

Abstract
Huntingtin (Htt) localizes to endosomes, but its role in the endocytic pathway is not established. Recently, we found that Htt is important for the activation of Rab11, a GTPase involved in endosomal recycling. Here we studied fibroblasts of healthy individuals and patients with Huntington's disease (HD), which is a movement disorder caused by polyglutamine expansion in Htt. The formation of endocytic vesicles containing transferrin at plasma membranes was the same in control and HD patient fibroblasts. However, HD fibroblasts were delayed in recycling biotin-transferrin back to the plasma membrane. Membranes of HD fibroblasts supported less nucleotide exchange on Rab11 than did control membranes. Rab11-positive vesicular and tubular structures in HD fibroblasts were abnormally large, suggesting that they were impaired in forming vesicles. We used total internal reflection fluorescence imaging of living fibroblasts to monitor fluorescence-labeled transferrin-carrying transport intermediates that emerged from recycling endosomes. HD fibroblasts had fewer small vesicles and more large vesicles and long tubules than did control fibroblasts. Dominant active Rab11 expressed in HD fibroblasts normalized the recycling of biotin-transferrin. We propose a novel mechanism for cellular dysfunction by the HD mutation arising from the inhibition of Rab11 activity and a deficit in vesicle formation at recycling endosomes.
Polyglutamine expansion in huntingtin (Htt) causes Huntington's disease (HD). Htt localizes to the cytoplasm and associates with vesicles (12) and subcellular organelles (51), including endosomes. Cells with mutant Htt have deficits in the transport of different cargo-carrying vesicles and subcellular organelles including mitochondria and early endosomes (15, 21, 29, 36, 44, 45). The molecular basis for these deficits is thought to involve aberrant interactions of mutant Htt with HAP1 and the motor proteins dynactin and kinesin (14, 31, 35). Htt interacts with components of the molecular machinery for clathrin-coated vesicles, leading to speculation that Htt might also be involved in the formation of clathrin-coated vesicles. However, the endocytosis of transferrin, a process that involves the packaging of transferrin and its receptor into clathrin-coated vesicles at the plasma membrane, is unaffected by the presence of mutant Htt (46). A recent study reported that mutant Htt does not affect the formation of clathrin-coated vesicles at the trans-Golgi network (11).
Endocytic recycling is a process whereby internalized receptors and other molecules are returned to the cell surface for reuse. This process is required for the maintenance of the plasma membrane and cellular homeostasis. The critical organelles in endocytic recycling are the sorting endosomes (short-loop recycling) and the recycling endosomes (long-loop recycling). Rab4 regulates the recycling events at sorting endosomes, whereas Rab11 acts at recycling endosomes by regulating vesicle formation (19, 34, 43, 47, 55). Recently, we found that the absence of wild-type Htt impaired the activity of Rab11 (32). We hypothesized that mutant Htt might have an effect on Rab11 activity and thereby interfere with vesicle formation at recycling endosomes. In this study, we investigated Rab11 activation and recycling in the endocytic pathway in human fibroblasts from healthy controls and HD patients. Primary human HD fibroblasts are ideal cells for identifying the effects of mutant Htt, because expanded full-length human Htt is expressed at endogenous levels from its own promoter, and cells homozygous for the HD gene can be studied. Changes that have been identified in HD fibroblasts include the slowed movement of early endosomes (36) and altered levels of catalase associated with the regulation of oxidative stress (10). HD fibroblasts also have deficits in regulatory functions that are seen in neural tissues, including impaired proteasome activity (41) and abnormal cholesterol synthesis (48). Other nonneuronal cells of peripheral tissues, including muscle (9) and lymphocytes (1, 50), and astrocytes and microglia in brain (33) are altered by the presence of the HD gene. Thus, although the neuron is the primary cell type most affected by the HD mutation, deficits in HD fibroblasts and other cells may forecast disease phenotypes in neurons.
Our findings for HD fibroblasts support deficits in Rab11 activity and in vesicle budding from recycling endosomes. Based on these data, we propose a novel mechanism for HD pathogenesis resulting from a deficiency in post-endocytic-membrane recycling.
MATERIALS AND METHODS
SDS-PAGE and Western blot analysis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blot analysis were performed as described previously (28). In brief, cell lysates or postnuclear supernatants (S1) were boiled in SDS-PAGE sample buffer for 5 min and loaded onto a Tris-glycine gel. After electrophoresis, proteins were transferred onto nitrocellulose blots. The blots were blocked with 5% nonfat milk in phosphate-buffered saline (PBS) containing 0.05% Tween 20 and incubated with antibodies or ABC reagents (ABC kit; Vector Labs) for detecting biotin-transferrin. The primary antisera used for Western blot analysis were anti-Htt1-17 Ab1 (1 μg ml−1), anti-human transferrin receptor (1:1,000; Zymed), anti-Rab11 (1:1,000; Chemicon), antiactin (1:500; Sigma-Aldrich), and anti-tubulin-alpha (1:5,000; Sigma-Aldrich). Peroxidase-conjugated secondary antibodies (Jackson Laboratories) were diluted at 1:5,000. Blots were developed using enhanced ECL (Pierce). Films were scanned for measuring band densities using NIH ImageJ software and quantitative analysis. The two-tailed Student t test was used to determine statistical significance.
Human fibroblast cell lines and tissue culture methods.
Primary human fibroblasts were obtained from the Coriell Cell Repository. The cell lines (and ages and CAG repeats in parentheses) for three HD lines used were GM04857 (23 years; 39/49CAG), GM04855 (11 years; 22/49CAG), and GM04281 (20 years; 17/70CAG). Control lines used were GM08399 (19 years), GM03440 (20 years), and GM08402 (32 years). Culture media and corresponding supplements were purchased from Invitrogen, Inc., and fetus bovine serum was obtained from Atlanta Biochemicals. Primary human fibroblast cells were maintained in complete Dulbecco's modified Eagle's medium supplemented with nonessential amino acids, biotin, 20 mM HEPES-K (pH 7.4), and 15% fetus bovine serum at 37°C in an incubator (Thermo) with a humid 5% CO2 atmosphere. The number of passages was fewer than 15 for all fibroblast lines.
Transferrin receptor recycling.
An assay of transferrin uptake was performed as previously described (42) with minor modifications except where indicated. Biotin-transferrin and holotransferrin were purchased from Sigma-Aldrich. Prior to loading biotin-transferrin, cells were washed in PBS and cultured in serum-free medium for 1 h. To study transferrin recycling, cells were loaded with biotin-transferrin at a final concentration of 5 μg ml−1 for cumulative uptake at 37°C for 30 min, collected, washed, divided into equal aliquots, and chased in the presence of 10 μg ml−1 holotransferrin at 37°C for different times. Cells from each time point were collected by centrifugation and lysed in sample buffer for SDS-PAGE and blot analysis. The density of each biotin-transferrin-positive band was measured. Data were represented as the percentage of retained transferrin. The 0-min chase was set at 100%.
To investigate if the elevation in levels of Rab11 activity can improve endocytic recycling in HD fibroblasts, homozygous HD fibroblasts (GM04857) and control fibroblasts (GM08399) were infected with lentivirus expressing enhanced green fluorescent protein (EGFP) or lentivirus bicistronically expressing dominant active Rab11Q70L and EGFP for 3 days. After verifying expression by the observation of EGFP signals, virus-treated fibroblasts were used for transferrin receptor recycling as described above.
Uptake of transferrin into sealed vesicles and determination of cell surface levels of transferrin receptor.
