

Abstract
Group I CD1 proteins are specialized antigen-presenting molecules that present both microbial and self lipid antigens to CD1-restricted α/β T lymphocytes. The production of high levels of gamma interferon and lysis of infected macrophages by lipid-specific T lymphocytes are believed to play pivotal roles mainly in the defense against mycobacterial infections. We previously demonstrated that Mycobacterium tuberculosis and bacillus Calmette-Guérin (Mycobacterium bovis BCG) induce human monocytes to differentiate into CD1− dendritic cells (DC), which cannot present lipid antigens to specific T cells. Here, we show that in human monocytes mycobacteria trigger phosphorylation of p38 mitogen-activated protein kinase to inhibit CD1 expression in DC derived from infected monocytes. Pretreatment with a specific p38 inhibitor renders monocytes insensitive to mycobacterial subversion and allows them to differentiate into CD1+ DC, which are fully capable of presenting lipid antigens to specific T cells. We also report that one of the pathogen recognition receptors triggered by BCG to activate p38 is complement receptor 3 (CR3), as shown by reduced p38 phosphorylation and partial reestablishment of CD1 membrane expression obtained by CR3 blockade before infection. In conclusion, we propose that p38 signaling is a novel pathway exploited by mycobacteria to affect the expression of CD1 antigen-presenting cells and avoid immune recognition.
CD1 molecules are nonpolymorphic glycoproteins with structural homology to major histocompatibility complex (MHC) class I molecules (21). They are classified into three groups. Group I molecules (CD1a, CD1b, and CD1c) are expressed on the surface of a limited set of antigen-presenting cells (APC), including Langerhans cells (27), dendritic cells (DC), and granulocyte-macrophage colony-stimulating factor (GM-CSF)-exposed macrophages (14). Group II includes CD1d, which is more widely expressed on hematopoietic and nonhematopoietic cells, whereas group III (CD1e) is restricted to myeloid DC (1).
Group I and group II CD1 molecules are specialized antigen-presenting molecules that bind and present microbial, environmental, and self lipids to αβ and γδ T cells and thus participate in the immune response during infectious, autoimmune, or allergic diseases (3). Group I CD1-restricted T cells have been investigated mostly in mycobacterial infections as the majority of microbial lipids, which form immunogenic complexes with CD1 molecules, are constituents of the Mycobacterium tuberculosis cell wall and membrane (22). The finding that CD1-restricted T lymphocytes specific for mycobacterial glycolipids are present in individuals previously infected with M. tuberculosis provided strong evidence that the CD1-restricted T-cell response has an effective role in host defense against mycobacteria (19, 32). Moreover, CD1b-restricted T cells specific for a mycobacterial diacylated sulfoglycolipid kill intracellular bacteria and are detected in M. tuberculosis-infected individuals (12). Since CD1 molecules are essential for lipid antigen recognition by specific T cells, it has been proposed that M. tuberculosis evolved strategies to inhibit CD1 expression in infected host cells (31). Consistent with this hypothesis, in vitro experiments have shown that exposure of monocytes to M. tuberculosis, to bacillus Calmette-Guérin (Mycobacterium bovis BCG), or to α-glucan, a polysaccharide that forms the outermost layer of the M. tuberculosis cell wall, leads to inhibition of CD1 molecule expression (10, 11, 18). Nevertheless, the molecular mechanisms exploited by mycobacteria to regulate CD1 expression have not been identified.
The aim of this study was to investigate the intracellular events involved in the blockade of CD1 molecule expression on DC derived from mycobacterium-infected monocytes.
MATERIALS AND METHODS
Reagents.
Recombinant interleukin-4 (IL-4) was purchased from R&D Systems (Minneapolis, MN), and GM-CSF was purchased from Gentaur (Brussels, Belgium). Lipopolysaccharide (LPS) from Escherichia coli was obtained from Sigma-Aldrich (St. Louis, MO). RPMI 1640 (Euroclone, Celbio Spa, Milan, Italy) was supplemented with 100 U/ml kanamycin, 1 mM glutamine, 1 mM sodium pyruvate, 1% nonessential amino acids, and 10% fetal bovine serum (HyClone, Logan, UT) to obtain a complete medium. Phosphate-buffered saline (PBS) was obtained from Euroclone.
The p38 inhibitor SB203580 and the extracellular signal-regulated kinase (ERK) inhibitor PD98059 were obtained from Calbiochem Biochemicals (San Diego, CA), and purified sulfatide was obtained from Fluka (Buchs, Switzerland).
Growth of mycobacteria.
M. tuberculosis H37Rv and BCG strain ATCC 27291 were grown with gentle agitation (80 rpm) in Middlebrook 7H9 broth (Difco, BD Diagnostics, Heidelberg, Germany) supplemented with 0.05% Tween 80 (Sigma-Aldrich) and 10% Middlebrook ADC enrichment (Becton Dickinson). Logarithmically growing cultures were washed twice in RPMI 1640. Mycobacteria were resuspended in RPMI 1640 containing 10% fetal bovine serum and then stored at −80°C. Vials were thawed, and bacterial viability was determined by counting the number of CFU on Middlebrook 7H10 agar plates. All preparations were analyzed for LPS contamination by the Limulus lysate assay (BioWhittaker Europe, Verviers, Belgium) and contained less than 10 pg/ml of LPS.
