

Abstract
Cockayne syndrome and xeroderma pigmentosum–Cockayne syndrome complex are rare autosomal recessive disorders with poorly understood biology. They are characterized by profound postnatal brain and somatic growth failure and by degeneration of multiple tissues resulting in cachexia, dementia, and premature aging. They result in premature death, usually in childhood, exceptionally in adults. This study compares the clinical course and pathology of a man with Cockayne syndrome group A who died at age 31½ years with 15 adequately documented other adults with Cockayne syndrome and 5 with xeroderma pigmentosum–Cockayne syndrome complex. Slowing of head and somatic growth was apparent before age 2 years, mental retardation and slowly progressive spasticity at 4 years, ataxia and hearing loss at 9 years, visual impairment at 14 years, typical Cockayne facies at 17 years, and cachexia and dementia in his twenties, with a retained outgoing personality. He experienced several transient right and left hemipareses and two episodes of status epilepticus following falls. Neuropathology disclosed profound microencephaly, bilateral old subdural hematomas, white-matter atrophy, tigroid leukodystrophy with string vessels, oligodendrocyte proliferation, bizarre reactive astrocytes, multifocal dystrophic calcification that was most marked in the basal ganglia, advanced atherosclerosis, mixed demyelinating and axonal neuropathy, and neurogenic muscular atrophy. Cellular degeneration of the organ of Corti, spiral and vestibular ganglia, and all chambers of the eye was severe. Rarely, and for unexplained reasons, in some patients with Cockayne syndrome the course is slower than usual, resulting in survival into adulthood. The profound dwarfing, failure of brain growth, cachexia, selectivity of tissue degeneration, and poor correlation between genotypes and phenotypes are not understood. Deficient repair of DNA can increase vulnerability to oxidative stress and play a role in the premature aging, but why patients with mutations in xeroderma pigmentosum genes present with the Cockayne syndrome phenotype is still not known.
Cockayne syndrome is an autosomal recessive disorder characterized by profound postnatal decline of somatic and brain growth resulting in dwarfing and limited cognition without affecting sociability.1–3 It is also characterized by cachexia, premature aging, dementia, worsening vision and hearing, cutaneous photosensitivity without propensity to cancer, endocrinopathies, progressive spasticity, ataxia, peripheral neuropathy, weakness, osteopenia, kyphosis, and joint contractures. The facies is distinctive, with sunken eyes and cheeks, decreased tearing and corneal scarring, miotic pupils, a sharp nose, a jutting chin, thin lips, inadequate salivation, and dental caries. Cataracts, pigmentary retinopathy, and optic atrophy contribute to visual loss.4 Neuropathology shows a small cerebrum and cerebellum with enlarged ventricles, cerebral atherosclerosis and microvasculopathy, basal ganglia and cerebellar calcification, so-called “tigroid” leukoencephalopathy, cerebellar atrophy, and a demyelinating peripheral neuropathy.
Among 140 cases Nance and Berry reviewed, the mean age at death in 37 patients was 12 years, of whom only 22% died at 20 years of age or older.3 They assigned patients with Cockayne syndrome to three clinical subtypes. Clinical subtype I corresponds to classic Cockayne syndrome and accounted for about 85% of cases. An extremely severe phenotype was assigned to clinical subtype II and included about 20 children with onset in infancy—sometimes even prenatally—who died in the first few years of life. Subtype II had intrauterine growth failure, congenital cataracts, or other structural eye abnormalities, and infants developed the typical facies and other Cockayne features very early. Clinical subtype III was limited to four patients who presented with mild or atypical phenotypes because of normal intelligence, linear growth, or reproductive capacity.
Genetic studies have defined two Cockayne syndrome complementation groups: complementation group A owing to mutations of the CSA (CKN1, ERCC8) gene on chromosome 5q12,5,6 which accounts for 20% of Cockayne cases,7 and complementation group B, owing to mutations of the CSB (ERCC6) gene on chromosome 10q11,8–10 which accounts for 80% of cases and at least four other clinically distinct disorders as well.11 Exceptionally, mutations of genes for xeroderma pigmentosum (XPB, XPD, and XPG) result in a Cockayne syndrome phenotype (xeroderma pigmentosum–Cockayne syndrome complex),12 although, thus far, only xeroderma pigmentosum B–Cockayne syndrome complex and xeroderma pigmentosum G–Cockayne syndrome complex have been associated with survival to adulthood.13–17 Clinical severity, length of survival, and clinical subtypes map imperfectly onto the Cockayne and xeroderma pigmentosum–Cockayne syndrome complementation groups.
In this report, we summarize the clinical and pathologic features of a 31½-year-old man with clinical subtype I and genetic complementation group A and compare him with 20 adequately described cases of either Cockayne syndrome or xeroderma pigmentosum–Cockayne syndrome complex who lived to 20 years of age or older.
CASE REPORT
This cachectic male, whose weight at death (11.3 kg) was average for a 15-month-old boy, weighed 3.6 kg at birth. Failure to grow was evident by 24 months, when his height fell to the 3rd percentile. His maximum weight was 17.2 kg (50th percentile for age 4 years) at 13 years. The head circumference was at the 3rd percentile at 4 months and below the 3rd percentile by 16 months and reached its maximum, 47 cm (50th percentile for age 12 months), in his teens. He developed facial, pubic, and axillary hair in adolescence but no deepening of the voice or growth of the penis or testes. Serial photographs (Figure 1) show that the Cockayne facies, barely discernible at 9 years, became more obvious at age 17 years. Dr Alan M. Aron diagnosed Cockayne syndrome at 20 years. Easy sunburning was reported at 24 years and freckling and tanning at 30 years. There was early baldness but no graying of the hair or excessive dental decay. He walked unsupported at 15 months. “Irregular gait” at 2 years was followed by spasticity, which was first noted at 42 months and required heel cord lengthening at 7 years; onset of joint contractures occurred at 20 years. Hyperactive tendon stretch reflexes were not supplanted by absent ankle jerks until age 30 years. Intention tremor and staggering were documented at 9 years. There were frequent falls in his twenties; titubation of the head at age 27 years, inability to maintain the trunk and head erect at 30 years, and mild cogwheel rigidity and a resting tremor at 26 years of age. Wheelchair-bound at 25 years, he remained able to stand and to take a few steps with support. Toilet-trained at 4 years, he became incontinent at age 30 years. Although unable to chew after age 30 years, swallowing remained possible.
Figure 1.

Patient at ages 9 (A), 13 (B), 17 (C), and 28 years (D). Note the cheerful expression and that the characteristic Cockayne facies (sunken eyes, sharp nose, jutting chin, emaciation) was not striking, even at age 17 years.
He was an alert, cheerful, interactive person. He developed poorly articulated words at 2 years, spoke simple sentences, and recognized a few written words by midchildhood. He sorted objects in a sheltered workshop until age 27 years. A high-tone hearing loss, documented at age 9 years, progressed to total deafness at age 25 years and mutism at age 28 years. Optic atrophy was described at age 14 years, retinitis pigmentosa at age 17 years, small unreactive pupils at age 20 years, and corneal and lens opacities at age 23 years. He underwent unilateral tarsorrhaphy at age 24 years for corneal ulcers. Vision became limited to light perception at age 28 years, and he was blind a year later.
In his early twenties, he had several unexplained 15-minute episodes of left or right hemiparesis and experienced several brief seizures, some related to falls. In his midtwenties, he suffered two fall-triggered episodes of status epilepticus lasting several hours, followed by severe right or left hemiparesis and impaired consciousness, with slow recovery over several months. Computed tomography at age 27 years revealed marked ventricular dilatation, profound thinning of the white matter, severe enlargement of the ventricles, cerebral and cerebellar atrophy, and calcification of the basal ganglia, without focal lucency or atrophy suggestive of an ischemic stroke (Figure 2). None of several interictal electroencephalograms (EEGs) were epileptiform, but all showed diffuse and focal slowing. Twice, attempts at lumbar punctures yielded no fluid. Arterial hypertension occurred only in the context of fever or status epilepticus. Tests for lipid profile, renal function, antithrombin III, proteins C and S, factor V Leiden, antiphospholipid antibodies, anticardiolipin antibodies, antinuclear antibodies, and anti-neutrophil cytoplasmic antibodies were normal.
