

Abstract
Transforming growth factor β (TGF-β) and platelet-derived growth factor A (PDGFΑ) play a central role in tissue morphogenesis and repair, but their interplay remain poorly understood. The nuclear factor I C (NFI-C) transcription factor has been implicated in TGF-β signaling, extracellular matrix deposition, and skin appendage pathologies, but a potential role in skin morphogenesis or healing had not been assessed. To evaluate this possibility, we performed a global gene expression analysis in NFI-C−/− and wild-type embryonic primary murine fibroblasts. This indicated that NFI-C acts mostly to repress gene expression in response to TGF-β1. Misregulated genes were prominently overrepresented by regulators of connective tissue inflammation and repair. In vivo skin healing revealed a faster inflammatory stage and wound closure in NFI-C−/− mice. Expression of PDGFA and PDGF-receptor alpha were increased in wounds of NFI-C−/− mice, explaining the early recruitment of macrophages and fibroblasts. Differentiation of fibroblasts to contractile myofibroblasts was also elevated, providing a rationale for faster wound closure. Taken together with the role of TGF-β in myofibroblast differentiation, our results imply a central role of NFI-C in the interplay of the two signaling pathways and in regulation of the progression of tissue regeneration.
The mammalian nuclear factor I (NFI) family of transcription-replication factors is encoded by four genes, NFI-A, NFI-B, NFI-C, and NFI-X, whose polypeptide products form homo- or heterodimers that bind similar palindromic DNA sequences to regulate gene expression (18, 40). Members of the NFI protein family are expressed in almost every organ and tissue (17). Because of their similar DNA-binding specificities, it has been difficult to attribute specific molecular functions to individual NFI proteins. However, mice with disruptions in each of the four NFI genes were found to display different phenotypes. NFI-A-deficient mice develop neurological defects and die shortly after birth (10), while the absence of NFI-B provokes a severe lung hypoplasia and neurological abnormalities (19, 48). NFI-X deficiency causes brain malformation and severe skeletal defects (13), and NFI-C is required for proper tooth development and normal growth of the newborn (47). The distinct phenotypes and developmental defects of knockout mice indicated specific and distinct functions for each NFI subtype.
Previous studies based on cultured cell lines have implicated NFI-C in the activation or repression of the expression of numerous cellular or viral genes (17, 25). Moreover, NFI-C activity was proposed to be controlled by growth factors such as transforming growth factor β (TGF-β) and tumor necrosis factor α, thereby regulating the expression of the α2(I) collagen (1, 39). NFI binding sites and TGF-β-responsive elements have been colocalized in the promoters of other human extracellular matrix (ECM) genes, including fibronectin (2), perlecan (24), and plasminogen activator inhibitor (37). Consistently, a TGF-β-responsive domain has been located in the transcriptional activation domain of the NFI-C isoform, and this sequence was also found to mediate interactions with histone H3 in a murine fibroblast cell line (1). Chromatin immunoprecipitation experiments indicated that the NFI-C histone-binding domain may prevent the propagation of a silent chromatin structure along the DNA and thereby protect genes from silencing (15, 16). Thus, NFI-C may be a mediator of TGF-β-regulated gene expression and chromatin dynamics.
TGF-β is a multifunctional cytokine known to affect a remarkable range of biological activities, including embryonic development, the cell cycle, and cell differentiation. It is a potent inducer of connective tissue accumulation in tissue remodeling as well as in fibrotic diseases (41, 52, 54). TGF-β, together with the platelet-derived growth factors (PDGFs), is also a key mediator of the skin healing process, as it is involved in each stage of wound repair, namely, the inflammation, proliferation, ECM formation, and remodeling stages (56). After injury, the wounded area is filled with blood clot and inflammatory cells. Neutrophils are attracted within a few minutes of wounding, followed by macrophages and lymphocytes. In addition to their defense function, inflammatory cells are also an important source of cytokines and growth factors. Secreted mainly by platelets and macrophages, PDGF acts as a potent mitogen and chemoattractant of fibroblasts and monocytes (38). PDGF isoforms, such as PDGFA, bind the PDGFα and β receptor homo- or heteromultimers on dermal fibroblasts to activate their migration and proliferation (20). Fibroblasts migrate to the wound site, where they deposit large amounts of ECM proteins to form granulation tissue and to differentiate into myofibroblasts. The differentiation of myofibroblasts is characterized by the onset of expression of the alpha smooth muscle actin (αSMA) contractile protein in response to TGF-β. The contraction of myofibroblasts then contributes to the closure of the injured area (11, 21). Finally, a transition from granulation tissue to a mature scar occurs, which results in tissue remodeling occurring from both the synthesis and catabolism of collagen. Thus, in the dermal compartment, TGF-β acts mainly to stimulate fibroblast proliferation, myofibroblast differentiation, and matrix deposition. However, TGF-β signaling has been implicated in both activating and inhibitory effects on mouse skin healing, depending on the experimental context (56). For instance, TGF-β added early after skin wounding improves healing, while a later treatment of the injury rather impairs reepithelialization by keratinocytes (49). At present, the molecular events that mediate these paradoxical effects of TGF-β remain mostly unknown.
In the present study, we hypothesized that NFI-C may play a role in the skin repair processes because of its proposed implication in TGF-β signaling and ECM gene regulation. Gene expression profiling studies of primary wild-type and NFI-C-null embryonic murine fibroblasts led to the identification of genes whose proper regulation by TGF-β1 required NFI-C. A prominent overrepresentation of genes involved in connective tissue remodeling and repair led us to perform skin wound healing experiments, revealing an accelerated early inflammatory phase and a concomitant increase in the recruitment of macrophage and myofibroblast in NFI-C−/− animals. PDGFA and its Pdgfrα receptor were upregulated in NFI-C−/− mouse skin during the inflammatory phase, providing a molecular explanation for the increased migration and proliferation of fibroblasts and the differentiation of the latter to myofibroblasts, and therefore for the faster healing kinetics. We conclude that NFI-C plays a central role in the regulation of connective tissue development and that it may interface the PDGF and TGF-β signaling pathways during tissue repair.
MATERIALS AND METHODS
Animal care and experimentation.
The NFI-C knockout mice generated by removal of the second exon from the NFI-C gene were described previously (47), and NFI-C-null animals were obtained by breeding heterozygous littermates. All mice in this study have a C57BL/6 background and were individually caged after the wounding, housed in a temperature-controlled room (23°C) on a 12-h dark/12-h light cycle. Animals were fed with standard rodent chow diet and water ad libitum. As knockout animals have brittle teeth, a ground standard rodent chow was provided to all animals (Kliba Nafag) three times a week after weaning at day 21 postbirth (P21).
Cell culture.
Primary mouse embryonic fibroblasts (MEFs) were isolated from 14-day-old embryos generated by the crossbreeding of heterozygotes. Briefly, the head and liver were removed from each embryo, and the remaining embryonic tissues were finely minced and digested by incubation in 0.05% trypsin-EDTA solution (Invitrogen). After incubation for 20 min at 37°C with agitation, trypsin was inactivated by adding complete medium (Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, Invitrogen). Isolated cells were harvested and cultured at 37°C and 5% CO2. MEFs between the second and fifth cell culture passage were used in all experiments. Animals and MEFs were genotyped by PCR as described previously (47). For the preparation of the mRNA samples used in the microarray experiments, fibroblasts were isolated from three wild-type and three knockout mice belonging to the same litter pairwise. Each type of cell was cultured in triplicate, and after incubation for 4 to 6 h with Dulbecco's modified Eagle's medium supplemented with 1% AlbuMAX II, 1% insulin-transferrin-selenium mix (Invitrogen), MEFs were incubated with 12.5 ng/ml of TGF-β1 (PeproTech EC) for 1 h or 10 h or were left untreated.
RNA extraction and real-time PCR analysis.
Total RNA was extracted from MEFs or from homogenized whole-thickness skin explants using TRIzol (Invitrogen) according to the manufacturer's instructions. Further purification of RNA was performed using RNeasy mini kits (Qiagen). RNA integrity was assessed either by a Nano Chip bioanalyzer (Agilent 2100) or in a 1% agarose gel after ethidium bromide staining. Reverse transcription (RT) was performed using an RT-PCR master mix (GE Healthcare). Briefly, 1 μg of total RNA was reverse transcribed with oligo(dT) primers, and first-strand reverse transcribed cDNA was diluted 1:200 in water before use. Real-time PCR was carried out with a real-time quantitative PCR (qPCR) core kit for Sybr green (Eurogentec) in an ABI Prism 7700 system (Applied Biosystems), as recommended in the manufacturer's instructions. Relative gene expression was calculated using the equation 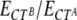 , where E represents the efficiency, CT the threshold cycle, B the endogenous reference, and A the gene of interest. The efficiency was estimated with the LinReg method, as described previously (26). As the endogenous reference, we used the geometric mean of gene expression of ribosomal protein L27 (RPL27) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for MEF studies and of eukaryotic elongation factor 1 alpha 1 and ribosomal protein S9 (RPS9) for the wound healing experiments. Sequences of primers used in real-time PCR are listed in Table S1 in the supplemental material. Statistical significance of differences in gene expression was examined using a two-tailed, paired t test for MEF analyses and a two-tailed, two-sample equal variance t test for wound healing studies.