Monocyte isolation, infection, and DC generation.
In vitro human studies were reviewed and approved by the Istituto Superiore di Sanità Ethical Committee (http://www.iss.it/coet/index.php?lang=1).
Peripheral blood mononuclear cells were purified from heparinized blood obtained from healthy donors (Blood Bank of University “La Sapienza,” Rome, Italy). Monocytes were then positively sorted using anti-CD14-labeled magnetic beads (Miltenyi, Bergisch Gladbech, Germany). In all experiments monocytes were infected with single-cell suspensions of BCG and M. tuberculosis at multiplicities of infection (ratios of bacteria to monocytes) of 6:1 and 3:1, respectively. The efficiency of infection or phagocytosis was determined by counting intracellular mycobacteria in cells stained by the Kinyoun method. DC were generated by culturing infected or noninfected monocytes for 5 days in complete medium containing 50 ng/ml GM-CSF and 1,000 U/ml IL-4 (conditioned culture). In some experiments the p38 inhibitor SB203580 or the ERK inhibitor PD98059 at a concentration of 3 μM was added 30 min before monocyte infection or to noninfected cells and was not removed during the 5 days of culture.
Fluorescence-activated cell sorting analysis.
All monoclonal antibodies (MAbs) were obtained from BD/Pharmingen (San Diego, CA). A phycoerythrin-conjugated goat anti-mouse MAb from Southern Biotech (Birmingham, AL) was used in association with MAb anti-CD1c. Cells were harvested and washed in PBS containing 1% fetal bovine serum and 0.1% NaN3 (staining buffer) and were stained using the MAbs mentioned above or appropriate isotype controls for background determination.
Stained cells were analyzed using a FACScan cytometer equipped with Cellquest software (Becton Dickinson).
Quantitative reverse transcription-PCR for detection of CD1 mRNA expression.
Total RNA was extracted from 106 monocytes, control DC, or DC derived from infected monocytes using an RNeasy mini kit (Qiagen, Milan, Italy), treated with DNase, and reverse transcribed using ImProm II (Promega Italia, Milan, Italy). cDNAs were amplified using primers specific for human β-actin (sense primer TCCTTCCTGGGCATGGAGTC and antisense primer CAGGAGGAGCAATGATCTTGATC), CD1a (sense primer ACAGCAATTCCAGCACCATCG and antisense primer AGCCTCCTGTCACCTGTATCTC), CD1b (sense primer GGTTGCTGAGTTAGAGGAGATATTC and antisense primer CCCTGCTGCCACCTTCTG), and CD1c (sense primer TGTATAATCTCATAAGAAGCACTTG and antisense primer TCAGCATTAGGAAGAATATCACC) in 20-μl mixtures with iQ SYBR green Supermix (Bio-Rad, Hercules, CA), using an iCycler iQ (Bio-Rad) for 40 cycles of 40 s at 95°C, 40 s at 57°C, and 60 s at 72°C. Cycle threshold data were converted to the number of molecules of CD1 mRNA per molecule of β-actin using standard curves established for each primer pair as a reference. Finally, data were expressed as increases relative to the levels of CD1 mRNA in freshly isolated CD1− monocytes.
Western blot analysis.
Monocytes (2 × 106 cells) were incubated with M. tuberculosis or BCG for different times and washed once with cold PBS. To detect phosphorylated and total p38 and ERK, total cell extracts were prepared from cellular pellets resuspended in 200 μl of 2× sodium dodecyl sulfate (SDS) sample buffer (20 mM dithiothreitol, 6% SDS, 0.25 M Tris [pH 6.8], 10% glycerol, 10 mM NaF, bromophenyl blue) and boiled for 5 min. Nuclear extracts were prepared to detect phosphorylated and total activating transcription factor 2 (ATF-2). Cells were resuspended in 200 μl hypotonic lysis buffer (10 mM HEPES [pH 7.8], 10 mM KCl, 1 mM MgCl2, 0.1 mM EGTA, 0.5 mM EDTA, 0.1% NP-40, 5% glycerol, 1% phenylmethylsulfonyl fluoride, 1% NaF, 1% NaVO4, 1% protease inhibitors) and kept on ice for 10 min. After centrifugation the nuclear pellets were extracted using 60 μl hypertonic lysis buffer (50 mM HEPES [pH 7.8], 400 mM NaCl, 1 mM MgCl2, 1 mM EGTA, 1 mM EDTA, 10% glycerol, 1% phenylmethylsulfonyl fluoride, 1% NaF, 1% NaVO4, 1% protease inhibitors) on ice for 40 min. Proteins were separated by SDS-polyacrylamide gel electrophoresis and blotted onto nitrocellulose membranes (Hybond C-Extra; Amersham Pharmacia Biotech, Uppsala, Sweden). Blots were incubated with anti-phosphorylated and total p38, ERK (R&D Systems), and ATF-2 (Cell Signaling Technology, Danvers, MA) rabbit antibodies. Then horseradish peroxidase-labeled anti-rabbit antibody was added, and the reaction was revealed using an ECL system (Amersham Pharmacia Biotech).
