly induced acidified-compartment formation after being taken up by the cellular digestive machinery, which may be a type of cellular stress that initiates lysosome-specific impairment. There was no difference observed regarding the process of entry of MVs between HeLa and IHGE cells. However, it still remains unclear whether cellular function is impaired by entry of P. gingivalis MVs.
In the present study, we analyzed cellular functional impairment involved with entry of P. gingivalis MVs into HeLa and IHGE cells. In addition, the role of gingipains in cellular impairment was also investigated.
MATERIALS AND METHODS
Bacterial strains.
P. gingivalis strain ATCC 33277, as well as the ATCC 33277-derived mutant strains KDP129 (Δkgp), KDP133 (ΔrgpA ΔrgpB), KDP136 (Δkgp ΔrgpA ΔrgpB) (44), and KDP150 (ΔfimA) (45), kindly provided by K. Nakayama (Nagasaki University, Japan), were used in this study. The organisms were anaerobically maintained in blood agar plates and grown in Trypticase soy broth (Nissui, Tokyo, Japan) supplemented with hemin (5 μg/ml; Wako Pure Chemical Industries, Osaka, Japan) and menadione (1 μg/ml; Sigma-Aldrich, St. Louis, MO), as described previously (36). The activities of Rgp and Kgp were determined using the synthetic substrates t-butyloxycarbonyl-l-leucylglycyl-l-arginine-4-methylcoumaryl-7-amide and t-butyloxycarbonyl-l-valyl-l-leucyl-l-lysine-4-methylcoumaryl-7-amide, respectively (Peptide Institute, Osaka, Japan), as described previously (37).
Reagents.
Cytochalasin D, nocodazole, and methyl-β-cyclodextrin (MβCD) were purchased from Sigma-Aldrich, and wortmannin was purchased from Wako Pure Chemical Industries. These reagents were dissolved in an aliquot of dimethyl sulfoxide (1.1 μg/ml) and added to the culture medium. Alexa Fluor-conjugated phalloidin and transferrin were purchased from Invitrogen (Carlsbad, CA). KYT-1 and KYT-36, proteinase inhibitors specific for gingipains (22), were purchased from Peptide Institute (Osaka, Japan).
Cell culture.
Human cervical epithelial HeLa cells (CCL-2) were obtained from the American Type Culture Collection (Manassas, VA). The cells were grown in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; JRH Biosciences Inc., Lenexa, KS), penicillin (100 U/ml), and streptomycin (50 μg/ml) at 37°C in 5% CO2. IHGE cells (31) were kindly provided by S. Murakami (Osaka University, Japan) and maintained in Humedia KB-2 (Kurabo, Osaka, Japan), as described previously (27).
Preparation of P. gingivalis MVs.
P. gingivalis MVs were prepared as described previously (49). MVs from the mutants KDP129, KDP133, and KDP136 were prepared following treatment of the bacterial cells with exogenous gingipains from strain ATCC 33277, as described previously (26). Briefly, 500 ml of ATCC 33277 cell culture was centrifuged at 10,000 × g for 20 min, and then the supernatant was filtrated through a filter with 0.2-μm pores (Nalge Nunc, Rochester, NY). Next, an MV-depleted supernatant containing soluble gingipains was obtained by another centrifugation at 100,000 × g for 50 min. The mutants were inoculated into fresh culture medium containing 50% MV-depleted supernatant. The organisms were grown to the logarithmic phase until the optical density at 600 nm reached 1.0, after which the MVs were prepared.
Drug treatments.
Drug treatment experiments were performed as described previously (49), with minor modifications. Briefly, HeLa and IHGE cells were washed twice with serum-free DMEM and incubated with the drugs listed below in DMEM at 37°C for 30 min prior to the assay. To disorganize the cytoskeletal architecture, cytochalasin D (1 μg/ml) and nocodazole (25 μM) were used. MβCD (10 mM; lipid raft disrupting agent) and wortmannin (100 nM; phosphoinositide-3-kinase inhibitor) were also used. All reagents were included in the media throughout all of the experiments.
Entry assay and fluorescence microscopy.
HeLa and IHGE cells (6 × 104 cells per well) were seeded onto glass coverslips in 24-well plates the day before the assay. The cells were washed twice, then incubated in serum-free DMEM for 15 min with MVs (30 μg/ml). After a wash with DMEM, the cells were further incubated in DMEM containing 10% FBS for various periods. For immunostaining, the cells were washed with phosphate-buffered saline (PBS), fixed with 3% paraformaldehyde in PBS for 15 min, and permeabilized with 0.1% Triton X-100 in PBS for 5 min. After two washes with PBS, the cells were incubated in blocking solution (0.1% gelatin in PBS) for 10 min and subsequently with primary antibodies diluted with blocking solution at room temperature for 1 h. MVs were probed with rabbit polyclonal antibodies against native fimbriae of P. gingivalis ATCC 33277, which were prepared as described previously (53). The cells were analyzed using a laser scanning confocal microscope (model LSM510; Carl Zeiss, Thornwood, NY) and Zeiss LSM Image Browser software at a magnification of ×630. The laser power and pinhole size were set based on the optimal trade-off between the z resolution and signal/noise ratio of the images in each experiment by LSM Image Browser software to avoid artifacts at different depths within the cells. To analyze the distribution of MVs that entered the cells, those located on the inside at a distance of 1 μm from the outermost actin filament were counted manually. At least 20 cells were analyzed in each experiment, with three independent experiments performed.
Immunoblotting.