, where E represents the efficiency, CT the threshold cycle, B the endogenous reference, and A the gene of interest. The efficiency was estimated with the LinReg method, as described previously (26). As the endogenous reference, we used the geometric mean of gene expression of ribosomal protein L27 (RPL27) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) for MEF studies and of eukaryotic elongation factor 1 alpha 1 and ribosomal protein S9 (RPS9) for the wound healing experiments. Sequences of primers used in real-time PCR are listed in Table S1 in the supplemental material. Statistical significance of differences in gene expression was examined using a two-tailed, paired t test for MEF analyses and a two-tailed, two-sample equal variance t test for wound healing studies.
Microarray.
Experiments were carried out with 3 μg of total RNA. cDNA synthesis and cRNA labeling were performed with the GeneChip expression 3′-amplification reagents two-cycle cDNA synthesis kit from Affymetrix. Complementary RNAs were hybridized to Affymetrix mouse genome 430 2.0 arrays. Quality controls and statistical analysis were performed using Affymetrix microarray suite 5.0 software and AIR software (Agilent Technology), respectively. All microarrays used in this study exhibited comparable distributions of signal intensities, fulfilling the requirement for performing gene expression comparison. Direct gene expression level comparison and paired t tests were carried out for statistical analysis. A gene was selected as having a significant change in expression level if the fold increase or decrease in expression was greater than 1.5 and the probability of a false positive was lower than 5% (P ≤ 0.05). Genes fulfilling these criteria were grouped based on their biological function using Ingenuity Pathway Analysis software (www.ingenuity.com).
Wound healing studies.
Wound healing experiments were performed as described previously (31) on six control mice (NFI-C+/+ and NFI-C+/−) and six NFI-C−/− mice that were 6-week-old females (two wounds per mouse). Offspring were generated from heterozygote breeding and by following all of the guidelines and rules of our veterinarian authorities. Briefly, hair follicle cycles were synchronized by shaving the back of the animal 10 days before starting the experiment. Mice were then anesthetized and shaved, and two full-thickness mid-dorsal wounds (25 mm2, square-shaped) were created by excising the skin and the underlying panniculus carnosus. To analyze the kinetics of wound closure, the area of the wound was measured daily, for 11 days, until complete wound closure. The wound area was expressed as the percentile of the initial wound size. The size at the midpoint of the wound was assessed by histomorphometry in sections of four wild-type and four knockout mice. The distance between the healthy tissues bordering the injured area was measured with ImageJ in milimeters. A two-tailed, two-sample equal variance t test was used to compare control and NFI-C-null groups. The healthy skin samples of day 0 and wounded skin samples of day 2 were collected for histology and immunostaining or snap-frozen in liquid nitrogen for gene expression analysis.
For the PDGF pathway blockade, imatinib mesylate (Gleevec) was dissolved in saline solution and administrated intraperitoneally at 75 mg/kg per day. The first injection was performed 24 h before wound excision. Three wild-type and three knockout mice were treated until day 3 after injury with imatinib or the saline solution alone. The kinetics of wound closure were measured until day 4 after wounding.
Protein extraction and ELISA.
Total proteins were isolated from the phenol-ethanol supernatant of TRIzol (Invitrogen)-treated skin samples according to the manufacturer's instructions. Protein pellets were dissolved in 9.5 M urea, 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) by pipetting. The concentration of protein contents was determined by the Bradford (Bio-Rad) and bicinchoninic acid protein (Pierce) assays. Levels of mouse PDGFA in skin protein samples were quantified using a Quantikine enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems), as recommended in the manufacturer's instructions. PDGFA concentrations were normalized to the total protein content. Statistical significance was assessed using a two-tailed, paired t test.
Immunohistochemistry.
Immunostaining was performed with a rat antibody against F4/80 (Abcam) and a mouse antibody against αSMA (Sigma) to distinguish macrophages and myofibroblasts, respectively, in the wounded area. Briefly, skin biopsies were prepared 2 days after wounding, fixed in 4% paraformaldehyde overnight, and embedded in paraffin. Histological sections (5 μm) were rehydrated, and the endogenous peroxidase activity was quenched with 5% hydrogen peroxide in phosphate buffer solution for 10 min. Slides were subjected to epitope retrieval by immersion in 10 mM citrate buffer adjusted to pH 6.0 and heating in a microwave oven set to 800 W for 3 min and 250 W for 5 min. Incubations with anti-F4/80 or anti-αSMA antibodies were performed overnight or for 2 h, respectively. Anti-F4/80 was diluted at 1/800 and detected with a goat anti-rat horseradish peroxidase-conjugated secondary antibody (Millipore). Color development was performed with the diaminobenzidine peroxidase substrate from Sigma. Anti-αSMA was diluted at 1/400 and detected with the M.O.M. immunodetection kit basic (Vector Laboratories), which blocks endogenous mouse immunoglobulin G and contains a biotinylated anti-mouse immunoglobulin G. Revelation was carried out with the M.O.M. immunodetection kit peroxidase (Vector Laboratories) followed by the peroxidase substrate (Sigma). All slides were counterstained with hematoxylin. After labeling, the F4/80- and αSMA-positive cells were quantified in the granulation tissue of five wild-type and five knockout mice. The number of positive cells was normalized to the wounded area expressed in mm2 with ImageJ. A two-tailed, two-sample equal variance t test was used to compare the control and NFI-C-null groups.
Microarray data accession number.
Microarray data were deposited in the GEO database (http://www.ncbi.nlm.nih.gov/projects/geo/) under GEO accession number GSE15871.
RESULTS
Identification of TGF-β1 and NFI-C target genes in primary embryonic fibroblasts.
Previous studies led to the proposal that TGF-β1-controlled gene expression may involve members of the NFI family of transcription factors; however, evidence remained limited to the assessment of synthetic constructs in cultured cell lines. In order to identify NFI-C and/or TGF-β1 target genes in primary cells, mRNAs were isolated from embryonic fibroblasts from three wild-type and three NFI-C knockout mice, and their levels were probed using microarrays. Prior to RNA extraction, fibroblast cultures were treated or not with TGF-β1 for 1 h to examine the immediate response to the growth factor or treated for 10 h to assess the delayed response. We first assessed the global effect of TGF-β1 by comparing the levels of mRNA extracted from the untreated cell cultures (basal expression) to those resulting from the 1 h or the 10 h TGF-β1 treatment. A total of 372 genes appeared to be differentially expressed upon the 1 h TGF-β1 treatment, according to the statistical analysis criteria described in Materials and Methods (see Tables S2 and S3 in the supplemental material). Among these, 117 mRNA levels displayed at least a twofold change. After the 10-h treatment, the number of differentially expressed genes increased to 4,519, with changes in expression ranging to over 20-fold. A number of TGF-β-regulated genes could be related to known functions of this signaling pathway, such as wound healing and tissue remodeling (see Tables S2 and S3 in the supplemental material). For instance, ECM proteins, such as tenascin C; regulators of ECM remodeling, such as serpine 1 and TIMP-3; and signaling proteins, such as TGF-β, Smad7, and PDGFA, were all induced by the growth factor, in agreement with previous studies (3, 32, 46, 53).
We next compared the gene expression profiles of untreated knockout and wild-type fibroblasts to identify NFI-C target genes. The expression of 62 genes was potentially affected in NFI-C deficient fibroblasts, with 60% of those being expressed at higher levels in knockout fibroblasts (Table 1; see Tables S2 and S4 in the supplemental material). Thus, expression of a limited set of genes appeared to be repressed by NFI-C in the absence of TGF-β1, and changes in expression were for the most part lower than twofold (Table 1). Interestingly, 47% of the differentially expressed genes in untreated knockout cells were also identified as TGF-β1 target genes. However, the basal effect of NFI-C in untreated cells appeared to be antagonistic to that of TGF-β1 for 66% of the coregulated genes, where NFI-C repressed TGF-β1-activated genes or vice versa. These included ECM components (Mfap4, Nov), remodeling enzymes (Serpin6a), and signaling molecules (Pdgfrα, Wnt5b), in which expression was affected similarly by the lack of NFI-C and by the TGF-β1 treatment (see Tables S3 and S4 in the supplemental material).
TABLE 1.