Complement receptor type 3 (CR3) blockade experiments.
Freshly isolated monocytes resuspended in complete medium were incubated at 4°C for 30 min before M. tuberculosis or BCG infection with 50 μg/ml purified mouse immunoglobulin G1 MAb anti-CD11b/Mac-1 (ICRF44 clone), with an appropriate isotype control MAb, or with purified anti-CD3 or anti-CD11c MAb (BD/Pharmingen). Noninfected or infected monocytes were analyzed for p38 and ATF-2 phosphorylation after 2 h of incubation at 37°C or for CD1 surface expression after 5 days of conditioned culture.
Antigen presentation assays.
A purified protein derivative-specific MHC class II-restricted T-cell clone and sulfatide-specific CD1a-restricted and (+)-2-palmitoyl- or 2-stearoyl-3-hydroxyphthioceranoyl-2′-sulfate-α-α′-d-trehalose (Ac2SGL)-specific CD1b-restricted T-cell clones were established and maintained as previously described (12). Control DC (MoDC) and DC derived from M. tuberculosis-infected monocytes (Mtb-MoDC) that were pretreated or not pretreated with the p38 inhibitor SB203580 (3 μM) were pulsed for 2 h at 37°C with sonicated sulfatide (10 μg/ml) before addition of a T-cell clone (3 × 104 cells/well in triplicate). MoDC and Mtb-MoDC (1 × 104 cells/well) pretreated or not pretreated with the p38 inhibitor SB203580 (3 μM) and MoDC infected at day 5 were cocultured with the CD1b-restricted T-cell clone. After 48 h supernatants were examined for gamma interferon (IFN-γ) production using commercially available enzyme-linked immunosorbent assay kits (R&D Systems) according to the manufacturer's instructions.
RESULTS
Mycobacterium-induced p38 phosphorylation inhibits CD1 surface expression on DC derived from infected monocytes.
Monocytes infected with M. tuberculosis or BCG and then cultured with GM-CSF and IL-4 differentiate into DC that lack CD1a, CD1b, and CD1c expression (11, 18). Since inactivation of the p38 mitogen-activated protein kinase (MAPK) accompanies differentiation of monocytes into DC (34), we sought to investigate whether p38 phosphorylation was involved in mycobacterium-induced inhibition of CD1 expression. Confirming previous results (24, 30), kinetic studies of MAPK phosphorylation following infection of human monocytes showed that M. tuberculosis induced both ERK phosphorylation and p38 phosphorylation, which were detectable 1 h after infection and persisted for at least 3 h after infection (Fig. 1A). To assess the role of MAPK phosphorylation in regulation of CD1 expression, monocytes were incubated with the specific p38 inhibitor SB203580 or with the ERK inhibitor PD98059 before infection and analyzed by immunofluorescence after 5 days of culture with GM-CSF and IL-4. Strikingly, pretreatment of monocytes with the p38 inhibitor rendered monocytes insensitive to the M. tuberculosis inhibitory activity and allowed their differentiation into CD1+ DC (Fig. 1B), while the ERK inhibitor had no effect. Similar results were obtained when BCG was used to infect monocytes (data not shown). Inhibitors did not interfere with the differentiation of control monocytes into DC and did not affect cell viability (data not shown). To the best of our knowledge, our results reveal an unexpected p38-mediated mechanism exploited by mycobacteria to regulate group I CD1 expression.
FIG. 1.

Mycobacterial infection of human monocytes causes their differentiation into CD1− DC by triggering p38 phosphorylation. (A) Monocytes were incubated with M. tuberculosis for the indicated times and treated as described in Materials and Methods to detect total ERK (t ERK), phosphorylated ERK (p ERK), total p38 (t p38), and phosphorylated p38 (p p38). Similar data were obtained in three independent experiments. (B) Monocytes were infected with M. tuberculosis and cultured with GM-CSF and IL-4 (Mtb-MoDC) for 5 days. Some of the monocytes were pretreated with the p38 inhibitor SB203580 (Mtb-MoDC+SB) or the ERK inhibitor PD98059 (Mtb-MoDC+PD) before infection. The markers excluded 95% of the events recorded with the appropriate isotype control, and the percentages indicate the percentages of positive cells. The results of one experiment that is representative of five experiments are shown.
ATF-2 is phosphorylated by mycobacterium-induced p38 activation.
To further explore intracellular signaling events upon mycobacterium-induced p38 activation, we focused on ATF-2, which has been shown to have a binding site in the promoter region of the CD1a molecule (5). We found that M. tuberculosis (Fig. 2A) and BCG (data not shown) induced phosphorylation of ATF-2 in a p38-dependent manner, as demonstrated by the inhibition of ATF-2 activation obtained with p38 inhibitor treatment before mycobacterial infection. Notably, activation of ATF-2 decreases CD1A transcription (5), and, in accordance with previous results (11, 18, 31), we found that M. tuberculosis infection of human monocytes strongly inhibits CD1a, CD1c, and, to a lesser extent, CD1b mRNA induction during the differentiation of the monocytes into DC, as determined by real-time PCR analysis at different time points (Fig. 2B). Similar results were obtained using BCG (data not shown). mRNA levels strictly correlate with surface expression of CD1 molecules on both infected and noninfected cells (data not shown), suggesting that CD1 transcriptional regulation is an important target of mycobacterial infection. Further studies will address the role of ATF-2 or other ATF/CREB family members in regulation of CD1b and CD1c expression.