Immunoblotting analysis was performed as described previously (37). Briefly, HeLa and IHGE cells (6.0 × 105 cells per six-well culture dish) were incubated in serum-free DMEM with MVs (30 μg/ml) for 15 min, then washed and incubated in DMEM for various periods. Cells incubated with MVs were lysed with passive lysis buffer (Promega, Madison, WI) containing Nα-p-tosyl-l-lysine chloromethyl ketone (10 mM; TLCK; Wako) and 1% complete protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Soluble fractions were collected by centrifugation.
Antibodies.
Mouse antipaxillin, focal adhesion kinase (FAK), and aldolase monoclonal antibodies were purchased from BD Biosciences (San Jose, CA). The mouse anti-β-actin monoclonal antibody was purchased from Sigma-Aldrich, and the mouse anti-transferrin receptor monoclonal antibody was purchased from Zymed Laboratories (South San Francisco, CA). Rabbit anti-Rgp polyclonal antibodies were generous gifts from T. Kadowaki (Kyushu University, Fukuoka, Japan). Horseradish peroxidase-conjugated secondary antibodies (horse anti-mouse immunoglobulin G [IgG] and goat anti-rabbit IgG), used for immunoblotting, were purchased from Cell Signaling Technology (Danvers, MA). Alexa Fluor-conjugated secondary antibodies (goat anti-mouse IgG and goat anti-rabbit IgG), used for fluorescence microscopy, were purchased from Invitrogen.
In vitro wound healing assay.
HeLa and IHGE cells (6.0 × 105 cells per six-well culture dish) were cultured until confluent. The cell layers were scratched using a plastic tip and washed three times with serum-free DMEM to remove debris, as described previously (21). The cells were incubated with MVs (30 μg/ml) for 2 h in serum-free DMEM at 37°C in 5% CO2. External nonadherent MVs were removed by washing the cells three times with DMEM, after which the cells were further incubated in 10% FBS-DMEM for 12 or 24 h. The migrated cells in the scratched areas were counted under a microscope, and the closure rate for each scratched area was determined using Image J. At least 10 fields were analyzed, and all assays were performed in triplicate on three separate occasions (n = 9).
Separation of basic subcellular fractions.
Separation of subcellular fractions was performed as described previously (12), with minor modifications. Briefly, HeLa cells (3.0 × 106 cells per 10-cm dish) were cultured in 10% FBS-DMEM until confluent. The cells were washed and incubated in serum-free DMEM with or without MVs (30 μg/ml) for 15 min, then washed and incubated in 10% FBS-DMEM at 37°C for 60 min. Next, the cells were treated with homogenizing buffer (220 mM mannitol, 80 mM sucrose, 20 mM HEPES [pH 7.4], protease inhibitor cocktail, and TLCK), and centrifuged at 1,000 × g to obtain supernatant S1 and pellet P1. The S1 fraction was further centrifuged at 7,000 × g for 10 min to obtain supernatant S7 and pellet P7. The S7 fraction was centrifuged at 20,000 × g for 10 min to obtain supernatant S20 and pellet P20. The S20 fraction was further buffered to 100 mM with KCl and centrifuged at 100,000 × g for 1 h to obtain supernatant S100 and pellet P100. S100 and P100 samples were loaded onto a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel for immunoblotting.
Statistical analyses.
All data are presented as the means ± standard deviations. Statistical analyses were performed using an unpaired Student t test. Multiple comparisons were performed by one-way analysis of variance and Scheffé's test using STAT View software (SAS Institute Inc., Cary, NC).
RESULTS
Degradation of TfR and paxillin by MVs.
First, we evaluated the effects of MV entry on cellular functional molecules, including transferrin receptor (TfR) and paxillin. TfR, a carrier protein for transferrin, is indispensable for iron metabolism. Integrin-related signaling molecules such as paxillin and FAK regulate focal adhesions involved in cellular anchorage and directed migration, which control wound healing and regeneration (41). P. gingivalis was previously reported to degrade paxillin and FAK (18, 27). As shown in Fig. 1A, MVs entered the cells at 60 min after addition, at which time both TfR and paxillin were found to be clearly degraded. It was surprising that the MVs were able to degrade these proteins, which are associated with the plasma membrane, despite their cellular uptake by the endosomal machinery. The effects of inhibitors against MV entry (MβCD, wortmannin, and cytochalasin D) (10) on the degradation of TfR and paxillin were further evaluated. Preliminary results confirmed that these reagents had no effects on the degradation of TfR or paxillin (data not shown), whereas they significantly prevented the degradation of TfR and paxillin by MVs, indicating that adhesion of MVs to the plasma membrane via lipid rafts is needed for degradation of these cellular proteins (Fig. 1B). In contrast, nocodazole (microtubule assembly inhibitor) had a negligible effect.
FIG. 1.

Degradation of TfR and paxillin in cells incubated with MVs. (A) Fluorescence microscopy findings for cells incubated with MVs. HeLa cells were incubated with MVs (30 μg/ml) at 37°C for 15 min in serum-free DMEM. After a wash with DMEM, the cells were incubated for 45 min in DMEM containing 10% FBS. For fluorescence microscopy, the cells were processed for staining for MVs (green) and TfR (white), as well as paxillin (white). Bars = 20 μm. (B) Immunoblotting of TfR and paxillin in cells incubated with MVs. HeLa and IHGE cells were separately incubated with MVs (30 μg/ml) at 37°C for 15 min in serum-free medium. After a wash, the cells were incubated in the presence of MβCD, cytochalasin D, wortmannin, nocodazole, or dimethyl sulfoxide (control) in serum-containing medium for the indicated time periods after addition of MVs. Immunoblotting was performed using antibodies against paxillin and TfR.