Figure 2.

Computed tomographic scans at age 25 years showing (A) thickening of the calvarium, severe panventricular enlargement, and calcification of the basal ganglia and (B) extreme thinning of the white matter, widened sulci, and thin bilateral subdural collections.
Molecular studies assigned him to the Cockayne syndrome type A complementation group (courtesy of Dr W. Vermeulen, Rotterdam). Polymerase chain reaction–restriction fragment length polymorphism analysis of DNA isolated from muscle was negative for point mutations in mitochondrial DNA at positions 3243, 8344, and 8993. Southern blot analysis showed no deletion in mitochondrial DNA. Mitochondrial enzyme activity of several respiratory chain complexes in muscle was nonspecifically mildly to moderately decreased, probably related to longstanding muscle atrophy.
PATHOLOGY
Methods
Tissues were fixed in formalin and processed using histologic and immunohistochemical methods. The peripheral nerve was examined with electron microscopy and nerve teasing. Frozen sections of skeletal muscle were stained using histochemical methods and compared with the muscle biopsy stained with the same methods when the patient was 27 years old. The eyes and temporal bones were examined using methods in subspecialty laboratories.18
General Autopsy
External examination revealed a small, bald male, 109 cm in length, weighing 11.3 kg, with a senile appearance, atrophic musculature, hyperostosis frontalis, and sunken eyes. The thickness of the panniculus at the umbilicus was markedly thinned at 0.1 cm. The genitalia were poorly developed. The viscera were small but without histologic abnormality. There was mild atherosclerosis in the aorta and coronary arteries and patchy bronchopneumonia. Transillumination of the eyes showed centrally located pinpoint pupils and numerous iris transillumination defects. There was posterior lentiglobus, a salt and pepper fundus, and optic atrophy.
Neuropathology
The calculated total fixed brain weight was 430 g (expected brain weight for a 31-year-old male: 1440 ± 20 g).19 There were bilateral chronic subdural hematoma membranes. The cerebral hemispheres had a normal configuration, without sulcal widening; the leptomeninges were diffusely opacified and fibrotic. The circle of Willis had patchy atherosclerosis (Figure 3A). The cortical ribbon was diffusely thin (2 mm or less in thickness) (Figure 3B). The cerebral white matter was markedly attenuated, and the ventricular system was diffusely dilated (see Figure 3B). The corpus callosum, optic nerves and chiasm, anterior commissure, and fornix were thin. The basal ganglia were atrophic, with brown discoloration of the globus pallidus and the pars reticularis of the substantia nigra. The pars compacta of the substantia nigra and the locus ceruleus had normal neuromelanin pigmentation. Both the vermis and lateral hemispheres of the cerebellum were extremely small (Figure 3C). The dentate nucleus had brown discoloration. The pons and medulla were unremarkable. There was discoloration of the spinal white matter. Minute petechial hemorrhages were scattered throughout the white matter of the cerebral hemispheres, brain stem, cerebellum, and spinal cord.
Figure 3.

A, Artery of the circle of Willis with 60% occlusive atherosclerosis of the lumen (L) (hematoxylin-eosin stain; original magnification, ×88). B, Coronal section of the cerebrum that parallels the earlier findings on computed tomography. C, Whole-mount section of the atrophic cerebellar vermis (hematoxylin-eosin stain).
The white matter of the centrum semiovale, optic nerves and tracts, major commissures, cerebellum, and spinal cord was markedly hypercellular (Figure 4A). The dominant cell type was consistent with proliferating oligodendrocytes with prominent vesicular nuclei, unusual, coarsely clumped chromatin, and scant cytoplasm (Figure 4B). There was also a reactive astrocytosis; the astrocytes had markedly enlarged atypical nuclei and copious cytoplasm. The Luxol fast blue stain showed extensive loss of myelin (see Figure 4A), whereas the Bodian method disclosed only partial loss of axons. The few preserved myelinated fibers stained strongly for myelin basic protein. The white-matter tracts of the brain stem, including the pontine white matter, were largely spared; pallor of the middle and lateral thirds of the cerebral peduncles was noted.
Figure 4.

A, Low-power view of white matter stained with Luxol fast blue–periodic acid–Schiff shows hypercellularity and is pale blue, indicative of demyelination (original magnification, ×347). B, High-power view of white matter stained with Luxol fast blue–periodic acid–Schiff shows pleomorphic oligodendrocytes (arrows) (original magnification, ×1375). C, Calcification (arrows) in the basal ganglia (hematoxylin-eosin stain; original magnification, ×602). D, Collagen IV immunostain demonstrates string vessels (arrow) (original magnification, ×400). E, Skeletal muscle showing small group atrophy (arrows) consistent with neurogenic atrophy (hematoxylin-eosin stain; original magnification, ×641). F, Toluidine blue–stained 1 micron–thick section of skeletal muscle showing marked nuclear atypia (arrows) (original magnification, ×1280).
The neocortex was thin but had a preserved architecture; a few neurons in the lower cortical layers were aberrantly positive for phosphorylated neurofilaments. There was marked neuronal loss and gliosis in CA1 of the hippocampus but not in the remaining sectors. No neurofibrillary tangles or Hirano bodies were detected. A few neurons in all sectors of the hippocampus, as well as the subiculum and entorhinal cortex, had phosphorylated neurofilament immunoreactivity. The lateral geniculate body showed neuronal pyknosis consistent with transneuronal degeneration. Calcification was present in the arteries, arterioles, and capillaries and in parenchymal calcospherites of the putamen, globus pallidus, thalamus, and deep cerebellar white matter (Figure 4C). There was marked pigment-spheroid degeneration in the globus pallidus, substantia nigra, caudate nucleus, and dentate nucleus of the cerebellum. The pigment was consistent with iron pigment by Prussian blue stain. The spheroids were swollen axons that were positive for ubiquitin. The substantia nigra was normocellular, with no neurofibrillary tangles or Lewy bodies. The ventral and dorsal cochlear nuclei were extremely small, with neuronal pyknosis. Fascicles of cranial nerves VII and VIII in the cerebellopontine angle were thin and fibrotic. The pyramidal tract was unremarkable. A few microglial nodules were present in the brainstem tegmentum. There was extensive loss of internal granular cells and Purkinje cells in both the cerebellar vermis and hemispheres, with proliferation and atypia of the Bergmann glia and of astrocytes in the molecular layer. The dentate nucleus had microvacuolation and gliosis. There was neuronal loss, chromatolysis, and gliosis of the anterior horns in the sacral cord, but this process was much less severe at the thoracic levels. The spinal roots showed fibrosis with loss of myelinated axons.
Collagen IV immunostaining of the cerebral and cerebellar white matter revealed obliterated microvessels consistent with so-called “string vessels” (Figure 4D).20 Counterstaining of collagen IV–immunostained sections with Luxol fast blue for myelin showed the prevalence of string vessels in demyelinated patches with normal-appearing microvessels in better myelinated areas. Occasional string vessels were present in the neocortex, diencephalon, and brain stem. There was a rough correlation between neuronal loss, gliosis, calcification, and the presence of string vessels in the putamen, caudate nucleus, cerebellum, and substantia nigra. The leptomeninges were thick and fibrotic, and the ependymal lining showed granular ependymitis. Bilateral chronic subdural hematomas showed recent rebleeding. Minute foci of perivascular hemorrhage with loss of myelin and axonal preservation were scattered throughout the neuraxis, consistent with acute hemorrhagic leukoencephalopathy of Weston Hurst, apparently a terminal event.