Genes whose expression is affected most in fibroblasts of NFI-C-null mice compared to fibroblasts of wild-type mice treated or not treated with TGF-β1
| Experimental condition and genea | Description | Fold changeb | P valuec |
|---|---|---|---|
| Basal | |||
| Fli1 | Friend leukemia integration 1 | −2.0 | 0.043 |
| Mfap4 | Microfibril-associated protein 4 | −2.0 | 0.010 |
| Ebf3 | Early B-cell factor 3 | −1.8 | 0.026 |
| Ahr | Aryl-hydrocarbon receptor | −1.7 | 0.002 |
| Meox1 | Mesenchyme homeobox 1 | −1.7 | 0.002 |
| Pdgfrα | PDGF receptor alpha | −1.7 | 0.038 |
| Ccl12 | Chemokine (C-C motif) ligand 12 | −1.7 | 0.033 |
| Ebf3 | Early B-cell factor 3 | −1.7 | 0.002 |
| Fdft1 | Farnesyl diphosphate farnesyl transferase 1 | −1.6 | 0.004 |
| Epha3 | Eph receptor A3 | −1.6 | 0.014 |
| Rgs5 | Regulator of G protein signaling 5 | 1.7 | 0.045 |
| Nnat | Neuronatin | 1.7 | 0.001 |
| Pax1 | Paired box gene 1 | 1.7 | 0.007 |
| Rgs4 | Regulator of G-protein signaling 4 | 1.7 | 0.001 |
| Mcpt8 | Mast cell protease 8 | 1.8 | 0.028 |
| Clca1/2 | Chloride channel calcium activated 1/2 | 1.8 | 0.001 |
| Chodl | Chondrolectin | 1.8 | 0.001 |
| Ppp1r3c | Protein phosphatase 1, regulatory subunit 3C | 1.9 | <0.001 |
| Tnfrsf11b | Tumor necrosis factor receptor superfamily, member 11b | 1.9 | 0.001 |
| Foxa1 | Forkhead box A1 | 2.1 | 0.004 |
| 1-h TGF-β1 treatment | |||
| Fli1 | Friend leukemia integration 1 | −2.1 | 0.005 |
| Foxg1 | Forkhead box G1 | −2.0 | 0.049 |
| Ctla2b | Cytotoxic T lymphocyte-associated protein 2 beta | −1.9 | 0.049 |
| Idi1 | Isopentenyl-diphosphate delta isomerase | −1.9 | 0.007 |
| Cd53 | CD53 antigen | −1.8 | 0.058 |
| Rab39b | RAB39B, member RAS oncogene family | −1.8 | 0.001 |
| Ahre | Aryl-hydrocarbon receptor | −1.7 | 0.010 |
| Tatdn2 | TatD DNase domain containing 2 | −1.7 | 0.003 |
| Atbf1 | AT motif binding factor 1 | −1.7 | 0.020 |
| Ifitm6 | Interferon-induced transmembrane protein 6 | −1.7 | 0.015 |
| Ctsf | Cathepsin F | 1.9 | 0.006 |
| Chodl | Chondrolectin | 1.9 | 0.004 |
| Akap9 | A kinase (PRKA) anchor protein (yotiao) 9 | 1.9 | 0.001 |
| Pax1 | Paired box gene 1 | 1.9 | 0.002 |
| Igsf4c | Immunoglobulin superfamily, member 4C | 1.9 | 0.001 |
| Sesn3 | Sestrin 3 | 2.0 | 0.001 |
| Ddx6 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 6 | 2.0 | 0.001 |
| Slc14a1 | Solute carrier family 14 (urea transporter), member 1 | 2.1 | 0.042 |
| Nnat | Neuronatin | 2.2 | <0.001 |
| Ppp1r3c | Protein phosphatase 1, regulatory subunit 3C | 2.4 | 0.001 |
| 10-h TGF-β1 treatment | |||
| Wisp2 | WNT1-inducible signaling pathway protein 2 | −2.0 | 0.026 |
| Foxg1 | Forkhead box G1 | −2.0 | 0.025 |
| Stc2 | Stanniocalcin 2 | −1.9 | 0.024 |
| Mafb | v-maf musculoaponeurotic fibrosarcoma oncogene family, protein B | −1.8 | 0.045 |
| Insig1 | Insulin induced gene 1 | −1.8 | 0.001 |
| Rab3il1 | RAB3A interacting protein (rabin3)-like 1 | −1.8 | 0.017 |
| Pdgfrα | PDGF receptor alpha polypeptide | −1.8 | 0.003 |
| Mfap4 | Microfibrillar-associated protein 4 | −1.8 | 0.002 |
| BC052328 | cDNA sequence BC052328 | −1.8 | 0.043 |
| 4930517K11Rik | RIKEN cDNA 4930517K11 gene | −1.8 | 0.014 |
| Nnat | Neuronatin | 1.8 | 0.002 |
| Perp | PERP, TP53 apoptosis effector | 1.9 | <0.001 |
| Gvin1 | GTPase, very large interferon inducible 1 | 1.9 | 0.016 |
| Steap2 | Six transmembrane epithelial antigen of prostate 2 | 1.9 | 0.041 |
| Olr1 | Oxidized low density lipoprotein (lectin-like) receptor 1 | 1.9 | 0.046 |
| Clca1 | Chloride channel calcium activated 1 | 1.9 | 0.002 |
| F3 | Coagulation factor III | 1.9 | 0.004 |
| C1s | Complement component 1, s subcomponent | 1.9 | 0.025 |
| Enpp5 | Ectonucleotide pyrophosphatase/phosphodiesterase 5 | 2.0 | 0.014 |
| Rgs5 | Regulator of G-protein signaling 5 | 2.1 | 0.001 |
The genes are listed according to the fold change values, with a negative fold change indicating a lower expression in knockout cells.
Fold change of mRNA levels in NFI-C knockout cells relative to wild-type cells, both treated and not treated with TGF-β1.
P values were derived from paired t test, as described in Materials and Methods.
To validate the mRNA profiling results, 12 potential NFI-C target genes were selected on the basis of their statistical significance and/or biological role. These included genes displaying the most prominent mRNA level changes when comparing knockout and wild-type fibroblasts (ClCa1/2, Fli1, Nnat, Perp, Ppp1r3c, and Tnfrsf11b) (Table 2). Other genes were selected to evaluate the role of NFI-C in the TGF-β1-mediated regulation of ECM deposition (Mfap4 and Col2a1). Genes implicated in fibrosis and ECM remodeling were also included, such as Wisp2, a member of the WNT/β-catenin pathway, Pdgfrα, one of the PDGF receptors, and Ahr, a ligand-activated transcriptional factor (6, 7, 9). Finally, Csf1 was also included, due to the identification of an NFI-C binding site in its promoter (27). Transcripts were extracted from four matched pairs of embryonic fibroblasts obtained from NFI-C knockout mice and their respective wild-type littermate controls. Each fibroblast culture was grown and processed in triplicate, and mRNA levels were quantified by RT-qPCR analysis. Misregulation was confirmed for 70%, 63%, and 50% of the genes selected from the microarray results, corresponding to untreated cells or to cells treated for 1 h or 10 h with TGF-β1, respectively, and reached statistical significance in the RT-qPCR assay (Table 2). Thus, most changes of mRNA levels as determined by microarray profiling could be validated experimentally using an independent assay method.
TABLE 2.
Validation of microarray RNA-profiling results using RT-qPCR
| Gene | Description | Experimental conditionc | Microarray |
RT-qPCR |
||
|---|---|---|---|---|---|---|
| Fold changea | P valueb | Fold changea | P valueb | |||
| Ahr | Aryl hydrocarbon receptor | Basal | −1.7 | 0.002 | −1.7 | 0.003 * |
| 1 h TGF-β1 | −1.7 | 0.010 | −1.6 | 0.011 * | ||
| 10 h TGF-β1 | −1.7 | 0.018 | −1.3 | 0.449 | ||
| ClCa1/2 | Chloride channel calcium activated 1/2 | Basal | 1.8 | <0.001 | 1.8 | 0.004 * |
| Col2a1 | Collagen type II, alpha 1 | Basal | 1.7 | 0.034 | 1.5 | 0.050 * |
| 1 h TGF-β1 | 1.8 | 0.014 | 1.3 | 0.175 | ||
| 10 h TGF-β1 | 1.6 | 0.019 | 1.5 | 0.018 * | ||
| Csf1 | Colony stimulating factor 1 | 10 h TGF-β1 | 1.5 | 0.045 | 2.3 | 0.163 |
| Fli1 | Friend leukemia virus integration 1 | Basal | −2.0 | 0.043 | −1.4 | 0.082 |
| 1 h TGF-β1 | −2.1 | 0.005 | 1.0 | |||
| 10 h TGF-β1 | −1.6 | 0.008 | 1.3 | 0.505 | ||
| Mfap4 | Microfibril-associated protein 4 | Basal | −2.0 | 0.010 | −2.4 | 0.001 * |
| 1 h TGF-β1 | −1.6 | 0.016 | −2.0 | 0.008 * | ||
| 10 h TGF-β1 | −1.8 | 0.002 | −1.4 | 0.003 * | ||
| Nnat | Neuronatin | Basal | 1.7 | <0.001 | 1.9 | <0.001 * |
| 1 h TGF-β1 | 2.2 | <0.001 | 2.0 | 0.009 * | ||
| 10 h TGF-β1 | 1.8 | 0.002 | 2.0 | 0.050 * | ||
| Pdgfrα | PDGF receptor alpha | Basal | −1.7 | 0.038 | −1.6 | 0.044 * |
| 1 h TGF-β1 | −1.6 | 0.007 | −1.4 | 0.026 * | ||
| 10 h TGF-β1 | −1.8 | 0.003 | −1.7 | 0.018 * | ||
| Perp | TP53 apoptosis effector | Basal | 1.6 | 0.001 | −1.1 | |
| 1 h TGF-β1 | 1.5 | 0.006 | 1.4 | 0.550 | ||
| 10 h TGF-β1 | 1.9 | <0.001 | 2.3 | 0.064 | ||
| Ppp1r3c | Protein phosphatase 1, regulatory (inhibitor) subunit 3C | Basal | 1.8 | <0.001 | 1.6 | 0.005 * |
| 1 h TGF-β1 | 2.4 | 0.001 | 2.3 | 0.050 * | ||
| Tnfrsf11b | Tumor necrosis factor receptor superfamily, member 11b | Basal | 1.9 | 0.001 | 1 | |
| Wisp2 | WNT1-inducible signaling pathway protein 2 | 10 h TGF-β1 | −2 | 0.02 | −1.8 | 0.026 * |
Fold change of inferred mRNA levels between knockout and wild-type fibroblasts. Negative fold change values indicate lower expression in knockout cells.
