

Abstract
A portion of the total cellular pool of the Legionella pneumophila chaperonin, HtpB, is found on the bacterial cell surface, where it can mediate invasion of nonphagocytic cells. HtpB continues to be abundantly produced and released by internalized L. pneumophila and may thus have postinvasion functions. We used here two functional models (protein-coated beads and expression of recombinant proteins in CHO cells) to investigate the competence of HtpB in mimicking early intracellular trafficking events of L. pneumophila, including the recruitment of mitochondria, cytoskeletal alterations, the inhibition of phagosome-lysosome fusion, and association with the endoplasmic reticulum. Microscopy and flow cytometry studies indicated that HtpB-coated beads recruited mitochondria in CHO cells and U937-derived macrophages and induced transient changes in the organization of actin microfilaments in CHO cells. Ectopic expression of HtpB in the cytoplasm of transfected CHO cells also led to modifications in actin microfilaments similar to those produced by HtpB-coated beads but did not change the distribution of mitochondria. Association of phagosomes containing HtpB-coated beads with the endoplasmic reticulum was not consistently detected by either fluorescence or electron microscopy studies, and only a modest delay in the fusion of TrOv-labeled lysosomes with phagosomes containing HtpB-coated beads was observed. HtpB is the first Legionella protein and the first chaperonin shown to, by means of our functional models, induce mitochondrial recruitment and microfilament rearrangements, two postinternalization events that typify the early trafficking of virulent L. pneumophila.
The gram-negative bacterium Legionella pneumophila is an intracellular parasite of amoebae (67) that has emerged as an accidental human pathogen capable of replicating in mononuclear phagocytes (38), primarily alveolar macrophages. Lung infection by L. pneumophila usually begins after the inhalation of contaminated water aerosol and manifests as an atypical pneumonia known as Legionnaires' disease (86).
The early events of L. pneumophila infection are well described at the cellular level. The first steps of infection are bacterial attachment to host cell receptors and subsequent internalization by conventional phagocytosis (27, 61, 75), coiling phagocytosis (12, 35), or macropinocytosis (84). Once internalized, L. pneumophila remains contained within a membrane-bound compartment, which is transformed into a specialized vacuole referred to as the Legionella-containing vacuole (LCV). The major cellular events of LCV conditioning include recruitment of vesicles and mitochondria (26, 33, 60), avoidance of both acidification and fusion with lysosomes (34, 37), and association with the endoplasmic reticulum (ER) (33, 42, 79, 80). Less known (or predictable) are the early changes in F-actin organization induced by L. pneumophila, which seem to be unrelated to the F-actin rearrangements required for its internalization (16, 44, 60, 78).
At the molecular level, at least five bacterial gene products, RtxA, EnhC (13, 14), LpnE (58), LvhB2 (65), and HtpB (25) are involved in cell entry, confirming L. pneumophila's flexibility in the use of alternate entry pathways. Conditioning of the LCV requires the Dot/Icm type IV secretion system of L. pneumophila, suggesting that the translocation of type IV-secreted effectors into host cells is essential for the recruitment of organelles and avoidance of acidification and fusion with lysosomes (3, 15, 53). Although a number of specific Dot/Icm effectors have been identified that mediate the sequestration of ER-derived vesicles to the LCV (19, 40, 48, 50, 63), no specific gene products have been linked to the recruitment of mitochondria or the inhibition of LCV-lysosome fusion.
It has been hypothesized that the factors that mediate the internalization of L. pneumophila are preformed and may also participate in the early conditioning of the LCV (see, for example, reference 41). This hypothesis is supported by the following observations: (i) conditioning of the LCV begins within minutes after L. pneumophila internalization (18, 42, 49, 68), and (ii) antibiotic-treated legionellae (incapable of de novo protein synthesis) are not affected in their ability to attach to or enter host cells (26, 39) and resume intracellular growth immediately after removal of the antibiotic (39). It seems that L. pneumophila is prearmed to deploy a sequence of coordinated events (which follow a precise timing) immediately after making contact with a host cell, a notion also suggested by detailed microscopy studies conducted in the genetically tractable amoeba Dictyostelium discoideum (49). Moreover, some of the infection steps clearly have a short duration (49), suggesting that L. pneumophila may transiently alter a number of cellular processes. The transient nature of such effects sometimes depends on the host cell being infected. For instance, in human macrophages avoidance of both LCV acidification and fusion with lysosomes persists throughout the intracellular growth cycle (70, 85), whereas in murine macrophages, LCVs acidify and fuse with lysosomes toward the end of the L. pneumophila replicative phase (76). In contrast, other LCV conditioning processes, such as the association with mitochondria and ER, are structurally maintained (regardless of the cellular host being infected) until the replicative phase of the intracellular growth cycle has been completed (26, 33, 60).
L. pneumophila HtpB is a member of the group 1 chaperonins, which includes the evolutionarily conserved and essential chaperonins of bacteria, mitochondria and plastids, with well-characterized roles in protein folding (28). The function of bacterial chaperonins, however, is not limited to protein folding. Chaperonins of bacteria can mediate adherence to mammalian cells (22), stabilize membrane lipids (81), paralyze insects (87), and activate eukaryotic signaling cascades (51, 88). The expression of HtpB is upregulated in the presence of L929 murine cells or human monocytes, and high levels of expression are maintained during the course of intracellular infection (21), leading to its accumulation in the lumen of the LCV, as shown in infected HeLa cells (24). The increased production of HtpB in L929 cells and monocytes correlates with virulence because spontaneous salt-tolerant, avirulent mutants of L. pneumophila are unable to upregulate the expression of HtpB upon contact with these host cells (21). In addition, HtpB is found in association with the L. pneumophila cytoplasmic membrane (5, 23), as well as on the bacterial cell surface (24), where it can mediate invasion of HeLa cells (25). As a L. pneumophila factor that mediates cell entry (25), and one that continues to be abundantly produced and released into the LCV after L. pneumophila internalization (24, 32), HtpB may participate in the early intracellular establishment of L. pneumophila. In the present study, we show that microbeads coated with purified HtpB (but not uncoated beads or beads coated with control proteins) are sufficient to attract mitochondria and transiently modify the organization of actin microfilaments in mammalian cells, two postinternalization events that typify the early trafficking of virulent L. pneumophila.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
L. pneumophila strain Lp02 and the Lp02dotA mutant JV309 (3, 83) were obtained from R. Isberg (Tufts University Medical School, Boston, MA). The Lp02dotB mutant JV303 (83) was obtained from J. Vogel (Washington University School of Medicine, St. Louis, MO). Lp02, Lp02dotA, and Lp02dotB were grown at 37°C on BCYE agar, or BYE broth, containing thymidine and streptomycin (both at 100 μg/ml) (3). Escherichia coli strains DH5α (cloning host) and JM109 (used to over produce GroEL and HtpB) were grown at 37°C on LB agar with or without antibiotics, as required (69). Bacterial medium components were from Difco-BD (Sparks, MD), EM Science (Gibbstown, MD), and (or) Sigma-Aldrich (St. Louis, MO).
GFP and RFP expression in L. pneumophila.
Plasmid pBH6119::htpAB, carrying the gfp gene under the control of the htpAB promoter (4), was a gift from K. Brassinga (University of Manitoba, Winnipeg, Manitoba, Canada). Plasmid pSW001 (52), encoding DsRed fluorescent protein (RFP), was a gift from H. Hilbi (ETH Zürich). Lp02 and Lp02dotB were transformed with pBH6119::htpAB or pSW001 by electroporation (4, 82), and green fluorescent protein (GFP)-expressing transformants were selected at 37°C on BCYE agar with streptomycin (100 μg/ml), and RFP-expressing transformants on BCYE agar with streptomycin and thymidine (100 μg/ml) and chloramphenicol (5 μg/ml).
Cell culture.
Wild-type, proline-requiring Chinese hamster ovary (CHO-wt) cells (29) were obtained from R. Gupta (McMaster University, Hamilton, Ontario, Canada). CHO-wt cells were cultured in minimal essential medium (MEM) supplemented with 5% fetal bovine serum (FBS), 100 U of penicillin/ml, and 100 μg of streptomycin/ml (all from Gibco-Invitrogen [Grand Island, NY], except for the FBS [HyClone, Logan, UT]). We also used CHO-htpB cells to compare the effects of HtpB-coated beads with the effects of cytoplasmic recombinant HtpB in the same cells. CHO-htpB cells are the stably transfected line of CHO-AA8 Tet-Off cells (Clontech-BD, Palo Alto, CA) containing the htpB gene cloned into pTRE2hyg (see below). CHO-htpB cells were grown at 37°C and 5% CO2 in αMEM supplemented with 5% FBS, 100 U of penicillin/ml, 100 μg of streptomycin/ml, 300 μg of Geneticin/ml, and 200 μg of hygromycin B/ml (all from Gibco except the FBS) and 10 ng of doxycycline (Clontech)/ml. Geneticin and hygromycin assured maintenance of the integrated plasmid, and doxycycline repressed HtpB expression. In the presence of 10 ng of doxycycline/ml, CHO-htpB and CHO-AA8 Tet-Off cells were similar in relation to morphology and permissiveness to L. pneumophila (10 ng of doxycycline/ml did not inhibit the growth of Lp02 or Lp02dotA). U937 cells were obtained from A. Issekutz (Dalhousie University) and grown in suspension in RPMI 1640 (Gibco) supplemented with 10% FBS, 2 mM l-glutamine (Gibco), 100 U of penicillin/ml, and 100 μg of streptomycin/ml at 37°C in 5% CO2. U937 cells differentiated into adherent, macrophagelike cells with 60 ng of phorbol 12-myristate 13-acetate (PMA; Sigma)/ml and plated at a density of 3 × 106 cells/well in six-well culture plates (Falcon-BD Biosciences Canada, Mississauga, Ontario, Canada). After PMA activation (24 h), the cells were washed once with RPMI 1640 supplemented with 5% FBS and 2 mM l-glutamine. Adherent U937-derived macrophages were further incubated for 24 to 48 h in the same medium prior to the addition of beads or bacteria.
Constructs for expression of recombinant proteins in CHO-AA8 Tet-Off cells.
Plasmid pTRE2hyg (PhCMV*-1, ColE1ori, Ampr Hygr; Clontech) was used for the expression of HtpB, GroEL, and DsRed. (i) The primers CHOhtpB forward and CHOhtpB reverse (Table 1) were used to PCR amplify htpB from plasmid pSH16 (31). The amplification product was T/A cloned into the EcoRV site of pBluescript KS (Stratagene, La Jolla, CA) to create pBS::htpB-1, from which a BamHI-ClaI fragment was subcloned into pTRE2hyg to generate pTRE2-htpB hyg. (ii) The primers groEL forward and groEL reverse (Table 1) were used to amplify groEL from E. coli DH5α chromosomal DNA. The PCR product was T/A cloned into the EcoRV site of pBluescript to create pBS::groEL, from which a NotI-SalI fragment was subcloned into pTRE2hyg to generate pTRE2-groEL hyg. Insertion of a Kozak sequence (45) and modification of the first leucine codon of the htpB sequence (Table 1) enhanced expression of the bacterial chaperonins in CHO cells. (iii) A BamHI-NotI fragment from plasmid pDsRed2 (pUCori, Plac, Ampr) encoding the RFP drFP583 from Discosoma sp. (Clontech) was subcloned into pTRE2hyg (using the same restriction sites) to generate pTRE2-Dsred2 hyg. Construct integrity was confirmed by DNA sequencing (DalGen; Dalhousie University). CHO-AA8 Tet-Off cells were transiently transfected by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Stably transfected CHO-htpB cells were isolated as single colonies of CHOAA8 Tet-Off cells carrying pTRE2-htpB hyg after dilution-cloning in medium with hygromycin B (200 μg/ml).
TABLE 1.
Sequences of PCR primers
| Primer | Sequence (5′-3′)a | Restriction site |
|---|---|---|
| CHOhtpB forward | AGTCTAGACAGCCGCCATGGCTAAAGAACTGCGTTTTGGTGATGA | None |
| CHOhtpB reverse | ACCTCTAGAAACTTACATCATTCCGCC | None |
| groEL forward | AGCGGCCGCCGCCATGGCAGCTAAAGACGTA | None |
| groEL reverse | GTGTCGACCACTTACATCATGCCGCCCAT | None |
| htpABP1 | CCCCGCGGCCGCTCAAGAGGTGTTGCTTCAGG | NotI |
| htpABP2 | CCCCGGATCCCCATACGACGAACAACAACG | BamHI |
| htpABP3 | CCCCGGATCCTGGGCGGAATGATGTAATTT | BamHI |
| htpABP4 | CCCCCTCGAGGGCACTGATTCCATATCAACTG | XhoI |
| htpB-Forward | GCCATTGCTCAAGTTGGAACTAT | None |
| htpB-Reverse | GCGTTGAAACCGTAGTTGTCTTT | None |
The boldfacing indicates a Kozak sequence, and the boldface italics indicates a modified leucine codon. The restriction sites are underlined.