Peripheral nerves exhibited both axonal pathology and demyelination with remyelination. The dense inclusions previously described in Cockayne syndrome21 were not observed.
The biopsy and autopsy specimens of skeletal muscle revealed neurogenic atrophy without myopathy or ragged red fibers (Figure 4E). The muscle nuclei were markedly pleomorphic and enlarged (Figure 4F). Mitochondrial DNA and histochemical studies were unremarkable.
There was multifocal absence of Bowman’s layer of the cornea. The chamber angle was open, but Schlemm’s canal was absent. The anterior iridic border and iris dilator muscle were atrophic. There was a mild lymphocytic infiltration of the iris stroma and ciliary body. The iris root contained numerous clumps of Koganei cells (melanophages). The ciliary processes were maldeveloped, short, and stubby. There were cataractous changes in the lens. There was patchy loss of melanin pigment granules of the retinal pigment epithelium, as well as areas of retinal pigment epithelial atrophy and hyperplasia (Figure 5). There was dropout of the ganglion cell layer of the retina, with atrophy and gliosis of the optic nerve.
Figure 5.

A, Low-power view of the anterior chamber of the eye demonstrating iris atrophy, a mioticpupil, and an abnormally flattened anterior surface of the cataractous lens (hematoxylin-eosinstain; original magnification, × 4); inset: higher-power view of the pupil showing a pigmented, fibrous pupillary membrane (arrow) and posterior synechiae to the lens (original magnification, ×40). B, Sclerosis of the ciliary body and ciliary processes (asterisks) (hematoxylin-eosin stain; original magnification, ×16); inset: pigmentary dispersion of the iris with melanophages studding the anterior iridic border (arrows) and absence of the iris dilator muscle (arrowhead) (hematoxylin-eosin stain, original magnification, ×63). C, Marked atrophy of the retina with wrinkling of the inner limiting membrane (asterisk), intraretinal pigment migration (arrow) and atrophy, and hyperplasia of the underlying retinal pigment epithelium (arrowheads) (hematoxylin-eosin stain; original magnification, × 160). D, Atrophy and gliosis of the optic nerve (hematoxylin-eosin stain; original magnification, × 12.5).
The most striking finding in the membranous labyrinth was severe and diffuse degeneration of the pars inferior (cochlea and saccule), including the hair cells, spiral ganglion cells, stria vascularis, and other supporting structures (Figure 6), without evidence of vascular degeneration. There was collapse of the endolymphatic duct of the pars inferior but not of the pars superior. There was severe loss of hair cells in the pars superior (utricle and semicircular canals) that affected all three cristae, with a few hair cells still present in the utricle. Neurons in Scarpa’s (vestibular) ganglion were markedly depleted, with loss of their peripheral dendritic processes. Dark cells of the vestibular labyrinth were still present. There was proliferation of connective tissue and neo-osteogenesis in the lateral semicircular canals. Minor developmental anomalies of the inner and middle ears included a scala communis between the apical and middle cochlear turns, a widened protympanum, thickened stapes crurae, and mesenchyme in the anterior epitympanum and round window niche. Cells of the geniculate ganglion appeared healthy and were present in appropriate numbers.
Figure 6.

Midmodiolar section of the cochlea. All cochlear turns show diffuse and severe degeneration of sensory, neural, and supporting structures, including loss of the organ of Corti, severe degeneration of the stria vascularis and spiral ligament, near-total loss of cochlear neurons (spiral ganglion cells), and collapse of the endolymphatic space (scala media). There is also severe atrophy of the saccular macula (hematoxylin-eosin stain; original magnification, ×19).
DISCUSSION
Comparison of This Case With Others in the Literature
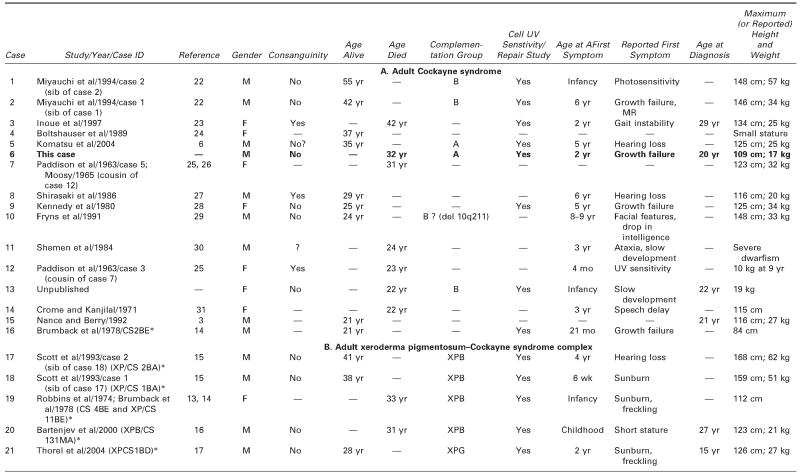
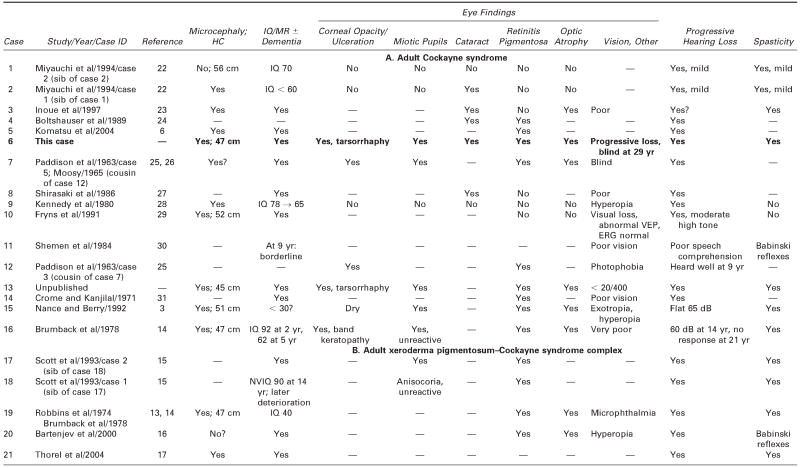
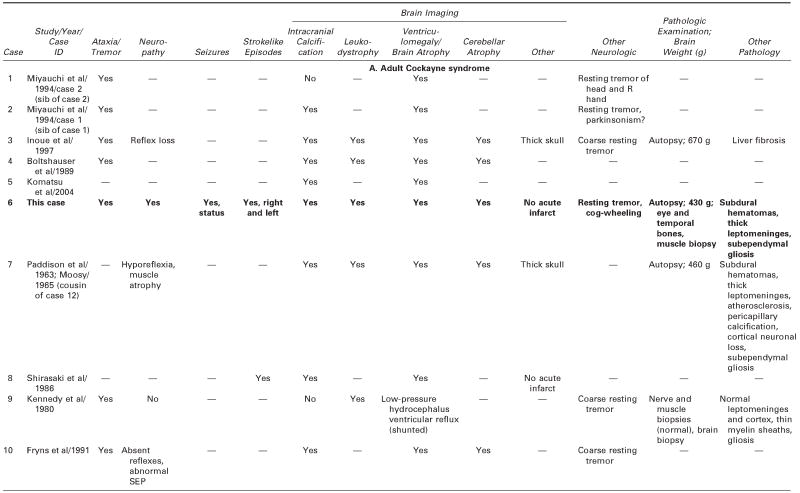
We reviewed all available clinical and pathologic files on the patient and correspondence with his parents. We searched the literature using the terms Cockayne syndrome, xeroderma pigmentosum, cachexia, premature aging, and DNA repair. We used information acquired at the National Institutes of Health–sponsored conference of May 2004: “Cockayne Syndrome and Related Disorders of DNA Repair and Transcription: From Bench to Bedside and Back.” This patient (case 6 in Tables 1 and 2) had all the classic symptoms of Cockayne syndrome clinical subtype I, but his course was slower and survival longer than typical in Cockayne syndrome. Our MEDLINE search yielded descriptions of only 20 other individuals with Cockayne syndrome clinical subtypes I or III who survived to age 20 years or beyond,3 15 individuals with Cockayne syndrome,3,6,14,22–31 and 5 individuals with xeroderma pigmentosum B–Cockayne syndrome complex.13–17 We did not include a 25-year-old man on whom only ophthalmologic information was provided,32 a doubtful case in a 41-year-old woman in whom questionable Cockayne syndrome was associated with neurofibromatosis,33 and one case described in Japanese.34 This small number is surprising because Nance and Berry reported that 8 (22%) of 37 cases died at 20 years or above,3 and the Cockayne syndrome parent newsletter (Share and Care Cockayne Syndrome Network) of December 2000 lists, in addition to 15 children with Cockayne syndrome whose mean age at death was 8½years (range 30 months to 15½years), 2 adults (13%) who died at ages 25½ and 26 years. The spotty clinical, pathologic, and genetic data on the 20 published cases and ours are summarized in Tables 1 and 2.3,6,13–17,22–31 We review available information to assist further clinical-genetic correlation.