To investigate if mutant Htt affects vesicle formation at the plasma membrane, we wanted to be sure that cells were at the same stage of endocytosis (synchronized) prior to the uptake of biotin-transferrin. We preincubated cells on ice for 15 min before treatment with biotin-transferrin on ice for 30 min. Incubation at 37°C allows the sequestration of biotin-transferrin into sealed vesicles. This can be observed by detecting the biotin-transferrin that remains in cells after an acidic-buffer wash (strip), which removes cell surface-bound biotin-transferrin. Under ideal conditions, biotin-transferrin should be present only in sealed vesicles when cells are incubated at 37°C for 0.5, 1, or 2 min and not under cold conditions (strip). However, we found that the 15-min preincubation on ice still allowed the entry of biotin-transferrin into sealed vesicles without incubation at 37°C (see Fig. S1 in the supplemental material). Therefore, we modified the protocol. After cells were extensively washed in ice-cold PBS as described above, they were preincubated on ice for 2 to 3 h and then treated with biotin-transferrin on ice for 30 min. The 2- to 3-h preincubation step significantly reduced the levels of biotin-transferrin observed after an acidic-buffer wash. The longer preincubation on ice successfully blocked the formation of sealed vesicles under cold conditions in control and HD cells (Fig. 1a). To block the reuptake of biotin-transferrin during incubation at 37°C, we added holotransferrin. After two washes in cold PBS, cells were collected and placed into prewarmed serum-free medium containing 10 μg ml−1 holotransferrin at 37°C for 0.5, 1, and 2 min or left on ice (as the total binding control or for analysis of steady-state levels of transferrin receptor at the cell surface). Each aliquot was transferred on ice, and 3 volumes of ice-cold acid buffer (0.2 M acetic acid, 0.5 M NaCl [pH 2.5]) were immediately added and mixed into the medium to stop the reaction for the indicated times. After mixing, cells were collected by centrifugation at full speed at 4°C for 30 s and resuspended in ice-cold acid buffer for 15 min at 4°C in order to remove surface-bound noninternalized biotin-transferrin. Cells were collected and lysed in sample buffer for SDS-PAGE and blotting analysis using an ABC kit (Vector Labs) to detect biotin-transferrin and tubulin antibody to detect tubulin. The density of each positive band was measured using NIH ImageJ software. The ratio of tubulin levels under each treatment condition (strip and 0.5, 1, and 2 min) relative to the level with no chase (0 min) was used to normalize the density of biotin-transferrin under each treatment condition. Without this tubulin normalization step, large variations occurred (data not shown). The biotin-transferrin signal under the “strip” condition was subtracted from the signal for each treatment period (0, 0.5, 1, and 2 min), and that value was used to calculate the percentage of internalized biotin-transferrin at each time point relative to 0 min.
FIG. 1.
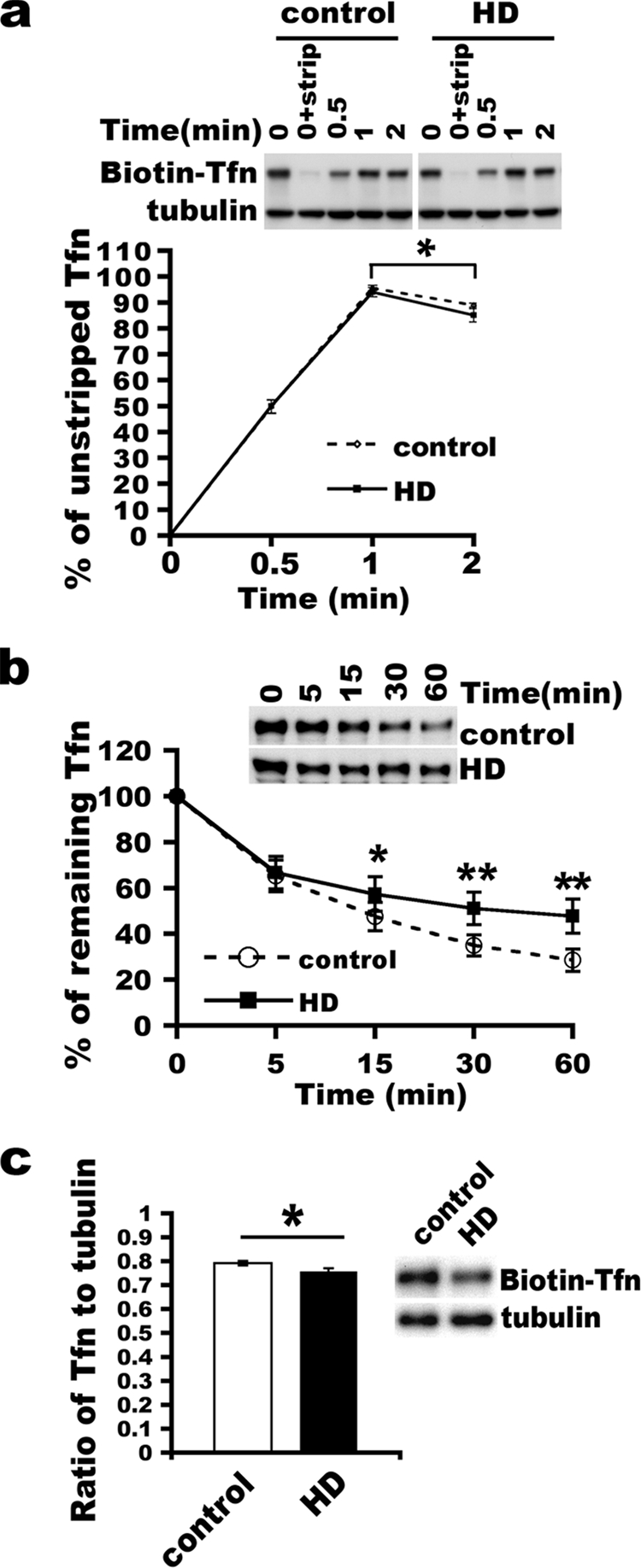
Mutant Htt does not affect vesicle formation at the plasma membrane but does inhibit receptor recycling. (a) Formation of sealed clathrin-coated vesicles at the plasma membrane occurs normally in HD cells. Single-round transferrin (Tfn) uptake was performed for the indicated times (0.5, 1, and 2 min). Noninternalized biotin-transferrin was stripped from cell surfaces using an acid buffer. 0 means no incubation at 37°C. 0+strip means no incubation at 37°C plus a wash with acid buffer. Cells were lysed in sample buffer for SDS-PAGE analysis and detection of biotin transferrin using an ABC kit. Note that the 0+strip condition shows a marked reduction in levels of biotin-transferrin, indicating a blockade of the entry of biotin-transferrin into sealed vesicles under cold conditions. The mean percentage of internalized (resistant to an acid wash) transferrin signal at each time was compared to the signal at 0 min (n = 6) (mean ± SD; *, P < 0.05; **, P < 0.01 [determined by the Student t test]). The top shows a representative blot. (b) Kinetics of transferrin recycling in control and HD fibroblasts. After the cumulative uptake of biotin-transferrin for 30 min at 37°C, cells were collected, washed, and incubated in the presence of unlabeled diferric transferrin for the indicated times. The percentage of retained biotin-transferrin after incubation for each time point was plotted, and the percentage at 0 min was set as 100%. Data shown were from three lines of control fibroblasts and three lines of HD fibroblasts (mean percentage of remaining transferrin ± SD; *, P < 0.01, determined by the Student t test). At the top is a representative blot. (c) Reduction of cell surface transferrin receptors at the basal state. After synchronized binding on ice, cells were washed and lysed for analysis. The ratio of bound biotin-transferrin to tubulin was graphed (n = 4) (mean ± SD; *, P < 0.05, determined by the Student t test). At the right is a representative blot.
Preparation of membranes by subcellular fractionation.
Homogenates were prepared in homogenization buffer (50 mM HEPES-Na [pH 7.0], 200 mM NaCl, 5 mM MaCl2, 1 mM dithiothreitol [DTT], 1 mM EDTA, and 0.25 M sucrose containing protease inhibitors) by passing cells through a 27½-gauge needle for 5 strokes on ice. Homogenates were centrifuged at 4°C at 14,000 rpm for 10 min. The postnuclear supernatant (S1) was further centrifuged at 4°C at 55,000 rpm for 1 h by using a TLA120.2 rotor (Beckman-Coulter). The supernatant (S2; cytosol) was discarded, and the pellet (P2′ total membranes) was resuspended in assay buffer supplemented with 1% Triton X-100 or directly in SDS-PAGE sample buffer for analysis. The P2 fraction was used as the source of guanine nucleotide exchange factor (GEF) in the GEF assay.
Guanine nucleotide exchange assays.