FIG. 2.
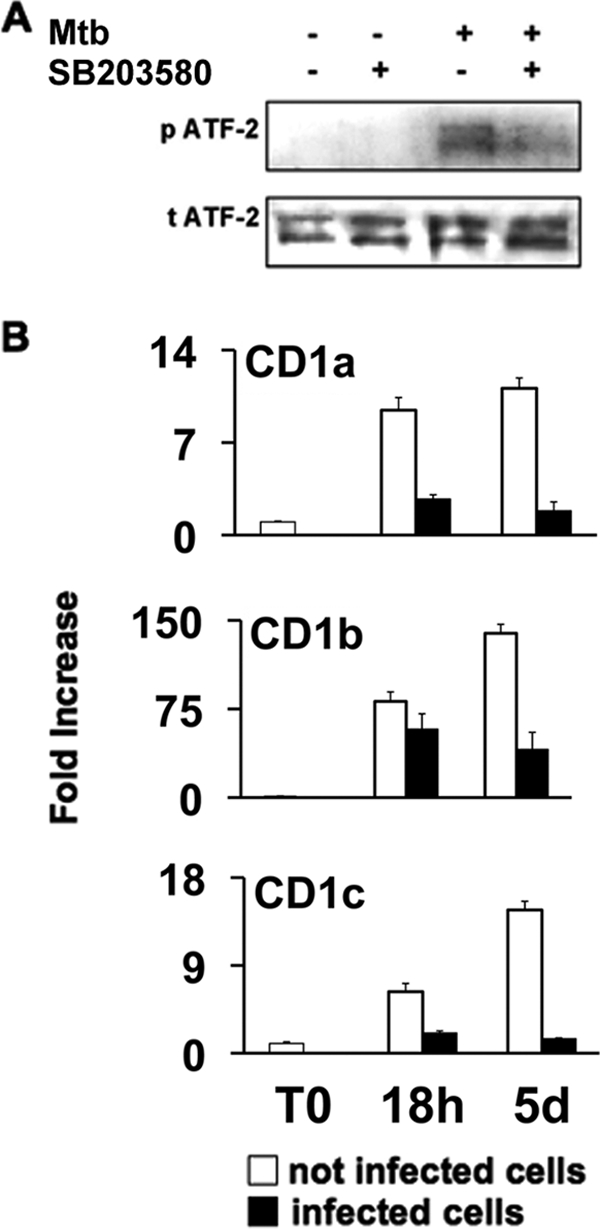
Mycobacteria activate ATF-2 through p38 phosphorylation and inhibit CD1 gene expression. (A) Monocytes were pretreated or not pretreated with p38 inhibitor (SB203580), and some of them were infected with M. tuberculosis (Mtb) for 2 h. Nuclear extracts were analyzed by immunoblotting with anti-phosphorylated ATF-2-specific antibody (p ATF-2) and anti-total ATF-2-specific antibody (t ATF-2). The results of one experiment that is representative of three experiments are shown. (B) CD1 gene expression was analyzed by quantitative reverse transcription-PCR. Untreated monocytes (open bars) and M. tuberculosis-infected monocytes (filled bars) were analyzed for CD1 mRNAs immediately after isolation (T0) and after 18 h (18h) and 5 days (5d) of culture with GM-CSF and IL-4. The mRNA levels are expressed as increases relative to the level of CD1 mRNA in freshly isolated CD1− monocytes, and the data are means and standard deviations of three replicates.
Inhibition of p38 restores the capacity of DC derived from M. tuberculosis-infected monocytes to present lipid antigens to CD1-restricted T cells.
Next we investigated whether p38 activation by mycobacteria may also affect lipid antigen presentation.
Mtb-MoDC did not present the lipid antigen sulfatide to a CD1a-restricted T-cell clone, and this correlated with low CD1a surface expression (Fig. 3A). Pretreatment with the p38 inhibitor restored both the CD1a expression by and the antigen-presenting capacity of Mtb-MoDC. Notably, similar results were obtained using a CD1b-restricted T-cell clone specific for Ac2SGL, a mycobacterial diacylated sulfoglycolipid that is presented by CD1 molecules following M. tuberculosis processing (12). As shown in Fig. 3B, control DC infected with M. tuberculosis on day 5 of culture stimulated the Ac2SGL-specific T-cell clone. In contrast, Mtb-MoDC had a drastically reduced capacity to present the same lipid antigen, in agreement with their low level of CD1b expression, although they were capable of presenting purified protein derivative to a specific MHC class II-restricted T-cell clone (data not shown). Pretreatment of M. tuberculosis-infected monocytes with the p38 inhibitor led to differentiation of CD1b+ Mtb-MoDC that were as efficient as control DC in presenting the lipid antigen. Together, these data indicate the pivotal role of p38 signaling in regulation of the CD1-restricted T-cell response during mycobacterial infections.