Table 1.
Adult Cockayne Syndrome and Xeroderma Pigmentosum–Cockayne Syndrome Complex: Medical Characteristics
 |
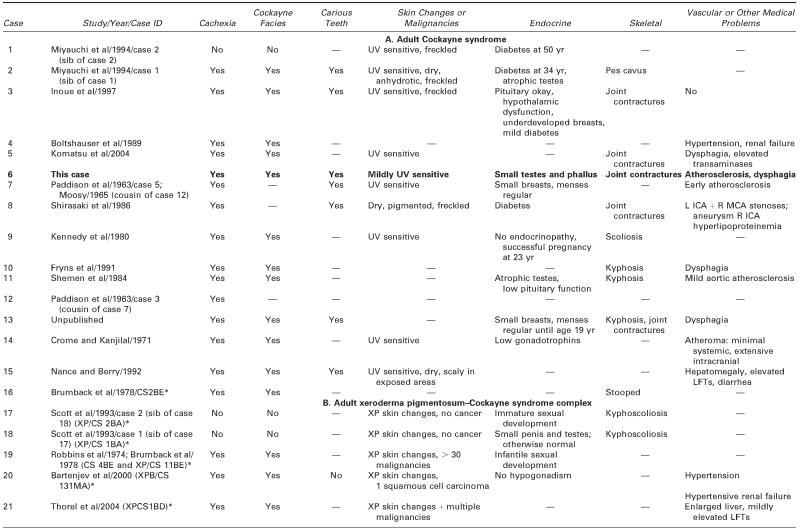 |
— = no information/does not apply; CS = Cockayne syndrome; ICA = internal carotic artery; LFTs = liver function tests; MCA = middle cerebral artery; MR = mental retardation; UV = ultraviolet; XP = xeroderma pigmentosum.
Identifying label for this case in the quoted paper and NIH registry.
Table 2.
Adult Cockayne Syndrome and Xeroderma Pigmentosum–Cockayne Syndrome Complex: Neurologic Findings, Vision, Hearing, and Pathology
 |
 |
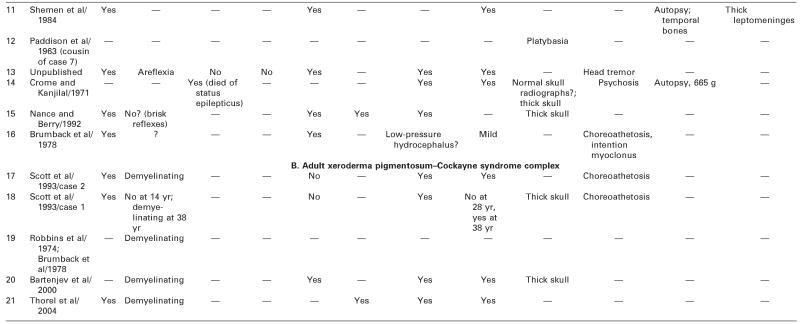 |
— = no information; ERG = electroretinogram; HC = head circumference; MR = mental retardation; NVIQ = nonverbal IQ; SEP =sensory-evoked potentials; VEP = visual-evoked potentials.
Genetics
Both genetic and phenotypic heterogeneity is notorious and poorly understood in the disorders of DNA repair.12,35 There is no clear parallelism between the Nance and Berry clinical subtypes I, II, and III3 and the Cockayne syndrome A and B complementation groups. A number of different locations and types of mutations in both CSA5,6,36 and CSB10,11,37 result in variably defective or absent gene products. Genotype-phenotype correlation is particularly poor in the case of CSB, which suggests that the phenotypes reflect the interaction of other genetic or environmental factors.38 Mutations of CSB are responsible for the great majority of classic Cockayne syndrome cases, including the two mildly affected, longest survivors (cases 1 and 2)22 and, presumably, case 10, who had a deletion in chromosome 10q11-q21.2 that encompassed the CSB gene locus.29
Mutations of CSB are associated with four other clinical disorders, the first two of which are characterized by very severe phenotypes and early death. The first is infantile Cockayne syndrome subtype II, which is symptomatic at birth and associated with serious eye abnormalities.4 The second is the clinically distinct cerebro-oculofacial-skeletal syndrome,39,40 which shares many features with severe subtype II Cockayne syndrome. The third is a disorder in one kindred41 resembling the severe DeSanctis-Cacchione variant of xeroderma pigmentosum neurologic disease.42,43 The fourth clinical disorder is a very mild disorder, UVsS, in which affected individuals have skin ultraviolet sensitivity and freckling, without proneness to skin cancers or the Cockayne syndrome phenotype.44
The clinical expression of XPB and XPG mutations is also heterogeneous. Besides xeroderma pigmentosum B cutaneous disease and xeroderma pigmentosum B-Cockayne syndrome complex phenotypes, other mutations of XPB produce some cases of the trichothiodystrophy phenotype.35 The mild or severe phenotypes that arise from different mutations of XPG were recently shown to depend on the amounts of XPG protein produced.17
Growth Failure and Cachexia
Dwarfism and cachexia are defining features of Cockayne syndrome and are often its presenting signs. Except for the severe infantile variants, growth is normal in utero because neonates with Cockayne syndrome, like case 6, are born with average or near-average lengths, weights, and head circumferences. Postnatal growth failure includes the brain. Microcephaly was mentioned in six cases, and in six others, the head circumference varied from mildly (52 cm) to extremely (47 cm) small. The brain weight was 50% or less of expectation in four autopsied cases. Only two cases (1 and 20)16,22 were not microcephalic (see Table 2). Normal growth in utero, followed by early postnatal growth failure without gross distortion of brain structure, suggests the possibility of a loss of a transplacental growth factor, but there is also clear evidence of progressive degeneration as measured by later severe atrophy. Whether widely postulated inadequate repair or enhanced susceptibility to oxidative DNA damage provides an adequate explanation for progressive brain atrophy remains to be determined. Given the brunt of the pathology in white matter, it suggests that brain atrophy might be largely due to leukoencephalopathy.
Somatic growth failure can be evident before age 1 year and is usually severe in typical Cockayne syndrome. The degree of dwarfing in adults is more variable, ranging from extreme (less than 135 cm [average for a 9½-year-old boy, range 84–134 cm] in 71% of cases) to mild, with short-average stature (135–168 cm) in cases 1 and 222 and 17 and 1815 (see Table 1). The cause of dwarfing in Cockayne syndrome is not clear. Nance and Berry reported variable levels of growth hormone in a few patients and a lack of response to growth hormone therapy in one case.3 Growth hormone levels were normal in cases 323 and 9.28
Cachexia is dramatic and progressive in typical Cockayne syndrome. It is usually obvious by the preschool years. Together with an intercurrent illness, it is the main cause of childhood death. In case 6, weight peaked at age 13 years, when it reached 17 kg, the size of an average 4 year old, and was down to 11 kg (average for age 15 months) at death. Weight was below 35 kg (average for an 11-year-old boy) in 80% of the adult cases with data (see Table 1). Although cachexia is a feature of aging, it occurs so early and is so profound in Cockayne syndrome as to suggest different pathophysiology from the cachexia of advanced aging.