An assay of [3H]GDP release was performed as described previously (27), with minor modifications. In brief, 0.5 μg of purified glutathione S-transferase (GST)-Rab11 or 0.3 μg of purified Rab5-His6 (a gift from David Lambright, University of Massachusetts Medical School) was immobilized onto beads and loaded with 20 pmol of [3H]GDP (11.9 Ci/mmol; Amersham) in a solution containing 20 mM HEPES (pH 7.2), 20 mM potassium acetate, 1 mM DTT, and 5 mM EDTA (the total volume was 10 μl) at 30°C for 30 min. Loaded Rab proteins were placed on ice, and MgCl2 was added to a final concentration of 10 mM. One hundred micrograms of total membranes was diluted in 40 μl of assay buffer (20 mM HEPES [pH 7.2], 5 mM magnesium acetate, 1 mM DTT, and 0.75 mM GTP/GDP). After the [3H]GDP-loaded Rab proteins were added, the GEF reaction was initiated by incubation at 30°C for indicated times and stopped by cooling the reaction mixtures on ice. After each reaction, beads were washed three times in cold wash buffer (20 mM Tris-Cl [pH 7.4], 20 mM NaCl, 5 mM MgCl2, 1 mM DTT) and transferred into vials for scintillation counting with Aquasol-2 (DuPont) scintillation fluid. The [3H]GTP uptake assay was performed similarly to the [3H]GDP release assay except that unlabeled GDP was first loaded onto GST-Rab11 and nucleotide exchange was conducted in the presence of 1 μCi [3H]GTP (GE Healthcare). The two-tailed Student t test was used to determine the statistical significance.
Immunohistochemistry and quantitative analysis.
After two washes in PBS, cells on coverslips were fixed in 4% paraformaldehyde-PBS at room temperature for 15 min, quenched in 50 mM NH4Cl containing 0.1% Triton X-100, washed twice in PBS containing 0.1% Triton X-100, blocked with 1% bovine serum albumin in PBS containing 0.05% Tween 20, and labeled with primary antibodies. Following the primary-antibody step, targeted proteins were visualized by incubation of cells on coverslips with secondary antibodies conjugated with Bodipy FL (Invitrogen, Inc.) or Cy-3 (Jackson Laboratories). Primary antibodies against Rab11 (1:100; Chemicon) and Htt Ab1 (1 μg ml−1) were used. Secondary antibodies including Hoechst 33258 (Invitrogen, Inc.) were diluted at 1:1,000. Individual images for each wavelength (405, 488, and 568 nm) were collected separately using a Bio-Rad laser scanning confocal microscope with krypton-argon and blue diode lasers through a 60× or 100× oil Nikon Plan Apo objective on an inverted Nikon Eclipse TE300 fluorescent microscope. The scanned images from each fluorescent channel were merged using PhotoShop.
The nonspherical Rab11-positive structures in fixed fibroblasts were selected for measurements of length using NIH ImageJ software. Pixels of the distance between the two ends of a nonspherical structure measured by NIH ImageJ were converted into micrometers based on the resolution of the confocal images. Forty-eight tubules from 12 control fibroblasts (GM08399) and 109 tubules from HD fibroblasts (GM04857) were analyzed. In principle, 3 to 5 structures per control fibroblast and 6 to 13 structures per HD cell were selected for measurement. Data are represented as average length per tubular structure. The two-tailed Student t test (length of tubular structures) and chi-square test (cell number) were performed to determine the statistical significance.
To quantify the degree of colocalization, we used ImageJ software and the JACoP plugin to determine the Pearson's correlation coefficient as described previously (5, 30, 53). Pearson's coefficient was expressed as the mean and the standard deviation (SD).
TIRF microscopy and quantitative analysis.
Total internal reflection fluorescence (TIRF) microscopy was used to evaluate trafficking in individual fibroblasts using an Apo 60× 1.45-numerical-aperture objective (Olympus) as previously described in detail (4). Human fibroblasts from control patients (GM08399) and from an HD homozygous patient (GM04857) were infected with lentivirus expressing EGFP and grown on thin glass coverslips. Coverslips were mounted onto the microscope within a heated environment that was maintained at 34.5°C to 35°C, and cells were focused by observing EGFP, washed once in image medium (serum-free HEPES-buffered Dulbecco's modified Eagle's medium without phenol red), and incubated in image medium containing 5 μg ml−1 Alexa568-transferrin (Invitrogen, Inc.) for 15 min. Cells were washed three times in imaging medium and incubated in fresh imaging medium containing 10 μg ml−1 unlabeled holotransferrin (Sigma-Aldrich) for 5 min, and 600 images were then recorded at 1 image per second using custom-built software (4). The illuminated depth was set at 100 nm. To optimize the consistency of the experimental conditions, at least one control fibroblast and one HD fibroblast were studied successively in each session as matched pairs for statistical analysis. The order in which the control and HD fibroblasts were selected was changed between sessions.
We quantified the density and the size distribution of vesicles after chase using video image 100. Video images (resolution of 320 by 240 pixels, 0.2 μm/pixel) were converted into tagged image file format. The brightness and contrast of the images were optimized to visualize individual vesicles. The imaged cell was outlined, and the region surrounding the cell was removed using NIH ImageJ software. NIH ImageJ reads transferrin-positive vesicles as particles (in the “analyze particles” function) (see Fig. S4 in the supplemental material). The original scale in square inches of particles was converted into square micrometers based on the real image resolution and the magnification on the computer screen. The software identified some small vesicles in close spatial proximity as a single large vesicle and did not count some individual vesicles that were too small or weak in signal. These errors were more frequent for control fibroblasts, which had higher densities of small vesicles than did the HD fibroblasts. Thus, the actual number of small vesicles in control fibroblasts may be greater than what we present in Fig. 4. To determine the density of vesicles per μm2, the cross-sectional area of imaged cells was calculated by tracking the edge of the cell with the cursor using NIH ImageJ software and converting the readout from square inches into square micrometers as described above.
FIG. 4.

Abnormal distribution of transferrin receptor in live HD fibroblasts viewed by TIRF. (a) Small vesicles are predominant carriers of recycling cargo transferrin and gradually disappear in control fibroblasts. (b) HD cells show transferrin concentrated in large vesicular structures. a and b show TIRF images recorded at 5:01, 10:00, and 13:30 min after washout of transferrin. Scale bar, 5 μm. (c) Density (mean ± SD) of transferrin (Tfn)-positive structures per μm2 in control and HD cells (n = 5 cells for each; *, P < 0.001, determined by the Student t test). (d) Size distribution of transferrin-positive structures. The density and the size of Alexa568-transferrin labeled structures were quantified after chase for 6 min 40 s: chase for 5 min before imaging and another chase for 1 min 40 s after the start of imaging (image 100 was used for quantification). Paired cells (means ± SD) (HD and control cells imaged at the same TIRF angle setting) were analyzed using NIH ImageJ software (n = 5 pairs) (*, P < 0.001, determined by the Student t test).
We defined a structure as being tubular if the length of the structure was at least three times the diameter of a typical small vesicle. Tubular structures that moved from one position to another, changed length, or fused with plasma membranes or with another vesicular tubular structure were defined as being tubular transport intermediates. All transferrin-labeled tubular structures were counted in the last 100 images from all imaged cells by two examiners who were blinded to cell types. Each moving tubular structure was counted only once. The longest length of a moving tubule was measured. The distance between two ends of a tubule was measured using NIH ImageJ analysis.
The two-tailed Student t test was used to determine the statistical significance.
RESULTS
Mutant Htt has no effect on the formation of clathrin-coated vesicles (endocytosis) at the plasma membrane but slows receptor recycling.
Since Htt interacts with proteins involved in the formation of clathrin-coated vesicles (22) and is located at plasma membrane-coated pits (51), we looked for effects of mutant Htt on vesicular budding at the plasma membrane. We performed a standard synchronized transferrin uptake assay (42) with fibroblasts from three control and three HD patients. The fibroblast lines used for the analysis had fewer than 15 passages, since HD fibroblasts of more than 15 passages have reduced levels of transferrin receptors (see Fig. S2 in the supplemental material). In the uptake assay, basal surface receptors were allowed to internalize with biotin-transferrin for up to 2 min, which is enough time for vesicle budding and for membrane recycling to occur directly from sorting endosomes (34). We monitored the formation of sealed vesicles containing biotin-transferrin after incubation at 37°C in the presence of holotransferrin for 0, 0.5, 1, and 2 min, followed by the removal (strip) of excess surface-bound biotin-transferrin with an acid buffer wash (0.5 M NaCl-0.2 N acetic acid [pH 2.5]). To prevent the entry of biotin-transferrin into sealed vesicles under cold conditions, cells were preincubated on ice for 2 to 3 h (Fig. 1a) instead of 15 min (see Materials and Methods, and see Fig. S1 in the supplemental material). Quantification of the acid-resistant biotin-transferrin (as a percentage of unstripped transferrin) showed that the kinetics of internalizing biotin-transferrin into sealed vesicles were the same in wild-type and HD fibroblasts, suggesting that the formation of clathrin-coated vesicles at the plasma membrane (endocytosis) occurs normally in the presence of mutant Htt (Fig. 1a). There was a similar drop in levels of acid-resistant biotin-transferrin detected between 1 min and 2 min in control and HD cells (P < 0.01). Thus, recycling at sorting endosomes in wild-type and HD cells occurs in the time normally required based on data from previous studies (34).