Statistical analysis was performed as mentioned in Materials and Methods. An asterisk indicates measurements for which a P value of smaller than 0.05 was obtained.
1 h TGF-β1, treatment with TGF-β1 for 1 h; 10 h TGF-β1, treatment with TGF-β1 for 10 h.
NFI-C contributes to the regulation of a subset of TGF-β-responsive genes.
To assess the relationship of TGF-β1 signaling and NFI-C function, we compared the gene expression changes elicited by the growth factor in knockout and wild-type cells. A 1-h TGF-β1 treatment yielded 182 mRNAs whose levels appeared to be altered in the NFI-C−/− fibroblasts, among which 76% were increased in knockout cells (see Tables S2 and S4 in the supplemental material). Similarly, 106 genes appeared to be misregulated in NFI-C-deficient cells after a 10-h TGF-β1 treatment, of which 67% had increased expression in knockout cells (see Tables S2 and S4 in the supplemental material). Differences in expression levels were moderate, with just a few displaying changes above a twofold ratio (Table 1). Interestingly, an increase in the NFI-A and/or NFI-B signals in knockout cells was observed for both comparisons, suggesting that the lack of NFI-C activated a compensatory mechanism leading to the increased expression of other family members (see Table S4 in the supplemental material). This compensatory effect may explain the relatively small transcriptional alterations that resulted from the lack of NFI-C. Nevertheless, a greater number of genes potentially controlled by NFI-C was detected in the presence of the growth factor than in the analysis of untreated cells (182 and 106 genes in the 1-h and 10-h TGF-β1-treated cells, respectively, in comparison to 62 genes in the nontreated cells) (see Table S2 in the supplemental material). Thus, additional potential NFI-C target genes were identified upon TGF-β1 treatment, as expected from a TGF-β1-mediated regulation of NFI-C activity.
A majority of the NFI-C target genes appeared to be overexpressed in NFI-C−/− fibroblasts treated by TGF-β1 (76% of the misregulated genes upon a 1-h treatment) (see Table S2 in the supplemental material). Eighteen genes whose expression was affected by the lack of NFI-C after a 1-h TGF-β1 treatment also belong to the group of TGF-β1-regulated genes (see Table S5 in the supplemental material). These genes were controlled in similar ways by the two regulators. For instance, 13 of these genes were repressed by NFI-C in the presence of TGF-β1, all of which were also downregulated by the 1-h TGF-β1 treatment. These included genes coding for transcription factors and ECM proteins, such as NFI-B and type III collagen. Analysis of the expression levels after a 10-h TGF-β1 treatment yielded similar conclusions. Among the 51 genes whose expression was affected by both the lack of NFI-C in cells treated with TGF-β1 and by the 10-h TGF-β1 treatment itself, 34 (70%) were repressed by NFI-C. Among these 34 genes, 24 (72%) were also repressed by the TGF-β1 treatment (see Table S5 in the supplemental material). Thus, if NFI-C basal activity suggestively antagonizes growth factor signaling, the converse is true in the presence of TGF-β1, where NFI-C may function for the most part to strengthen gene expression changes elicited by TGF-β1 signaling. Overall, these results are indicative of a functional link between TGF-β1 signaling and NFI-C, and they indicate that NFI-C acts mostly to repress gene expression in the presence of TGF-β1.
Global analysis of NFI-C target genes reveals links to ECM deposition and tissue healing.
A global functional analysis of the NFI-C and TGF-β1-regulated genes was performed using the Ingenuity Pathway Analysis software. These genes were sorted according to their biological role, and the percentage of genes assigned to each category was normalized to that of microarray probes pertaining to the considered category. Fifteen percent of all genes differentially expressed in untreated cells versus 1-h TGF-β1-induced cells fell into the connective tissue development and function class, while only 0.8% of all genes probed by the microarray belonged to this category, yielding an 18-fold overrepresentation of this gene function among early TGF-β1 targets (Fig. 1a; see Table S6 in the supplemental material). Consistently, prolonged TGF-β1 treatment also yielded genes of the connective tissue development and function category. This result is in concordance with the known role of the growth factor in connective tissue remodeling.
FIG. 1.

Specific biological functions ascribed to TGF-β1 and NFI-C potential target genes. The percentage of misregulated genes assigned to each biological category was normalized to that of all genes on the microarray pertaining to the considered category. Illustrated are the functions that were overrepresented more than 10-fold in the early (1 h) and late (10 h) TGF-β1 response (a) and more than 13-fold in the comparisons of NFI-C-null versus wild-type fibroblasts (b). D&F, “development and function”; KO, knockout; WT, wild type. Red and blue bars indicate function targeted by only TGF-β1 or only NFI-C, respectively, while the violet ones depict functions potentially regulated by both TGF-β1 and NFI-C. Gray bars indicate functions targeted by TGF-β1 and NFI-C but not by their combination, while orange bars implicate both regulatory molecules together but not either one alone.
Among the specific functions potentially assigned to NFI-C target genes, the connective tissue development and function class was again overrepresented in both nontreated and TGF-β1-treated cells. The embryonic development function also featured prominently in potential NFI-C target genes after TGF-β1 treatment (Fig. 1b). These findings are consistent with previous studies linking the various NFI isoforms to developmental defects (8) and with the colocalization of NFI binding sites and TGF-β-responsive elements on several ECM gene promoters (2, 24, 37). Consistently, several genes encoding ECM proteins, or regulating their deposition, were found in potential NFI-C target genes within the connective tissue development and function category (see Table S7 in the supplemental material). For instance, NFI-C deletion affected the expression of genes coding for the collagen-binding microfibril-associated protein 4 (Mfap4) and collagen type II alpha 1 (Col2a1), an isoform expressed prominently in cartilage and by mouse embryo fibroblasts (28, 42). In addition, NFI-C appeared to control the expression of numerous key regulatory genes, such as Wnt5b, which plays a role in WNT/β-catenin signaling, while the insulin-like growth factor 2 (Igf2) and the receptor for the aryl hydrocarbon receptor (Ahr) are involved in fibroblast growth and liver fibrosis (4, 14, 30, 34). The alpha subunit of the receptor for PDGF (Pdgfrα) regulates inflammation and cellular response during the tissue remodeling linked to the healing processes of mesenchyme-derived tissues (6). All these proteins are involved in a variety of signaling pathways implicated in connective tissue regulation. Overall, these results implicate NFI-C in the response to several signaling pathways that control ECM protein deposition as well as the proliferation of cells that contribute to tissue remodeling and/or wound healing.
NFI-C gene deletion accelerates early skin wound healing in vivo.
Immunofluorescence analysis of skin sections indicated that NFI-C is preferentially expressed in dermal fibroblasts (see Fig. S1 in the supplemental material). Fibroblasts and TGF-β1 are prominent actors in the repair of wounded tissues. Our finding of an association of NFI-C with TGF-β1-regulated genes in fibroblasts prompted us to assess skin wound healing in NFI-C control and knockout mice as an in vivo model of tissue repair and remodeling. Initial experiments did not indicate significant differences between NFI-C+/+ and NFI-C+/− mice, which is consistent with previous findings of the lack of phenotype in the heterozygotic mice (47 and data not shown). Thus, we compared the wound closure kinetics from six 6-week-old NFI-C−/− female mice to those of six controls (NFI-C+). This revealed that NFI-C−/− mice healed more rapidly than the NFI-C+ animals, as the wound surface of the homozygote knockout mice was significantly smaller during the first 3 days of healing (Fig. 2a). This result was corroborated by histomorphometry quantification of the distance between the edges of the wounds at day 2 (Fig. 2b and c). Histological analysis did not reveal differences in the hair follicle cycle or in the proliferation of the keratinocyte strand responsible for wound reepithelialization. Moreover, comparison of the wound closure kinetics of knockout and wild-type animals indicated statistically significant differences of slopes between day 0 and day 1 (Fig. 2a). This provided evidence for an altered early inflammatory stage of the wound healing in NFI-C−/− animals. Differences in the wounded areas between the two genotypes were no longer statistically significant 4 days after wounding, and the kinetics of wound closure in knockout mice slowed down to coincide with that of the control group at the time of complete closure. Faster wound closure kinetics thus appeared to occur at a very early step, indicating that NFI-C may be involved in the onset of tissue repair during the healing process.