The characteristic wizened Cockayne syndrome facies was not apparent until adolescence in cases 6 (see Figure 1), 11,30 and 15,3 and it apparently did not develop at all in cases 1,22 17,15 and 18,15 which helps explain late diagnosis in long-term survivors.
Premature Aging and Cardiovascular Disease
Premature aging, with its associated diseases, such as type 2 diabetes mellitus, cardiovascular disease, osteopenia, kyphosis, hair thinning, and skin atrophy, is another unexplained hallmark of Cockayne syndrome. Attributing the stooped posture or kyphoscoliosis mentioned in cases 10,29 11,30 13, 16,14 17,15 and 1815 and documented in many children with Cockayne syndrome to the osteopenia of premature aging begs the question of its pathophysiology. Egly (quoted in Bohr et al35) reported that mutation of XPD indirectly lowers the expression of reporter genes regulated by vitamin D, retinoic acid, and estrogen. It is also not clear whether the prominent joint contractures in cases 3,23 5,6 6, and 13 were caused by spasticity, neuropathic weakness, limited mobility, osteoporosis with degenerative changes in the spine and articulations, or all of them. Cases 56 and 6 were bald in their early thirties, and thinning of the hair is mentioned in many of the reports.
Early hypertension and atherosclerosis are other features of premature aging in Cockayne syndrome. Cases 424 and 2016 died of hypertensive nephrosclerosis, which, in case 20,16 developed in his late twenties. Hypertensive nephrosclerosis can occur much earlier, as in two brothers aged 14 and 15 years and a 4-year-old girl.45 Aortic atherosclerosis was found in case 6 and was also mentioned in cases 1130 and 1431 and in the 14-year-old boy just mentioned.45 Systemic atherosclerosis is not universal because it was not present at age 40 years in case 3.23
Cases 6, 7,25,26 and 14,31 but not 3,23 had premature cerebral atherosclerosis and arteriolosclerosis at autopsy, which can result in strokes. This was even reported in some children with Cockayne syndrome.46 Case 9,28 who had several risk factors for stroke (eg, diabetes and hyperlipidemia) and in whom cerebral angiography disclosed stenosis of the C1 and C2 portions of the left internal carotid artery, experienced at age 29 years a sudden right hemiparesis and dysarthria that receded 4 days after fibrinolytic therapy. Several episodes of transient hemiparesis in case 6 were not investigated; they might have followed unobserved seizures or been transient ischemic attacks. We investigated possible mitochondrial dysfunction that might have caused transient strokelike episodes akin to mitochondrial encephalopathy with lactic acidosis and strokelike episodes (MELAS),47 but this was uninformative. Explaining the two episodes of severe and lasting hemiparesis with impaired consciousness after mild falls is difficult. They followed prolonged status epilepticus but were too long-lasting (ie, 2 weeks) for transient postictal deficits. Subdural membranes noted at autopsy in case 6 might have been due to falls and relatively minor head trauma in a person with atrophy and fragile vessels. Neither falls nor epilepsy is mentioned in case 7,25,26 who also had subdural hematomas. Subdural hematomas without an underlying brain contusion are not typically associated with either prolonged coma and hemiparesis or seizures. Therefore, the cause of the episodes of prolonged hemiparesis in case 6 is uncertain.
Endocrine Status
The prevalence of diabetes mellitus can be increased in Cockayne syndrome, and it was reported in four Japanese adults (cases 1 and 2,22 3,23 and 827). Thorough endocrine investigation in case 323 indicated that pituitary function, including growth hormone levels and response, was normal. It was suggested that the basis of endocrine disturbance, in particular the ubiquitous hypogonadism, might be hypothalamic. Most individuals with Cockayne syndrome develop secondary sexual characteristics, including case 6, who had to be shaved and had pubic and axillary hair. He, like almost one third of male individuals with Cockayne syndrome,3 had underdeveloped testes and a small phallus, and it is unknown whether these men are fertile. The breasts of adolescents and women are often described as underdeveloped, with relatively large nipples (eg, Cockayne,2 Nance and Berry,3 and Soffer et al48; cases 725,26 and 13). Sexual development in case 19 with xeroderma pigmentosum B–Cockayne syndrome complex is described as infantile,13,14 and the ovaries of case 323 were atrophic, yet she and other women with Cockayne syndrome menstruated despite their emaciation. Surprisingly, case 9, who at 19 years had reached her maximal height of 125 cm and weight of 34 kg (both at the 50th percentile for age 10 years), carried a successful pregnancy at age 23.28 Another woman with mild Cockayne syndrome had two pregnancies terminated by cesarean sections; the first at age 17 years, with delivery at 23 weeks, was followed by neonatal death, and the second at age 18 years, carried to 34 weeks, resulted in the birth of a normal child.49
Cutaneous Abnormalities and Carcinogenesis
The hallmark of both Cockayne syndrome and xeroderma pigmentosum is hypersensitivity to killing by ultraviolet light in cell culture, but the disorders differ in that unscheduled DNA synthesis after ultraviolet exposure is reduced in xeroderma pigmentosum and normal in Cockayne syndrome. Recovery of ribonucleic acid (RNA) synthesis is severely reduced in xeroderma pigmentosum but is only mildly reduced in Cockayne syndrome.50–52 Both diseases are associated with an increased risk of sunburn and actinic skin atrophy. Although severe freckling and a 1000-fold increased risk of cancer in ultraviolet light-exposed tissues (ie, skin and eyes) are features of xeroderma pigmentosum and some cases of xeroderma pigmentosum–Cockayne syndrome complex, it is not of Cockayne syndrome, including the present group of long-term survivors. Among the five cases with xeroderma pigmentosum B–Cockayne syndrome complex, all of whom had classic xeroderma pigmentosum skin changes, neither of the brothers (cases 17 and 18) had developed cancer at ages 41 and 38 years.15 Case 1913,14 had multiple skin malignancies starting at age 18 years, and case 2016 had one squamous cell carcinoma on his eyelid. Both died of cardiovascular disease in their thirties.
Neurologic Abnormalities
Cockayne syndrome is characterized by both mental retardation—attributable to developmental microcephaly—and later-developing dementia associated with progressive loss of brain substance. The severity of cognitive impairment varies in Cockayne syndrome. Nance and Berry drew attention to rare patients without severe cognitive impairment who had mild or incomplete phenotypes, later onsets, and more benign and protracted courses.3 Lanning and Similä described a 7-year-old boy of normal intelligence,53 and Czeizel and Marchalko described a mildly affected subtype III teenage girl with an IQ of 128.54 She had the slender nose, sunken eyes, corneal infiltrates, hearing loss, sensitivity to sunlight, and low normal stature (158 cm) despite cachexia that started at age 3 years and resulted in a maximum weight of 30 kg.
Table 2 shows that essentially all of the adults had severe or progressively severe cognitive deficits. The age at onset of dementia is variable. Case 9, with an IQ of 78, graduated from high school and attended junior college despite dysarthria, bilateral sensorineural hearing loss, a demyelinating peripheral neuropathy, and progressively severe ataxia and tremor.28 Her IQ had declined to 65 by age 25 years, when she was shunted for presumed low-pressure hydrocephalus. Intellectual and physical decline was very slow in the oldest adults with Cockayne syndrome identified in this survey. Cases 1 and 2, Japanese brothers aged 42 and 55 years, had IQs of 70 and 60, respectively.22 Preserved personality in the face of such severe curtailment of brain growth and ongoing degeneration is a striking feature of this disease that begs for an explanation.