Next, we analyzed post-endocytic-receptor recycling using a biochemical assay. Transferrin receptors were first saturated by taking up biotin-transferrin and were then allowed to recycle in the presence of unlabeled transferrin to remove recycled iron-free biotin-transferrin. The recycling rate of transferrin receptor in three control fibroblast cell lines was in the range reported previously for other types of cells (mean percentages of biotin-transferrin retained in cells ± SD of 34.87% ± 4.63% and 28.47% ± 4.9% for 30 min and 60 min, respectively) (Fig. 1b) (38, 47, 54). The values for fibroblasts from HD patient were similar for the one homozygous and two heterozygous lines, so we grouped the data together for statistical analysis. The mean percentage of remaining biotin-transferrin was significantly higher in the three HD lines than in the three control fibroblast lines (mean percentages ± SD for control versus HD lines were 34.87% ± 4.63% versus 51.01% ± 7.05% for 30 min [P < 0.01, determined by the Student t test] and 28.47% ± 4.9% versus 47.75% ± 7.44% for 60 min [P < 0.01, determined by the Student t test]) (Fig. 1b). These results provided support that endocytic recycling was slowed in the presence of mutant Htt.
We had established that total levels of the transferrin receptor protein were the same in control and HD cells of fewer than 15 passages (see Fig. S2 in the supplemental material). We wanted to know if HD fibroblasts had a change in the distribution of transferrin receptors; a decline in numbers of receptors at the cell surface would be consistent with altered transferrin receptor recycling. To examine cell surface transferrin receptors, cells were incubated with biotin-transferrin on ice. After a washout of unbound biotin-transferrin in cold PBS, cells were lysed for analysis. The ratio of receptor-bound transferrin at the plasma membrane to tubulin in low-passage HD cells (<15 passages) was significantly reduced compared to that in control cells (mean ratio of bound transferrin to tubulin ± SD for control versus HD cells of 0.79 ± 0.01 versus 0.76 ± 0.01, respectively, [P < 0.05, determined by the Student t test]) (Fig. 1c). Therefore, in the HD fibroblasts, the return of transferrin receptors to the plasma membranes was delayed. These data strengthen the idea that an impairment of receptor recycling to the plasma membrane occurs in HD fibroblasts.
Rab11 activation is reduced in primary HD fibroblasts.
Since Rab11 activity is important for endocytic recycling (38, 47) and is deficient in Htt-null embryonic stem cells (32), we investigated if mutant Htt perturbs Rab11 activity. Like other Rab proteins, Rab11 undergoes cycles of nucleotide exchange between GDP-bound (inactive) and GTP-bound (active) states; activation occurs at membranes and requires a GEF (19, 40, 55). Since the GEF for Rab11 is unknown, we used total membranes from control and HD fibroblasts as a source of GEF. To measure GEF activity, we determined the amount of [3H]GDP released from purified GST-Rab11 or His6-Rab5 in the presence of membranes. After incubation of [3H]GDP-labeled Rab11 with HD fibroblast-derived total membranes for 30 min, the level of [3H]GDP remaining on GST-Rab11 in HD fibroblast-derived total membranes was significantly higher than the level of [3H]GDP remaining on GST-Rab11 after incubation with total membranes from control fibroblasts (mean percentage of remaining [3H]GDP on Rab11 ± SD for control versus HD fibroblasts of 43.63% ± 4.39% versus 64.99% ± 5.49% [P < 0.01, determined by the Student t test]) (Fig. 2a). Under the same conditions, levels of [3H]GDP remaining on Rab5, which functions at early endosomes, were not significantly different in the presence of total membranes from HD fibroblasts or control fibroblasts (mean percentage of remaining [3H]GDP on Rab5 ± SD for control versus HD fibroblasts of 50.8% ± 8.62% versus 55.47% ± 5.89% [P = 0.487, determined by the Student t test]) (Fig. 2a). Thus, compared to membranes of control fibroblasts, HD fibroblasts supported less GEF activity on Rab11 and similar GEF activity on Rab5.
FIG. 2.

Inhibition of nucleotide exchange on Rab11 in HD cells. One hundred micrograms of total membranes from primary control and HD fibroblasts was extracted with 1% Triton X-100 and centrifuged. The extracted supernatants were used to catalyze GDP release from [3H]GDP-GST-Rab11 and [3H]GDP-Rab5-His6 at 30°C for 30 min (a) or [3H]GTP uptake into GDP-GST-Rab11 at 30°C for indicated times (b). [3H]GDP-GST-Rab11, [3H]GDP-Rab5-His6, or [3H]GTP-GST-Rab11 on beads was collected by centrifugation, washed in cold washing buffer, and measured by scintillation counting. Data for panel a are represented as mean percentages ± SD for retaining [3H]GDP (n = 3; *, P < 0.01 by the Student t test). n/s, no significance. In panel b, the cpm value for each time point from each experiment was converted into picomoles, and the mean values in picomoles ± SD of [3H]GTP were graphed (n = 3; *, P < 0.01, determined by the Student t test).
An alternative way to assess GEF activity is to examine the uptake of [3H]GTP onto GST-Rab11. Total membranes of HD fibroblasts supported significantly less uptake of [3H]GDP than did total membranes from control fibroblasts (mean concentrations of [3H]GTP bound onto Rab11 ± SD for control versus HD cells of 0.154 ± 0.018 pmol versus 0.153 ± 0.025 pmol at 5 min [P = 0.986, determined by the Student t test], 0.402 ± 0.023 pmol versus 0.306 ± 0.019 pmol at 15 min [P < 0.01, determined by the Student t test], and 0.464 ± 0.187 pmol versus 0.37 ± 0.023 pmol at 30 min [P < 0.05, determined by the Student t test]) (Fig. 2b). Thus, based on measurements of GDP release and GTP incorporation in the in vitro assays, the presence of mutant Htt inhibits the activation of Rab11.
Mutant Htt colocalizes with Rab11 on endosomal structures that have an abnormal morphology.
We found that HD fibroblast membranes supported less activation of Rab11 than did control membranes. We wanted to know if endogenous mutant Htt in fibroblasts was associated with Rab11. In previous work, we found by immunoprecipitation assay that wild-type Htt is precipitated by GDP-bound Rab11 and interacts with a complex that has GEF activity toward Rab11 (32). Since Rab11 functions mainly at recycling endosomes (34, 38, 47, 54), we performed immunofluorescence labeling to determine if Htt and Rab11 colocalize at recycling endosomes. To ensure that we were detecting only mutant Htt in HD fibroblasts, we used a cell line from a patient who was homozygous for the HD mutation (39 and 49 CAG repeats). As previously described in other studies of human fibroblasts (36, 51), we observed immunoreactive endogenous Htt distributed diffusely in the cytoplasm and on punctate structures in control and HD fibroblasts (see Fig. S3 in the supplemental material). In accordance with observations reported previously for other cell types (17, 39, 47, 52, 54), Rab11 immunoreactivity in control and HD fibroblasts occurred diffusely in the cytoplasm and on punctate structures (see Fig. S3 in the supplemental material). These patterns of labeling were not detected if the primary anti-Htt and anti-Rab11 antisera were omitted from the labeling procedure (results not shown). Wild-type Htt in control fibroblasts and mutant Htt in HD fibroblasts were present in some of the Rab11-positive structures (Fig. 3a and see Fig. S3 in the supplemental material). Quantification of Pearson's correlation coefficient (r) showed a moderate degree of colocalization between Htt and Rab11 in both control and HD fibroblasts (n = 10 control and 10 HD cells; mean r ± SD of 0.2842 ± 0.09974 for control fibroblasts and 0.4947 ± 0.1991 for HD fibroblasts [P = 0.01, determined by the Student t test]). In some HD fibroblasts, the Htt- and Rab11-double-positive structures appeared larger than those in the control fibroblasts (Fig. 3a and see Fig. S3 in the supplemental material). Measurements showed that the Htt-Rab11-positive tubular structures in HD fibroblasts were significantly longer than those in control fibroblasts (3.95 ± 0.92 μm versus 1.67 ± 0.26 μm for HD versus control fibroblasts [P < 0.001, determined by the Student t test]) (Fig. 3a and b). These data suggested that recycling endosomes colabeled with Rab11 and mutant Htt had an abnormal morphology. This aberrant “enhanced tubular” morphology resembles the abnormal endosomes formed in cells that express a dominant negative Rab11 mutant (54).