Expression of recombinant proteins in CHOAA8 Tet-Off cells.
Expression from the pTRE2-hyg constructs (previous section) was induced in the absence of doxycycline and confirmed in cells grown on 22-by-22-mm coverslips placed inside six-well plates, by direct (DsRed2) or indirect (HtpB and GroEL) fluorescence microscopy using a BX61 Olympus microscope (Olympus Canada, Markham, Ontario, Canada). For indirect fluorescence microscopy, cells on coverslips were fixed in 4% paraformaldehyde for 10 min, permeabilized in 0.1% Triton X-100 for 5 min, and blocked in 2% bovine serum albumin (BSA) for 30 min, all prepared in phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 2 mM KH2PO4, 10 mM NaH2PO4 [pH 7.4]). Immunostaining was done with our rHtpB-specific polyclonal antibody (see below) diluted 1:500 and Alexa Fluor-546 secondary antibody (Molecular Probes-Invitrogen, Carlsbad, CA) diluted 1:200. All washes were done by passing the coverslips 3× in fresh PBS for 10 min. For stably transfected CHO-htpB cells, ectopic expression of HtpB (referred to as eHtpB) was assessed by fluorescence microscopy and immunoblotting at different concentrations of doxycycline. CHO-htpB cells seeded at a density of ∼5 × 105 cells/well into six-well plates (Falcon) were grown overnight with 10 ng of doxycycline/ml. Cells were washed and further cultured for 48 h in medium with different concentrations of doxycycline. Cells were then detached with 2 mg of trypsin (Sigma)/ml, harvested, and washed with PBS, before being counted in an improved Neubauer chamber (Hausser Scientific, Horsham, PA). 5× Laemmli sample buffer (5 μl) containing 10% 2-mercaptoethanol was added to ∼6 × 105 cells in 10 μl of PBS. Samples were boiled for 10 min and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting according to the procedures of Laemmli and Towbin et al. as reported previously (24). Proteins transferred to a PALL nitrocellulose membrane (VWR Canada, Mississauga, Ontario, Canada) were stained with Ponceau-S, and a reference TIFF image was obtained in an Epson ES 1200C scanner (Epson Canada, Toronto, Ontario, Canada). Membranes were then immunostained with a hybridoma supernatant of HtpB-specific monoclonal antibody GW2X4B8B2H6 (30) diluted 1:200, followed by alkaline phosphatase-conjugated anti-mouse immunoglobulin G (IgG; Cedarlane Laboratories, Hornby, Ontario, Canada) diluted 1:5,000. Color was developed with 5-bromo-4-chloro-3-indolylphosphate (BCIP) and nitroblue tetrazolium (Sigma), and a TIFF image of the stained membrane was produced using an Epson ES 1200C scanner. Stained bands were analyzed by densitometry (on the unprocessed TIFF images) using the single-band analysis tool of Gel-Pro Analyzer v.4.5 software (Media Cybernetics, Silver Spring, MD).
Purification of HtpB from L. pneumophila, recombinant HtpB from E. coli, and GroEL from E. coli.
Lp02 harboring the htpAB operon in RSF1010 (1) or E. coli JM109 harboring plasmid pSH16 (pUC19 with the htpAB operon insert [31]) were grown, respectively, at 30°C for 48 h, 150 rpm, in 2 liters of BYE broth with kanamycin (30 μg/ml) and at 30°C for 24 h and 150 rpm in 2 liters of LB broth with ampicillin (100 μg/ml), followed by heat shock at 42°C for 2 h. Cultures were centrifuged at 6,000 × g for 20 min, and pelleted bacterial cells were resuspended in 40 ml of column buffer (50 mM Tris [pH 7.6], 35 mM KCl, 25 mM NH4Cl, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 5 mM dithiothreitol). Crude bacterial lysates were made by passing the bacterial suspensions four times through a French press cell at 20,000 lb/in2. Sarkosyl (2% in water) was added (200 μl) to the lysate prior to sonication in three cycles, each for 1 min, plus 4 min of cooling on ice. Lysates were clarified by centrifugation at 8,800 × g for 10 min. HtpB was precipitated from the clarified lysate with (NH4)2SO4 (41% saturation), resolubilized in column buffer, and subjected to ion-exchange chromatography in a DE-52 (Whatman, Clifton, NJ) 50-ml column, eluting with 1 liter of a 0 to 0.4 M KCl gradient. Fractions containing HtpB (detected by dot immunoblot with monoclonal antibody GW2X4B8B2H6) were pooled and concentrated by ultrafiltration in a positive pressure 400-ml chamber (Amicon, Beverly, MA) using a 10,000 molecular weight cutoff, 70-mm Spectrapor membrane (Spectrum, Rancho Dominguez, CA). The concentrated sample was then sequentially run through a 0.6-liter G200 and a 0.4-liter G75 Sephadex column (Amersham-GE Healtcare Canada, Baie d'Urfé, Quebec, Canada). Fractions containing HtpB were pooled, dialyzed, and concentrated by ultrafiltration. The final protein concentration was determined by using a protein assay (Bio-Rad Canada, Mississauga, Ontario, Canada). Recombinant HtpB from E. coli contains GroEL and is referred to as rHtpB, whereas the HtpB from L. pneumophila is simply referred to as HtpB. GroEL was purified from E. coli JM109 harboring plasmid pTrcGroE (obtained from P. B. Sigler, Molecular Biophysics and Biochemistry, Yale University), which is pTrc99a carrying the E. coli groELS operon under the trc promoter. E. coli-pTrcGroE was grown at 37°C for ∼18 h at 150 rpm, in 2 liters of LB containing ampicillin (100 μg/ml) and then incubated for 1.5 h at 40°C with 5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to induce maximal production of GroEL. Bacterial cells were harvested and lysed, and GroEL purified from the crude bacterial lysate as described above for HtpB and rHtpB. Detection of GroEL in the various chromatography fractions was done by dot immunoblot with the GroEL-specific monoclonal antibody SPA-870 (StressGen-Assay Designs, Ann Arbor, MI). Our in-house GroEL should be distinguished from the commercial one (StressGen), referred to as GroEL. Reagents used in chaperonin purifications were all analytical grade from Sigma, Fisher Scientific (Fair Lawn, NJ), or BDH, Inc. (Toronto, Ontario, Canada).
Anti-rHtpB serum.
Purified rHtpB (100 μg) was emulsified in a total of 300 μl of complete Freund adjuvant (Sigma), and subcutaneously injected into three different sites (100 μl/site) on the back of a New Zealand White rabbit (Charles River Canada, Saint-Constant, Quebec, Canada). The initial injection was followed by two boosters (3 weeks apart) of 100 μg of rHtpB in 300 μl of incomplete Freund adjuvant (Sigma), subcutaneously injected on the back in three different sites (100 μl/site). The rabbit was euthanized 9 weeks after the initial injection (3 weeks after the second booster). Hyperimmune serum was recovered and kept frozen at −80°C for long-term storage or at 4°C for short-term storage. Rabbit was cared for following guidelines outlined by the Canadian Council for Animal Care, under a protocol approved by the Dalhousie University Committee on Laboratory Animals.
Protein-coated beads.
Polystyrene carboxylated beads (∼9 × 109), 1 μm in diameter, with blue (excitation, 365 nm/emission, 415 nm), yellow-green (505/515), or crimson red (625/645) fluorescence (Molecular Probes, Eugene, OR) were coated with 200 μg of BSA (Sigma), GroEL, in-house GroEL, rHtpB, or HtpB using a carbodiimide kit (Polysciences, Warrington, PA) in a total volume of 1 ml. Levels of bound proteins were equivalent between beads (11 ± 7 fg/bead, with GroEL [5 fg/bead] and in house GroEL [18.8 fg/bead] showing the lowest and highest average levels), as estimated by subtracting protein concentrations before and after reaction with carbodiimide-activated beads (Bio-Rad assay). Bound chaperonins were confirmed by dot immunoblot: 3 μl of coated beads (∼6 × 106 beads) was spotted onto nitrocellulose and stained with our rHtpB-specific polyclonal antibody at a 1:1,000 dilution, followed by goat anti-rabbit IgG conjugated to alkaline phosphatase (Cedarlane Laboratories) at 1:5,000 dilution, all in 0.1 M Tris-buffered saline (pH 8). Pretreatment of ∼108 beads with rHtpB-specific antiserum (diluted 1:100) was done at 37°C for 1 h in 200 μl of PBS with 0.2% BSA, followed by three washes with 1 ml of PBS. All beads were stored in PBS with 0.05% sodium azide (J. T. Baker, Phillipsburg NJ). Before use, all beads were washed three times with PBS and counted directly in a Petroff-Hausser chamber (Hausser Scientific).
Association of beads with cells.
CHO-wt and CHO-htpB cells seeded on 22-by-22 mm no. 0 glass coverslips (Fisher Scientific) in six-well plates (5 × 105 cells/coverslip) were cultured for 48 h in 3 ml of culture medium. Cells were washed three times with 1 ml of PBS, and differently coated blue fluorescent beads were added at a bead/cell ratio of 20 in culture medium. Plates were centrifuged at 1,000 × g for 5 min at 25°C, to enhance bead-cell contact, and incubated for different times at 37°C in 5% CO2. Unattached beads were removed by six washes with PBS. The cells were then stained for 30 min at 37°C in 5% CO2 with 5 μM 5-chloro-methyl-fluorescein diacetate (Molecular Probes) in 1 ml of medium with antibiotics, washed three times with PBS, and chased for 20 min with 1 ml of fresh medium. Cells were then fixed in 4% paraformaldehyde in PBS for 10 min and mounted with Vectashield (Vector Laboratories Canada, Burlington, Ontario, Canada) on glass slides. The number of beads/cell was determined by direct fluorescence microscopy counts in a BX61 Olympus microscope.
Beads and L. pneumophila association with mitochondria.
CHO-wt and CHO-htpB cells were seeded onto coverslips in six-well plates and incubated with differently coated blue fluorescent beads, as described in the previous section. L. pneumophila grown on BCYE for 3 days were harvested in 5 ml of distilled H2O (dH2O), and the bacterial suspension standardized to 1 optical density unit at 620 nm (∼109 bacterial cells/ml). An inoculum of ∼108 bacteria was prepared in 3 ml of αMEM without antibiotics, vortexed thoroughly, and added to each well. The plates were centrifuged at 1,000 × g for 5 min at 25°C to promote bead- or bacterium-cell contact and incubated for 1 h to allow internalization. Unattached beads or bacteria were removed with six washes with PBS and the mitochondria were stained (1 h samples), or the cells were further incubated for 2 h with fresh medium prior to staining (3 h samples). Staining was done with 300 nM MitoTracker Orange CMTMRos (Molecular Probes) diluted in prewarmed αMEM-5% FBS with antibiotics for 45 min at 37°C in 5% CO2, followed by three PBS washes, and a 10-min chase in fresh medium. Association with mitochondria was quantified in a BX61 Olympus fluorescence microscope equipped with an Evolution QET monochrome camera (Media Cybernetics), and images were taken and processed by using Image Pro Plus (version 5.0.1; Media Cybernetics).
Isolation and immunolabeling of phagosomes containing beads.