All adults with Cockayne syndrome and adequate clinical information had the characteristic motor abnormalities of childhood Cockayne syndrome, including spasticity, ataxia, and peripheral neuropathy. Demyelinating or mixed peripheral neuropathy was documented in cases 6 and 1029 and the five cases of xeroderma pigmentosum B–Cockayne syndrome complex,13–17 and it was presumably present in case 323 who lost their reflexes or developed muscle atrophy. The age at symptomatic onset is variable. Case 18 had no sign of neuropathy at 14 years of age but did at 38 years.15 Case 15 had brisk reflexes at 21 years,3 and nerve conduction velocity, electromyography, and muscle biopsy were normal in case 9 at age 25 years.28 The progressive leukodystrophy is the likely neuropathologic correlate of spasticity and gait and limb ataxia.
Surprisingly, in the face of sometimes striking basal ganglia calcification, Nance and Berry described movement disorders in only 4 (3%) of 140 cases reviewed.3 These might be more frequent among adults, with evidence of movement disorders in 9 of 21 adult cases. The two brothers with xeroderma pigmentosum B–Cockayne syndrome complex15 and case 1614 were stated to have choreoathetosis. A resting tremor suggestive of parkinsonism was reported in six cases (6 and 13, 1 and 2,22 9,28 and 1029).
Nance and Berry reported epilepsy in only 5% to 10% of their 140 cases, and it was infrequent in the adults.3 Case 6 might have had brief undocumented seizures without interictal epileptiform EEGs, as well as the two bouts of status epilepticus just discussed. Status epilepticus was not unique to him because case 14 reportedly died of status epilepticus.31
Cockayne syndrome is characterized by both failure of brain development and loss of brain volume attributable in part to progressive leukoencephalopathy. Failure of brain growth explains the worsening early microcephaly, reported in all but 1 (case 122) of the 12 individuals with data (see Table 2), but not the ubiquitously enlarged ventricles (hydrocephalus ex vacuo), severe cerebellar atrophy, or thickened skull. Besides atrophy, normal-pressure hydrocephalus was diagnosed in three cases (cases 9,28 16,14 and 1913,14), as well as in two children.14 It was attributable to leptomeningeal fibrosis and granular ependymitis impeding the flow of cerebrospinal fluid (pathologic findings documented in cases 6, 7,25,26 and 1130) as well as in several childhood autopsies (eg, Rowlatt55 and Leech et al56). These patients had accelerating cognitive decline, gait instability and festination, and urinary incontinence, and a 10-year-old girl had four unexplained episodes of headache, vomiting, and hallucinations.14 Those who were imaged had panventricular dilatation without sulcal widening. Cisternography documented ventricular radionuclide reflux and stagnation consistent with low-pressure hydrocephalus. Case 9 was shunted, with alleged stabilization of her deficits.28 Proof of this diagnosis in patients who have clearly progressive brain atrophy requires further documentation.
Vision and Ocular Findings
All of the adults with Cockayne syndrome and xeroderma pigmentosum–Cockayne syndrome complex, even those mildly affected, had abnormal eye findings, but descriptions of the ocular pathology in Cockayne syndrome are sparse.4,32,57,58 Clinically, small pupils and corneal ulceration or clouding owing to impaired lacrimation are common. Cataracts, retinal degeneration, and optic atrophy are also frequent, although not present in mildly affected cases 1 and 222 and 10.29 Calcific band keratopathy and corneal inflammation can result from corneal exposure,3 which required tarsorrhaphy in cases 6 and 13. The iris transillumination defect observed in case 6 correlates with pigmentary dispersion of the anterior chamber. The miotic pupils—lacking in case 2016—are due to the absence of the iris dilator muscle and the presence of a pupillary membrane. In older patients with Cockayne syndrome, intraretinal pigment migration is found to be consistent with a more advanced stage of retinal dystrophy.3 The posterior lentiglobus in the present case has not been described in other reports of Cockayne syndrome.
Hearing and Vestibular System Abnormalities
Although progressive sensorineural hearing loss of variable severity, reported in essentially every case in this series, is a defining feature of Cockayne syndrome, we identified only two other patients beside case 6 in whom the temporal bones had been examined: case 11, a 24-year-old man,30 and a 17-year-old adolescent.59 In neither of these two cases were audiometric data available. In case 11, there was loss of outer and inner hair cells and loss of spiral ganglion neurons limited to the basal turn of the cochlea but no other abnormalities of the temporal bones.30 Degeneration of the basal turn of the cochlea indicates high-tone hearing loss, which was reported in both case 1130 and his affected younger sisters. Case 6 started out with a symmetric, bilateral high-tone loss that eventually progressed to profound deafness. In the adolescent, there was complete absence of the sensory epithelium of the organ of Corti, marked attenuation or even absence of the stria vascularis, and severe depletion of spiral ganglion neurons, similar to the findings in case 6.59 The vestibular maculae and cristae were atrophic in both cases, making it likely that vestibular involvement might have contributed to ataxia and falls. Quantitative study in the adolescent revealed transneuronal degeneration of the ventral and dorsal cochlear nuclei with pyknotic neurons,59 findings also noted in case 6. Degeneration did not affect higher relays of the auditory system in the 17-year-old patient because the size of neurons approached norms in the medial geniculate and primary auditory cortex.
The greater vulnerability of the pars inferior (cochlea and saccule), which is the phylogenetically newer part of the inner ear, compared with the pars superior (utricle and semicircular canals), is provocative and is similar to that seen in some other developmental disorders, such as Scheibe dysplasia.18 The cellular and molecular basis for this selective vulnerability remains unknown.
Neuropathology
The main neuropathologic features of cases 3,23 6, 7,25,26 9,28 and 1431 were severe loss of both central and peripheral myelin, cerebellar atrophy, and basal ganglia calcification without evidence of gross brain malformation or major architectural alteration. The basis for thinning of the neocortex remains to be investigated. Cerebellar atrophy is very severe in typical Cockayne syndrome, in which there is neuronal loss in all cell layers, astrocytic and microglial proliferation, and severe distortion of Purkinje cell apical dendrites.48 Kohji et al observed TUNEL (terminal dUTP nick end labeling)-positive granular neurons of two of four children and adolescents with Cockayne syndrome, suggesting that apoptosis can contribute to the severe neuronal loss.60
Reasons for the selective vulnerability of oligodendroglia and Schwann cells resulting in inadequate myelin maintenance and loss of central and peripheral myelin—and perhaps dysmyelination, as suggested by a brain biopsy in case 928—are not understood. Classically, myelin is stated, for unknown reasons, to be relatively spared around blood vessels, which has suggested the name “tigroid leukodystrophy.” An intriguing possibility is that some of the white-matter pathology in Cockayne syndrome might be related to microvascular pathology because better myelinated patches had preserved microvessels, whereas areas with demyelination had increased numbers of string vessels. The latter are obliterated microvessels that have been postulated to contribute to the frequent white-matter pathology detected in the aged brain.61 Further studies of microvascular pathology in Cockayne syndrome are warranted, although this is a feature of this disorder, along with meningeal fibrosis, dystrophic calcification of the basal ganglia, and cerebral arteriosclerosis, that is common in the elderly62 and that leads to consideration of Cockayne syndrome as a progeria.63
Pathophysiology
Contemporary research on material from patients, mouse models, and tissue culture is starting to elucidate the cellular roles of CSA, CSB, XPB, XPG, proteins and other xeroderma pigmentosum gene products in the pathophysiology of the complex disorders of DNA repair.17,35,38,64,65 Cockayne syndrome and xeroderma pigmentosum cells are characterized by markedly increased sensitivity to killing by ultraviolet radiation and defective excision of damaged bases in the strands of DNA that are actively being transcribed into RNA (transcription-coupled repair). Defective DNA repair stalls RNA polymerase transcription, explaining why Cockayne syndrome and xeroderma pigmentosum cells fail to recover normally after ultraviolet irradiation or other causes of transcription block.