FIG. 2.

Acceleration of skin wound healing in NFI-C−/− mice. (a) Kinetics of wound closure in wild-type and NFI-C knockout mice. The surfaces of wounds were recorded daily and plotted as percentage of the initial wound area at day 0, and they are represented as the mean of the surface of 12 wounds ± the standard error of the mean (SEM). (b and c) Hematoxylin and eosin staining of wild-type (b) and NFI-C-null (c) wounds at day 2. Yellow dashed lines border the injured tissue. The distance between healthy dermises (double arrow) was measured in sections of four wild-type and four knockout mice, and the average value ± SEM is indicated in each panel. *, P ≤ 0.05; **, P ≤ 0.01. Bar, 500 μm.
NFI-C gene deletion exacerbates activation of the PDGFA pathway.
To unravel the molecular mechanisms of the faster early healing step, we assessed the expression levels of NFI-C-regulated genes belonging to the connective tissue family, focusing on Col2α1, Ahr, Mfap4, and Pdgfrα. In parallel, to assess the expression level of key regulators of the early inflammatory response during tissue repair (56), we evaluated the expression of cytokines, such as Il1β and Tnfα, and of growth factors comprising Tgf-β1, Csf1, and PdgfA. Skin samples were taken at the time of wounding (day 0), which corresponds to healthy skin, and 2 days after wounding, when the differences of wound closure were most significant. We did not find significant differences in the expression of Col2α1 and Ahr when comparing NFI-C+ and NFI-C−/− skin samples (data not shown). In contrast, Mfap4 expression decreased 2 days after wounding, while its expression was slightly but significantly increased in knockout mice, as expected from the faster rearrangement of the skin connective tissue (Fig. 3a). Quantification of the cytokine and growth factor mRNAs indicated a clear increase in the expression of Il1β, Tnfα, Csf1, and Tgf-β1 at day 2, but this occurred to a similar extent in NFI-C-null mice and control mice, indicating a normal onset of inflammation in knockout mice (data not shown). These molecules are known to be secreted by a number of inflammatory cells recruited to the wound site but also by noninflammatory cells such as keratinocytes (56). In contrast, the expression of PdgfA was increased in knockout animals, at both day 0 and day 2 (Fig. 3b). Interestingly, we also found an increased expression of its receptor alpha in the wounded NFI-C−/− mouse skin compared to that of NFI-C+ mice (2.1-fold versus 1.3-fold) (Fig. 3c), indicating that the whole PDGFA pathway may be overactive in the skin of NFI-C−/− animals. To confirm this result at the protein level, we quantified by ELISA the amount of PDGFA in skin biopsies at days 0 and 2 of healing. As expected, knockout mice produced a significantly higher level of PDGFA than control littermates (Fig. 3d). Imatinib mesylate, an inhibitor of tyrosine kinase receptors, was shown to be a potent repressor of PDGF signaling (36). Consistently, treating mice with imatinib significantly impaired healing, and it abolished significant differences in the wound closure kinetics of wild-type compared to knockout animals (Fig. 3e). This result further supports the interpretation that the improved healing of NFI-C−/− mice may result from the overactivation of the PDGF pathway.
FIG. 3.

Expression of PDGF signaling intermediates and Mfap4 in the wounded skin of NFI-C-null and wild-type mice. RT-qPCR assays were performed on mRNAs extracted from normal (day 0) or wounded (day 2) skin samples to evaluate gene expression of Mfap4 (a), PdgfA (b), and Pdgfrα (c). The level of expression was normalized to endogenous reference genes as described in Materials and Methods. Values represent the means of five mice ± the standard deviation. (d) Quantification of PDGFA in skin protein extracts was determined by ELISA, and values were normalized to the total protein content. Values represent the means of four mice ± the standard error of the means (SEM). (e) Quantification of wound closure after injury in wild-type and knockout mice treated with a saline solution or with imatinib mesylate (Gleevec). Values represent the means of three mice ± SEM. *, P ≤ 0.05; **, P ≤ 0.01.
The first phase of wound healing consists of clot formation and of the recruitment of monocytic cells, such as macrophages (56). Because of the chemotactic function of PDGFA on macrophages at the wound bed, we looked at the expression of the EGFR-like F4/80 mRNA in the wounded tissue as a marker of the recruitment of macrophages (23). We found that its expression was significantly higher in knockout animals 2 days after wounding than at day 0 or in the control group at day 2 (4.5- and 2-fold, respectively) (Fig. 4a). To confirm this finding, immunostaining of F4/80 was performed on sections of wounded skin from both genotypes (Fig. 4b and c), and the amount of F4/80-positive cells per wound area was measured at day 2 of healing (Fig. 4d). This revealed more macrophages infiltrating the early granulation tissue of knockout mice, as may be expected from the increased chemotactic response elicited by the elevated PDGFA levels. While this may lead to an increased immune response, this did not readily explain faster wound closure, as PDGF-activated macrophages have not been implicated in this process (44, 55).
FIG. 4.

Increased infiltration of macrophages in the wounds of NFI-C-deficient mice. (a) RT-qPCR assays of mRNA extracted from skin samples were performed to evaluate the expression of a macrophage marker, F4/80 (EGF-like module and mucin-like, hormone receptor-like sequence), in NFI-C−/− mice and control mice at day 0 and day 2 postwounding. The level of expression was normalized to endogenous reference genes as described in Materials and Methods. Values represent the means of five mice ± the standard deviation. F4/80 protein levels in injured skin was assessed by the immunostaining of granulation tissues at day 2 postwounding in wild-type (b) and knockout (c) animals. The arrowheads indicate all the F4/80-positive cells in the subsected fields. Tissues were counterstained with hematoxylin. Staining of red blood cells (rbc) is due to the endogenous presence of peroxidase. Bar, 50 μm. (d) F4/80-positive cells were quantified in five control (black column) and five knockout (gray column) mice (mean ± the standard error of the mean) and normalized to the wounded area as described in Materials and Methods. *, P ≤ 0.05.
NFI-C gene deletion results in accelerated myofibroblast differentiation in vivo.
Wound closure has been attributed in part to the contraction of myofibroblasts within the wound bed tissue. PDGF is also chemotactic and mitogenic for fibroblasts (44), which express the PDGF receptor alpha on their surfaces (6). We observed a small but significant increase in the proliferation in primary cultures of NFI-C−/− dermal fibroblasts compared to cultures from wild-type littermates when recombinant PDGFA was added to the medium. In contrast, no significant difference was found in the presence of 10% fetal calf serum (see Fig. S2 in the supplemental material). This result and the increased expression of Pdgfrα in knockout animals thus suggested an elevated fibroblast proliferation at the wound area, where they may express markers of contractile cells such as the αSMA and differentiate to myofibroblasts (22, 44). TGF-β1 is a major promoter of myofibroblast differentiation, as it induces αSMA expression (22). However, an even earlier marker of myofibroblastic differentiation is the expression of a spliced isoform of fibronectin termed ED-A FN, which precedes αSMA induction by TGF-β1 (43). RT-qPCR analysis of skin mRNA samples showed an increase in ED-A-domain splice variant relative to total fibronectin mRNA at day 2 of wound healing. The increase in the ED-A splice variant was increased in knockout animals compared to that in wild-type mice, suggesting earlier myofibroblast differentiation (1.4-fold) (Fig. 5a). Consistently, the αSMA mRNA was also increased in knockout animals compared to wild-type mice at day 2 after wounding (NFI-C+, 0.096 ± 0.013; NFI-C−/−, 0.132 ± 0.019; P = 0.016). The elevated expression of αSMA was then confirmed by immunostaining, revealing a significantly higher density of αSMA-positive cells in the granulation tissues of knockout animals (Fig. 5b to f). These results indicated that an earlier differentiation of myofibroblasts occurred in the absence of NFI-C, whereas the staining of other cells, such as that of the vascular smooth muscle cells, was not affected. Taken together, these results imply that the exacerbated activation of PDGF pathway observed in the absence of NFI-C may have resulted in an increased density of macrophages and fibroblasts in the wounds of NF-C-null mice, where the upregulation of TGF-β1 target genes may lead to increased myofibroblast differentiation and enhanced tissue repair at the early stage of healing.
FIG. 5.