Proteolytic activity of Rgp required for MV entry.
We also evaluated the roles of bacterial fimbriae and gingipains (Kgp and Rgp) on MV entry. Results of our previous study suggested that the interaction of P. gingivalis fimbriae with cellular α5β1 integrin promotes bacterial adhesion to epithelial cells, independent of lipid rafts, while the subsequent process of entry by the bacterium requires lipid raft components (50). Rgp is an enzyme that processes precursor fimbria proteins (45); thus, Rgp-null mutants lack mature fimbriae on their bacterial surfaces. Indeed, MVs from an Rgp-null mutant were found to negligibly adhere to epithelial cells (data not shown). However, we previously developed a novel method to express mature fimbriae on the bacterial surfaces of Rgp-null mutants using bacterial culture supernatants containing soluble Rgp (26). Gingipain-null mutant strains (Kgp, Rgp, and Kgp/Rgp null) were grown with exogenous gingipains, as described in Materials and Methods, and MVs prepared from these treated gingipain-null mutants were shown to possess mature fimbriae (Fig. 2A). Exogenous gingipain treatment restored the ability of the Rgp- and Kgp/Rgp-null MVs to bind to epithelial cells (data not shown), which was previously reported (26). Finally, the amounts of fimbria proteins and levels of gingipain activity were measured to ensure consistency among the different MVs (Fig. 2A and B).
FIG. 2.

Maturation of precursor fimbria proteins of gingipain-null MVs. (A) Immunoblotting of fimbria protein (fimbrillin, a subunit protein of fimbriae). MVs from gingipain-null mutants KDP129 (Δkgp), KDP133 (ΔrgpA ΔrgpB), and KDP136 (Δkgp ΔrgpA ΔrgpB) were prepared following treatment of the bacterial cells with exogenous gingipains of strain ATCC 33277, as described in Materials and Methods. A culture supernatant sample containing soluble gingipains without MVs was prepared from the wild strain as an exogenous gingipain fraction. Treated MVs from the gingipain-null mutant strains with the exogenous gingipain fraction were loaded onto sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and probed with antifimbria antibodies. (B) Gingipain activities of MVs. The activities of Rgp and Kgp are described in Materials and Methods.
The fimbriated Rgp-null MVs bound to the cellular plasma membrane, though they significantly lost their ability to enter HeLa and IHGE cells (Fig. 3A and B). Kgp/Rgp double-null MVs also failed to enter the cells. In contrast, Kgp-null MVs showed only a 30% reduction of entry efficiency. These results suggest that Rgp plays a major role in mediating intracellular entry via lipid rafts. The proteinase inhibitors specific for gingipains (KYT-36 for Kgp and KYT-1 for Rgp) were further analyzed to determine whether the proteolytic ability of Rgp was involved in MV entry. Exogenous KYT-1 clearly inhibited the entry of MVs into the cells at 30 min, whereas KYT-36 did not (Fig. 3C). However, the inhibitory effect became negligible at 120 min. These results suggest that the proteolytic activity of Rgp is a major factor in mediating MV entry, though another factor(s), such as the adhesive ability of Rgp, is also likely involved.
FIG. 3.

Loss of Rgp activity prevented MV entry into cells. (A) HeLa and IHGE cells were incubated separately with MVs prepared from strains ATCC 33277 (wild strain), KDP129 (Δkgp), KDP133 (ΔrgpA ΔrgpB), and KDP136 (Δkgp ΔrgpA ΔrgpB) at 37°C for 15 min in serum-free medium. After a wash, the cells were incubated in serum-containing medium for 120 min after the addition of MVs. For fluorescence microscopy, the cells were processed for staining for MVs (green) and actin (Alexa Fluor 568-conjugated phalloidin red). (B) Quantification of MV entry into cells. The percentages of mutant MVs that entered in comparison to wild-type MVs are shown. At least 20 cells were analyzed in each experiment, with three independent experiments performed. *, P < 0.01. (C) Wild-type MVs were incubated separately with KYT-1 (10 μM; inhibitor of Rgp) and KYT-36 (10 μM; inhibitor of Kgp) in serum-free DMEM at 37°C for 30 min, after which they were incubated with HeLa cells at 37°C for 15 min. After a wash with DMEM, the cells were incubated for the indicated time periods after addition of MVs in serum-containing medium. Bars (A and C) = 20 μm.
Kgp is responsible for degradation of integrin-related molecules, while Rgp is responsible for TfR degradation.
Next, we examined if gingipains were involved in the degradation of TfR, paxillin, and FAK using gingipain-null MVs and quantitatively evaluated that degradation over time using immunoblotting (Fig. 4). Kgp-null MVs degraded TfR as effectively as wild-type MVs, while paxillin and FAK were negligibly degraded. Unexpectedly, Rgp-null MVs, which only slightly entered the cells, degraded paxillin and FAK, though less effectively than the wild-type MVs. On the other hand, TfR was not degraded by Rgp-null MVs, and double-mutant Rgp/Kgp-null MVs failed to degrade any of the proteins. In addition, using fimbria-null MVs that had lost their ability to adhere to the cells, we confirmed that adhesion of MVs to epithelial cells via fimbriae was necessary for these degradations (data not shown).
FIG. 4.