FIG. 3.

Inhibition of p38 restores the capacity of Mtb-MoDC to present lipid antigens to CD1-restricted T cells. The APC function of Mtb-MoDC was compared to that of control MoDC, DC derived from uninfected monocytes pretreated with the p38 inhibitor SB203580 (MoDC+SB), or M. tuberculosis-infected monocytes pretreated with the p38 inhibitor SB203580 (Mtb-MoDC+SB). In some experiments MoDC were infected with M. tuberculosis (MoDC+Mtb) at the end of the differentiation culture (day 5). (A) APC pulsed or not pulsed with sulfatide were cocultured with a sulfatide-specific CD1a-restricted T-cell clone. (B) A CD1b-restricted T-cell clone specific for Ac2SGL was used to test the capacity of infected cells to process mycobacteria and present lipid antigens. The bars indicate the amounts of IFN-γ secreted by responder T-cell clones, and the lines indicate the percentages of CD1 molecule expression on APC. The amounts of IFN-γ (mean ± standard deviation of three replicates) were measured after 48 h of culture. The results of one experiment that is representative of three experiments are shown. An asterisk indicates that there is a significant difference in IFN-γ secretion (P < 0.05) between M. tuberculosis-infected cells treated with SB203580 and nontreated cells.
CR3 is involved in the inhibition of CD1 expression by BCG.
Inhibition of CD1 expression on mycobacterium-infected monocytes occurs at an early step during their differentiation into DC, while later infections have no effect (11). In an attempt to identify a putative receptor involved in this phenomenon, we analyzed the kinetics of expression of the phagocytic receptors known to bind mycobacterial polysaccharides, as we previously showed that cell wall-associated α-glucan mimics the effects of the whole bacterium (10). Freshly isolated monocytes expressed CR3, while the mannose receptor (29) and DC-specific intercellular adhesion molecule 3-grabbing nonintegrin (6) were induced only after 12 and 24 h of culture with GM-CSF and IL-4, respectively (Fig. 4). These data suggested that we should focus on CR3, which is involved in the nonopsonic phagocytosis of mycobacteria.
FIG. 4.

Kinetics of phagocytic receptor appearance at the onset of monocyte differentiation into DC. Surface expression of CR3, mannose receptor (MR), and DC-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN) was analyzed using freshly isolated monocytes (Mo T0) and monocytes after 12 and 24 h of culture with GM-CSF and IL-4 (Mo 12h and Mo 24h, respectively). The dotted lines indicate data for the appropriate isotype control MAbs. Data from one experiment that is representative of three experiments are shown.
Interestingly, monocytes pretreated with the anti-CR3 MAb ICRF44 before BCG infection differentiated into DC with increased expression of CD1a, CD1c, and, to a lesser extent, CD1b compared to untreated infected cells (Fig. 5A). Treatment with an isotype control MAb, anti-CD3 MAb, or anti-CD11c MAb before infection did not restore CD1 expression, and anti-CR3 MAb alone did not have any effect (data not shown). Notably, CR3 blockade also caused a decrease in BCG-induced p38 and ATF-2 phosphorylation (Fig. 5B and C). However, blocking CR3 on monocytes before M. tuberculosis infection barely restored CD1 molecule expression and did not prevent p38 and ATF-2 phosphorylation (data not shown).
FIG. 5.

BCG triggers CR3 to phosphorylate p38 and ATF-2 and to inhibit CD1 expression on DC derived from BCG-infected monocytes (BCG-MoDC). (A) Monocytes were incubated or not incubated with an anti-CR3 MAb (clone ICRF44) (αCR3) or control isotype before BCG infection. Group I CD1 expression was analyzed by flow cytometry on day 5 of culture with GM-CSF and IL-4. The dot plots show that BCG infection did not interfere with the differentiation of monocytes into CD14− DC. The markers in the histogram plots were set to exclude 95% of events recorded with the appropriate isotype control. The percentages indicate the percentages of positive cells. Data from one experiment that is representative of five experiments are shown. (B) Monocytes were stimulated with BCG for 2 h. Cells were pretreated with an anti-CR3 MAb (ICRF44) or an isotype control before BCG infection. Cell lysates were analyzed by immunoblotting with a specific anti-phosphorylated p38 antibody (p p38) and a specific anti-total p38 antibody (t p38). Similar data were obtained in three independent experiments. (C) Monocytes were pretreated with p38 inhibitor or with the anti-CR3 MAb ICRF44 and then stimulated with BCG for 2 h. Cell lysates were analyzed by immunoblotting with a specific anti-phosphorylated ATF-2 antibody (p ATF-2) and a specific anti-total ATF-2 antibody (t ATF-2). Similar data were obtained in three independent experiments.