Early myofibroblast differentiation accompanies enhanced wound healing in knockout animals. The expression levels of two genes involved in the myofibroblastic phenotype are shown as follows. (a) The spliced variant of fibronectin (ED-A FN) was assayed by RT-qPCR performed on mRNA extracted from skin samples of NFI-C−/− and control mice at day 0 and day 2 postwounding. The level of expression was normalized to the total fibronectin mRNA as described in Materials and Methods. Values represent the means of five mice ± the standard deviation. (b) αSMA-positive cells were labeled and quantified in five control mice (black column) and five knockout mice (gray column) (means ± standard error of the mean) and normalized to the wounded area as described in Materials and Methods. *, P ≤ 0.05. Immunostaining of the myofibroblastic marker αSMA in the granulation tissue of wild-type (c) and knockout (d) animals (panels e and f are enlargements of panels c and d, respectively) at day 2 postwounding. The arrowheads indicate the αSMA-positive cells in a subset of the fields. Vascular muscle cells (vmc) and red blood cells (rbc) are indicated to show similar staining of αSMA vessels in normal or knockout mice. Tissues were counterstained with hematoxylin. Bars, 25 μm.
DISCUSSION
TGF-β regulates numerous classes of genes involved in the cell cycle, apoptosis, cell differentiation, and ECM deposition, and its misregulation leads to physiological disorders that include fibrosis and abnormal wound healing. Regulation of ECM and fibrosis disease-related genes by TGF-β has been proposed to involve binding sites for members of the CTF/NFI family of transcription factors in fibroblastic cell lines (1, 2, 24, 37, 39). However, whether NFI-C mediates TGF-β signaling in vivo remains unknown. In this work, we performed an mRNA profiling analysis of primary MEFs expressing or not expressing NFI-C that were treated or not with TGF-β1. This led to the identification of NFI-C target genes that prominently featured functions associated with tissue remodeling and development. Given the involvement of tissue remodeling in wound healing, we next analyzed NFI-C functions in this latter process and identified an accelerated early wound healing phenotype in knockout mice that we have linked to abnormal PDGF signaling and faster myofibroblast differentiation.
Upon the treatment of fibroblasts with TGF-β1, the number of genes that were found to be potential targets of NFI-C increased strikingly. This was particularly noticeable after 1 h of treatment, as threefold more genes were misregulated in knockout cells than in untreated cells. This finding is consistent with the observations of Alevizopoulos et al. (1), who demonstrated that TGF-β1 potentiates transcriptional regulation by NFI-C. Furthermore, when comparing the transcriptional activity of NFI-C in the presence and in the absence of TGF-β1, we noticed an increase in the number of genes that were downregulated by NFIC in TGF-β1-treated cells, and the expression of most of these genes was also inhibited by TGF-β1. This indicates that NFI-C acts mainly as a repressor of genes whose expression is controlled by TGF-β1. Indeed, NFI proteins have been proposed to complex with the Ski oncogene (50), a repressor of TGF-β signaling that binds to Smad3/4 and represses the expression from promoters that associate with Smad3/4 complexes (29). As NFI has also been shown to recruit of Ski at promoters (50), the two mechanisms may act in concert to downregulate the expression of TGF-β target genes.
Analysis of the biological function of misregulated genes suggested that TGF-β1 and NFI-C control genes contribute to the development and function of connective tissues. However, regulation of the expression of ECM genes per se, as previously suggested by low-throughput approaches, does not appear to be the sole function of NFI-C. Analysis of the specific functions of the misregulated genes revealed intermediates of a signaling pathway known to control ECM deposition, tissue remodeling, and/or cell proliferation, such as the aryl hydrocarbon receptor, the PDGF receptor, and the insulin-like growth factor. Therefore, our results implicate NFI-C in the control of signaling pathways that not only mediate ECM deposition but also coordinate more general connective tissue remodeling and repair. This conclusion is supported by the lack of abnormal ECM protein deposition or fibrosis during wound healing in NFI-C knockout mice. Our study revealed an acceleration of the early signaling events that lead to cell recruitment and differentiation within the inflammation phase of wound healing. Indeed, formation of granulation tissue and its later remodeling stage per se were not affected during the healing process, but it is the timing of these events that was altered.
Previous studies have shown that TGF-β1 may act to promote early steps of wound healing, while abnormal signaling was linked to either accelerated or delayed healing (56). For instance, treatment of an injury with TGF-β1 at the day of wounding was shown to trigger an early infiltration of immune cells and an increase in myofibroblast differentiation, resulting in an accelerated healing and tissue repair (49). Closure of the wound has been attributed to tissue contraction mediated by myofibroblasts. Differentiation into this contractile form of fibroblasts depends on the de novo expression of the ED-A fibronectin variant (43). The ED-A variant establishes new interaction with integrins α9β1 and α4β1, and it enhances the TGF-β1-dependent expression of αSMA in fibroblasts (22). TGF-β is therefore a key regulator of wound closure, as it promotes ED-A FN alternative splicing, myofibroblast differentiation, and αSMA expression (5, 11, 21).
The faster early healing process of NFI-C knockout mice is accompanied by the increased recruitment of monocytic cells as well as by a higher expression of ED-A FN and αSMA in the early granulation tissue. Furthermore, immunostaining analyses revealed a higher number of myofibroblasts in the wounds of NFI-C-null animals, explaining the enhancement of contraction at the early stage of healing. Later during healing, the kinetics of wound closure decreased in knockout animals, and full closure of the wound coincided with that of the wild-type animals. Thus, the lack of NFI-C displays opposite effects on early or late healing steps, and these effects are similar to those elicited by an early or late topical treatment of the wound with TGF-β1, which accelerates or delays healing, respectively (49). The finding that, in the absence of the growth factor, NFI-C acts mainly as an antagonist to the changes in gene expression elicited by TGF-β1 readily provides an explanation for this phenomenon. Early in the healing process, just after wounding when TGF-β1 levels are relatively low, the lack of NFI-C will tend to derepress TGF-β1 target genes and thereby accelerate the healing process, as does the ectopic addition of the growth factor. Later in the wound healing process, TGF-β1 expression is increased in mice of both genotypes. Our results indicate that at this stage, NFI-C acts as an agonist of the signaling pathway in the presence of the growth factor. This fits well with the observation that wound closure of wild-type animals catches up with the slowed-down closure of knockout mice later in the healing process, when both the ectopic addition of TGF-β1 and the presence of NFI-C decrease the kinetics of healing. Thus, the dual antagonistic or agonistic role of NFI-C on healing closely parallels the effect of the ectopic addition of TGF-β1, strengthening our conclusion of an implication of the two regulators in a pathway that controls proper timing of this tissue repair process.
While our study cannot formally rule out the possibility of high local concentrations of active TGF-β proteins at the wounds of knockout animals, we rather noted an increase in its expression at later healing stages, which was not altered by the lack of NFI-C. Consistently, the gene expression profiling study provided alternative genes whose altered expression may explain faster healing of the wound of NFI-C-deficient mice. Accelerated wound healing was found to be accompanied by an increase in the expression of both PDGFA and one of its receptors, Pdgfrα, in the wounded skin of knockout animals. Increased PdgfA mRNA and protein levels were observed in the skin of knockout mice, even in the normal unwounded skin tissue, indicating that it may be the primary event mediating an early onset of healing. The regulation of PdgfA expression by the NFI family in skin fibroblasts is consistent with the previous observation of the regulation of PdgfA gene transcription by the interplay of NFI-X and SP1 in muscle cells. NFI-X was shown to interact with SP1 and to antagonize SP1 binding and activation of the PdgfA gene promoter (35). Whether a similar mechanism may also operate to control PdgfA expression in the skin remains to be ascertained.
The implication of PDGF in the enhancement of an early step of NFI-C−/− skin wound healing is supported by preliminary data indicating that treatment with an inhibitor of PDGF receptors abolished detectable differences of wound closure by wild-type and knockout animals. Thus, the improved healing observed in null animals may critically depend on the overactivation of the PDGF pathway. PDGF is known to play a central role in the early healing process as a chemotactic signal for monocytes (12), and the finding of a higher expression of a macrophage marker and an increased macrophage infiltration in the early granulation tissue of knockout animals is consistent with increased signaling by this regulatory cascade. As macrophages are known to secrete PDGFA (38), increased signaling and an accelerated recruitment of these cells at the site of injury should trigger the induction of a positive feedback loop, resulting from the de novo synthesis of PDGF and from its autocrine action on cells within the granulation tissue.
In addition to its role on the recruitment of monocytes, PDGFA has also been implicated in the acceleration of the recruitment and proliferation of fibroblasts at the site of injury (44), which in turn may improve repair. Pdgfrα expression was shown to be under the control of TGF-β1 in a complex fashion. For instance, TGF-β1 downregulated Pdgfrα in cultures of human fibroblasts, while it increased its expression in sclerodermal fibroblasts (45, 57). Similarly, our results indicate that the lack of NFI-C increases the expression of PDGFA and of its receptor alpha in wound biopsies in vivo, while Pdgfrα expression was reduced in cultured fibroblasts. The reasons for such differences have remained unclear, but they may stem from the different behavior of the fibroblasts depending on their origin and/or on the cellular context. In any case, these observations support our conclusion of an antagonistic link between NFI-C and TGF-β signaling in the skin in vivo.