Four 25-cm2 cell culture flasks (Falcon) were seeded with 107 PMA-activated U937 cells in 7 ml of fresh RPMI 1640, and incubated for 2 days. After the nonadherent cells were washed with RPMI, each flask contained ∼5 × 106 U937-derived macrophages, which were incubated with red fluorescent Lp02, or beads, at a ratio of ∼1,000 bacteria/cell or ∼100 beads/cell. U937 cells did not efficiently take up Lp02dotB, and thus formalin-killed red fluorescent Lp02 (10% formalin in PBS for 10 min, followed by three washes with PBS) was used as a negative control. Flasks were centrifuged at 500 × g for 10 min at 25°C in a swing-out no. 1622 rotor of a Universal 32R centrifuge (Hettich, Beverly, MA) and subsequently incubated at 37°C for 3 h with hourly centrifugations (as described above) to maximize bacterium- or bead-cell contact. Phagosomes containing bacteria or beads were then isolated by the method of Chakraborty et al. (9) using siliconized polypropylene microcentrifuge tubes (Sigma). The cells were washed three times with 5 ml of PBS at 37°C and scraped into 1 ml of cold homogenization buffer (250 mM sucrose, 20 mM HEPES, 0.5 mM EGTA, and 0.1% gelatin, adjusted to pH 7.0 with 6 N KOH). Scraped cells were lysed by 12 forceful passages through a 27-gauge needle, and the lysate was centrifuged at 400 × g for 10 min at 4°C to pellet the cell debris and unbroken cells. The supernatant (∼1 ml) was centrifuged at 2,000 × g for 20 min at 4°C to pellet phagosomes, which were either processed for electron microscopy (see below) or fixed for 15 min at 4°C in 2% paraformaldehyde in 1 ml of PBS. Fixed phagosomes were pelleted by centrifugation at 12,000 × g for 3 min and resuspended in 1 ml of PBS with 0.1% Triton X-100 for 10 min at room temperature. Immunolabeling with an anti-cytochrome c mouse monoclonal (0.5 mg/ml; Zymed-Invitrogen, Carlsbad, CA) and Alexa Fluor 546-conjugated goat anti-mouse IgG (Molecular Probes) was performed as follows, in a volume of 200 μl: step 1, 2% BSA in PBS added to phagosomes for 1 h at room temperature; step 2, monoclonal antibody diluted 1:600 in PBS with 0.2% BSA for 1 h at 37°C; and step 3, anti-mouse IgG diluted 1:1,000 in PBS with 0.2% BSA for 1 h at 37°C. All steps were followed by two washes in PBS with centrifugation at 12,000 × g for 3 min at room temperature in a Hettich MICRO 20 microcentrifuge. Immunolabeled phagosomes were resuspended in 0.4 ml of PBS and run in a FACScalibur (BD Biosciences Canada, Mississauga, Ontario, Canada) to score the colocalization of red fluorescence (bacteria or beads) and green fluorescence (mitochondria). Ten thousand events were acquired per sample with the forward scatter acquisition set at E02, and data from the FL1 and FL4 channels were saved and plotted.
Effect of beads, L. pneumophila, and ectopic expression of HtpB on cytoskeletal organization.
CHO-htpB cells, grown on 22-by-22-mm coverslips in six-well plates (as described above), were either incubated with green fluorescent beads for up to 3 h, infected with L. pneumophila for up to 3 h, or washed three times with fresh αMEM without doxycycline and further cultured in this medium for 48 h. Cells were then fixed for 10 min in 4% paraformaldehyde in αMEM, permeabilized for 5 min in 0.1% Triton X-100 in PBS, and blocked for 30 min in 2% BSA in PBS. F-actin was stained for 20 min with 5 U of Alexa Fluor 546-conjugated phalloidin (Molecular Probes)/ml. Tubulin was stained with monoclonal antibody DM1A (a gift from Tom MacRae, Dalhousie University) at a 1:500 dilution, followed by Alexa Fluor 546-conjugated goat anti-rabbit antibody (Molecular Probes) diluted at 1:200, all in PBS. F-actin and tubulin organization were assessed by using an LSM 510 confocal microscope equipped with argon 458/488-nm and helium/neon 548-nm lasers, and images were captured and processed by using 3D for LSM and the LSM-5 image browser, respectively (Carl Zeiss Canada, Toronto, Ontario, Canada).
Bead and L. pneumophila association with lysosomes in CHO-htpB cells.
The lysosomal network of CHO-htpB cells grown on coverslips was prelabeled for 1 h at 37°C in 5% CO2 with 100 μg of Texas Red ovalbumin (TrOv; Molecular Probes)/ml in 1 ml of αMEM. Prelabeled cells were washed three times with PBS and chased for 1 h in fresh αMEM. Differently coated blue fluorescent beads or GFP-legionellae were added for 1 h to the prelabeled cells as described in previous sections. Cells were washed six times with PBS, fixed in paraformaldehyde-periodate-lysine-5% sucrose (55), and mounted with ProLong Gold Antifade (Molecular Probes) (1-h samples) or further incubated for 2 h in fresh αMEM at 37°C in 5% CO2 (3-h samples) prior to fixation, mounting, and analysis by fluorescence microscopy. Phagosome-lysosome fusion was also assessed using LAMP-1 as a marker. Differently coated blue beads or Lp02 and Lp02dotB were added to CHO-htpB cells for 1 h. Cells were either washed six times with PBS and extracellular beads labeled for 1 min with 0.5 mg of Texas Red-dextran (Molecular Probes)/ml in αMEM at room temperature or washed six times with PBS and further incubated for 2 h in fresh αMEM at 37°C in 5% CO2 before extracellular bead labeling. Cells were then fixed with paraformaldehyde-periodate-lysine-5% sucrose (55), permeabilized for 10 s in methanol, and blocked 30 min in 2% BSA in PBS. LAMP-1 was immunostained with monoclonal antibody UH1 hybridoma supernatant (Developmental Studies Hybridoma Bank, Iowa City, IA) diluted 1:2 and Alexa Fluor 488-conjugated goat anti-mouse IgG (Molecular Probes) diluted 1:200. Lp02 and Lp02dotB were immunostained with rabbit anti-MOMP serum (8) diluted 1:2,000, followed by Alexa Fluor 546-conjugated goat anti-rabbit IgG (Molecular Probes) diluted 1:1,000. Coverslips mounted on glass slides with ProLong Gold Antifade (Molecular Probes) were analyzed by fluorescence microscopy, and only intracellular beads (lacking a red rim of Texas Red-dextran) were scored.
Association of beads with the ER in CHO-htpB cells.
Cells were incubated with differently coated yellow-green fluorescent beads as described in previous sections. The ER was stained for 15 min at 37°C in 5% CO2 with 1 μM ER-Tracker Blue-White DPX (Molecular Probes) in αMEM, followed by three PBS washes, and a 10-min chase in αMEM. Cells were fixed and mounted, as described above, for fluorescence microscopy. In addition, CHO-htpB cells were incubated with blue fluorescent beads, fixed and permeabilized as described in the previous section for TrOv staining, and blocked for 30 min in PBS containing 2% goat serum (PBS-G). The ER was then immunostained with a BiP-specific rabbit antibody (Affinity Bioreagents, Golden, CO) diluted 1:250 in PBS-G, followed by Oregon Green 488-conjugated goat anti-rabbit IgG (Molecular Probes) diluted 1:1,000 in PBS-G. Coverslips were mounted on glass slides as described above and analyzed by fluorescence microscopy.
Transmission electron microscopy (TEM).
CHO-htpB cells or U937-derived macrophages in 6-well plates were incubated with differently coated blue fluorescent beads or infected with L. pneumophila as described above. The bead/cell ratios were adjusted to 20 for CHO-htpB cells and 10 for macrophages. CHO-htpB cells were infected with L. pneumophila at a bacterium/cell ratio of ∼200. Macrophages were infected with Lp02 at a ratio of 20 or with Lp02dotB (which was internalized less efficiently) at a ratio of 200. Cells were then gently scraped, pelleted at 500 × g for 5 min at room temperature, fixed overnight at 4°C with 2.5% glutaraldehyde in 0.1 M sodium cacodylate pH 7, and washed three times in 0.1 M cacodylate at room temperature.
Pelleted phagosomes containing beads were fixed for 2 h at room temperature (without resuspending or disturbing the pellet) in cacodylate buffer containing 2.5% glutaraldehyde. Phagosomes were washed three times with cacodylate buffer, without disturbing the pellet. The pellets of fixed cells and phagosomes were postfixed with 1% osmium tetroxide in double-distilled H2O for 1 h at 4°C. They were then en bloc stained with aqueous uranyl acetate, dehydrated in acetone, embedded in epoxy resin, ultrathin sectioned, and stained with uranyl acetate and lead salts as described elsewhere (26). Sectioned specimens were observed in a JEOL JEM 1230 transmission electron microscope equipped with a high-resolution Hamamatsu ORCA-HR digital camera.
Invasion and intracellular growth assays.
CHO-htpB cells were seeded at 2 × 105cells/well into 24-well plates and grown for 48 h. When expression of eHtpB was needed, cells were grown for 48 h in αMEM without doxycycline. Immediately before the assays, cells were washed three times with PBS and replenished with αMEM without antibiotics (except for 10 ng of doxycycline/ml when needed). Lp02 or Lp02dotA grown on BCYE for 3 days were harvested in dH2O, and the bacterial suspensions were standardized to an optical density at 620 nm of 1 U (∼109 legionellae/ml). Inocula of ∼5 × 107 bacteria (to provide a bacterium/cell ratio of ∼200) or mixed inocula of bacteria (as described above) and 5 × 106 beads (to provide a bead/cell ratio of ∼25) were added in αMEM (with or without doxycycline) in triplicate. Plates were centrifuged at 1,000 × g for 5 min at 25°C to promote bead- and (or) bacterium-cell contact and further incubated at 37°C in 5% CO2 for 1 h. Monolayers were washed six times with PBS, treated for 1.5 h with αMEM-5% FBS containing 100 μg of gentamicin/ml and washed three times with PBS. Cells were then either (i) lysed immediately in 1 ml of dH2O to determine both invasion rate and time zero counts for intracellular growth assays or (ii) replenished with fresh αMEM (and 10 ng of doxycycline/ml when needed) and incubated for an additional 24, 48, or 72 h to obtain bacterial cell counts. All CFU/ml (equal to CFU/well) counts were performed in dH2O-lysed cells (final volume, 1 ml), using dH2O as a diluent, and samples were plated onto BCYE agar.
Statistics.
Unless otherwise stated, statistical significance was assessed with a one-way analysis of variance (http://www.physics.csbsju.edu/stats/anova.html). For multiple comparisons, one-way analysis of variance and the Bonferroni post test (http://graphpad.com/quickcalcs/posttest1.cfm) were used, and for pairwise comparisons of proportions the Fisher exact test was applied using Minitab software (v15.1.30.0; Minitab, Inc., State College, PA).
Attempt to delete htpB in L. pneumophila Lp02.
Allelic replacement of htpB with a kanamycin resistance (Kmr) cassette was attempted using the counterselectable plasmid pBRDX (47), which consists of the pBOC20 backbone (carrying cat and sacB) with an added rdxA gene of Helicobacter pylori, encoding a nitroreductase capable of activating metronidazole (7). Briefly, 707 bp of upstream and 620 bp of downstream flanking DNA sequences of the htpAB operon of L. pneumophila Lp02 were amplified by PCR using the primers htpABP1 (forward) and htpABP2 (reverse) (for the upstream or F1 region) and htpABP3 (forward) and htpABP4 (reverse) (for the downstream or F2 region) (Table 1). The F1 product containing NotI and BamHI restriction sites and the F2 product containing BamHI and XhoI restriction sites were ligated sequentially into pBluescript KS to create pBS-htpABF1F2. Digestion of pBS-htpABF1F2 with BamHI and subsequent ligation of the Kmr cassette restricted with BamHI from plasmid p34S-Km3 (17) between F1 and F2 resulted in the construct htpABKan, which was then subcloned from pBluescript into the NotI and XhoI sites of pBRDX and electroporated (4, 82) into L. pneumophila Lp02. Transformants were selected on BCYE agar with 40 μg of kanamycin/ml, and potential allelic recombinants were counterselected for the absence of rdxA, sacB, and cat by replica plating Kmr transformants onto BCYE with 5% (wt/vol) sucrose, BCYE with 20 μg of metronidazole/ml, or BCYE with 10 μg of chloramphenicol/ml. The presence of htpB was probed by PCR using htpB-forward and htpB-reverse primers (Table 1) and by immunoblotting (see above).
RESULTS
CHO cells associate well with differently coated microbeads.
Even though our previous work with HtpB-coated beads was done in HeLa cells (25), here we used CHO cells for their experimental advantages: (i) they support the intracellular growth of L. pneumophila (42, 43), (ii) genetic tools for the ectopic expression of recombinant proteins are available, (iii) CHO cells show a spread distribution of nonfilamentous mitochondria, and (iv) CHO cells show a better association than HeLa cells with microbeads. At a bead/cell ratio of 20, we previously determined that HeLa cells associated, in average, with <1 BSA-coated bead/cell (25), whereas CHO-wt cells (at the same bead/cell ratio) associated with two BSA-coated beads/cell (Table 2). All beads tested associated similarly with CHO-wt and CHO-htpB cells not expressing eHtpB, and differences in the number of beads per cell (which fluctuated between two and five) were not statistically significant for any type of bead (Table 2).
TABLE 2.