Degradation of TfR and integrin-related molecules (paxillin and FAK) by MVs. HeLa and IHGE cells were incubated with wild-type and gingipain-null MVs at 37°C for 15 min in serum-free medium. After a wash, the cells were incubated in serum-containing medium for the indicated time periods after addition of MVs. The degradation of TfR, paxillin, and FAK was analyzed at each time point by an immunoblotting method.
Fluorescence microscopy analysis was also performed to observe the degradation of TfR and paxillin. As expected, TfR was clearly degraded by the Kgp-null MVs, whereas paxillin was not (Fig. 5A). Furthermore, Rgp-null MVs were localized on the cellular plasma membrane without marked entry; however, paxillin on the cellular plasma membrane was degraded, while TfR was not. Since TfR was apparently degraded by wild-type MVs, its effect on the cellular uptake of transferrin was examined. As shown in Fig. 5B, cells incubated with wild-type and Kgp-null MVs failed to take up transferrin in an extracellular manner, whereas such impairment was not observed in cells incubated with Rgp-null MVs. These results indicate that Rgp is involved in the degradation of TfR, resulting in impaired uptake of extracellular transferrin. However, it is unclear whether the negligible degradation of TfR by Rgp-null MVs was due to direct involvement of Rgp or the inaccessibility of intracellular TfR molecules to the Rgp-null MVs.
FIG. 5.

Fluorescence microscopy analysis of TfR and paxillin in cells incubated with MVs. (A) HeLa cells were incubated with wild-type and gingipain-null MVs (green) at 37°C for 15 min in serum-free medium. After a wash, the cells were incubated for 45 min in serum-containing medium. Distribution of TfR (white) and paxillin (white) was analyzed by confocal microscopy. (B) Following incubation with MVs for 15 min in serum-free medium, the cells were washed and incubated with Alexa Fluor 594-conjugated transferrin (white) at 37°C for 45 min in serum-containing medium. For fluorescence microscopy, cells were processed for staining with DAPI (4′,6-diamidino-2-pheylindole; blue). At least 20 cells were analyzed in each experiment, with three independent experiments performed. Bars (A and B) = 20 μm.
Coexistence of intracellular gingipains with fimbriae on MVs.
In our previous study, MV fimbriae were shown to be associated with intracellular MVs (10). In addition, another study found that purified gingipains in a single group were sorted to perinuclear regions by unknown machinery, following entry into HeLa cells (42). Thus, herein we verified the possibility that gingipains become detached from MVs and escape endocytic compartments to the cytoplasm for degradation of cellular proteins. As shown in Fig. 6, intracellular Rgp coexisted with fimbriae within insoluble fractions, indicating that gingipains were retained in the endocytic compartments with MVs. In addition, fimbria-null MVs did not demonstrate any degradation of TfR or paxillin, suggesting a negligible release of gingipains from MVs in our assay system (data not shown). Thus, the degradation of cellular proteins is likely dependent on MV-associated gingipains.
FIG. 6.
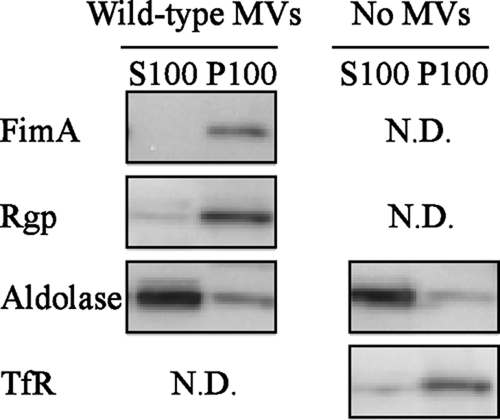
Coexistence of Rgp and fimbriae in the cellular plasma membrane fraction. HeLa cells were incubated with or without wild-type MVs at 37°C for 15 min in serum-free medium. After a wash, the cells were incubated at 37°C for 45 min in serum-containing medium. Separation of basic subcellular fractions was performed, and each was labeled separately with antibodies against fimbriae, Rgp, TfR (marker of insoluble fraction), and aldolase (marker of soluble fraction). The cellular homogenate was separated into supernatants (S100) and pellets (P100), as described in Materials and Methods.
Inhibition of cellular migration and proliferation by MVs.
Cellular migration and proliferation are critical functions for wound healing and tissue regeneration of periodontal tissues destroyed by periodontal pathogens (15), and P. gingivalis has been reported to inhibit those functions (18, 27). Thus, we examined if MVs inhibit cellular migration and proliferation using an in vitro wound healing assay (41). After HeLa and IHGE cell monolayers were scratched, the cells proliferated and migrated to fill the scratched areas in a time-dependent manner (Fig. 7). Scratch closure in HeLa cells was significantly inhibited at 12 and 24 h after the addition of wild-type MVs, with similar results seen with IHGE cells. These inhibitory effects were not clearly seen with the Rgp- and Rgp/Kgp-null MVs, whereas Kgp-null MVs inhibited scratch closure as effectively as the wild-type MVs. Furthermore, cells did not recover from this impairment after 24 h in the serum-containing condition, suggesting that the initial cellular damage caused by the MVs persisted over a long period. These results also indicate that Kgp, which degrades paxillin and FAK, is not significantly involved in inhibition of cellular migration and proliferation, whereas depletion of intracellular iron seems to be one of the causative factors. As for other important findings, it is possible that MV entry into cells is necessary to impair cellular migration and proliferation, together with Rgp activity.
FIG. 7.