FIG. 3.

Abnormality of Rab11-positive endosomal structures in HD fibroblasts. (a) Primary fibroblasts were cultured on coverslips overnight and processed for immunofluorescence labeling. Both wild-type and mutant Htt (red) are localized to Rab11 (green)-positive structures (yellow). Shown are representative confocal images of human fibroblasts. Arrows indicate Rab11-positive tubular structures, and arrowheads point to punctate structures. Scale bar, 10 μm. (b) Comparison of Rab11-positive structures in low-passage HD and age-matched control fibroblasts. The length of Rab11-positive structures colabeled with Htt was measured in confocal images using NIH ImageJ software. Data are represented as mean lengths of Rab11-positive structures analyzed in control (n = 12 cells) and HD (n = 12 cells) fibroblasts (*, P < 0.001, determined by a two-tailed Student's t test). (c) Comparison of levels of Rab11 and Htt in low-passage control and HD fibroblasts. Twenty-five micrograms of cell lysates from control (GM08399) and HD (GM04857) fibroblasts was analyzed by SDS-PAGE and Western blotting with antibodies against Htt, Rab11, and tubulin. Films were scanned, and the density of each band was measured using NIH ImageJ software. Data are represented as mean percentages of the control ± SD (n = 4) (significance determined by the Student t test). n/s, no significance. A Western blot from one of the experiments is shown at the right.
We noticed that the fluorescent signals for immunoreactive mutant Htt and Rab11 on endosomes in HD fibroblasts were stronger than those for wild-type Htt and Rab11 on endosomes in control fibroblasts (Fig. 3a). Western blot analysis of lysates from the same control and HD cell lines used for immunohistochemistry showed that the levels of Rab11 were comparable in control and HD fibroblasts (the Rab11 content in HD fibroblasts was 99.98% ± 4.29% of the Rab11 in control fibroblasts). The level of mutant Htt in the HD fibroblasts was similar to that of wild-type Htt in the control fibroblasts (99.34% ± 8.62% of control fibroblasts) (Fig. 3c). A reasonable explanation for the increase in the immunoreactive signal for mutant Htt and Rab11 detected in HD fibroblasts compared to controls was that there was a redistribution of mutant Htt and Rab11 from the cytoplasm to endosomal structures.
Imaging of transferrin-labeled transport intermediates in living HD fibroblasts reveals reduced vesicle formation at recycling endosomes.
The finding that Rab11-positive recycling endosomes were longer in HD fibroblasts than in control fibroblasts suggested that vesicle formation from these recycling endosomes was impaired. We wanted to establish that the abnormal recycling endosomes seen in the HD fibroblasts were involved in transferrin receptor recycling and gave rise to transport intermediates with transferrin-containing cargo. Therefore, we undertook recordings in real time of the trafficking of labeled transferrin in fibroblasts using TIRF microscopy. TIRF microscopy records dynamic events inside the cell within 100 to 200 nm from the plasma membrane. Fibroblasts are ideal for use in TIRF microscopy because they grow very flat and are adherent. The idea here was to observe Alexa568-transferrin vesicles (transport intermediates) that have emerged from recycling endosomes. The recycling of transferrin via recycling endosomes has a half-life of ∼12 min (and a half-life of ∼2 min at sorting endosomes) (34). We observed a recycling rate of transferrin in control fibroblasts (Fig. 1b) that was similar to that in other cell types (38, 47, 54). Hence, we used a labeling protocol to maximize the possibility that the vesicles detected by TIRF microscopy were recycling ones and to minimize detection of newly formed endocytic vesicles from the plasma membrane: after taking up Alexa568-transferrin for 15 min, the cells were washed extensively to remove uninternalized Alexa568-transferrin and incubated in fresh imaging medium with unlabeled transferrin for 5 min prior to imaging for 10 min. This 5-min incubation in imaging medium before recording allowed any endocytic vesicles that were newly formed from the plasma membrane to move out of the TIRF field. Thus, the only transferrin-positive vesicles visible by TIRF microscopy should be those that are transport intermediates returning to the plasma membrane. We cannot fully exclude the possible detection of some endosomes that locate near the plasma membrane.
TIRF images revealed that control fibroblasts had numerous small vesicles spread throughout the cytoplasm with an average density of 4.5 transferrin-positive structures per μm2 (Fig. 4a and c and 5a; also see Movies S1 and S2 in the supplemental material). Under the same conditions, HD fibroblasts had 0.5 transferrin-positive structures within the same area (Fig. 4c). The proportion of small vesicles (<0.065 μm2) was significantly reduced in HD fibroblasts (mean percentage ± SD for HD versus control fibroblasts of 14.6% ± 1.4% versus 56.1% ± 15.7% [P < 0.001, determined by the Student t test]) (Fig. 4d). On the other hand, HD cells had an increased proportion of large vesicular structures (>0.32 μm2) compared with control fibroblasts (mean percentages ± SD for HD versus control fibroblasts of 19.3% ± 3.8% versus 5.9% ± 2.4% for 0.32 to 0.65 μm2 and 34.1% ± 4.2% versus 2.8% ± 3.1% for >0.65 μm2 [P < 0.001, determined by the Student t test]) (Fig. 4b and d and 5b and see Movies S3 and S4 in the supplemental material). These real-time data reveal a switch from small to large vesicles in HD fibroblasts, supporting an inhibition in the formation of small vesicular intermediates in HD cells.
FIG. 5.

Long tubular intermediates formed in living HD fibroblasts viewed by TIRF. (a) Short tubular and small vesicular intermediates in control fibroblasts. White arrows point to a short tubule emerging and moving, whereas the oval surrounds two vesicles that fused with the plasma membrane and disappeared in the next image. The low signal is due to the small size of transferrin (Tfn)-positive structures. (b) Long tubules serve as transport intermediates in HD fibroblasts. Images are shown 4 s apart in order to show the change of tubular intermediates. Open arrows point to a tubule gradually fusing with the plasma membrane and becoming shorter and shorter, while the solid arrow indicates a growing tubule that becomes longer and longer. Scale bars, 10 μm. (c) Increase in the mean average length of tubules in HD fibroblasts ± SD (n = 72 tubules from five control cells and 72 tubules from five HD cells; *, P < 0.001, determined by the Student t test). (d) Quantification of the mean number of tubular intermediates per cell observed in the last 100 images from each cell ± standard error of the mean (10 control and 10 HD cells) imaged by TIRF microscopy (*, P < 0.05, determined by paired Student t test).
TIRF microscopy showed that transferrin-positive tubular structures occurred in control fibroblasts (Fig. 5a; also see Movies S1 and S2 in the supplemental material) and HD fibroblasts (Fig. 5b and see Movie S4 in the supplemental material). However, the tubular structures were significantly longer in HD fibroblasts than those in control fibroblasts (mean length ± SD for HD versus control fibroblasts of 5.4 ± 0.1 μm versus 1.7 ± 0.3 μm [P < 0.001, determined by the Student t test]) (Fig. 5c). Moreover, we observed some very long tubules emanating from transferrin-positive punctate structures in HD fibroblasts (see Movie S4 in the supplemental material). This dynamic relationship between labeled structures provided evidence that the emerging tubules were transport intermediates. Quantitative analysis in the last 100 TIRF images revealed that the mean number of tubular transport intermediates was significantly greater in HD fibroblasts than in control fibroblasts (mean number ± standard error of the mean for HD versus control fibroblasts of 26.2 ± 2.3 versus 12.7 ± 3.1 [P < 0.05, determined by the Student t test]) (Fig. 5d; also see Movies S1 to S4 in the supplemental material). These data show that transferrin-containing transport intermediates and the recycling endosomes from which they emerge have an abnormal tubular morphology similar to that seen for mutant Htt-Rab11-positive structures observed in fixed HD fibroblasts. Altogether, the findings obtained by TIRF microscopy suggest that vesicle formation at endosomes is inhibited in the presence of mutant Htt.
Dominant active Rab11 normalizes biotin-transferrin recycling in HD fibroblasts.