DISCUSSION
The interference of pathogens with the regulation of antigen-presenting molecules is a well-defined escape mechanism that has been studied mainly in relation to MHC- or CD1d-restricted immune responses (20, 25, 33, 35). In contrast, regulation of group 1 CD1 molecules and the pathogen-dependent interference with their membrane expression are poorly understood. In this work we found that p38 signaling is the intracellular pathway exploited by mycobacteria to inhibit CD1 molecule expression on DC derived from infected monocytes. Activation of the MAPK signaling pathway upon mycobacterial infection has been demonstrated to inhibit several antimicrobial mechanisms (13). Mycobacteria exploit p38 to arrest phagosome maturation in murine macrophages, and pharmacological blockade of p38 activity enhances phagosome acidification (9). Moreover, activation of MAPK signaling by an M. tuberculosis 19-kDa lipoprotein has been demonstrated to inhibit class II transactivator and, as a consequence, class II MHC expression by murine macrophages (20). Thus, our findings for p38-mediated regulation of both CD1 molecule expression and lipid antigen presentation confirm the pivotal role of MAPK signaling in regulation of the host defense against mycobacterial infection. Of the several known downstream targets of p38, we chose to examine ATF-2, since this molecule has been shown to bind the CD1A promoter region and to decrease CD1A transcription (5). ATF-2 was phosphorylated in a p38-dependent manner in mycobacterium-infected monocytes, suggesting that its activation may be involved in mycobacterium-induced inhibition of CD1A gene transcription. However, additional transcription factors and/or other p38-dependent posttranscriptional mechanisms involved in the regulation of CD1b and CD1c expression have to be investigated.
CR3 plays a pivotal role in the opsonic and nonopsonic phagocytosis of mycobacteria (16, 28). A lectin-like domain in the α subunit of CR3 mediates the nonopsonic binding of mycobacterial outer capsular carbohydrates, such as α-glucan, which has been shown to inhibit CD1 expression (7, 10). We found that CR3 blockade before infection resulted in reduced p38 and ATF-2 phosphorylation and increased expression of CD1 molecules in DC derived from BCG-infected monocytes. These data strongly suggest that BCG causes inhibition of CD1 expression through CR3-dependent p38 phosphorylation. Unexpectedly, the CR3 blockade effects were only marginal in DC derived from M. tuberculosis-infected monocytes. Since M. tuberculosis and BCG share the outermost structures of the cell wall (10), they should interact with the same cellular receptor(s). Thus, it is not clear why anti-CR3 MAb does not inhibit the consequences of M. tuberculosis infection as efficiently as it inhibits the consequences of BCG infection for CD1 expression. Infection, in contrast to treatment with isolated components, challenges monocytes with several mycobacterial cell wall components, which may lead to simultaneous engagement of different pathogen recognition receptors, including Toll-like receptors 2 and 4. Both of these receptors induce p38 phosphorylation (13) and could contribute in different ways to group I CD1 regulation by BCG or M. tuberculosis together with CR3. However, previously, Toll-like receptor 2 stimulation by M. tuberculosis or an M. tuberculosis product was associated with weak induction of CD1 molecule expression on human monocytes (15, 26). These different results might have been due to the diverse in vitro systems studied, which may reproduce different stages of the disease. The use of monocytes cultured in the absence of added cytokines may reproduce monocyte-M. tuberculosis interactions that occur during primary tuberculosis (TB). Our method, which uses DC differentiation stimuli, mimics postprimary TB, when monocytes are recruited together with memory T lymphocytes into inflammatory sites where M. tuberculosis is actively replicating. In fact, we have previously shown that activated lymphocytes secrete cytokines, such as GM-CSF, which drive differentiation of monocytes into DC (17).
Findings concerning the protective role of group I CD1-restricted T cells in infectious diseases are complicated by the absence of animal models (2). Moreover, there have been only a few conflicting reports on group I CD1 expression on APC in lungs and/or lymph nodes from patients affected by TB (4, 8, 23). Thus, if group I CD1-restricted T lymphocytes contribute to the effective defense against M. tuberculosis (12), the inhibition of CD1 expression might allow M. tuberculosis to effectively evade T-cell surveillance during infection in vivo. Along these lines, we have shown that the functional consequence of mycobacterium-induced CD1 inhibition is reduced activation of CD1-restricted and lipid antigen-specific T cells. In conclusion, our findings show that M. tuberculosis has evolved a strategy to hide its lipid antigens to T cells by blocking the CD1 lipid-presenting molecules on inflammatory APC. Restoration of lipid-specific T-cell responses in vitro by pharmacological inhibition of p38 may provide interesting new perspectives for intervention against TB based on blockade of the M. tuberculosis-dependent modulation of CD1 expression.
Acknowledgments
We thank Federica Sallusto for critical reading of the manuscript.
This work was supported in part by the EC FP6 project “MILD-TB” (contract 037326) and by the collaborative ISS-NIH Project (grant 5303).
We have no financial conflicts of interest.
Editor: J. L. Flynn
Footnotes
Published ahead of print on 31 August 2009.