Inhibition of migration and proliferation of epithelial cells by MVs. Confluent layers of HeLa and IHGE cells were scratched with a plastic tip, then incubated with wild-type and gingipain-null MVs at 37°C for 2 h in serum-free medium. Next, the cells were washed twice and allowed to proliferate at 37°C for 12 or 24 h in serum-containing medium, during which time they migrated to fill the scratched areas. The rates of wound closure were determined from assays performed in triplicate on three separate occasions (n = 9) and are shown beneath the images. *, P < 0.05.
DISCUSSION
Delivery of extracellular bacteria or bacterial products often occurs initially via endocytosis into the lumen of the host endocytic compartment. Thereafter, the invaded components occasionally move to the host cell cytoplasm through lysis of the endocytic compartment or delivery of proteins across the endocytic membrane via sophisticated machinery, such as the type III secretion system (4, 11). In previous studies, MVs secreted from enterotoxigenic E. coli and P. aeruginosa MVs were found to escape from the cellular digestive machinery for successful delivery of their virulence factors (5, 28). However, in a different study, P. gingivalis MVs failed to escape from endocytic compartments after entry and were finally sorted to lytic compartments (10). In the present study, we found that P. gingivalis MVs impaired cellular functions in HeLa and IHGE cells, as MV-associated gingipains degraded cellular proteins such as TfR, paxillin, and FAK, which resulted in depletion of intracellular transferrin and inhibition of cellular migration. P. gingivalis is harbored within the microbial biofilm (dental plaque) that develops on gingival margins (33). Based on the pathological features of chronic periodontitis caused by P. gingivalis, daily and continuous attacks by MVs, sometimes referred to as bacterial bombs, from the biofilm seem to destroy periodontal tissue. Thus, it has been suggested that P. gingivalis MVs are a chronic offensive weapon in the longstanding battle between the bacterial biofilm and the host.
Although MVs are not able to escape from endocytic compartments (10), P. gingivalis MVs degrade cellular functional proteins, which results in cellular functional impairment. Thus, it is important to determine how MVs are able to access TfR, paxillin, and FAK when enwrapped within endocytic compartments. We found that MVs swiftly degraded paxillin localized on the plasma membranes of HeLa and IHGE cells (Fig. 1A and 5A). Thus, MVs likely degrade paxillin while they are attached to and localized on the plasma membrane immediately before being isolated by endocytic compartments. In contrast, TfR is constantly internalized via endocytic vesicles that fuse with the early endosome and returns to the plasma membrane through the recycling endosome (1). P. gingivalis MVs entered via a lipid raft-dependent endocytic pathway and were subsequently routed to the early endosome, followed by sorting to lysosomal compartments (10). Localization of TfR on both the plasma membrane and interior faces of the endocytic compartments enwrapping MVs likely enables its degradation by the MVs. Indeed, several inhibitors of MV entry prevented the degradation of TfR (Fig. 4B). In contrast, TfR in the recycling pathway seems to be inaccessible to MVs. Indeed, paxillin was nearly completely degraded at 30 min after the addition of MVs, whereas TfR was not degraded at that time point, indicating that it takes a longer period of time to degrade TfR in motion (Fig. 4).
It is also important to consider why Rgp-null MVs, which only slightly entered the cells, are able to degrade paxillin. As noted above, paxillin is predominantly expressed on the cellular plasma membrane. We found that Rgp-null MVs adhered to that membrane (Fig. 5A) and that then Kgp from the MVs degraded paxillin, possibly due to direct access. These mechanisms may explain the phenomenon, though additional experiments are necessary.
In the present experiments, Rgp was necessary for efficient entry of MVs, indicating the possibility that Rgp exposes cellular cryptic ligands in a proteolytic manner and promotes the entry of MVs into host cells. Indeed, it was previously reported that treatment of matrix proteins with Rgp enhanced the binding of P. gingivalis fimbriae to those proteins, including collagen and fibronectin (29). Furthermore, bacterial invasion of gingival epithelial cells by P. gingivalis was reportedly inhibited by protease inhibitors (32). Thus, it is possible that a cellular cryptic ligand(s) on lipid rafts is exposed by Rgp, which is used by the cells to interact with fimbriae and/or other adhesins of P. gingivalis.
Our previous results showing that a Kgp-null mutant strain, KDP129, degraded paxillin and suggesting that the degradation occurred through the activity of Rgp (37) are contrary to the present results. Therefore, we performed the same assay using Kgp-null MVs with and without pretreatment with exogenous gingipains. In both conditions, MVs entered the epithelial cells with similar efficiencies. However, MVs without treatment degraded paxillin, whereas those with treatment did not, which was unexpected and indicated that pretreatment with exogenous gingipains provides contrasting results (data not shown). Rgp and Kgp were previously shown to process premature forms of various surface-associated proteins, including fimbriae, RgpA, RgpB, and Kgp (23); hemagglutinins (16); hemoglobin receptor protein (38); Tlr (47); HmuR (46); IhtA and IhtB (6); and RagA and RagB (17). Therefore, we think that the premature forms of RgpA and other premature proteins are related to the proteolysis of paxillin by an unknown mechanism, whereas the mature proteins are not, though further investigation is required. Nevertheless, we believe that treatment of Rgp- and Kgp-null mutants with exogenous gingipains, which maturate surface-associated proteins of gingipain mutants, is necessary to examine the definitive roles of Rgp and Kgp.