Gene repair is dependent on 30 or more proteins. XPB, like XPD, is a helicase that unwinds the damaged DNA for excision and repair, whereas XPG is an endonuclease that cleaves the damaged DNA in preparation for repair. XPB is at the core of the transcription factor IIH (TFIIH) complex, which is involved in two critical pathways: DNA repair and RNA polymerase II transcription. CSB, XPG, and presumably the other proteins responsible for the disorders of DNA repair have other biologic roles as well. These other functions are beginning to help explain the complex phenotypes in Cockayne syndrome. Different XPG mutations or deletions result in different amounts of XPG protein produced, which, in turn, correlates with longevity.17 Much remains to be elucidated before the pathophysiology of phenotypic variability and specific tissue vulnerability are understood.
CSA and CSB proteins are not components of the same complex; their interaction is not strong; and they do not colocalize in nuclear matrix preparations. Their role is to assist RNA polymerase III in dealing with transcription blocks caused by DNA damage and then in recruiting the nuclear excision proteins to remove the damage. After ultraviolet irradiation, the CSA complex associates with RNA polymerase II and thus influences one of the pathways for transcription-coupled repair, the other of which is mediated by CSB.
Although CSA is translocated to the nuclear matrix after damage in a CSB-dependent manner, CSB is not a damage-recognition factor on its own.37 CSB stimulates global genome base excision repair indirectly and prevents ultraviolet-induced apoptosis through its pivotal role in DNA repair. It also removes a key player in transcription, RNA polymerase II, by ubiquitination and proteasomal degradation and can stimulate adenosine triphosphatase activity by binding to DNA.37 Thus, the roles of these proteins are not limited to DNA repair. CSB interacts with many other genetically controlled proteins, which might explain the extreme variability of its phenotypes and which supports the contention that they are polygenically influenced.
Defective CSA and CSB gene products in DNA transcription and repair and defective genome maintenance might be responsible for premature aging and atherosclerosis.66 Increased sensitivity to ultraviolet light explains why the skin, hair, and eyes of patients with Cockayne syndrome and xeroderma pigmentosum are vulnerable to sun exposure. Unlike xeroderma pigmentosum, however, Cockayne syndrome does not increase the risk of skin and other cancers. This lack of carcinogenicity in Cockayne syndrome might be due to inhibition of cellular growth because accelerated aging increases apoptosis of DNA-damaged cells and decreased cell division of senescent cells.63 Thus, lack of carcinogenicity and cachexia might share a common underlying mechanism.
Aging is considered to be caused by the accumulation of damage to nuclei and mitochondria from various sources, such as reactive oxygen species—the products of normal oxidative energy metabolism—and exogenous damage, such as ultraviolet or gamma radiation and other toxins.66 Even non-ultraviolet radiation-exposed, nondividing cells in the nervous system and inner ear are subjected to oxidative stress. Faulty repair of oxidative damage from normal metabolism can result in an accumulation of defective residues that can contribute to premature cell death by apoptosis.60,64,67–69 Cockayne syndrome cells respond poorly to oxidative stress.70
Whether mitochondrial dysfunction plays a role in hearing loss, retinitis pigmentosa, and other manifestations of Cockayne syndrome remains to be determined. Mitochondrial function seems not to have been studied thus far in Cockayne syndrome; it was normal in the muscle of case 6. Mitochondrial dysfunction has been shown to play a role in neurodegenerative disorders associated with aging (eg, Parkinson disease).71 Another mechanism invoked to explain premature aging and vulnerability to oxidative stress is inadequate targeting of proteins to the mitochondria and other intracellular organelles.72
CONCLUSIONS
This review has demonstrated the clinical heterogeneity of Cockayne syndrome, which it shares with other disorders of DNA repair.35 Understanding the molecular mechanisms underlying these disorders is progressing rapidly, but many questions remain with respect to correlations between genotypes and phenotypes. Further study of these rare disorders might help elucidate mechanisms that are relevant to pervasive problems of aging, including cachexia, carcinogenesis, myelin maintenance, and progressive neuronal degeneration, because “the ultimate answers depend on the wealth of information to be gleaned from medical records and biological samples obtained from patients.”11
Acknowledgments
We express our gratitude to the parents of the patient for allowing us to study their son longitudinally and report his case and furnishing photographs and details of his life; to Dr Herbert Cole, who diligently cared for him; to Dr Alan M. Aron, who made the initial diagnosis and provided clinical information; to Dr David Bregman for performing the general autopsy; to Ms Anja Raams and Dr Wim Vermeulen for molecular studies; to Dr Virginia Kimonis for referring case 13 and sharing clinical details and results of complementation testing; to Dr Stuart Clarkson for supplying clinical details on case 20; to Dr Salvatore DiMauro for discussion of the potential contribution of mitochondrial dysfunction in Cockayne syndrome; and to Dr Kenneth H. Kraemer for bringing the Bartenjev et al16 and Thorel et al17 articles to our attention and for reviewing the manuscript. We thank Jorge Bermudez and Barry Mordin for valuable technical assistance.
Footnotes
The majority of the work was done at Albert Einstein College of Medicine and Montefiore Medical Center, Bronx, NY. Small parts of the work were done at Columbia University College of Physicians and Surgeons, New York, NY, and Harvard Medical School, Boston, MA.
References
- 1.Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child. 1946;21:52–54. [PubMed] [Google Scholar]
- 2.Cockayne EA. Dwarfism with retinal atrophy and deafness. Arch Dis Child. 1936;11:1–8. doi: 10.1136/adc.11.61.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nance MA, Berry SA. Cockayne syndrome: Review of 140 cases. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- 4.Dollfus H, Porto F, Caussade P, et al. Ocular manifestation in the inherited DNA repair disorders. Surv Ophthalmol. 2003;48:107–122. doi: 10.1016/s0039-6257(02)00400-9. [DOI] [PubMed] [Google Scholar]
- 5.Henning KA, Li L, Iyer N, et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell. 1995;82:555–564. doi: 10.1016/0092-8674(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 6.Komatsu A, Suzuki S, Inagaki T, et al. A kindred with Cockayne syndrome caused by multiple splicing variants of the CSA gene. Am J Med Genet A. 2004;128:67–71. doi: 10.1002/ajmg.a.30087. [DOI] [PubMed] [Google Scholar]
- 7.Stefanini M, Fawcett H, Botta E, et al. Genetic analysis of twentytwo patients with Cockayne syndrome. Hum Genet. 1996;97:418–423. doi: 10.1007/BF02267059. [DOI] [PubMed] [Google Scholar]
- 8.Troelstra C, Landsvater RM, Weigant J, et al. Localization of the nucleotide excision repair gene ERCC6 to human chromosome 10q11–q21. Genomics. 1992;12:745–749. doi: 10.1016/0888-7543(92)90304-b. [DOI] [PubMed] [Google Scholar]