REFERENCES
- 1.Angenieux, C., J. Salamero, D. Fricker, J. P. Cazenave, B. Goud, D. Hanau, and H. de La Salle. 2000. Characterization of CD1e, a third type of CD1 molecule expressed in dendritic cells. J. Biol. Chem. 275:37757-37764. [DOI] [PubMed] [Google Scholar]
- 2.Behar, S. M., and S. A. Porcelli. 2007. CD1-restricted T cells in host defense to infectious diseases. Curr. Top. Microbiol. Immunol. 314:215-250. [DOI] [PubMed] [Google Scholar]
- 3.Bricard, G., and S. A. Porcelli. 2007. Antigen presentation by CD1 molecules and the generation of lipid-specific T cell immunity. Cell Mol. Life Sci. 64:1824-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buettner, M., C. Meinken, M. Bastian, R. Bhat, E. Stossel, G. Faller, G. Cianciolo, J. Ficker, M. Wagner, M. Rollinghoff, and S. Stenger. 2005. Inverse correlation of maturity and antibacterial activity in human dendritic cells. J. Immunol. 174:4203-4209. [DOI] [PubMed] [Google Scholar]
- 5.Colmone, A., S. Li, and C. R. Wang. 2006. Activating transcription factor/cAMP response element binding protein family member regulated transcription of CD1A. J. Immunol. 177:7024-7032. [DOI] [PubMed] [Google Scholar]
- 6.Curtis, B. M., S. Scharnowske, and A. J. Watson. 1992. Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc. Natl. Acad. Sci. USA 89:8356-8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cywes, C., H. C. Hoppe, M. Daffe, and M. R. Ehlers. 1997. Nonopsonic binding of Mycobacterium tuberculosis to complement receptor type 3 is mediated by capsular polysaccharides and is strain dependent. Infect. Immun. 65:4258-4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flores-Batista, V. C., N. Boechat, P. M. Lago, L. C. Lazzarini, L. R. Pessanha, A. S. Almeida, T. T. Mafort, A. L. Kritski, J. L. Ho, and J. R. Lapa-e-Silva. 2007. Low expression of antigen-presenting and costimulatory molecules by lung cells from tuberculosis patients. Braz. J. Med. Biol. Res. 40:1671-1679. [DOI] [PubMed] [Google Scholar]
- 9.Fratti, R. A., J. Chua, and V. Deretic. 2003. Induction of p38 mitogen-activated protein kinase reduces early endosome autoantigen 1 (EEA1) recruitment to phagosomal membranes. J. Biol. Chem. 278:46961-46967. [DOI] [PubMed] [Google Scholar]
- 10.Gagliardi, M. C., A. Lemassu, R. Teloni, S. Mariotti, V. Sargentini, M. Pardini, M. Daffe, and R. Nisini. 2007. Cell wall-associated alpha-glucan is instrumental for Mycobacterium tuberculosis to block CD1 molecule expression and disable the function of dendritic cell derived from infected monocyte. Cell. Microbiol. 9:2081-2092. [DOI] [PubMed] [Google Scholar]
- 11.Gagliardi, M. C., R. Teloni, S. Mariotti, E. Iona, M. Pardini, L. Fattorini, G. Orefici, and R. Nisini. 2004. Bacillus Calmette-Guerin shares with virulent Mycobacterium tuberculosis the capacity to subvert monocyte differentiation into dendritic cell: implication for its efficacy as a vaccine preventing tuberculosis. Vaccine 22:3848-3857. [DOI] [PubMed] [Google Scholar]
- 12.Gilleron, M., S. Stenger, Z. Mazorra, F. Wittke, S. Mariotti, G. Bohmer, J. Prandi, L. Mori, G. Puzo, and G. De Libero. 2004. Diacylated sulfoglycolipids are novel mycobacterial antigens stimulating CD1-restricted T cells during infection with Mycobacterium tuberculosis. J. Exp. Med. 199:649-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jo, E. K., C. S. Yang, C. H. Choi, and C. V. Harding. 2007. Intracellular signalling cascades regulating innate immune responses to mycobacteria: branching out from Toll-like receptors. Cell. Microbiol. 9:1087-1098. [DOI] [PubMed] [Google Scholar]
- 14.Kasinrerk, W., T. Baumruker, O. Majdic, W. Knapp, and H. Stockinger. 1993. CD1 molecule expression on human monocytes induced by granulocyte-macrophage colony-stimulating factor. J. Immunol. 150:579-584. [PubMed] [Google Scholar]
- 15.Krutzik, S. R., M. T. Ochoa, P. A. Sieling, S. Uematsu, Y. W. Ng, A. Legaspi, P. T. Liu, S. T. Cole, P. J. Godowski, Y. Maeda, E. N. Sarno, M. V. Norgard, P. J. Brennan, S. Akira, T. H. Rea, and R. L. Modlin. 2003. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat. Med. 9:525-532. [DOI] [PubMed] [Google Scholar]
- 16.Le Cabec, V., S. Carreno, A. Moisand, C. Bordier, and I. Maridonneau-Parini. 2002. Complement receptor 3 (CD11b/CD18) mediates type I and type II phagocytosis during nonopsonic and opsonic phagocytosis, respectively. J. Immunol. 169:2003-2009. [DOI] [PubMed] [Google Scholar]