The present results also indicate that loss of intracellular TfR due to MVs causes serious impairment of cellular migration and proliferation. Fundamental cellular operations, including DNA synthesis and ATP generation, require iron, while transferrin-TfR complexes are internalized and ferric iron is released from transferrin at endosomal pH levels (1). This is the first known report to show that TfR degradation by P. gingivalis causes impairment of cellular functions, and it is notable that TfR is a target molecule of the bacterium.
Cells treated with MVs significantly lost paxillin/FAK proteins, which might be expected to evoke detachment of the cells from their substrate after short periods of incubation. However, the cells were not detached. This observation is consistent with the phenotype of paxillin- and FAK-deficient cells, which exhibited delayed spreading and migration in previous experiments but did not detach from the substrate (14, 20). In addition, Rgp-null MVs which degraded paxillin/FAK did not significantly inhibit cellular migration, suggesting that paxillin and FAK are not largely involved in inhibition of cellular migration and proliferation, whereas depletion of intracellular iron is likely one of the causative factors (Fig. 7). We intend to conduct additional studies to elucidate the influence of paxillin/FAK degraded by invading MVs.
Collectively, our findings suggest a molecular basis for host cell impairment by P. gingivalis MVs. Previous reports have shown that MVs possess various virulent abilities (9, 13, 25, 39, 40, 43, 48), while our results indicate a new aspect of cellular function impairment by P. gingivalis MVs.
Acknowledgments
This research was supported in part by a grant from the 21st Century Center of Excellence program entitled Origination of Frontier BioDentistry, held at Osaka University Graduate School of Dentistry, as well as grants-in-aid for fundamental research (B), exploratory research, and scientific research on innovative areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Editor: A. Camilli
Footnotes
Published ahead of print on 8 September 2009.
REFERENCES
- 1.Ajioka, R. S., J. D. Phillips, and J. P. Kushner. 2006. Biosynthesis of heme in mammals. Biochim. Biophys. Acta 1763:723-736. [DOI] [PubMed] [Google Scholar]
- 2.Amano, A., I. Nakagawa, N. Okahashi, and N. Hamada. 2004. Variations of Porphyromonas gingivalis fimbriae in relation to microbial pathogenesis. J. Periodontal Res. 39:136-142. [DOI] [PubMed] [Google Scholar]
- 3.Bauman, S. J., and M. J. Kuehn. 2006. Purification of outer membrane vesicles from Pseudomonas aeruginosa and their activation of an IL-8 response. Microbes Infect. 8:2400-2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanke, S. R. 2006. Portals and pathways: principles of bacterial toxin entry into host cells. Microbe 1:26-32. [Google Scholar]
- 5.Bomberger, J. M., D. P. Maceachran, B. A. Coutermarsh, S. Ye, G. A. O'Toole, and B. A. Stanton. 2009. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 5:e1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dashper, S. G., A. Hendtlass, N. Slakeski, C. Jackson, K. J. Cross, L. Brownfield, R. Hamilton, I. Barr, and E. C. Reynolds. 2000. Characterization of a novel outer membrane hemin-binding protein of Porphyromonas gingivalis. J. Bacteriol. 182:6456-6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.del Castillo, F. J., S. C. Leal, F. Moreno, and I. del Castillo. 1997. The Escherichia coli K-12 sheA gene encodes a 34-kDa secreted haemolysin. Mol. Microbiol. 25:107-115. [DOI] [PubMed] [Google Scholar]
- 8.Demuth, D. R., D. James, Y. Kowashi, and S. Kato. 2003. Interaction of Actinobacillus actinomycetemcomitans outer membrane vesicles with HL60 cells does not require leukotoxin. Cell. Microbiol. 5:111-121. [DOI] [PubMed] [Google Scholar]
- 9.Duncan, L., M. Yoshioka, F. Chandad, and D. Grenier. 2004. Loss of lipopolysaccharide receptor CD14 from the surface of human macrophage-like cells mediated by Porphyromonas gingivalis outer membrane vesicles. Microb. Pathog. 36:319-325. [DOI] [PubMed] [Google Scholar]
- 10.Furuta, N., K. Tsuda, H. Omori, T. Yoshimori, F. Yoshimura, and A. Amano. 2009. Porphyromonas gingivalis outer membrane vesicles enter human epithelial cells via an endocytic pathway and are sorted to lysosomal compartments. Infect. Immun. 77:4187-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerlach, R. G., and M. Hensel. 2007. Protein secretion systems and adhesins: the molecular armory of gram-negative pathogens. Int. J. Med. Microbiol. 297:401-415. [DOI] [PubMed] [Google Scholar]
- 12.Gjoen, T., T. O. Berg, and T. Berg. 1997. Subcellular fractionation of rat liver parenchymal cells by differential centrifugation, p. 179-180. In J. Graham and D. Rickwood (ed.), Subcellular fractionation: a practical approach. Oxford University Press, Oxford, England.