Fluorescence microscopy quantification, by direct counts, of the number of differently coated beads associated with CHO-wt and CHO-htpB cells not expressing eHtpB
| Type of bead coating | Mean no. of beads per cell ± SDa |
|||
|---|---|---|---|---|
| CHO-wt cells |
CHO-htpB cellsb |
|||
| 1 h | 3 h | 1 h | 3 h | |
| BSA | 1.6 ± 0.6 | 2 ± 0.9 | 2 ± 0.4 (NS) | 2.1 ± 0.4 (NS) |
| HtpB | 2.7 ± 0.1 | 2.8 ± 1.9 | 2 ± 0.2 | 3 ± 0.8 |
| rHtpB | 2.2 ± 0.4 | 2 ± 0.6 | ND | 3.1 ± 1.0 (NS) |
| GroEL | ND | ND | 3.1 ± 0.2 (NS) | 5 ± 0.8 (NS) |
| In-house GroEL | 2.7 ± 0.7 | 2.4 ± 0.6 | ND | 4.3 ± 0.2 (NS) |
| None | 5.9 ± 1.3 | 5 ± 3.8 | 3.1 ± 0.6 (NS) | 5 ± 1.7 (NS) |
| HtpB + PAbc | 1.5 (n = 1) | 1.7 ± 0.3 | ND | 2.6 ± 1.2 (NS) |
The results are the means of two experiments for CHO-wt cells and three experiments for CHO-htpB cells, with 50 to 100 scored cells/experiment.
eHtpB was repressed in the presence of 10ng of doxycycline/ml. P values, calculated only for CHO-htpB cells (n = 3) in relation to HtpB-coated beads, indicated no significant differences (NS) at 1 and 3 h.
Beads were treated with rHtpB-specific antiserum (PAb) before they were added to the cells.
Phagosomes containing L. pneumophila and HtpB-coated beads attract mitochondria.
In agreement with the original observation of Horwitz that nascent phagosomes containing L. pneumophila rapidly become surrounded by small vesicles and mitochondria (33), ∼80% of phagosomes containing L. pneumophila strain Lp02 were observed by TEM to be surrounded by mitochondria and/or small vesicles in infected CHO-htpB cells not expressing eHtpB (Fig. 1A) and in U937-derived macrophages (Fig. 1E). In contrast, Lp02dotB phagosomes did not recruit mitochondria or vesicles (data not shown). At 1 h after the addition of beads to CHO-htpB cells, ∼70% of phagosomes containing HtpB-coated beads were surrounded with mitochondria (Fig. 1B) and/or vesicles (not shown), as Lp02 did, whereas ∼80% of phagosomes containing the various control beads were not (Fig. 1C and D). These observations were replicated in U937-derived macrophages (Fig. 1F to H). The interaction of phagosomes containing Lp02 or HtpB-coated beads and mitochondria persisted at later times, as shown in TEM specimens fixed 3, 6, and 24 h after inoculation (Fig. 1I to K).
FIG. 1.

TEM shows that Lp02 and HtpB-coated beads attract mitochondria. (A) CHO-htpB cells infected with L. pneumophila Lp02 for ∼20 min prior to processing. (B to D) CHO-htpB cells incubated with HtpB (B)-, BSA (C)-, or GroEL (D)-coated beads for 1 h. CHO-htp cells did not express eHtpB. (E to H) In U937-derived macrophages (fixed 1 h after infection or addition of beads), Lp02 (E) and HtpB-coated beads (F) attracted mitochondria and portions of smooth ER, whereas BSA (G)- or GroEL (H)-coated beads showed no effect. For panels A to H, black arrows indicate sectioned bacteria or beads, black arrowheads indicate mitochondria, small white arrows indicate rough ER, and white arrowheads indicate smooth ER. Bars, 0.5 μm. Between 30 and 50 phagosomes were scored per cell type. Lp02 (I) and HtpB-coated beads (J and K) in CHO-htpB cells not expressing eHtpB were fixed at 6 h (J) and 24 h (I and K) postinoculation. At these later times, some phagosomes containing HtpB-coated beads were surrounded by a cloud of mitochondria (J and K). “B”, examples of bacterial cells in LCVs; “L”, latex beads. White arrows indicate examples of mitochondria.
HtpB-coated beads are significantly more capable than other beads to associate with mitochondria in CHO cells.
Using quantitative fluorescence microscopy, we determined that 1 and 3 h after the addition of beads to CHO-wt cells, significantly more HtpB- and rHtpB-coated beads (P < 0.01) were closely surrounded by mitochondria in relation to the other beads tested (Fig. 2A). Pretreatment of HtpB-coated beads with an rHtpB-specific antibody caused the levels of mitochondrial association to drop from 70 to 75% to 40 to 45%, suggesting specificity (Fig. 2A). Given the possible contamination of purified HtpB with L. pneumophila components, it is important to particularly compare the effects mediated by HtpB-coated beads to those mediated by beads coated with rHtpB and in-house GroEL. The latter two protein preparations were obtained (via the same purification method) from an E. coli whole-cell lysate and likely carried similar E. coli contaminants, but, most importantly, rHtpB-coated beads carried no L. pneumophila components other than HtpB.
FIG. 2.

Quantitative fluorescence microscopy analysis of mitochondria recruitment by L. pneumophila Lp02 and HtpB-coated beads. (A) Bar graph showing the percentages (means + 1 SD, n = 3 experiments, 100 scored cells/experiment) of differently coated beads associated with mitochondria at 1 and 3 h postinoculation in CHO-wt cells. P values were calculated in relation to HtpB-coated beads. **, (P < 0.01); ns, no significance. (B) Bar graph as in panel A, but with CHO-htpB cells not expressing eHtpB. P values were calculated in relation to HtpB-coated beads. **, P < 0.01; *, P < 0.05; ns, no significance. (C and D) Representative composite images of CHO-htpB cells incubated with blue fluorescent HtpB (C)- or GroEL (D)-coated beads for 1 h and stained with red MitoTracker. Mitochondria within one bead radius around each bead were considered as one positive event. Arrows point to positive beads, and arrowheads point to beads not associated with mitochondria. As a size reference, the diameter of the beads is 1 μm.
We confirmed the results obtained with CHO-wt cells in CHO-htpB cells not expressing eHtpB (Fig. 2B) and derived further data from the use of additional controls. The commercially available GroEL was not significantly more capable of mediating association with mitochondria than our in-house GroEL. The level of mitochondrial association of Lp02dotB (used as negative control) was no different from that of the control beads and, similarly, the mitochondrial association levels of HtpB-coated beads and L. pneumophila Lp02 (used as positive control) were not significantly different. Finally, pretreatment of HtpB-coated beads with antibodies significantly reduced their mitochondrial association levels in CHO-htpB cells, but antibody pretreatment of GroEL-coated beads had no significant effect. We concluded that inert beads coated with HtpB or rHtpB were as competent as virulent, live Lp02 at attracting mitochondria.
Two representative fluorescence microscopy images of CHO-htpB cells stained with MitoTracker are shown in Fig. 2C and D as examples of how fluorescent blue coated beads were scored as associated, or not, with mitochondria. It should be noted that the number of mitochondria associated with L. pneumophila or the differently coated beads was not determined. Therefore, a bacterium or a bead closely surrounded by or in contact with mitochondria was scored as one positive event (Fig. 2), regardless of how many mitochondria were involved. Consequently, the effect of HtpB may have been underestimated.
Phagosomes containing HtpB-coated beads, purified from U937-derived macrophages, closely interact with mitochondria.
The resolution of MitoTracker staining in U937-derived macrophages was low because these cells did not stretch thinly, and their mitochondria appeared filamentous. Therefore, we quantified the association of phagosomes with mitochondria by TEM in whole U937-derived macrophages and by flow cytometry in preparations of isolated phagosomes.
TEM provided excellent resolution in single planes (ultrathin sections) and allowed us to accurately determine any mitochondrion-phagosome contacts, as well as the number of mitochondria in contact with each phagosome (e.g., Fig. 1F). The results presented in Table 3 confirmed that phagosomes containing HtpB-coated beads were as competent as Lp02 LCVs at recruiting mitochondria and (on average) ∼2.3-fold more likely to contact mitochondria than phagosomes containing BSA- or GroEL-coated beads, as previously shown in CHO-htpB cells not expressing eHtpB (Fig. 2). Moreover, the total number of mitochondria per scored phagosomes was significantly higher for Htp-coated beads in relation to control beads (Table 3).
TABLE 3.
TEM quantification, by direct counts, of the number of mitochondria found in contact with phagosomes containing L. pneumophila or differently coated beads, in U937-derived macrophages at 3 h postinoculation
| Phagosome content | Events scoreda |
|||||
|---|---|---|---|---|---|---|
| No. of cells observed | No. of phagosomes (+/-) | Total no. of phagosomes | P value 1 | Total MT/total no. of phagosomes | P value 2 | |
| Bacteriab | ||||||
| Lp02 | 528 | 16/28 | 44 | 0.828 | 23/44 | 0.675 |
| Beads coated with: | ||||||
| HtpB | 678 | 16/32 | 48 | NA | 28/48 | NA |
| GroEL | 564 | 8/36 | 44 | 0.153 | 10/44 | 0.001 |
| BSA | 1,008 | 4/30 | 34 | 0.036 | 6/34 | <0.001 |
Only 3.4 to 8.3% of the cell sections observed showed phagosomes. The “+” phagosomes refer to those in contact with mitochondria; “−” phagosomes were not in contact with mitochondria. Association with vesicles or ER was not scored. “Total MT” refers to the total number of mitochondria observed in contact with phagosomes, and the “total number of phagosomes” refers to all of the phagosomes scored. P value 1 was calculated for differences in ratios of “+” phagosomes to total phagosomes in relation to HtpB-coated beads, using the Fisher exact test. P value 2 was calculated for differences in ratios of the total MT to the total phagosomes in relation to HtpB-coated beads, using the Fisher exact test. NA, not applicable.
We did not find enough phagosomes containing Lp02dotB to provide a meaningful quantification. Only two phagosomes were observed, with no associated mitochondria.
By flow cytometry, isolated LCVs containing red fluorescent Lp02 cells were 4.3-fold more likely to colocalize with mitochondria than phagosomes containing formalin-killed fluorescent Lp02 cells, validating the usefulness of flow cytometry in detecting interactions between isolated phagosomes and mitochondria. In our experiments with isolated phagosomes containing beads, we found an intrinsic variability in the net number of colocalization events between experiments, but a consistent ratio of events between bead preparations was maintained. Therefore, we calculated the fold values in relation to preparations of BSA-coated beads, which showed both the lowest variability between and within experiments and the lowest colocalization numbers per 10,000 events in all experiments (110 ± 129, n = 4). Representative flow cytometry experiments are shown in Fig. 3A and B. Isolated phagosomes containing HtpB-coated beads were (2 ± 0.5)-fold (n = 4) more likely to colocalize with mitochondria than phagosomes containing BSA-coated beads. This was consistent with the ∼2-fold percent ratio and the ∼2.3-fold percent ratio observed in CHO cells by fluorescence microscopy and in U937 macrophages by TEM between HtpB- or rHtpB-coated beads and the control beads with mitochondria (shown in Fig. 2 and Table 3, respectively). The colocalization of mitochondria with purified phagosomes containing GroEL-coated beads was (1.3 ± 0.3)-fold higher (n = 4) than the basal level of BSA-coated beads, and the fold increase observed for phagosomes with rHtpB-coated beads was 3.0 ± 2.1 (n = 4). To confirm whether the colocalization of red and green fluorescence represented a true interaction of phagosomes and mitochondria, or simple particle proximity, we used TEM.
FIG. 3.

Quantitative analysis by flow cytometry and corresponding TEM analysis of mitochondrial recruitment by LCVs or phagosomes containing HtpB-coated beads in U937 macrophages. (A and B) Representative flow cytometry plots (in arbitrary fluorescence intensity units) showing colocalization of isolated LCVs containing red fluorescent Lp02 (A) or isolated phagosomes containing BSA-, GroEL-, and HtpB-coated red fluorescent beads (B) with green fluorescent immunostained mitochondria. The clustering of events (in the form of bands) along the y axis of the plots in panel B likely represent groups of one, two, and three beads simultaneously detected. Colocalization events (the number given on each plot) were those within the upper right rectangle of each plot in relation to a total count of 10,000 events/plot. (C) TEM image of a sectioned phagosome containing one HtpB-coated bead and surrounding mitochondrial sections. The arrowhead points to a representative mitochondrion section. Bar, 200 nm. (D) TEM image showing that mitochondria sometimes appeared morphologically “empty” (area marked by the asterisk) as also seen in panel E. Bar, 100 nm. (E) Detail of a close interaction between a mitochondrion and a phagosome containing one HtpB-coated bead. Notice the deformed mitochondrial membrane at the mitochondrion-phagosome interface. An arrowhead points at the mitochondrial outer membrane, a white arrow points at the phagosomal membrane, and black arrow points at the bead's surface. Notice how tightly the phagosomal membrane surrounds the contained bead. Size bar, 100 nm.
Approximately 50 to 60% of the beads in TEM sections did not have a surrounding membrane, perhaps explaining the low number of colocalization events quantified by flow cytometry. All beads within phagosomes were tightly surrounded by a membrane, as previously reported (25). About 60% of HtpB-coated beads in phagosomes showed sectioned mitochondria in the immediate vicinity (Fig. 3C). Occasionally, we had the opportunity to see perpendicular sections of the phagosome-mitochondrion interface (Fig. 3D and E), showing an intimate interaction that caused the mitochondrial membrane to deform. Although mitochondria were also seen in the vicinity of ∼25% of in-house GroEL-coated beads (free or in phagosomes), intimate contact was not observed. These results indicated that only phagosomes containing HtpB-coated beads closely interacted with mitochondria and suggested that most positive events observed by flow cytometry with GroEL-coated beads likely represented particle proximity rather than close contact interactions.