This proposed negative modulation of TGF-β signaling by the basal activity of NFI-C may also explain another phenotype observed in NFI-C−/− mice: the failure of molar tooth root development and the formation of aberrantly short roots. During this aberrant root development, cells that normally remain on the surface of the dentin matrix become trapped within the matrix, forming a material morphologically similar to osteodentin, a form of dentin deposited during the repair of deep caries (33). A similar short-root- and osteodentin-forming phenotype is seen in mice overexpressing TGF-β1 from the odontoblast-specific dentin sialophosphoprotein gene promoter (51). Our model that the loss of NFI-C may potentiate the signaling by low levels of TGF-β1 would indeed predict enhanced TGF-β signaling in the NFI-C−/− mice that could directly result in the tooth root phenotype seen in these animals.
At present, the mechanisms that regulate the progression of skin wound healing are still poorly understood. PDGF and TGF-β are consistently found at injury sites where they may directly or indirectly control migration, proliferation, and differentiation of macrophages and fibroblasts, but the precise relationships and molecular determinants of these regulatory pathways have remained unclear. Overall, our results shed light on some of these complex regulatory interactions, as they indicate that NFI-C contributes to and modulates the response elicited by both the TGF-β and the PDGFA signaling pathways. Thus, NFI-C may coordinate and interconnect the actions of these two signaling pathways during the wound healing process.
In addition to its role on tissue development and repair, our gene ontology analysis suggested that NFI-C may regulate embryogenesis as well as liver function and hepatic diseases. Consistently, the Ahr gene that is downregulated in NFI-C-null fibroblasts has been linked to hepatic fibrosis in Ahr−/− mice (34). Healing of the skin is a repair process that resembles the autoregenerative process observed after partial hepatectomy, whereas an exaggerated healing process leads mainly to fibrosis. Other ongoing studies have also implicated NFI-C in the proper timing of another cyclic regenerative process: the hair follicle regrowth during the first hair cycle after birth (G. Plasari, A. Calabrese, and N. Mermod, unpublished data). We did not notice any difference in the hair follicle stage between wild-type and knockout animals at the time of wound excisions, which were performed later after birth. This implies that the wound healing phenotype reported here does not result from differences in hair follicle cycling. Taken together, these findings imply that NFI-C may play a more general function in pathways that regulate tissue regeneration, either after injury or as part of a normal degenerative-regenerative cycle.
Supplementary Material
Acknowledgments
We thank Keith Harshman, Alexandra Paillusson, and Sylvain Pradervand at the University of Lausanne DNA Array Facility for expert assistance and statistical analyses of gene expression profiling experiments and Simone Edelmann for improving the manuscript. We thank Virginie Philippe for expert assistance with animal work and Catherine Roger for advice on immunostaining. We thank Bertrand Rochat from CHUV for providing Gleevec tablets. We thank George Steele-Perkins for providing primary fibroblasts and knockout mice and Ione Gutscher, Florence Högger, and Armindo Teixeira for excellent assistance.
This work has been supported by grants from the Swiss NSF to N.M. and by the University of Lausanne.
Footnotes
Published ahead of print on 14 September 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Alevizopoulos, A., Y. Dusserre, M. Tsai-Pflugfelder, T. von der Weid, W. Wahli, and N. Mermod. 1995. A proline-rich TGF-beta-responsive transcriptional activator interacts with histone H3. Genes Dev. 9:3051-3066. [DOI] [PubMed] [Google Scholar]
- 2.Alonso, C. R., C. G. Pesce, and A. R. Kornblihtt. 1996. The CCAAT-binding proteins CP1 and NF-I cooperate with ATF-2 in the transcription of the fibronectin gene. J. Biol. Chem. 271:22271-22279. [DOI] [PubMed] [Google Scholar]
- 3.Arici, A., P. C. MacDonald, and M. L. Casey. 1996. Modulation of the levels of transforming growth factor beta messenger ribonucleic acids in human endometrial stromal cells. Biol. Reprod. 54:463-469. [DOI] [PubMed] [Google Scholar]
- 4.Baker, J., J. P. Liu, E. J. Robertson, and A. Efstratiadis. 1993. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75:73-82. [PubMed] [Google Scholar]
- 5.Balza, E., L. Borsi, G. Allemanni, and L. Zardi. 1988. Transforming growth factor beta regulates the levels of different fibronectin isoforms in normal human cultured fibroblasts. FEBS Lett. 228:42-44. [DOI] [PubMed] [Google Scholar]
- 6.Bonner, J. C. 2004. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 15:255-273. [DOI] [PubMed] [Google Scholar]
- 7.Bowley, E., D. B. O'Gorman, and B. S. Gan. 2007. Beta-catenin signaling in fibroproliferative disease. J. Surg. Res. 138:141-150. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhry, A. Z., G. E. Lyons, and R. M. Gronostajski. 1997. Expression patterns of the four nuclear factor I genes during mouse embryogenesis indicate a potential role in development. Dev. Dyn. 208:313-325. [DOI] [PubMed] [Google Scholar]
- 9.Corchero, J., G. Martin-Partido, S. L. Dallas, and P. M. Fernandez-Salguero. 2004. Liver portal fibrosis in dioxin receptor-null mice that overexpress the latent transforming growth factor-beta-binding protein-1. Int. J. Exp. Pathol. 85:295-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.das Neves, L., C. S. Duchala, F. Tolentino-Silva, M. A. Haxhiu, C. Colmenares, W. B. Macklin, C. E. Campbell, K. G. Butz, and R. M. Gronostajski. 1999. Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. Proc. Natl. Acad. Sci. USA 96:11946-11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desmoulière, A., A. Geinoz, F. Gabbiani, and G. Gabbiani. 1993. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122:103-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deuel, T. F., R. M. Senior, J. S. Huang, and G. L. Griffin. 1982. Chemotaxis of monocytes and neutrophils to platelet-derived growth factor. J. Clin. Investig. 69:1046-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Driller, K., A. Pagenstecher, M. Uhl, H. Omran, A. Berlis, A. Grunder, and A. E. Sippel. 2007. Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol. Cell. Biol. 27:3855-3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elizondo, G., P. Fernandez-Salguero, M. S. Sheikh, G. Y. Kim, A. J. Fornace, K. S. Lee, and F. J. Gonzalez. 2000. Altered cell cycle control at the G2/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol. Pharmacol. 57:1056-1063. [PubMed] [Google Scholar]
- 15.Esnault, G., S. Majocchi, D. Martinet, N. Besuchet-Schmutz, J. S. Beckmann, and N. Mermod. 2009. Transcription factor CTF1 acts as a chromatin domain boundary that shields human telomeric genes from silencing. Mol. Cell. Biol. 29:2409-2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrari, S., K. C. Simmen, Y. Dusserre, K. Muller, G. Fourel, E. Gilson, and N. Mermod. 2004. Chromatin domain boundaries delimited by a histone-binding protein in yeast. J. Biol. Chem. 279:55520-55530. [DOI] [PubMed] [Google Scholar]
- 17.Gronostajski, R. M. 2000. Roles of the NFI/CTF gene family in transcription and development. Gene 249:31-45. [DOI] [PubMed] [Google Scholar]
- 18.Gronostajski, R. M., S. Adhya, K. Nagata, R. A. Guggenheimer, and J. Hurwitz. 1985. Site-specific DNA binding of nuclear factor I: analyses of cellular binding sites. Mol. Cell. Biol. 5:964-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gründer, A., T. T. Ebel, M. Mallo, G. Schwarzkopf, T. Shimizu, A. E. Sippel, and H. Schrewe. 2002. Nuclear factor I-B (Nfib) deficient mice have severe lung hypoplasia. Mech. Dev. 112:69-77. [DOI] [PubMed] [Google Scholar]
- 20.Hart, C. E., and D. F. Bowen-Pope. 1990. Platelet-derived growth factor receptor: current views of the two-subunit model. J. Investig. Dermatol. 94:53S-57S. [DOI] [PubMed] [Google Scholar]
- 21.Hinz, B. 2007. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 127:526-537. [DOI] [PubMed] [Google Scholar]