- 13.Grenier, D., and D. Mayrand. 1987. Functional characterization of extracellular vesicles produced by Bacteroides gingivalis. Infect. Immun. 55:111-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagel, M., E. L. George, A. Kim, R. Tamimi, S. L. Opitz, C. E. Turner, A. Imamoto, and S. M. Thomas. 2002. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell. Biol. 22:901-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hakkinen, L., V. J. Uitto, and H. Larjava. 2000. Cell biology of gingival wound healing. Periodontol. 2000 24:127-152. [PubMed] [Google Scholar]
- 16.Han, N., J. Whitlock, and A. Progulske-Fox. 1996. The hemagglutinin gene A (hagA) of Porphyromonas gingivalis 381 contains four large, contiguous, direct repeats. Infect. Immun. 64:4000-4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanley, S. A., J. Aduse Opoku, and M. A. Curtis. 1999. A 55-kilodalton immunodominant antigen of Porphyromonas gingivalis W50 has arisen via horizontal gene transfer. Infect. Immun. 67:1157-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hintermann, E., S. K. Haake, U. Christen, A. Sharabi, and V. Quaranta. 2002. Discrete proteolysis of focal contact and adherens junction components in Porphyromonas gingivalis-infected oral keratinocytes: a strategy for cell adhesion and migration disabling. Infect. Immun. 70:5846-5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hynes, S. O., J. I. Keenan, J. A. Ferris, H. Annuk, and A. P. Moran. 2005. Lewis epitopes on outer membrane vesicles of relevance to Helicobacter pylori pathogenesis. Helicobacter 10:146-156. [DOI] [PubMed] [Google Scholar]
- 20.Ilic, D., Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada, and T. Yamamoto. 1995. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377:539-544. [DOI] [PubMed] [Google Scholar]
- 21.Inaba, H., S. Kawai, K. Nakayama, K., N. Okahashi, and A. Amano. 2004. Effect of enamel matrix derivative on periodontal ligament cells in vitro is diminished by Porphyromonas gingivalis. J. Periodontol. 75:858-865. [DOI] [PubMed] [Google Scholar]
- 22.Kadowaki, T., A. Baba, N. Abe, R. Takii, M. Hashimoto, T. Tsukuba, S. Okazaki, Y. Suda, T. Asao, and K. Yamamoto. 2004. Suppression of pathogenicity of Porphyromonas gingivalis by newly developed gingipain inhibitors. Mol. Pharmacol. 66:1599-1606. [DOI] [PubMed] [Google Scholar]
- 23.Kadowaki, T., K. Nakayama, F. Yoshimura, K. Okamoto, N. Abe, and K. Yamamoto. 1998. Arg-gingipain acts as a major processing enzyme for various cell surface proteins in Porphyromonas gingivalis. J. Biol. Chem. 273:29072-29076. [DOI] [PubMed] [Google Scholar]
- 24.Kadowaki, T., R. Takii, K. Yamatake, T. Kawakubo, T. Tsukuba, and K. Yamamoto. 2007. A role for gingipains in cellular responses and bacterial survival in Porphyromonas gingivalis-infected cells. Front. Biosci. 12:4800-4809. [DOI] [PubMed] [Google Scholar]
- 25.Kamaguchi, A., K. Nakayama, S. Ichiyama, R. Nakamura, T. Watanabe, M. Ohta, H. Baba, and T. Ohyama. 2003. Effect of Porphyromonas gingivalis vesicles on coaggregation of Staphylococcus aureus to oral microorganisms. Curr. Microbiol. 47:485-491. [DOI] [PubMed] [Google Scholar]
- 26.Kato, T., T. Tsuda, H. Omori, T. Kato, T. Yoshimori, and A. Amano. 2007. Maturation of fimbria precursor protein by exogenous gingipains in Porphyromonas gingivalis gingipain-null mutant. FEMS Microbiol. Lett. 273:96-102. [DOI] [PubMed] [Google Scholar]
- 27.Kato, T., S. Kawai, K. Nakano, H. Inaba, M. Kuboniwa, I. Nakagawa, K. Tsuda, H. Omori, T. Ooshima, T. Yoshimori, and A. Amano. 2007. Virulence of Porphyromonas gingivalis is altered by substitution of fimbria gene with different genotype. Cell. Microbiol. 9:753-765. [DOI] [PubMed] [Google Scholar]
- 28.Kesty, N. C., K. M. Mason, M. Reedy, S. E. Miller, and M. J. Kuehn. 2004. Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J. 23:4538-4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kontani, M., S. Kimura, I. Nakagawa, and S. Hamada. 1997. Adherence of Porphyromonas gingivalis to matrix proteins via a fimbrial cryptic receptor exposed by its own arginine-specific protease. Mol. Microbiol. 24:1179-1187. [DOI] [PubMed] [Google Scholar]
- 30.Kuehn, M. J., and N. C. Kesty. 2005. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 19:2645-2655. [DOI] [PubMed] [Google Scholar]
- 31.Kusumoto, Y., H. Hirano, K. Saitoh, S. Yamada, M. Takedachi, T. Nozaki, Y. Ozawa, Y. Nakahira, T. Saho, H. Ogo, Y. Shimabukuro, H. Okada, and S. Murakami. 2004. Human gingival epithelial cells produce chemotactic factors interleukin-8 and monocyte chemoattractant protein-1 after stimulation with Porphyromonas gingivalis via toll-like receptor 2. J. Periodontol. 75:370-379. [DOI] [PubMed] [Google Scholar]