HtpB-coated beads alter the organization of the actin cytoskeleton in CHO cells.
Because cytoskeleton integrity is crucial in organelle trafficking (6, 57, 77) and L. pneumophila induces cytoskeletal rearrangements in infected cells (16, 44, 78), we examined whether HtpB-coated beads had an effect on the cytoskeleton of CHO cells.
Approximately 80% of untreated (no beads) CHO-wt cells showed fine stress fibers (such as those shown in Fig. 4A for CHO-htpB cells). However, cells associated with HtpB-coated beads (82%) or rHtpB-coated beads (70%) showed what we called the “altered actin” phenotype (Table 4), which consisted of no obvious stress fibers and a strong staining of peripheral F-actin bundles. Similar results were obtained with CHO-htpB cells, although the basal level of altered actin was higher than in CHO-wt cells. F-actin rearrangement was not bead/cell ratio dependent, e.g., more BSA-coated beads per cell did not induce the effect, and some cells with only one HtpB-coated bead attached still showed the altered actin phenotype (data not shown).
FIG. 4.

Confocal microscopy analysis of the F-actin rearrangements induced by L. pneumophila Lp02 and HtpB-coated beads in CHO-htpB cells not expressing eHtpB. (A to E) Representative single-slice overlay images of untreated CHO-htpB control cells (A), cells infected for 1 h with GFP-expressing Lp02 (B) or GFP-expressing Lp02dotB (D), or cells incubated with green fluorescent HtpB (C)- or GroEL (E)-coated beads for 1 h. F-actin was labeled with phalloidin-Alexa Fluor 546 (red). Size bars, 10 μm. The arrow in panel A points at cytoplasmic stress fibers, and the arrow in panel B points at peripheral F-actin bundles. The altered actin phenotype was characterized by the loss of stress fibers and the accentuation of peripheral F-actin bundles (clearly seen in panels B and C). (F) Bar graph showing percentages (means + 1 SD, n = 3 independent experiments) of CHO-htpB cells associated with bacteria or beads that show the altered actin phenotype at 1 and 3 h after infection or bead addition. P values were calculated in relation to HtpB-coated beads. **, P < 0.01; and *, P < 0.05.
TABLE 4.
Fluorescence microscopy quantification, by direct counts, of CHO cells that showed an altered actin microfilaments network after a 2-h exposure to differently coated beads
| Bead coating | Mean % cells with an altered actin phenotype ± SDa |
|
|---|---|---|
| CHO-wt cellsb | CHO-htpBc | |
| None | 23.4 ± 4.7 | 34.5 ± 8.3 |
| HtpB | 81.8 ± 0.4 | 62.4 ± 14.4 |
| rHtpB | 69.5 ± 12.5 | 72.7 ± 4.2 |
| In-house GroEL | 21.0 ± 4.4 | 32.7 ± 15.1 |
The results are the means of two experiments, with 100 scored cells/experiment.
The percentage of CHO-wt cells with no beads and altered actin was 23.2 ± 4.9 (n = 8).
CHO-htpB cells in these experiments did not express eHtpB. The percentage of CHO-htpB cells with no beads and altered actin was 36.4 ± 16.7 (n = 8).
Immunostaining with a tubulin-specific antibody showed no changes in the microtubule network of CHO-htpB cells infected with Lp02 or incubated with HtpB-coated beads in relation to untreated cells. All cells showed an intact network of microtubules radiating from a central region (data not shown). We thus concluded that HtpB presented on the surface of microbeads specifically and strongly mediates a rearrangement of F-actin, which is not induced by GroEL-coated beads and does not involve the microtubule network.
Ectopic expression of eHtpB in the cytoplasm of CHO-htpB cells has an effect on the actin cytoskeleton similar to that of externally added HtpB-coated beads.
To directly compare the effects of HtpB added from without (in the form of beads) to the effects of ectopic HtpB (eHtpB produced from within) in the same cells, we conducted a more thorough assay with CHO-htpB cells not expressing eHtpB that included additional bead controls and live L. pneumophila (Fig. 4). Infection of CHO-htpB cells with Lp02 induced the same altered actin phenotype produced by HtpB-coated beads (Fig. 4A to C), albeit to a lesser extent (Fig. 4F). However, infection with Lp02dotB, or incubation with GroEL-coated beads induced no change (Fig. 4D and E). The quantification of cells with altered actin microfilaments at 1 and 3 h (Fig. 4F) indicated that (i) HtpB-coated beads were more able than Lp02 cells to induce the altered actin phenotype; (ii) pretreatment of HtpB-coated beads with antibody eliminated the effect, whereas the antibody did not have an effect on GroEL-coated beads; and (iii) the induced effect was clearly transient for both live Lp02 and HtpB-coated beads. The basal level of untreated cells showing the altered actin phenotype fluctuated between 28% ± 5% and 36% ± 5%.
Expression of eHtpB in the cytoplasm of CHO-htpB cells was effectively controlled by doxycycline (Fig. 5A and B) but shown to be variable between cells, particularly at low doxycycline concentrations, e.g., 0.01 ng/ml (Fig. 5C). We thus chose not to regulate intermediate eHtpB levels and decided to unambiguously induce HtpB expression in 0 ng of doxycycline/ml and repress it with 10 ng of doxycycline/ml in an on/off fashion. We also included recombinant GroEL and DsRed2 as control proteins expressed in transiently transfected cells. Induced CHO-htpB cells and transiently transfected cells expressing eHtpB showed a significant alteration of the actin cytoskeleton that was not present in cells expressing the control recombinant proteins (Fig. 5D and E). The similarity of this effect to that produced by HtpB-coated beads was striking but also puzzling in terms of understanding how a protein presented from both sides of the plasma membrane had the same effect. Of importance is that cells expressing eHtpB did not show alterations in shape, size, ultrastructure, growth rate, microtubule organization, and overall distribution of the mitochondria and ER (not shown).
FIG. 5.

Expression of eHtpB in CHO-htpB cells and its effect on the actin cytoskeleton. (A) Reference image (Ponceau-S stained) for the immunostained nitrocellulose membrane shown in panel B. Loading equivalence, ∼6 × 105 CHO-htpB cells/lane. (B) Immunoblot analysis of eHtpB in cells incubated for 48 h in various concentrations of doxycycline. The fold increase shown is relative to the eHtpB produced in 1 ng of doxycycline/ml. An arrowhead points at full-size eHtpB (∼60 kDa) within the box. Strongly immunostained bands under the box likely represent eHtpB degradation products. Lane Std, protein standards (the masses are indicated at the far left in kilodaltons). (C) Representative images, in differential interference contrast (DIC) or epifluorescence (Anti-rHtpB PAb) modes, showing eHtpB expression at different doxycycline (Dox) concentrations. The bar in the top right micrograph indicates 10 μm and applies to all of the micrographs. (D) Representative fluorescence confocal microscopy grayscale images showing the effect of various recombinant proteins (R-protein) upon the actin cytoskeleton of CHO-AA8 Tet-Off cells (F-actin). CHO-htpB cells were observed after 48 h in doxycycline-free medium, and transiently transfected AA8 cells were observed 48 h after transfection. The bar in the top right micrograph indicates 10 μm and applies to all of the micrographs. (E) Bar graph showing percentages (means ± 1 SD from three independent experiments) of cells with altered F-actin after ectopic expression of proteins in transiently transfected CHO-AA8 cells (left) and stably transfected CHO-htpB cells (right). Background levels of altered actin were provided by nontransfected cells (AA8) and noninduced CHO-htpB cells, respectively. P values in the transient group were calculated in relation to cells expressing HtpB. **, P < 0.01.
HtpB-coated beads do not recruit ER or alter the lysosomal network.
Using quantitative fluorescence microscopy, we found that differently coated beads did not conclusively associate with the ER network of CHO-htpB cells, either through immunostaining of BiP or through direct staining with ER-Tracker Blue-White DPX. Likewise, ribosome studding and rough ER association with phagosomes containing HtpB-coated beads was not consistently observed by TEM at 1 h (e.g., Fig. 1B and F) or 6- and 24-h incubations (e.g., Fig. 1J and K), indicating that HtpB does not mediate interactions with the ER.
Maturation of LCVs or bead-containing phagosomes along the endocytic pathway was first evaluated by scoring the colocalization with LAMP-1, a protein of late endosomes and lysosomes. At 1 and 3 h after infection, Lp02 LCVs seldom colocalized with LAMP-1, but phagosomes containing the dotB mutant more frequently acquired LAMP-1 staining (Table 5). Regardless of the type of coating, phagosomes containing beads all showed positive LAMP-1 staining at significantly higher frequencies in relation to Lp02 LCVs or even the dotB mutant-containing phagosomes (P < 0.05, Table 5).
TABLE 5.
Fluorescence microscopy quantification, by direct counts, of colocalization events between differently coated beads or bacteria and LAMP-1-positive compartments (late endosomes and lysosomes) in CHO-htpB cells not expressing eHtpB
| Type of particlea | Mean % particles colocalized with LAMP-1 ± SD (P) atb: |
|
|---|---|---|
| 1 h postinoculation | 3 h postinoculation | |
| BSA-coated beads | 71 ± 2 (n.s) | 77 ± 7 (NS) |
| HtpB-coated beads | 67 ± 4 | 75 ± 10 |
| GroEL-coated beads | 68 ± 9 (NS) | 76 ± 4 (NS) |
| Uncoated beads | 71 ± 8 (NS) | 74 ± 5 (NS) |
| HtpB-coated beads + PAb* | 74 ± 9 (NS) | 76 ± 10 (NS) |
| GroEL-coated beads + PAb* | 71 ± 7 (NS) | 76 ± 12 (NS) |
| L. pneumophila Lp02 | 8 ± 0 (<0.01) | 13 ± 3 (<0.01) |
| Lp02dotB mutant | 25 ± 2 (<0.05) | 39 ± 4 (<0.05) |
*, Beads were preincubated with rHtpB-specific rabbit polyclonal antiserum (PAb), before inoculation of the CHO-htpB cells. PAb cross-reacts with GroEL.
Results are the means of three experiments, with 50 beads or bacterial cells scored per experiment. P values, calculated as multiple comparisons, are shown in parentheses in relation to HtpB-coated beads. NS, not significant.
We next assessed phagosome-lysosome fusion in CHO-htpB cells using TrOv. In agreement with the findings of Joshi et al. (41), phagosomes containing Lp02 or the dotB mutant rarely colocalized with TrOv at either 1 h or 3 h postinfection (Table 6). In contrast, phagosomes containing beads (regardless of the type of coating) all colocalized with TrOv at significantly higher frequencies in relation to live L. pneumophila (P < 0.01, Table 6). However, among all bead types used, HtpB-coated beads consistently showed, at 1 h postinoculation, the lowest frequency of colocalization with TrOv (Table 6). Collectively, these results indicate that whereas HtpB modestly delays the delivery of TrOv to phagosomes containing beads, it cannot block phagosome-lysosome fusion.
TABLE 6.
Fluorescence microscopy quantification, by direct counts, of colocalization events between differently coated beads or bacteria and TrOv-labeled lysosomes in CHO-htpB cells not expressing eHtpB
| Type of particlea | Mean % particles colocalized with TrOv ± SD (P) atb: |
|
|---|---|---|
| 1 h postinoculation | 3 h postinoculation | |
| BSA-coated beads | 33 ± 3 (<0.05) | 40 ± 4 (<0.05) |
| HtpB-coated beads | 23 ± 2 | 32 ± 3 |
| GroEL-coated beads | 29 ± 1 (<0.05) | 34 ± 2 (NS) |
| Uncoated beads | 32 ± 1 (<0.05) | 37 ± 1 (NS) |
| HtpB-coated beads + PAb* | 31 ± 4 (<0.05) | 38 ± 2 (NS) |
| GroEL-coated beads + PAb* | 41 ± 1 (<0.05) | 34 ± 3 (NS) |
| L. pneumophila Lp02 | <1 ± 0 (<0.01) | <1 ± 0 (<0.01) |
| Lp02dotB mutant | <1 ± 0 (<0.01) | <1 ± 0 (<0.01) |
*, Beads were preincubated with rHtpB-specific rabbit polyclonal antiserum (PAb) before inoculation of the CHO-htpB cells. PAb cross-reacts with GroEL.
The results are the means of three experiments, with 100 scored beads or bacteria/experiment. P values, calculated as multiple comparisons, are given in parentheses in relation to HtpB-coated beads. NS, not significant.
Cell invasion and intracellular growth of Lp02dotA is not changed in the presence of HtpB-coated beads or eHtpB.