- 22.Hinz, B., G. Celetta, J. J. Tomasek, G. Gabbiani, and C. Chaponnier. 2001. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 12:2730-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirsch, S., J. M. Austyn, and S. Gordon. 1981. Expression of the macrophage-specific antigen F4/80 during differentiation of mouse bone marrow cells in culture. J. Exp. Med. 154:713-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iozzo, R. V., J. Pillarisetti, B. Sharma, A. D. Murdoch, K. G. Danielson, J. Uitto, and A. Mauviel. 1997. Structural and functional characterization of the human perlecan gene promoter. Transcriptional activation by transforming growth factor-beta via a nuclear factor 1-binding element. J. Biol. Chem. 272:5219-5228. [DOI] [PubMed] [Google Scholar]
- 25.Jones, K. A., J. T. Kadonaga, P. J. Rosenfeld, T. J. Kelly, and R. Tjian. 1987. A cellular DNA-binding protein that activates eukaryotic transcription and DNA replication. Cell 48:79-89. [DOI] [PubMed] [Google Scholar]
- 26.Karlen, Y., A. McNair, S. Perseguers, C. Mazza, and N. Mermod. 2007. Statistical significance of quantitative PCR. BMC Bioinformatics 8:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konicek, B. W., X. L. Xia, and M. A. Harrington. 1995. Binding of a CTF/NF1-like protein to the mouse colony-stimulating factor-1 gene promoter. DNA Cell Biol. 14:961-969. [DOI] [PubMed] [Google Scholar]
- 28.Lengner, C. J., C. Lepper, A. J. van Wijnen, J. L. Stein, G. S. Stein, and J. B. Lian. 2004. Primary mouse embryonic fibroblasts: a model of mesenchymal cartilage formation. J. Cell Physiol. 200:327-333. [DOI] [PubMed] [Google Scholar]
- 29.Liu, X., Y. Sun, R. A. Weinberg, and H. F. Lodish. 2001. Ski/Sno and TGF-beta signaling. Cytokine Growth Factor Rev. 12:1-8. [DOI] [PubMed] [Google Scholar]
- 30.Logan, C. Y., and R. Nusse. 2004. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20:781-810. [DOI] [PubMed] [Google Scholar]
- 31.Michalik, L., B. Desvergne, N. S. Tan, S. Basu-Modak, P. Escher, J. Rieusset, J. M. Peters, G. Kaya, F. J. Gonzalez, J. Zakany, D. Metzger, P. Chambon, D. Duboule, and W. Wahli. 2001. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J. Cell Biol. 154:799-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagarajan, R. P., J. Zhang, W. Li, and Y. Chen. 1999. Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J. Biol. Chem. 274:33412-33418. [DOI] [PubMed] [Google Scholar]
- 33.Park, J. C., Y. Herr, H. J. Kim, R. M. Gronostajski, and M. I. Cho. 2007. Nfic gene disruption inhibits differentiation of odontoblasts responsible for root formation and results in formation of short and abnormal roots in mice. J. Periodontol. 78:1795-1802. [DOI] [PubMed] [Google Scholar]
- 34.Puga, A., C. R. Tomlinson, and Y. Xia. 2005. Ah receptor signals cross-talk with multiple developmental pathways. Biochem. Pharmacol. 69:199-207. [DOI] [PubMed] [Google Scholar]
- 35.Rafty, L. A., F. S. Santiago, and L. M. Khachigian. 2002. NF1/X represses PDGF A-chain transcription by interacting with Sp1 and antagonizing Sp1 occupancy of the promoter. EMBO J. 21:334-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajkumar, V. S., X. Shiwen, M. Bostrom, P. Leoni, J. Muddle, M. Ivarsson, B. Gerdin, C. P. Denton, G. Bou-Gharios, C. M. Black, and D. J. Abraham. 2006. Platelet-derived growth factor-beta receptor activation is essential for fibroblast and pericyte recruitment during cutaneous wound healing. Am. J. Pathol. 169:2254-2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riccio, A., P. V. Pedone, L. R. Lund, T. Olesen, H. S. Olsen, and P. A. Andreasen. 1992. Transforming growth factor beta 1-responsive element: closely associated binding sites for USF and CCAAT-binding transcription factor-nuclear factor I in the type 1 plasminogen activator inhibitor gene. Mol. Cell. Biol. 12:1846-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross, R., E. W. Raines, and D. F. Bowen-Pope. 1986. The biology of platelet-derived growth factor. Cell 46:155-169. [DOI] [PubMed] [Google Scholar]
- 39.Rossi, P., G. Karsenty, A. B. Roberts, N. S. Roche, M. B. Sporn, and B. de Crombrugghe. 1988. A nuclear factor 1 binding site mediates the transcriptional activation of a type I collagen promoter by transforming growth factor-beta. Cell 52:405-414. [DOI] [PubMed] [Google Scholar]
- 40.Roulet, E., P. Bucher, R. Schneider, E. Wingender, Y. Dusserre, T. Werner, and N. Mermod. 2000. Experimental analysis and computer prediction of CTF/NFI transcription factor DNA binding sites. J. Mol. Biol. 297:833-848. [DOI] [PubMed] [Google Scholar]
- 41.Schiller, M., D. Javelaud, and A. Mauviel. 2004. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J. Dermatol. Sci. 35:83-92. [DOI] [PubMed] [Google Scholar]
- 42.Schlosser, A., T. Thomsen, J. M. Shipley, P. W. Hein, F. Brasch, I. Tornoe, O. Nielsen, K. Skjodt, N. Palaniyar, W. Steinhilber, F. X. McCormack, and U. Holmskov. 2006. Microfibril-associated protein 4 binds to surfactant protein A (SP-A) and colocalizes with SP-A in the extracellular matrix of the lung. Scand. J. Immunol. 64:104-116. [DOI] [PubMed] [Google Scholar]
- 43.Serini, G., M. L. Bochaton-Piallat, P. Ropraz, A. Geinoz, L. Borsi, L. Zardi, and G. Gabbiani. 1998. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J. Cell Biol. 142:873-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Serini, G., and G. Gabbiani. 1999. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 250:273-283. [DOI] [PubMed] [Google Scholar]
- 45.Soma, Y., M. Mizoguchi, K. Yamane, N. Yazawa, M. Kubo, H. Ihn, K. Kikuchi, and K. Tamaki. 2002. Specific inhibition of human skin fibroblast chemotaxis to platelet-derived growth factor A-chain homodimer by transforming growth factor-beta1. Arch. Dermatol. Res. 293:609-613. [DOI] [PubMed] [Google Scholar]
- 46.Starksen, N. F., G. R. Harsh IV, V. C. Gibbs, and L. T. Williams. 1987. Regulated expression of the platelet-derived growth factor A chain gene in microvascular endothelial cells. J. Biol. Chem. 262:14381-14384. [PubMed] [Google Scholar]
- 47.Steele-Perkins, G., K. G. Butz, G. E. Lyons, M. Zeichner-David, H. J. Kim, M. I. Cho, and R. M. Gronostajski. 2003. Essential role for NFI-C/CTF transcription-replication factor in tooth root development. Mol. Cell. Biol. 23:1075-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steele-Perkins, G., C. Plachez, K. G. Butz, G. Yang, C. J. Bachurski, S. L. Kinsman, E. D. Litwack, L. J. Richards, and R. M. Gronostajski. 2005. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol. Cell. Biol. 25:685-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan, N. S., L. Michalik, B. Desvergne, and W. Wahli. 2005. Genetic- or transforming growth factor-beta 1-induced changes in epidermal peroxisome proliferator-activated receptor beta/delta expression dictate wound repair kinetics. J. Biol. Chem. 280:18163-18170. [DOI] [PubMed] [Google Scholar]
- 50.Tarapore, P., C. Richmond, G. Zheng, S. B. Cohen, B. Kelder, J. Kopchick, U. Kruse, A. E. Sippel, C. Colmenares, and E. Stavnezer. 1997. DNA binding and transcriptional activation by the Ski oncoprotein mediated by interaction with NFI. Nucleic Acids Res. 25:3895-3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thyagarajan, T., T. Sreenath, A. Cho, J. T. Wright, and A. B. Kulkarni. 2001. Reduced expression of dentin sialophosphoprotein is associated with dysplastic dentin in mice overexpressing transforming growth factor-beta 1 in teeth. J. Biol. Chem. 276:11016-11020. [DOI] [PubMed] [Google Scholar]
- 52.Uitto, J., and D. Kouba. 2000. Cytokine modulation of extracellular matrix gene expression: relevance to fibrotic skin diseases. J. Dermatol. Sci. 24(Suppl. 1):S60-S69. [DOI] [PubMed] [Google Scholar]
- 53.Verrecchia, F., M. L. Chu, and A. Mauviel. 2001. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 276:17058-17062. [DOI] [PubMed] [Google Scholar]
- 54.Verrecchia, F., and A. Mauviel. 2007. Transforming growth factor-beta and fibrosis. World J. Gastroenterol. 13:3056-3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vyalov, S., A. Desmouliere, and G. Gabbiani. 1993. GM-CSF-induced granulation tissue formation: relationships between macrophage and myofibroblast accumulation. Virchows Arch. B 63:231-239. [DOI] [PubMed] [Google Scholar]
- 56.Werner, S., and R. Grose. 2003. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 83:835-870. [DOI] [PubMed] [Google Scholar]
- 57.Yamakage, A., K. Kikuchi, E. A. Smith, E. C. LeRoy, and M. Trojanowska. 1992. Selective upregulation of platelet-derived growth factor alpha receptors by transforming growth factor beta in scleroderma fibroblasts. J. Exp. Med. 175:1227-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.