We found that transferrin receptor recycling was slowed in HD fibroblasts compared to control fibroblasts (Fig. 1b), which coincides with defective Rab11 activation in HD cells (Fig. 2). To test if the elevation in levels of Rab11 activity can improve transferrin recycling, we infected HD fibroblasts with lentivirus expressing EGFP or lentivirus bicistronically expressing dominant active Rab11Q70L and EGFP. The results showed that transferrin recycling in HD fibroblasts expressing Rab11Q70L was significantly improved at 60 min compared to that in uninfected HD cells (mean percentage of biotin-transferrin retained in cells ± SD for no virus versus Rab11Q70L of 46.9% ± 7.4% versus 30.7% ± 8.6% for 60 min [P < 0.05, determined by the Student t test]) (Fig. 6a) or HD fibroblasts expressing EGFP alone (mean percentages of biotin-transferrin retained in cells ± SD for EGFP versus Rab11Q70L of 60.3% ± 5.4% versus 38.9% ± 9.4% for 30 min [P < 0.05, determined by the Student t test] and 53.1% ± 5% versus 30.7% ± 8.6% for 60 min [P < 0.01, determined by the Student t test]) (Fig. 6a). HD cells expressing EGFP alone did not significantly differ from HD cells with no virus in percentages of remaining biotin-transferrin at 60 min. Thus, our findings suggest that in the presence of the HD mutation, dominant active Rab11 rescues a deficit in transferrin receptor recycling. Control fibroblasts expressing Rab11Q70L were significantly delayed in biotin-transferrin recycling compared to that of uninfected cells or EGFP-expressing fibroblasts (see Fig. S5 in the supplemental material). The inhibitory effect of Rab11Q70L on recycling under normal conditions is noteworthy for other cells (25).
FIG. 6.

The delay in recycling of biotin-transferrin is rescued by expressing dominant active Rab11 in HD fibroblasts, and long tubules are detectable in fixed HD cells. (a) Primary HD fibroblasts (GM04857) were infected with lentivirus expressing Rab11Q70L and EGFP under a bicistronic promoter or lentivirus expressing EGFP alone for 3 days and processed for biochemical assay of biotin-transferrin recycling as described in the legend of Fig. 1b. The data contributed by the same HD cell line from results shown in Fig. 1b were graphed for comparison to the no-virus infection condition (n = 4) (mean percentage of remaining transferrin ± SD) (*, P < 0.05 for Rab11Q70L relative to no virus, determined by the Student t test). At top is a representative blot. (b) Accumulation of Alexa568-transferrin in Rab11-positive tubular structures in the periphery of HD fibroblasts. After the synchronized uptake of Alexa568-transferrin was performed for 30 min at 37°C, cells were fixed for immunofluorescence labeling with an antibody against Rab11 (green). Shown are representative confocal images. Scale bar, 5 μm. Signals of Alexa568-transferrin are undetectable in control fibroblasts after a 30-min chase, as expected. HD fibroblasts show the presence of Alexa568-transferrin long tubules and large puncta in the cell periphery. Arrows in the insets at bottom point to the site where immunoreactive Rab11 is localized to the Alexa568-transferrin-positive tubule.
We noticed that at 5 min of incubation, EGFP expression in HD fibroblasts delayed receptor recycling compared to that for no virus infection (Fig. 6a). This difference did not occur in control fibroblasts (see Fig. S5 in the supplemental material). We wanted to rule out an effect of EGFP on the formation of tubular intermediates detected by TIRF, which requires the use of EGFP to identify cells (Fig. 5). Therefore, we examined the synchronized uptake of Alexa568-transferrin. After incubation at 37°C for 30 min in the presence of unlabeled holotransferrin, signals of Alexa568-transferrin dramatically declined to nearly undetectable levels (Fig. 6b, top). These data show an efficient recycling of Alexa568-transferrin in control fibroblasts. However, under the same conditions, some of the HD fibroblasts manifested Alexa568-transferrin-positive long tubules and/or large puncta (Fig. 6b, bottom). The long tubules and large puncta were colabeled with Rab11 (Fig. 6b, bottom). The presence of transferrin-positive long tubules and large puncta in HD fibroblasts that lack EGFP expression strengthens our conclusion that mutant Htt causes a deficit in vesicle formation at the recycling endosome.
DISCUSSION
Vesicle-mediated trafficking encompasses vesicle formation at donor membranes, the targeting and fusion of vesicles with acceptor membranes, and the molecular machinery that regulates these events. The processes involved in trafficking are considered separate from those of vesicle transport along cytoskeletons, although these two processes are tightly coordinated (6, 8, 37). In this study, we provide support for a trafficking defect in HD. We show that the activation of Rab11 is impaired in the context of the HD mutation, as shown by a reduction in nucleotide exchange on Rab11. On the other hand, the activation of Rab5, a critical GTPase for endocytosis (7), was unaffected by the presence of mutant Htt. The formation of clathrin-coated vesicles at the plasma membrane (clathrin-dependent endocytosis) in HD fibroblasts was similar to that in control fibroblasts, consistent with findings that mutant Htt does not affect the formation of clathrin-coated vesicles at the trans-Golgi network (11). Based on what is known about the role of Rab11, a deficit in Rab11 activity would be expected to impair vesicle formation at recycling endosomes (Fig. 7). The prediction was verified experimentally. Accordingly, HD fibroblasts were deficient in the recycling of transferrin receptors back to the plasma membrane and had morphological changes (extended tubules) consistent with an impaired formation of vesicles from recycling endosomes. Moreover, expressing dominant active Rab11 in HD fibroblasts normalized the recycling deficit. Altogether, our findings support a novel mechanism in HD resulting from the loss of Rab11 activity.
FIG. 7.

Schematic representations of Rab11 activation in control and HD cells and functional consequences on cellular homeostasis. (Left) Normal Rab11 activity on endosomal membranes and downstream consequences. In the cytoplasm, GDP-bound Rab11 partners with rabGDP dissociation inhibitor. To fulfill functions, Rab11GDP is recruited onto endosomal membranes for activation by a GEF and converted to Rab11GTP, which recruits a cohort of effectors. The GEF for Rab11 is not known, but Htt functions in this process. The consequence of normal Rab11 activity is the formation of small vesicles and short tubules at recycling endosomes and the maintenance of a normal level of receptors and transporters at cell surfaces. (Right) Proposed model in which mutant Htt interferes with Rab11 activity. The presence of mutant Htt inhibits the conversion of Rab11GDP into Rab11GTP, as indicated by the dashed arrow. The consequence of insufficient Rab11 activity is the formation of long tubules and large vesicles at recycling endosomes and a reduced level of receptors and transporters at cell surfaces, leading to cellular dysfunction and atrophy.
The mechanism by which mutant Htt inhibits nucleotide exchange from Rab11GDP to Rab11GTP is not established. Knowledge of the GEF for Rab11 in mammalian cells will be required to understand the mechanism. Our previous studies suggested that wild-type Htt functions in a protein complex that acts as a Rab11 GEF (32). Mutant Htt may inhibit Rab11 activity by interfering with the normal function of Htt in a Rab11 GEF complex. Mutant Htt was previously reported to disrupt functions related to other Rab proteins, namely, Rab5 and Rab8, which interact with Htt via HAP40 (36) and FIP2/optineurin (23), respectively. These deficits have been shown to involve interference by mutant Htt with Rab effector proteins and not by direct interference with the activity of Rab proteins.
Immunofluorescence microscopy and TIRF imaging in living HD cells reveal aberrant dynamics at recycling endosomes.
By immunofluorescence microscopy, Rab11-positive recycling endosomes in HD fibroblasts had an extended tubular morphology that resembles that of endosomes seen in cells expressing a dominant negative Rab11 mutant (47, 54). Recycling endosomes elongate further in HeLa cells that express a dominant negative Rab11 mutant if microtubules are depolymerized with nocodazole, suggesting that tubulation stems from the inhibition of vesicle formation (54). Quantitative electron microscopy has been the main method to study vesicle formation based on counts of the numbers of formed vesicles. The development of time-lapse microscopic imaging and fluorescence tags enables the direct visualization of vesicle formation in live cells. However, due to an abundance of labeled structures, real-time imaging of the recycling vesicles with traditional epifluorescence microscopy does not allow the discrimination of different endocytic compartments, including endosomes (early, sorting, and recycling endosomes) and inward and outward transport intermediates generated from these endosomes. TIRF imaging, on the other hand, reduces the noise because it has a resolution of 100 to 200 nm, in the range of a thin section in the electron microscope (40 to 60 nm). TIRF imaging has been used to detect the dynamics of clathrin-coated vesicles at the plasma membrane (20).