Intracellular growth of a dotA avirulent mutant in macrophages can be restored when the mutant is cointernalized into the same phagosome with wild-type L. pneumophila (15). Therefore, we set out to determine whether HtpB-coated beads or eHtpB could restore the intracellular growth of Lp02dotA or enhance the invasiveness and growth of Lp02 in CHO-htpB cells.
TEM of CHO-htpB cells infected with Lp02 in the presence of BSA-coated or HtpB-coated beads showed that bacteria and beads did not colocalize in the same phagosome (Fig. 6A and B). However, ∼50% of the internalized dotA mutants colocalized with BSA-coated or HtpB-coated beads (Fig. 6C and D). We concluded that Lp02 and Lp02dotA, despite having similar invasion rates in CHO-htpB cells not expressing eHtpB (0.28% ± 0.04% and 0.30% ± 0.07%, respectively, expressed as means ± the standard deviation [SD; n = 6] of the percent intracellular bacteria/bacterial inoculum, after a 3-h incubation), they clearly followed different pathways of internalization. The invasion rates of Lp02 and Lp02dotA did not change in the presence of beads, regardless of their coating. Phagosomes containing Lp02dotA alone did not show associated mitochondria and, even though HtpB-coated beads attracted mitochondria to the phagosomes shared with Lp02dotA (e.g., Fig. 6D), they did not rescue the intracellular growth of the mutant (Fig. 7A). However, when we examined the effect of the differently coated beads on the intracellular growth of Lp02, there was a slight but consistent improvement of early growth in the presence of HtpB-coated beads (Fig. 7B).
FIG. 6.
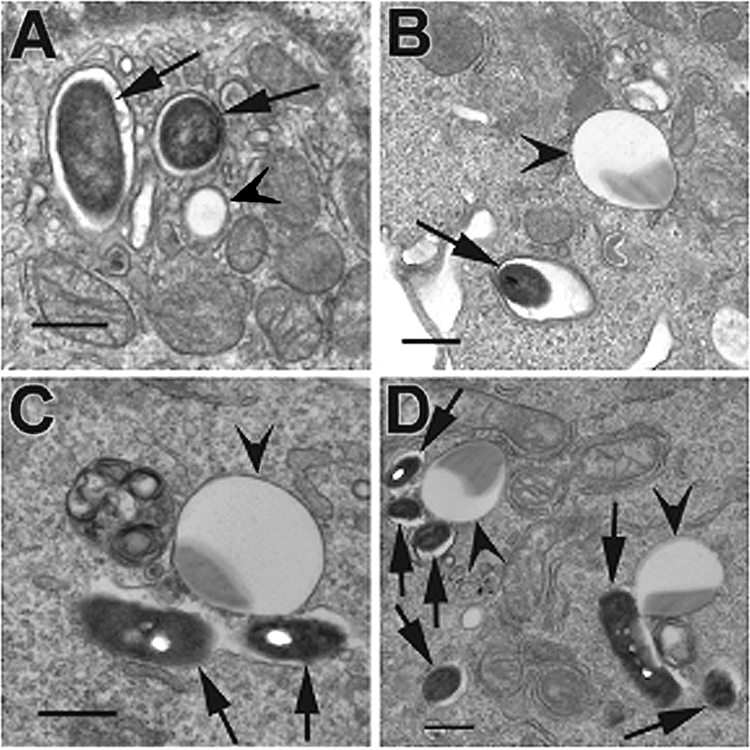
Electron micrographs of L. pneumophila internalized by CHO-htpB cells not expressing eHtpB in the presence of coated beads. CHO-htpB cells were infected for 1 h with Lp02 plus BSA-coated beads (A), Lp02 plus HtpB-coated beads (B), Lp02dotA plus BSA-coated beads (C), or Lp02dotA plus HtpB-coated beads (D). Colocalization of bacteria (arrows) and coated beads (arrowheads) in the same phagosomes is presented in panels C and D. Bars, 0.5 μm. At least 50 cell sections (∼100 phagosomes) were scored per experiment.
FIG. 7.

Invasiveness and intracellular growth of L. pneumophila in CHO-htpB cells in the presence of coated beads or eHtpB. (A and B) Graphs showing the growth or survival curves of L. pneumophila, presented as mean CFU/ml ± 1 SD from two independent experiments run in triplicate with either Lp02dotA (A) or Lp02 (B) in the presence of differently coated beads. P values were calculated in relation to growth in the presence of HtpB-coated beads. *, P < 0.05. (C) Bar graph showing percentages (means ± 1 SD from two independent experiments run in triplicate) of bacterial invasion for Lp02 or Lp02dotA in CHO-htpB cells expressing (+eHtpB) or not (-eHtpB) endogenous HtpB. Invasion was calculated as the percent bacteria that survived a gentamicin treatment in relation to the bacterial inoculum. (D) Graph showing the growth and survival curves of Lp02 and Lp02dotA, respectively, presented as the mean CFU/ml ± 1 SD (n = 3 experiments) in CHO-htpB cells expressing (+eHtpB) or not expressing endogenous HtpB.
In invasion assays with Lp02 and Lp02dotA in CHO-htpB cells expressing eHtpB, no differences were detected in relation to invasion of cells not producing eHtpB (Fig. 7C). In addition, the intracellular growth of Lp02 in CHO-htpB cells was very similar in the presence or absence of eHtpB, and the production of eHtpB did not restore the intracellular growth of Lp02dotA (Fig. 7D). Collectively, these results indicated that while HtpB-coated beads might promote the growth of virulent L. pneumophila in trans, they cannot compensate for all of the signals and type IV secreted factors missing from the dotA mutant, which would be necessary for LCV maturation and initiation of intracellular growth.
Failure to delete htpB from L. pneumophila Lp02.
To directly investigate the role of HtpB in Legionella pathogenesis, we attempted to delete htpB by allelic replacement and characterize the mutant's ability to alter mitochondrial trafficking and microfilament organization. Hundreds of Kmr Lp02 transformants grew on BCYE with kanamycin and, upon replica plating, we were able to counterselect potential allelic recombinants that were kanamycin, metronidazole, and sucrose resistant but chloramphenicol sensitive. However, the presence of htpB was confirmed by PCR, and HtpB was detected by immunoblotting, in all of the potential ΔhtpB clones. We have been unable to fully explain these results, which suggest that genetic rearrangements occurred to avoid deletion of an essential gene, wherein the Kmr transformants were forced to grow in a highly selective medium.
DISCUSSION
Shortly after internalization of virulent L. pneumophila by host cells, LCVs associate with mitochondria, which then remain associated with the LCVs for different lengths of time, as shown by ultrastructural studies of infected monocytes (33), MRC-5 cells (60), and HeLa cells (26). Phagosomes containing HtpB-coated beads consistently recruited mitochondria in CHO cells and U937-derived macrophages, and mitochondria remained associated with phagosomes in CHO cells for up to 24 h, thereby demonstrating that, when presented on the surface of inert beads, HtpB is sufficient to emulate this L. pneumophila trait.
The conclusions derived from the application of our two functional models are restricted to the impact of purified or recombinant HtpB on cultured cells, out of the context of a natural L. pneumophila infection. Therefore, it remains to be determined whether HtpB has a virulence-related role of importance to L. pneumophila pathogenesis. One way to provide evidence for such a role would be to delete htpB from the L. pneumophila genome. However, we could not delete htpB by allelic replacement with pBRDX, a methodology that we and others have successfully used to delete at least six L. pneumophila genes (47, 56; unpublished results), suggesting that it is the essential nature of htpB and not the method used that impeded the deletion. The essential nature of htpB has also been suggested by others (20). These investigators could not delete the stress regulator gene rpoH from the genome of L. pneumophila or replace the rpoH promoter by a controllable IPTG-inducible promoter, and although they observed a reduced amount of htpB transcripts in clones carrying antisense sequences for rpoH or htpB the levels of HtpB were not reduced (20). Although temperature-sensitive htpB mutants could in principle be created, they would likely show secondary defects arising from defective folding of many proteins, and phenotypic changes in these mutants could be equivocally assigned to the defective HtpB. We have hypothesized that a way to overcome the barrier imposed by htpB's essentiality and avoid the ambiguity of temperature-sensitive mutants would be to create an htpAB deletion mutant after inserting the groELS operon from E. coli in the L. pneumophila chromosome. GroELS could thus provide the presumed essential chaperonin functions, while GroEL (being unable to attract mitochondria when presented on microbeads) would likely have no contribution to the recruitment of mitochondria in the htpB deletion mutant. Finally, an alternate approach would be to determine whether HtpB mediates mitochondrial recruitment when expressed in a surrogate intracellular bacterial pathogen that does not naturally recruit mitochondria. For the latter, however, it would be necessary to demonstrate that HtpB can be secreted and properly localized to the cell surface of the surrogate pathogen.
In spite of the high similarity that exists between bacterial chaperonins, the fact that HtpB's ability to recruit mitochondria and alter stress fibers was not shared with GroEL suggests that specific HtpB domains are responsible for these effects. That few amino acid changes could result in new chaperonin functions is a baffling but not unprecedented issue, since Yoshida et al. (87) unequivocally established that only 4 amino acid changes transformed the GroEL from E. coli into a potent insect toxin. Future work could thus be focused on the use of truncated or amino acid-substituted forms of HtpB in our functional models, as an experimental tool to identify such specific domains.
Recruitment of host mitochondria to the LCV in human monocytes depends on a functional Dot/Icm system, as retrospectively inferred from experiments with a spontaneous L. pneumophila mutant unable to recruit mitochondria (36), later shown to regain a wild-type phenotype by genetic complementation with the dot/icm locus (53). By reporting that dotA and dotB mutants were unable to attract mitochondria in CHO cells, we have provided further evidence for the need of a functional Dot/Icm system in mitochondrial recruitment. These results imply that a potential link exists between HtpB and the Dot/Icm system, a link also suggested by the observation that dot/icm mutants accumulate HtpB in the periplasm (1). Either HtpB is secreted by the Dot/Icm system, or Dot/Icm effectors are also required (besides HtpB) for optimal mitochondrial recruitment (functional redundancy).
Recruitment of mitochondria to the LCV is a feature shared with the parasitophorous vacuoles of Chlamydia psittaci (54, 62), Toxoplasma gondii (74), and Encephalitozoon microsporidia (71). As far as we know, the molecular mechanism of recruitment has been described only in the case of T. gondii, where the N terminus of ROP2, a T. gondii protein localized to the parasitophorous vacuole membrane, has characteristics of a mitochondrial targeting signal sequence and is exposed to the host cytosol (73). ROP2 inserts into the mitochondrial membrane tethering the organelle to the vacuole. HtpB is abundantly released into the lumen of the LCV in infected HeLa cells (24, 32) and possesses the conserved glycine- and methionine-rich C terminus responsible for the membrane-targeted lipochaperonin activity previously reported for GroEL (81). However, even if HtpB could associate with the LCV membrane, the lack of both a predicted mitochondrial targeting signal and an overall homology to ROP2 suggest that HtpB recruits mitochondria by a novel, yet-to-be-described mechanism.
The actin rearrangement mediated by HtpB was not related to the actin polymerization changes required for internalization. That is, CHO cells similarly internalized all beads, but only HtpB-coated beads induced the disappearance of stress fibers. In addition, Lp02dotA invaded CHO cells but did not alter stress fibers. Based on our results with CHO-htpB cells, which indicated that HtpB induces the disappearance of stress fibers when it is presented from either side of the plasma membrane, we propose that this postinvasion rearrangement of F-actin is the result of an HtpB-mediated signaling event. HtpB may get inserted into the plasma and/or LCV membrane, emulating a lipochaperonin activity (81), from where it could directly or indirectly initiate a signaling process. Alternatively, HtpB could interact with plasma membrane surface signaling molecules (e.g., Ras) that also signal from endomembranes while in transit to the plasma cell membrane (10, 11). Evidence to support an HtpB-mediated signaling mechanism includes the following: (i) the striking morphological resemblance between our altered actin phenotype and that induced in porcine aortic endothelial cells by the constitutive activation of the signaling molecule Rnd1 (2); (ii) bacterial chaperonins, in general, modulate cell signaling pathways (reviewed in reference 51) and, in particular, can activate extracellular signal-regulated kinase 1/2-mitogen-activated protein kinase pathways when added to the cell culture medium (88, 89); (iii) HtpB modulates cytokine mRNA levels and interleukin-1 secretion in macrophages via a protein kinase C-dependent signaling pathway (64); and (iv) HtpB has the ability to deliver an intracellular signal in Saccharomyces cerevisiae that activates two signaling pathways controlled by Ras2p and results in pseudohyphal growth (66). Moreover, the fact that Lp02 and HtpB-coated beads did not colocalize in the same phagosome (Fig. 6) suggests that the beneficial effect of HtpB-coated beads on the early growth of Lp02 (Fig. 7B) was indirect, perhaps mediated through a signaling mechanism.