- 17.Mariotti, S., V. Sargentini, C. Marcantonio, E. Todero, R. Teloni, M. C. Gagliardi, A. R. Ciccaglione, and R. Nisini. 2008. T-cell-mediated and antigen-dependent differentiation of human monocyte into different dendritic cell subsets: a feedback control of Th1/Th2 responses. FASEB J. 22:3370-3379. [DOI] [PubMed] [Google Scholar]
- 18.Mariotti, S., R. Teloni, E. Iona, L. Fattorini, F. Giannoni, G. Romagnoli, G. Orefici, and R. Nisini. 2002. Mycobacterium tuberculosis subverts the differentiation of human monocytes into dendritic cells. Eur. J. Immunol. 32:3050-3058. [DOI] [PubMed] [Google Scholar]
- 19.Moody, D. B., T. Ulrichs, W. Muhlecker, D. C. Young, S. S. Gurcha, E. Grant, J. P. Rosat, M. B. Brenner, C. E. Costello, G. S. Besra, and S. A. Porcelli. 2000. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature 404:884-888. [DOI] [PubMed] [Google Scholar]
- 20.Pennini, M. E., R. K. Pai, D. C. Schultz, W. H. Boom, and C. V. Harding. 2006. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J. Immunol. 176:4323-4330. [DOI] [PubMed] [Google Scholar]
- 21.Porcelli, S. A. 1995. The CD1 family: a third lineage of antigen-presenting molecules. Adv. Immunol. 59:1-98. [DOI] [PubMed] [Google Scholar]
- 22.Puzo, G. 1990. The carbohydrate- and lipid-containing cell wall of mycobacteria, phenolic glycolipids: structure and immunological properties. Crit. Rev. Microbiol. 17:305-327. [DOI] [PubMed] [Google Scholar]
- 23.Randhawa, P. S. 1990. Lymphocyte subsets in granulomas of human tuberculosis: an in situ immunofluorescence study using monoclonal antibodies. Pathology 22:153-155. [DOI] [PubMed] [Google Scholar]
- 24.Reiling, N., A. Blumenthal, H. D. Flad, M. Ernst, and S. Ehlers. 2001. Mycobacteria-induced TNF-alpha and IL-10 formation by human macrophages is differentially regulated at the level of mitogen-activated protein kinase activity. J. Immunol. 167:3339-3345. [DOI] [PubMed] [Google Scholar]
- 25.Renukaradhya, G. J., T. J. Webb, M. A. Khan, Y. L. Lin, W. Du, J. Gervay-Hague, and R. R. Brutkiewicz. 2005. Virus-induced inhibition of CD1d1-mediated antigen presentation: reciprocal regulation by p38 and ERK. J. Immunol. 175:4301-4308. [DOI] [PubMed] [Google Scholar]
- 26.Roura-Mir, C., L. Wang, T. Y. Cheng, I. Matsunaga, C. C. Dascher, S. L. Peng, M. J. Fenton, C. Kirschning, and D. B. Moody. 2005. Mycobacterium tuberculosis regulates CD1 antigen presentation pathways through TLR-2. J. Immunol. 175:1758-1766. [DOI] [PubMed] [Google Scholar]
- 27.Salamero, J., H. Bausinger, A. M. Mommaas, D. Lipsker, F. Proamer, J. P. Cazenave, B. Goud, H. de la Salle, and D. Hanau. 2001. CD1a molecules traffic through the early recycling endosomal pathway in human Langerhans cells. J. Investig. Dermatol. 116:401-408. [DOI] [PubMed] [Google Scholar]
- 28.Schlesinger, L. S., C. G. Bellinger-Kawahara, N. R. Payne, and M. A. Horwitz. 1990. Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J. Immunol. 144:2771-2780. [PubMed] [Google Scholar]
- 29.Schlesinger, L. S., S. R. Hull, and T. M. Kaufman. 1994. Binding of the terminal mannosyl units of lipoarabinomannan from a virulent strain of Mycobacterium tuberculosis to human macrophages. J. Immunol. 152:4070-4079. [PubMed] [Google Scholar]
- 30.Schorey, J. S., and A. M. Cooper. 2003. Macrophage signalling upon mycobacterial infection: the MAP kinases lead the way. Cell. Microbiol. 5:133-142. [DOI] [PubMed] [Google Scholar]
- 31.Stenger, S., K. R. Niazi, and R. L. Modlin. 1998. Down-regulation of CD1 on antigen-presenting cells by infection with Mycobacterium tuberculosis. J. Immunol. 161:3582-3588. [PubMed] [Google Scholar]
- 32.Ulrichs, T., D. B. Moody, E. Grant, S. H. Kaufmann, and S. A. Porcelli. 2003. T-cell responses to CD1-presented lipid antigens in humans with Mycobacterium tuberculosis infection. Infect. Immun. 71:3076-3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vaessen, R. T., A. Houweling, and A. J. van der Eb. 1987. Post-transcriptional control of class I MHC mRNA expression in adenovirus 12-transformed cells. Science 235:1486-1488. [DOI] [PubMed] [Google Scholar]
- 34.Xie, J., J. Qian, J. Yang, S. Wang, M. E. Freeman III, and Q. Yi. 2005. Critical roles of Raf/MEK/ERK and PI3K/AKT signaling and inactivation of p38 MAP kinase in the differentiation and survival of monocyte-derived immature dendritic cells. Exp. Hematol. 33:564-572. [DOI] [PubMed] [Google Scholar]
- 35.Yuan, W., A. Dasgupta, and P. Cresswell. 2006. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat. Immunol. 7:835-842. [DOI] [PubMed] [Google Scholar]