In this study, we used TIRF imaging of labeled transferrin in living fibroblasts to detect transport intermediates that have emerged from recycling endosomes. Our findings for control fibroblasts using TIRF fit with predictions based on traditional microscopic methods showing that recycling endosomes generate transport intermediates of spherical and/or tubular shapes for the recycling of cargos back to the plasma membrane (13, 26, 34, 47, 54). If Rab11 dysfunction in HD cells was sufficient to cause a deficit in vesicle formation at recycling endosomes, we would expect to see the number of transport intermediates reduced by TIRF imaging. Our findings confirm this idea. The majority of transferrin-labeled structures observed in control fibroblasts were small vesicles, whereas HD fibroblasts had fewer small vesicular intermediates and more tubular intermediates than did control fibroblasts. Moreover, the tubular intermediates in HD fibroblasts were much longer than those in control fibroblasts. Similar long tubules were also detected in fixed HD fibroblasts after the synchronized uptake of Alexa568-transferrin for 30 min.
Relevance of Rab11 dysfunction to HD pathogenesis.
Endocytic recycling is the primary way by which cells maintain plasma membrane constituents (18). By inhibiting Rab11 activation and slowing the formation of transport intermediates that carry key regulatory proteins back to the plasma membrane, mutant Htt may cause a broad range of adverse effects downstream of endocytic recycling (Fig. 7). We observed a small decrease in surface levels of transferrin receptors, consistent with a deficit in endocytic recycling. Since HD is a slowly progressing disease, a small decrease in levels of surface transferrin receptors at steady state could have cumulative effects on iron homeostasis and impair energy metabolism. Mutant Htt could disrupt other endocytic recycling-dependent events. These include signal transduction (recycling of ion channels exemplified by the Ca2+ channels TRPV5 and TRPV6 [49]), management of oxidative stress (recycling of transporters exemplified by EAAC1 that is required for the synthesis of glutathione [16]), cholesterol trafficking (25), and maintenance of the cell size through plasma membrane replenishment. The impact on different homeostatic systems may be cell specific and cumulative.
Recycling endosomes in polarized cells are also key stations for sorting newly synthesized secretory proteins to the apical domain through a process called transcytosis (2, 3, 24, 43). Thus, in neurons, the reduced activity of Rab11 at recycling endosomes could affect both trafficking within somatodendritic regions and the sorting of critical receptors or transporters to axonal regions through transcytosis.
In summary, we identify an inhibitory effect of mutant Htt on the activation of Rab11, which causes a deficit in vesicle formation from recycling endosomes and a slowing in recycling cargo and plasma membrane constituents from recycling endosomes back to the cell surface. A dysfunction in Rab11 activity and recycling endosomes in HD could have implications for the proper trafficking of many critical proteins besides transferrin and for effects in both somatodendritic sites and axons of neurons. Those critical proteins that rely on Rab11 activity are potential therapeutic targets for HD.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (grant NS R01-38194 to N.A., Philip D. Zamore, and M.D.), the DANA Foundation (to N.A. and M.D.), the High Q Foundation (to M.D.), and the Huntington's Disease Society of America (to M.D.). Core resources supported by Diabetes Endocrinology Research Center grant DK P30-32520 (University of Massachusetts Medical School [UMMS]) were also used. X.-L. is supported by a John J. Wasmuth postdoctoral fellowship from the Hereditary Disease Foundation and DANA Foundation. We declare no conflict of interests.
We thank David Lambright (UMMS) for his advice and reagents and Silvia Corvera (UMMS) for helpful discussions.
Footnotes
Published ahead of print on 14 September 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Almeida, S., A. B. Sarmento-Ribeiro, C. Januario, A. C. Rego, and C. R. Oliveira. 2008. Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington's disease patients. Biochem. Biophys. Res. Commun. 374:599-603. [DOI] [PubMed] [Google Scholar]
- 2.Apodaca, G., L. A. Katz, and K. E. Mostov. 1994. Receptor-mediated transcytosis of IgA in MDCK cells is via apical recycling endosomes. J. Cell Biol. 125:67-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barroso, M., and E. S. Sztul. 1994. Basolateral to apical transcytosis in polarized cells is indirect and involves BFA and trimeric G protein sensitive passage through the apical endosome. J. Cell Biol. 124:83-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellve, K. D., D. Leonard, C. Standley, L. M. Lifshitz, R. A. Tuft, A. Hayakawa, S. Corvera, and K. E. Fogarty. 2006. Plasma membrane domains specialized for clathrin-mediated endocytosis in primary cells. J. Biol. Chem. 281:16139-16146. [DOI] [PubMed] [Google Scholar]
- 5.Bolte, S., and F. P. Cordelieres. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224:213-232. [DOI] [PubMed] [Google Scholar]
- 6.Bonifacino, J. S., and B. S. Glick. 2004. The mechanisms of vesicle budding and fusion. Cell 116:153-166. [DOI] [PubMed] [Google Scholar]
- 7.Bucci, C., R. G. Parton, I. H. Mather, H. Stunnenberg, K. Simons, B. Hoflack, and M. Zerial. 1992. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70:715-728. [DOI] [PubMed] [Google Scholar]
- 8.Caviston, J. P., and E. L. Holzbaur. 2006. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 16:530-537. [DOI] [PubMed] [Google Scholar]
- 9.Ciammola, A., J. Sassone, L. Alberti, G. Meola, E. Mancinelli, M. A. Russo, F. Squitieri, and V. Silani. 2006. Increased apoptosis, huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington's disease subjects. Cell Death Differ. 13:2068-2078. [DOI] [PubMed] [Google Scholar]
- 10.del Hoyo, P., A. Garcia-Redondo, F. de Bustos, J. A. Molina, Y. Sayed, H. Alonso-Navarro, L. Caballero, J. Arenas, and F. J. Jimenez-Jimenez. 2006. Oxidative stress in skin fibroblasts cultures of patients with Huntington's disease. Neurochem. Res. 31:1103-1109. [DOI] [PubMed] [Google Scholar]
- 11.Del Toro, D., J. Alberch, F. Lazaro-Dieguez, R. Martin-Ibanez, X. Xifro, G. Egea, and J. M. Canals. 2009. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol. Biol. Cell 20:1478-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DiFiglia, M., E. Sapp, K. Chase, C. Schwarz, A. Meloni, C. Young, E. Martin, J. P. Vonsattel, R. Carraway, S. A. Reeves, et al. 1995. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 14:1075-1081. [DOI] [PubMed] [Google Scholar]
- 13.D'Souza-Schorey, C., E. van Donselaar, V. W. Hsu, C. Yang, P. D. Stahl, and P. J. Peters. 1998. ARF6 targets recycling vesicles to the plasma membrane: insights from an ultrastructural investigation. J. Cell Biol. 140:603-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelender, S., A. H. Sharp, V. Colomer, M. K. Tokito, A. Lanahan, P. Worley, E. L. Holzbaur, and C. A. Ross. 1997. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum. Mol. Genet. 6:2205-2212. [DOI] [PubMed] [Google Scholar]
- 15.Gauthier, L. R., B. C. Charrin, M. Borrell-Pages, J. P. Dompierre, H. Rangone, F. P. Cordelieres, J. De Mey, M. E. MacDonald, V. Lessmann, S. Humbert, and F. Saudou. 2004. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118:127-138. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez, M. I., B. T. Susarla, K. M. Fournier, A. L. Sheldon, and M. B. Robinson. 2007. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J. Neurochem. 103:1917-1931. [DOI] [PubMed] [Google Scholar]
- 17.Green, E. G., E. Ramm, N. M. Riley, D. J. Spiro, J. R. Goldenring, and M. Wessling-Resnick. 1997. Rab11 is associated with transferrin-containing recycling compartments in K562 cells. Biochem. Biophys. Res. Commun. 239:612-616. [DOI] [PubMed] [Google Scholar]
- 18.Griffiths, G., R. Back, and M. Marsh. 1989. A quantitative analysis of the endocytic pathway in baby hamster kidney cells. J. Cell Biol. 109:2703-2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grosshans, B. L., D. Ortiz, and P. Novick. 2006. Rabs and their effectors: achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. USA 103:11821-11827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Groves, J. T., R. Parthasarathy, and M. B. Forstner. 2008. Fluorescence imaging of membrane dynamics. Annu. Rev. Biomed. Eng. 10:311-338. [DOI] [PubMed] [Google Scholar]