Mitochondrial movement primarily occurs along microtubule tracks in eukaryotic cells (57, 77), and disruption of actin microfilaments results in increased mitochondrial movement along microtubule tracks in sea urchin coelomocytes (46). In addition, the transient clustering of mitochondria around the nucleus of PtK2 cells infected with vaccinia virus happens to be a response to virus-mediated changes in F-actin organization, which in turn depends on intact microtubules (72). Because the microtubule network remained unaffected during both L. pneumophila infection and exposure to HtpB-coated beads, we propose that the transient HtpB-mediated perturbation of the actin cytoskeleton (as a putative signaling response) might, in fact, constitute the mechanism of mitochondrial recruitment to the LCV. Disruption of stress fibers and recruitment of mitochondria were two robust effects induced by the same protein attached onto beads. This causal convergence, in addition to the common dependence of these effects on type IV secretion during infection with live L. pneumophila, provides further support to the argument that the transient L. pneumophila-induced changes in F-actin organization could result in mitochondrial recruitment to the LCV.
Possible explanations for the failure of eHtpB in altering mitochondrial trafficking might include the lack of a directional signal and the sustained alteration of the actin cytoskeleton. In induced CHO-htpB cells, eHtpB was found throughout the cytoplasm, and even in the nucleus (Fig. 5). If the signaling hypothesis is correct, a cell expressing eHtpB would potentially receive multiple signals from everywhere, whereas internalized Lp02 or HtpB-coated beads would deliver localized, directional signals. Moreover, the altered actin phenotype of induced CHO-htpB cells persisted for days, as opposed to that resulting from the interaction with Lp02 or HtpB-coated beads, which only lasted hours. Although the transient nature of the latter effect could be explained by the breakdown of bead-bound HtpB within phagosomes, we determined by dot immunoblotting that beads recovered from U937-derived macrophages 3 h after addition still tested positive for the presence of chaperonins (data not shown). Therefore, the delivery of a localized signal with transient effects might be crucial to redirecting the trafficking of mitochondria to the LCV, after which a tight interaction between the vacuolar and mitochondrial membranes (as suggested by Fig. 3E) may assure a persistent association throughout the intracellular growth cycle of L. pneumophila. The molecular basis for this tight interaction is currently unknown, but its strength was further suggested by the fact that it persisted throughout the phagosome isolation procedure. Alternative explanations to the signaling hypothesis are that (i) HtpB might physically recruit (bind) host factors responsible for the observed effects, as reported for other L. pneumophila factors that recruit host effectors to the LCV and regulate its trafficking (18, 43), and (ii) HtpB may directly interact with F-actin. An example of an effector that binds to actin and induces F-actin rearrangements is SipA, a Salmonella factor involved in bacterial internalization (90).
The colocalization of HtpB-coated beads and bacteria in the same phagosome or the presence of eHtpB in the host cell cytoplasm of CHO-htpB cells did not rescue the intracellular growth of Lp02dotA. This is not surprising because HtpB alone was unable to induce recruitment of ER to the LCV or effectively inhibit fusion with lysosomes. The establishment of the LCV involves multiple Dot/Icm translocated effectors with apparently redundant functions (reviewed in reference 59). In this respect, the study of L. pneumophila's intracellular biology is unlikely to involve, at this point, the simultaneous deletion of all genes encoding redundant effectors (some of which may be essential). Therefore, the complexity of functional redundancy in L. pneumophila could be alternatively addressed by using protein functional models. Our studies constitute an example of how the application of two functional models to the study of an essential L. pneumophila protein has unfolded two seemingly unique functions of purified and recombinant HtpB and a novel facet of chaperonin biology.
Acknowledgments
We thank Michele Swanson (University of Michigan at Ann Arbor) for generously hosting A.C. in her lab and providing training, as well as the means to conduct BiP-labeling and TrOv-labeling experiments. Her discussions in relation to the role of HtpB in L. pneumophila pathogenesis, as well as her critical reading of an early precursor to the manuscript, are also greatly appreciated. The generosity of Karen Brassinga (a former Dalhousie University colleague), Paul Sigler, and Hubert Hilbi, for donating plasmids, as well as that of Ralph Isberg and Joe Vogel, for donating strains, is also acknowledged. We thank Paul S. Hoffman (a former Dalhousie University colleague) for monoclonal antibody GW2X4B8B2H6, MOMP-specific antiserum, and plasmids pBRDX and pSH16; Andrew Issekutz for U937 cells; and Radhey Gupta for CHO cells. Mary Ann Trevors and Kaitlyn Carson are gratefully acknowledged for their help in processing TEM specimens and statistical analyses, respectively. The UH1 LAMP-1-specific antibody developed by S. Uthayakumar and B. L. Granger was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City.
This study was funded by a Discovery grant to R.A.G. from the Natural Sciences and Engineering Research Council of Canada. R.A.G. holds an NSERC Canada Research Chair in food- and waterborne bacterial pathogens.
Editor: J. B. Bliska
Footnotes
Published ahead of print on 17 August 2009.
REFERENCES
- 1.Allan, D. S. 2002. Secretion of Hsp60 chaperonin (GroEL) homologs by Legionella pneumophila. M.Sc. thesis. Dalhousie University, Halifax, Nova Scotia, Canada.
- 2.Aspenstrom, P., A. Fransson, and J. Saras. 2004. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem. J. 377:327-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger, K. H., J. J. Merriam, and R. R. Isberg. 1994. Altered intracellular targeting properties associated with mutations in the Legionella pneumophila dotA gene. Mol. Microbiol. 14:809-822. [DOI] [PubMed] [Google Scholar]
- 4.Berk, S. G., G. Faulkner, E. Garduño, M. C. Joy, M. A. Ortiz-Jiménez, and R. A. Garduño. 2008. Packaging of live Legionella pneumophila into pellets expelled by Tetrahymena spp. does not require bacterial replication and depends on a Dot/Icm-mediated survival mechanism. Appl. Environ. Microbiol. 74:2187-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blander, S. J., and M. A. Horwitz. 1993. Major cytoplasmic membrane protein of Legionella pneumophila, a genus common antigen and member of the hsp60 family of heat shock proteins, induces protective immunity in a guinea pig model of Legionnaires' disease. J. Clin. Investig. 91:717-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boldogh, I. R., and L. A. Pon. 2006. Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta 1763:450-462. [DOI] [PubMed] [Google Scholar]
- 7.Brassinga, A. K. C., M. A. Croxen, C. J. Shoemaker, M. G. Morash, J. J. LeBlanc, and P. S. Hoffman. 2006. Novel use of Helicobacter pylori nitroreductase (rdxA) as a counterselectable marker in allelic vector exchange to create Legionella pneumophila Philadelphia-1 mutants, p. 339-342. In N. P. Cianciotto, Y. Abukwaik, P. H. Edelstein, B. S. Fields, D. F. Geary, T. G. Harrison, C. A. Joseph, R. M. Ratcliff, J. E. Stout, and M. S. Swanson (ed.), Legionella: state of the art 30 years after its recognition. ASM Press, Washington, DC.
- 8.Butler, C. A., and P. S. Hoffman. 1990. Characterization of a major 31-kilodalton peptidoglycan-bound protein of Legionella pneumophila. J. Bacteriol. 172:2401-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakraborty, P., S. Sturgill-Koszycki, and D. G. Russell. 1994. Isolation and characterization of pathogen-containing phagosomes. Methods Cell Biol. 45:261-276. [DOI] [PubMed] [Google Scholar]
- 10.Chiu, V. K., T. Bivona, A. Hach, J. B. Sajous, J. Silletti, H. Wiener, R. L. Johnson, A. D. Cox, and M. R. Philips. 2002. Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 4:343-350. [DOI] [PubMed] [Google Scholar]
- 11.Choy, E., V. K. Chiu, J. Silletti, M. Feoktistov, T. Morimoto, D. Michaelson, L. E. Ivanov, and M. R. Philips. 1999. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98:69-80. [DOI] [PubMed] [Google Scholar]
- 12.Cirillo, J. D., S. L. Cirillo, L. Yan, L. E. Bermudez, S. Falkow, and L. S. Tompkins. 1999. Intracellular growth in Acanthamoeba castellanii affects monocyte entry mechanisms and enhances virulence of Legionella pneumophila. Infect. Immun. 67:4427-4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cirillo, S. L., L. E. Bermudez, S. H. El-Etr, G. E. Duhamel, and J. D. Cirillo. 2001. Legionella pneumophila entry gene rtxA is involved in virulence. Infect. Immun. 69:508-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cirillo, S. L., J. Lum, and J. D. Cirillo. 2000. Identification of novel loci involved in entry by Legionella pneumophila. Microbiology 146:1345-1359. [DOI] [PubMed] [Google Scholar]
- 15.Coers, J., C. Monahan, and C. R. Roy. 1999. Modulation of phagosome biogenesis by Legionella pneumophila creates an organelle permissive for intracellular growth. Nat. Cell Biol. 1:451-453. [DOI] [PubMed] [Google Scholar]
- 16.Coxon, P. Y., J. T. Summersgill, J. A. Ramirez, and R. D. Miller. 1998. Signal transduction during Legionella pneumophila entry into human monocytes. Infect. Immun. 66:2905-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dennis, J. J., and G. J. Zylstra. 1998. Improved antibiotic-resistance cassettes through restriction site elimination using Pfu DNA polymerase PCR. BioTechniques 25:772-776. [DOI] [PubMed] [Google Scholar]
- 18.Derre, I., and R. R. Isberg. 2004. Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect. Immun. 72:3048-3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derre, I., and R. R. Isberg. 2005. LidA, a translocated substrate of the Legionella pneumophila type IV secretion system, interferes with the early secretory pathway. Infect. Immun. 73:4370-4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ewann, F., and P. S. Hoffman. 2006. Modulation of rpoH expression using an antisense strategy, p. 336-338. In N. P. Cianciotto, Y. AbuKwaik, P. H. Edelstein, B. S. Fields, D. F. Geary, T. G. Harrison, C. A. Joseph, R. M. Ratcliff, J. E. Stout, and M. S. Swanson (ed.), Legionella-State of the art 30 years after its recognition. ASM Press, Washington, DC.