- 32.Lamont, R. J., A. Chan, C. M. Belton, K. T. Izutsu, D. Vasel, and A. Weinberg. 1995. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect. Immun. 63:3878-3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamont, R. J., and H. F. Jenkinson. 1998. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62:1244-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayrand, D., and S. C. Holt. 1988. Biology of asaccharolytic black-pigmented Bacteroides species. Microbiol. Rev. 52:134-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayrand, D., and D. Grenier. 1989. Biological activities of outer membrane vesicles. Can. J. Microbiol. 35:607-613. [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa, I., A. Amano, M. Kuboniwa, T. Nakamura, S. Kawabata, and S. Hamada. 2002. Functional differences among FimA variants of Porphyromonas gingivalis and their effects on adhesion to and invasion of human epithelial cells. Infect. Immun. 70:277-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakagawa, I., H. Inaba, T. Yamamura, T. Kato, S. Kawai, T. Ooshima, and A. Amano. 2006. Invasion of epithelial cells and proteolysis of cellular focal adhesion components by distinct types of Porphyromonas gingivalis fimbriae. Infect. Immun. 74:3773-3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okamoto, K., K. Nakayama, T. Kadowaki, N. Abe, D. B. Ratnayake, and K. Yamamoto. 1998. Involvement of a lysine-specific cysteine proteinase in hemoglobin adsorption and heme accumulation by Porphyromonas gingivalis. J. Biol. Chem. 273:21225-21231. [DOI] [PubMed] [Google Scholar]
- 39.Pham, K., D. Feik, B. F. Hammond, T. E. Rams, and E. J. Whitaker. 2002. Aggregation of human platelets by gingipain-R from Porphyromonas gingivalis cells and membrane vesicles. Platelets 13:21-30. [DOI] [PubMed] [Google Scholar]
- 40.Qi, M., H. Miyakawa, and H. K. Kuramitsu. 2003. Porphyromonas gingivalis induces murine macrophage foam cell formation. Microb. Pathog. 35:259-267. [DOI] [PubMed] [Google Scholar]
- 41.Ren, X. D., W. B. Kiosses, and M. A. Schwartz. 1999. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J. 18:578-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scragg, M. A., A. Alsam, M. Rangarajan, J. M. Slaney, P. Shepherd, D. M. Williams, and M. A. Curtis. 2002. Nuclear targeting of Porphyromonas gingivalis W50 protease in epithelial cells. Infect. Immun. 70:5740-5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma, A., E. K. Novak, H. T. Sojar, R. T. Swank, H. K. Kuramitsu, and R. J. Genco. 2000. Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activators of murine platelets. Oral Microbiol. Immunol. 15:393-396. [DOI] [PubMed] [Google Scholar]
- 44.Shi, Y., D. B. Ratnayake, K. Okamoto, N. Abe, K. Yamamoto, and K. Nakayama. 1999. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of Porphyromonas gingivalis. Construction of mutants with a combination of rgpA, rgpB, kgp, and hagA. J. Biol. Chem. 274:17955-17960. [DOI] [PubMed] [Google Scholar]
- 45.Shoji, M., M. Naito, H. Yukitake, K. Sato, E. Sakai, N. Ohara, and K. Nakayama. 2004. The major structural components of two cell surface filaments of Porphyromonas gingivalis are matured through lipoprotein precursors. Mol. Microbiol. 52:1513-1525. [DOI] [PubMed] [Google Scholar]
- 46.Simpson, W., T. Olczak, and C. A. Genco. 2000. Characterization and expression of HmuR, a TonB-dependent hemoglobin receptor of Porphyromonas gingivalis. J. Bacteriol. 182:5737-5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slakeski, N., S. G. Dashper, P. Cook, P., C. Poon, C. Moore, and E. C. Reynolds. 2000. A Porphyromonas gingivalis genetic locus encoding a heme transport system. Oral Microbiol. Immunol. 15:388-392. [DOI] [PubMed] [Google Scholar]
- 48.Srisatjaluk, R., G. J. Kotwal, L. A. Hunt, and D. E. Justus. 2002. Modulation of gamma interferon-induced major histocompatibility complex class II gene expression by Porphyromonas gingivalis membrane vesicles. Infect. Immun. 70:1185-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsuda, K., A. Amano, K. Umebayashi, H. Inaba, I. Nakagawa, Y. Nakanishi, and T. Yoshimori. 2005. Molecular dissection of internalization of Porphyromonas gingivalis by cells using fluorescent beads coated with bacterial membrane vesicle. Cell Struct. Funct. 30:81-91. [DOI] [PubMed] [Google Scholar]
- 50.Tsuda, K., N. Furuta, H. Inaba, S. Kawai, K. Hanada, T. Yoshimori, and A. Amano. 2008. Functional analysis of α5β1 integrin and lipid rafts in invasion of epithelial cells by Porphyromonas gingivalis using fluorescent beads coated with bacterial membrane vesicles. Cell Struct. Funct. 33:123-132. [DOI] [PubMed] [Google Scholar]
- 51.Wai, S. N., B. Lindmark, T. Soderblom, A. Takade, M. Westermark, J. Oscarsson, J. Jass, A. Richter Dahlfors, Y. Mizunoe, and B. F. Uhlin. 2003. Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell 115:25-35. [DOI] [PubMed] [Google Scholar]
- 52.Williams, R. C., and S. Offenbacher. 2000. Periodontal medicine: the emergence of a new branch of periodontology. Periodontol. 2000 23:9-12. [DOI] [PubMed] [Google Scholar]
- 53.Yoshimura, F., T. Takasawa, M. Yoneyama, T. Yamaguchi, H. Shiokawa, and T. Suzuki. 1985. Fimbriae from the oral anaerobe Bacteroides gingivalis: physical, chemical, and immunological properties. J. Bacteriol. 163:730-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou, L., R. Srisatjaluk, D. E. Justus, and R. J. Doyle. 1998. On the origin of membrane vesicles in gram-negative bacteria. FEMS Microbiol. Lett. 163:223-228. [DOI] [PubMed] [Google Scholar]