- 21.Gunawardena, S., L. S. Her, R. G. Brusch, R. A. Laymon, I. R. Niesman, B. Gordesky-Gold, L. Sintasath, N. M. Bonini, and L. S. Goldstein. 2003. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40:25-40. [DOI] [PubMed] [Google Scholar]
- 22.Harjes, P., and E. E. Wanker. 2003. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem. Sci. 28:425-433. [DOI] [PubMed] [Google Scholar]
- 23.Hattula, K., and J. Peranen. 2000. FIP-2, a coiled-coil protein, links huntingtin to Rab8 and modulates cellular morphogenesis. Curr. Biol. 10:1603-1606. [DOI] [PubMed] [Google Scholar]
- 24.Hemery, I., A. M. Durand-Schneider, G. Feldmann, J. P. Vaerman, and M. Maurice. 1996. The transcytotic pathway of an apical plasma membrane protein (B10) in hepatocytes is similar to that of IgA and occurs via a tubular pericentriolar compartment. J. Cell Sci. 109(Pt. 6):1215-1227. [DOI] [PubMed] [Google Scholar]
- 25.Holtta-Vuori, M., K. Tanhuanpaa, W. Mobius, P. Somerharju, and E. Ikonen. 2002. Modulation of cellular cholesterol transport and homeostasis by Rab11. Mol. Biol. Cell 13:3107-3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hopkins, C. R., A. Gibson, M. Shipman, D. K. Strickland, and I. S. Trowbridge. 1994. In migrating fibroblasts, recycling receptors are concentrated in narrow tubules in the pericentriolar area, and then routed to the plasma membrane of the leading lamella. J. Cell Biol. 125:1265-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones, S., C. J. Richardson, R. J. Litt, and N. Segev. 1998. Identification of regulators for Ypt1 GTPase nucleotide cycling. Mol. Biol. Cell 9:2819-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kegel, K. B., M. Kim, E. Sapp, C. McIntyre, J. G. Castano, N. Aronin, and M. DiFiglia. 2000. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 20:7268-7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, W. C., M. Yoshihara, and J. T. Littleton. 2004. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc. Natl. Acad. Sci. USA 101:3224-3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewkowicz, E., F. Herit, C. Le Clainche, P. Bourdoncle, F. Perez, and F. Niedergang. 2008. The microtubule-binding protein CLIP-170 coordinates mDia1 and actin reorganization during CR3-mediated phagocytosis. J. Cell Biol. 183:1287-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, S. H., C. A. Gutekunst, S. M. Hersch, and X. J. Li. 1998. Interaction of huntingtin-associated protein with dynactin P150Glued. J. Neurosci. 18:1261-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li, X., E. Sapp, A. Valencia, K. B. Kegel, Z. H. Qin, J. Alexander, N. Masso, P. Reeves, J. J. Ritch, S. Zeitlin, N. Aronin, and M. Difiglia. 2008. A function of huntingtin in guanine nucleotide exchange on Rab11. Neuroreport 19:1643-1647. [DOI] [PubMed] [Google Scholar]
- 33.Lobsiger, C. S., and D. W. Cleveland. 2007. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 10:1355-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maxfield, F. R., and T. E. McGraw. 2004. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5:121-132. [DOI] [PubMed] [Google Scholar]
- 35.McGuire, J. R., J. Rong, S. H. Li, and X. J. Li. 2006. Interaction of huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J. Biol. Chem. 281:3552-3559. [DOI] [PubMed] [Google Scholar]
- 36.Pal, A., F. Severin, B. Lommer, A. Shevchenko, and M. Zerial. 2006. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington's disease. J. Cell Biol. 172:605-618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pfeffer, S. R. 2007. Unsolved mysteries in membrane traffic. Annu. Rev. Biochem. 76:629-645. [DOI] [PubMed] [Google Scholar]
- 38.Ren, M., G. Xu, J. Zeng, C. De Lemos-Chiarandini, M. Adesnik, and D. D. Sabatini. 1998. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc. Natl. Acad. Sci. USA 95:6187-6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riggs, B., W. Rothwell, S. Mische, G. R. Hickson, J. Matheson, T. S. Hays, G. W. Gould, and W. Sullivan. 2003. Actin cytoskeleton remodeling during early Drosophila furrow formation requires recycling endosomal components nuclear-fallout and Rab11. J. Cell Biol. 163:143-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seabra, M. C., and C. Wasmeier. 2004. Controlling the location and activation of Rab GTPases. Curr. Opin. Cell Biol. 16:451-457. [DOI] [PubMed] [Google Scholar]
- 41.Seo, H., K. C. Sonntag, and O. Isacson. 2004. Generalized brain and skin proteasome inhibition in Huntington's disease. Ann. Neurol. 56:319-328. [DOI] [PubMed] [Google Scholar]
- 42.Sever, S., H. Damke, and S. L. Schmid. 2000. Dynamin:GTP controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J. Cell Biol. 150:1137-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheff, D. R., E. A. Daro, M. Hull, and I. Mellman. 1999. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J. Cell Biol. 145:123-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szebenyi, G., G. A. Morfini, A. Babcock, M. Gould, K. Selkoe, D. L. Stenoien, M. Young, P. W. Faber, M. E. MacDonald, M. J. McPhaul, and S. T. Brady. 2003. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40:41-52. [DOI] [PubMed] [Google Scholar]
- 45.Trushina, E., R. B. Dyer, J. D. Badger II, D. Ure, L. Eide, D. D. Tran, B. T. Vrieze, V. Legendre-Guillemin, P. S. McPherson, B. S. Mandavilli, B. Van Houten, S. Zeitlin, M. McNiven, R. Aebersold, M. Hayden, J. E. Parisi, E. Seeberg, I. Dragatsis, K. Doyle, A. Bender, C. Chacko, and C. T. McMurray. 2004. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 24:8195-8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trushina, E., R. D. Singh, R. B. Dyer, S. Cao, V. H. Shah, R. G. Parton, R. E. Pagano, and C. T. McMurray. 2006. Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum. Mol. Genet. 15:3578-3591. [DOI] [PubMed] [Google Scholar]
- 47.Ullrich, O., S. Reinsch, S. Urbe, M. Zerial, and R. G. Parton. 1996. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 135:913-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valenza, M., D. Rigamonti, D. Goffredo, C. Zuccato, S. Fenu, L. Jamot, A. Strand, A. Tarditi, B. Woodman, M. Racchi, C. Mariotti, S. Di Donato, A. Corsini, G. Bates, R. Pruss, J. M. Olson, S. Sipione, M. Tartari, and E. Cattaneo. 2005. Dysfunction of the cholesterol biosynthetic pathway in Huntington's disease. J. Neurosci. 25:9932-9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van de Graaf, S. F., Q. Chang, A. R. Mensenkamp, J. G. Hoenderop, and R. J. Bindels. 2006. Direct interaction with Rab11a targets the epithelial Ca2+ channels TRPV5 and TRPV6 to the plasma membrane. Mol. Cell. Biol. 26:303-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varani, K., M. P. Abbracchio, M. Cannella, G. Cislaghi, P. Giallonardo, C. Mariotti, E. Cattabriga, F. Cattabeni, P. A. Borea, F. Squitieri, and E. Cattaneo. 2003. Aberrant A2A receptor function in peripheral blood cells in Huntington's disease. FASEB J. 17:2148-2150. [DOI] [PubMed] [Google Scholar]
- 51.Velier, J., M. Kim, C. Schwarz, T. W. Kim, E. Sapp, K. Chase, N. Aronin, and M. DiFiglia. 1998. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp. Neurol. 152:34-40. [DOI] [PubMed] [Google Scholar]
- 52.Wakabayashi, Y., J. Lippincott-Schwartz, and I. M. Arias. 2004. Intracellular trafficking of bile salt export pump (ABCB11) in polarized hepatic cells: constitutive cycling between the canalicular membrane and rab11-positive endosomes. Mol. Biol. Cell 15:3485-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, Y., Y. Azuma, D. Moore, N. Osheroff, and K. L. Neufeld. 2008. Interaction between tumor suppressor adenomatous polyposis coli and topoisomerase IIalpha: implication for the G2/M transition. Mol. Biol. Cell 19:4076-4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilcke, M., L. Johannes, T. Galli, V. Mayau, B. Goud, and J. Salamero. 2000. Rab11 regulates the compartmentalization of early endosomes required for efficient transport from early endosomes to the trans-Golgi network. J. Cell Biol. 151:1207-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zerial, M., and H. McBride. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2:107-117. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.