- 21.Fernandez, R. C., S. M. Logan, S. H. Lee, and P. S. Hoffman. 1996. Elevated levels of Legionella pneumophila stress protein Hsp60 early in infection of human monocytes and L929 cells correlate with virulence. Infect. Immun. 64:1968-1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frisk, A., C. A. Ison, and T. Lagergard. 1998. GroEL heat shock protein of Haemophilus ducreyi: association with cell surface and capacity to bind to eukaryotic cells. Infect. Immun. 66:1252-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabay, J. E., and M. A. Horwitz. 1985. Isolation and characterization of the cytoplasmic and outer membranes of the Legionnaires' disease bacterium (Legionella pneumophila). J. Exp. Med. 161:409-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garduno, R. A., G. Faulkner, M. A. Trevors, N. Vats, and P. S. Hoffman. 1998. Immunolocalization of Hsp60 in Legionella pneumophila. J. Bacteriol. 180:505-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garduno, R. A., E. Garduno, and P. S. Hoffman. 1998. Surface-associated Hsp60 chaperonin of Legionella pneumophila mediates invasion in a HeLa cell model. Infect. Immun. 66:4602-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garduno, R. A., F. D. Quinn, and P. S. Hoffman. 1998. HeLa cells as a model to study the invasiveness and biology of Legionella pneumophila. Can. J. Microbiol. 44:430-440. [PubMed] [Google Scholar]
- 27.Gibson, F. C., III, A. O. Tzianabos, and F. G. Rodgers. 1994. Adherence of Legionella pneumophila to U-937 cells, guinea-pig alveolar macrophages, and MRC-5 cells by a novel, complement-independent binding mechanism. Can. J. Microbiol. 40:865-872. [DOI] [PubMed] [Google Scholar]
- 28.Gupta, R. S. 1995. Evolution of the chaperonin families (Hsp60, Hsp10, and Tcp-1) of proteins and the origin of eukaryotic cells. Mol. Microbiol. 15:1-11. [DOI] [PubMed] [Google Scholar]
- 29.Gupta, R. S., T. K. W. Ho, M. R. K. Moffat, and R. Gupta. 1982. Podophyllotoxin-resistant mutants of Chinese hamster ovary cells: alteration in a microtubule-associated protein. J. Biol. Chem. 257:1071-1078. [PubMed] [Google Scholar]
- 30.Helsel, L. O., W. F. Bibb, C. A. Butler, P. S. Hoffman, and R. M. McKinney. 1988. Recognition of a genus-wide antigen of Legionella by a monoclonal antibody. Curr. Microbiol. 16:201-208. [Google Scholar]
- 31.Hoffman, P. S., C. A. Butler, and F. D. Quinn. 1989. Cloning and temperature-dependent expression in Escherichia coli of a Legionella pneumophila gene coding for a genus-common 60-kilodalton antigen. Infect. Immun. 57:1731-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffman, P. S., L. Houston, and C. A. Butler. 1990. Legionella pneumophila htpAB heat shock operon: nucleotide sequence and expression of the 60-kilodalton antigen in L. pneumophila-infected HeLa cells. Infect. Immun. 58:3380-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horwitz, M. A. 1983. Formation of a novel phagosome by the Legionnaires' disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 158:1319-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horwitz, M. A. 1983. The Legionnaires' disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J. Exp. Med. 158:2108-2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horwitz, M. A. 1984. Phagocytosis of the Legionnaires' disease bacterium (Legionella pneumophila) occurs by a novel mechanism: engulfment within a pseudopod coil. Cell 36:27-33. [DOI] [PubMed] [Google Scholar]
- 36.Horwitz, M. A. 1987. Characterization of avirulent mutant Legionella pneumophila that survive but do not multiply within human monocytes. J. Exp. Med. 166:1310-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horwitz, M. A., and F. R. Maxfield. 1984. Legionella pneumophila inhibits acidification of its phagosome in human monocytes. J. Cell Biol. 99:1936-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horwitz, M. A., and S. C. Silverstein. 1980. Legionnaires' disease bacterium (Legionella pneumophila) multiples intracellularly in human monocytes. J. Clin. Investig. 66:441-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horwitz, M. A., and S. C. Silverstein. 1983. Intracellular multiplication of Legionnaires' disease bacteria (Legionella pneumophila) in human monocytes is reversibly inhibited by erythromycin and rifampin. J. Clin. Investig. 71:15-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ingmundson, A., A. Delprato, D. G. Lambright, and C. R. Roy. 2007. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450:365-369. [DOI] [PubMed] [Google Scholar]
- 41.Joshi, A. D., S. Sturgill-Koszycki, and M. S. Swanson. 2001. Evidence that Dot-dependent and -independent factors isolate the Legionella pneumophila phagosome from the endocytic network in mouse macrophages. Cell Microbiol. 3:99-114. [DOI] [PubMed] [Google Scholar]
- 42.Kagan, J. C., and C. R. Roy. 2002. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4:945-954. [DOI] [PubMed] [Google Scholar]
- 43.Kagan, J. C., M. P. Stein, M. Pypaert, and C. R. Roy. 2004. Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J. Exp. Med. 199:1201-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khelef, N., H. A. Shuman, and F. R. Maxfield. 2001. Phagocytosis of wild-type Legionella pneumophila occurs through a wortmannin-insensitive pathway. Infect. Immun. 69:5157-5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozak, M. 1987. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol. 196:947-950. [DOI] [PubMed] [Google Scholar]
- 46.Krendel, M., G. Sgourdas, and E. M. Bonder. 1998. Disassembly of actin filaments leads to increased rate and frequency of mitochondrial movement along microtubules. Cell Motil. Cytoskeleton 40:368-378. [DOI] [PubMed] [Google Scholar]
- 47.LeBlanc, J. J., R. J. Davidson, and P. S. Hoffman. 2006. Compensatory functions of two alkyl hydroperoxide reductases in the oxidative defense system Legionella pneumophila. J. Bacteriol. 188:6235-6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu, Y., and Z. Q. Luo. 2007. The Legionella pneumophila effector SidJ is required for efficient recruitment of endoplasmic reticulum proteins to the bacterial phagosome. Infect. Immun. 75:592-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu, H., and M. Clarke. 2005. Dynamic properties of Legionella-containing phagosomes in Dictyostelium amoebae. Cell Microbiol. 7:995-1007. [DOI] [PubMed] [Google Scholar]
- 50.Machner, M. P., and R. R. Isberg. 2006. Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev. Cell 11:47-56. [DOI] [PubMed] [Google Scholar]
- 51.Maguire, M., A. R. Coates, and B. Henderson. 2002. Chaperonin 60 unfolds its secrets of cellular communication. Cell Stress Chaperones 7:317-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mampel, J., T. Spirig, S. S. Weber, J. A. J. Haagensen, S. Molin, and H. Hilbi. 2006. Planktonic replication is essential for biofilm formation by Legionella pneumophila in a complex medium under static and dynamic flow conditions. Appl. Environ. Microbiol. 72:2885-2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marra, A., S. J. Blander, M. A. Horwitz, and H. A. Shuman. 1992. Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. Proc. Natl. Acad. Sci. USA 89:9607-9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsumoto, A., H. Bessho, K. Uehira, and T. Suda. 1991. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. J. Electron. Microsc. 40:356-363. [PubMed] [Google Scholar]
- 55.McLean, I. W., and P. K. Nakane. 1974. Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J. Histochem. Cytochem. 22:1077-1083. [DOI] [PubMed] [Google Scholar]
- 56.Morash, M. G., A. K. Brassinga, M. Warthan, P. Gourabathini, R. A. Garduño, S. D. Goodman, and P. S. Hoffman. 2009. Reciprocal expression of integration host factor and HU in the developmental cycle and infectivity of Legionella pneumophila. Appl. Environ. Microbiol. 75:1826-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morris, R. L., and P. J. Hollenbeck. 1995. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 131:1315-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newton, H. J., F. M. Sansom, V. Bennett-Wood, and E. L. Hartland. 2006. Identification of Legionella pneumophila-specific genes by genomic subtractive hybridization with Legionella micdadei and identification of lpnE, a gene required for efficient host cell entry. Infect. Immun. 74:1683-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ninio, S., and C. R. Roy. 2007. Effector proteins translocated by Legionella pneumophila: strength in numbers. Trends Microbiol. 15:372-380. [DOI] [PubMed] [Google Scholar]
- 60.Oldham, L. J., and F. G. Rodgers. 1985. Adhesion, penetration and intracellular replication of Legionella pneumophila: an in vitro model of pathogenesis. J. Gen. Microbiol. 131:697-706. [DOI] [PubMed] [Google Scholar]
- 61.Payne, N. R., and M. A. Horwitz. 1987. Phagocytosis of Legionella pneumophila is mediated by human monocyte complement receptors. J. Exp. Med. 166:1377-1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peterson, E. M., and L. M. de la Maza. 1988. Chlamydia parasitism: ultrastructural characterization of the interaction between the chlamydial cell envelope and the host cell. J. Bacteriol. 170:1389-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ragaz, C., H. Pietsch, S. Urwyler, A. Tiaden, S. S. Weber, and H. Hilbi. 2008. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell Microbiol. 10:2416-2433. [DOI] [PubMed] [Google Scholar]
- 64.Retzlaff, C., Y. Yamamoto, S. Okubo, P. S. Hoffman, H. Friedman, and T. W. Klein. 1996. Legionella pneumophila heat-shock protein-induced increase of interleukin-1 beta mRNA involves protein kinase C signalling in macrophages. Immunology 89:281-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ridenour, D. A., S. L. Cirillo, S. Feng, M. M. Samrakandi, and J. D. Cirillo. 2003. Identification of a gene that affects the efficiency of host cell infection by Legionella pneumophila in a temperature-dependent fashion. Infect. Immun. 71:6256-6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Riveroll, A. L. 2005. The Legionella pneumophila chaperonin: an investigation of virulence-related roles in a yeast model. Ph.D. thesis. Dalhousie University, Halifax, Nova Scotia, Canada.
- 67.Rowbotham, T. J. 1986. Current views on the relationships between amoebae, legionellae and man. Isr. J. Med. Sci. 22:678-689. [PubMed] [Google Scholar]
- 68.Roy, C. R., K. H. Berger, and R. R. Isberg. 1998. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol. Microbiol. 28:663-674. [DOI] [PubMed] [Google Scholar]
- 69.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 70.Sauer, J. D., J. G. Shannon, D. Howe, S. F. Hayes, M. S. Swanson, and R. A. Heinzen. 2005. Specificity of Legionella pneumophila and Coxiella burnetii vacuoles and versatility of Legionella pneumophila revealed by coinfection. Infect. Immun. 73:4494-4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scanlon, M., G. J. Leitch, G. S. Visvesvara, and A. P. Shaw. 2004. Relationship between the host cell mitochondria and the parasitophorous vacuole in cells infected with Encephalitozoon microsporidia. J. Eukaryot. Microbiol. 51:81-87. [DOI] [PubMed] [Google Scholar]
- 72.Schepis, A., B. Schramm, C. A. de Haan, and J. K. Locker. 2006. Vaccinia virus-induced microtubule-dependent cellular rearrangements. Traffic 7:308-323. [DOI] [PubMed] [Google Scholar]
- 73.Sinai, A. P., and K. A. Joiner. 2001. The Toxoplasma gondii protein ROP2 mediates host organelle association with the parasitophorous vacuole membrane. J. Cell Biol. 154:95-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sinai, A. P., P. Webster, and K. A. Joiner. 1997. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: a high-affinity interaction. J. Cell Sci. 110:2117-2128. [DOI] [PubMed] [Google Scholar]
- 75.Steinert, M., M. Ott, P. C. Luck, E. Tannich, and J. Hacker. 1994. Studies on the uptake and intracellular replication of Legionella pneumophila in protozoa and in macrophage-like cells. FEMS Microbiol. Ecol. 15:299-308. [Google Scholar]
- 76.Sturgill-Koszycki, S., and M. S. Swanson. 2000. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J. Exp. Med. 192:1261-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Summerhayes, I. C., D. Wong, and L. B. Chen. 1983. Effect of microtubules and intermediate filaments on mitochondrial distribution. J. Cell Sci. 61:87-105. [DOI] [PubMed] [Google Scholar]
- 78.Susa, M., and R. Marre. 1999. Legionella pneumophila invasion of MRC-5 cells induces tyrosine protein phosphorylation. Infect. Immun. 67:4490-4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Swanson, M. S., and R. R. Isberg. 1995. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63:3609-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tilney, L. G., O. S. Harb, P. S. Connelly, C. G. Robinson, and C. R. Roy. 2001. How the parasitic bacterium Legionella pneumophila modifies its phagosome and transforms it into rough ER: implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 114:4637-4650. [DOI] [PubMed] [Google Scholar]
- 81.Törok, Z., I. Horvath, P. Goloubinoff, E. Kovacs, A. Glatz, G. Balogh, and L. Vigh. 1997. Evidence for a lipochaperonin: association of active protein-folding GroESL oligomers with lipids can stabilize membranes under heat shock conditions. Proc. Natl. Acad. Sci. USA 94:2192-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Viswanathan, V. K., and N. P. Cianciotto. 2000. Electroporation of Legionella species, p. 203-211. In Electro-transformation of bacteria. Springer Verlag Publications, Heidelberg, Germany.
- 83.Vogel, J., C. Roy, and R. R. Isberg. 1996. Use of salt to isolate Legionella pneumophila mutants unable to replicate in macrophages. Ann. N. Y. Acad. Sci. 797:271-272. [DOI] [PubMed] [Google Scholar]
- 84.Watarai, M., I. Derre, J. Kirby, J. D. Growney, W. F. Dietrich, and R. R. Isberg. 2001. Legionella pneumophila is internalized by a macropinocytotic uptake pathway controlled by the Dot/Icm system and the mouse Lgn1 locus. J. Exp. Med. 194:1081-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wieland, H., F. Goetz, and B. Neumeister. 2004. Phagosomal acidification is not a prerequisite for intracellular multiplication of Legionella pneumophila in human monocytes. J. Infect. Dis. 189:1610-1614. [DOI] [PubMed] [Google Scholar]
- 86.Winn, W. C., Jr. 1988. Legionnaires disease: historical perspective. Clin. Microbiol. Rev. 1:60-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yoshida, N., K. Oeda, E. Watanabe, T. Mikami, Y. Fukita, K. Nishimura, K. Komai, and K. Matsuda. 2001. Protein function: chaperonin turned insect toxin. Nature 411:44. [DOI] [PubMed] [Google Scholar]
- 88.Zhang, L., S. L. Pelech, D. Mayrand, D. Grenier, J. Heino, and V. J. Uitto. 2001. Bacterial heat shock protein-60 increases epithelial cell proliferation through the ERK1/2 MAP kinases. Exp. Cell Res. 266:11-20. [DOI] [PubMed] [Google Scholar]
- 89.Zhang, L., S. Pelech, and V. J. Uitto. 2004. Long-term effect of heat shock protein 60 from Actinobacillus actinomycetemcomitans on epithelial cell viability and mitogen-activated protein kinases. Infect. Immun. 72:38-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou, D., M. S. Mooseker, and J. E. Galan. 1999. Role of the Salmonella typhimurium actin binding protein SipA in bacterial internalization. Science 283:2092-2095. [DOI] [PubMed] [Google Scholar]