- 9.Troelstra C, van Gool A, deWit J, et al. ERCC6, a member of a sub-family of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell. 1992;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- 10.Mallery DL, Tanganelli B, Colella S, et al. Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am J Hum Genet. 1998;62:77–85. doi: 10.1086/301686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spivak G. The many faces of Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101:15273–15274. doi: 10.1073/pnas.0406894101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH. Cockayne syndrome and xeroderma pigmentosum: DNA repair disorders with overlaps and paradoxes. Neurology. 2000;55:1442–1449. doi: 10.1212/wnl.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbins JH, Kraemer KH, Lutzner MA, et al. Xeroderma pigmentosum. An inherited disease with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med. 1974;80:221–248. doi: 10.7326/0003-4819-80-2-221. [DOI] [PubMed] [Google Scholar]
- 14.Brumback RA, Yoder FW, Andrews AD, et al. Normal pressure hydrocephalus. Recognition and relationship to neurological abnormalities in Cockayne’s syndrome. Arch Neurol. 1978;35:337–345. doi: 10.1001/archneur.1978.00500300011002. [DOI] [PubMed] [Google Scholar]
- 15.Scott RJ, Itin P, Kleijer WJ, et al. Xeroderma pigmentosum–Cockayne syndrome complex in two patients: Absence of skin tumors despite severe deficiency of DNA excision repair. J Am Acad Dermatol. 1993;29:883–889. doi: 10.1016/0190-9622(93)70263-s. [DOI] [PubMed] [Google Scholar]
- 16.Bartenjev M, Butina R, Potocnik M. Rare case of Cockayne syndrome with xeroderma pigmentosum. Acta Derm Venereol. 2000;80:213–214. doi: 10.1080/000155500750043032. [DOI] [PubMed] [Google Scholar]
- 17.Thorel F, Constantinou A, Dunand-Sauthier I, et al. Definition of a short regions of XPG necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol Cell Biol. 2004;24:10670–10680. doi: 10.1128/MCB.24.24.10670-10680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuknecht HF. Pathology of the Ear. 2. Philadelphia: Lea and Febiger; 1993. [Google Scholar]
- 19.Dekaban A, Sadowsky D. Changes in brain weights during the span of human life: Relation of brain weights to body heights and body weights. Ann Neurol. 1978;4:345–356. doi: 10.1002/ana.410040410. [DOI] [PubMed] [Google Scholar]
- 20.Johnson PC, Nielsen SL, Richardson EP., Jr Enlarged oligodendroglial nuclei in Cockayne’s syndrome, abstract. J Neuropathol Exp Neurol. 1980;39:364. [Google Scholar]
- 21.Grunnet ML, Zimmerman AW, Lewis RA. Ultrastructure and electrodiagnosis of peripheral neuropathy in Cockayne’s syndrome. Neurology. 1983;33:1606–1609. doi: 10.1212/wnl.33.12.1606. [DOI] [PubMed] [Google Scholar]
- 22.Miyauchi H, Horio T, Akaeda T, et al. Cockayne syndrome in two adult siblings. J Am Acad Dermatol. 1994;30:329–335. doi: 10.1016/s0190-9622(94)70034-6. [DOI] [PubMed] [Google Scholar]
- 23.Inoue T, Sano N, Ito Y, et al. An adult case of Cockayne syndrome without sclerotic angiopathy. Intern Med. 1997;36:565–570. doi: 10.2169/internalmedicine.36.565. [DOI] [PubMed] [Google Scholar]
- 24.Boltshauser E, Yalcinkaya C, Wichmann W, et al. MRI in Cockayne syndrome type I. Neuroradiology. 1989;31:276–277. doi: 10.1007/BF00344359. [DOI] [PubMed] [Google Scholar]
- 25.Paddison RM, Moossy J, Derbes VJ, Kloepfer W. Cockayne’s syndrome. A report of five new cases with biochemical, chromosomal, dermatologic, genetic and neuropathologic observations. Derm Trop Ecol Geogr. 1963;15:195–203. [PubMed] [Google Scholar]
- 26.Moosy J. The neuropathology of Cockayne’s syndrome. J Neuropathol Exp Neurol. 1965;26:654–660. doi: 10.1097/00005072-196710000-00010. [DOI] [PubMed] [Google Scholar]
- 27.Shirasaki N, Hayashi M, Handa Y, et al. Cockayne’s syndrome presenting cerebral ischemic attack: Case report. No To Shinkei. 1986;38:871–875. [PubMed] [Google Scholar]
- 28.Kennedy RM, Rowe VD, Kepes JJ. Cockayne syndrome: An atypical case. Neurology. 1980;30:1268–1272. doi: 10.1212/wnl.30.12.1268. [DOI] [PubMed] [Google Scholar]
- 29.Fryns JP, Bulcke J, Verdu P, et al. Apparent late-onset Cockayne syndrome and interstitial deletion of the long arm of chromosome 10 (del(10)(q11.23q21.2)) Am J Med Genet. 1991;40:343–344. doi: 10.1002/ajmg.1320400320. [DOI] [PubMed] [Google Scholar]
- 30.Shemen LJ, Mitchell DP, Farkashidy J. Cockayne’s syndrome—An audiologic and temporal bone analysis. Am J Otol. 1984;5:300–307. [PubMed] [Google Scholar]
- 31.Crome L, Kanjilal GC. Cockayne’s syndrome: Case report. J Neurol Neurosurg Psychiatry. 1971;32:171–178. doi: 10.1136/jnnp.34.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Traboulsi EI, DeBecker I, Maumenee IH. Ocular findings in Cockayne syndrome. Am J Ophthalmol. 1992;114:579–583. doi: 10.1016/s0002-9394(14)74486-1. [DOI] [PubMed] [Google Scholar]
- 33.Felgenhauer W-R, Ammann F. Syndrome de Cockayne fruste associé à la neurofibromatose de Recklinghausen. J Génét Hum. 1967;16:6–24. [PubMed] [Google Scholar]
- 34.Yoshida K, Takeda S, Shoji S, et al. An adult case of Cockayne syndrome with protracted clinical course—A study of ultraviolet irradiation test using cultured skin fibroblasts. Rhinsho Shinkeigaku. 1998;28:609–615. [PubMed] [Google Scholar]
- 35.Bohr VA, Sander M, Kraemer KH. Rare diseases provide rare insights into nucleotide excision repair, transcription-coupled repair, TFIIH, aging and cancer. DNA Repair. 2005;4:293–302. doi: 10.1016/j.dnarep.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 36.Ren Y, Saijo M, Nakatsu Y, et al. Three novel mutations responsible for Cockayne syndrome group A. Genes Genet Syst. 2003;78:93–102. doi: 10.1266/ggs.78.93. [DOI] [PubMed] [Google Scholar]
- 37.Licht CL, Stevnsner T, Bohr VA. Cockayne syndrome group B. Cellular and biochemical functions. Am J Hum Genet. 2003;73:1217–1239. doi: 10.1086/380399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lehrmann AR. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie. 2003;85:1101–1111. doi: 10.1016/j.biochi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 39.Pena SDJ, Shokeir MHK. Autosomal recessive cerebro-oculo-facio-skeletal (COFS) syndrome. Clin Genet. 1979;5:285–293. doi: 10.1111/j.1399-0004.1974.tb01695.x. [DOI] [PubMed] [Google Scholar]
- 40.Meira LB, Graham JM, Jr, Greenberg CR, et al. Manitoba aboriginal kindred with original cerebro-oculo-facio-skeletal syndrome has a mutation in the Cockayne syndrome group B (CSB) gene. Am J Clin Genet. 2000;66:1221–1228. doi: 10.1086/302867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collela S, Nardo T, Botta E, et al. Identical mutations in the CSB gene associated with either Cockayne syndrome or the DeSanctis-Cacchione variant of xeroderma pigmentosum. Hum Mol Genet. 2000;9:1171–1175. doi: 10.1093/hmg/9.8.1171. [DOI] [PubMed] [Google Scholar]
- 42.Robbins JH. Xeroderma pigmentosum. Defective DNA repair causes skin cancer and neurodegeneration. JAMA. 1988;260:384–388. doi: 10.1001/jama.260.3.384. [DOI] [PubMed] [Google Scholar]
- 43.Robbins JH, Brumback RA, Mendiones M, et al. Neurological disease in xeroderma pigmentosum. Documentation of a late onset type of the juvenile onset form. Brain. 1991;114:1335–1361. doi: 10.1093/brain/114.3.1335. [DOI] [PubMed] [Google Scholar]
- 44.Horibata K, Iwamaoto Y, Kuraoka I, et al. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101:15410–15415. doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higginbottom MC, Griswold WR, Lyons Jones K, et al. The Cockayne syndrome: An evaluation of hypertension and studies of renal pathology. Pediatrics. 1979;64:929–934. [PubMed] [Google Scholar]
- 46.Fujimoto WY, Greene ML, Seegmiller JE. Cockayne’s syndrome: Report of a case with hyperlipoproteinemia, hyperinsulinemia, renal disease, and normal growth hormone. J Pediatr. 1969;75:881–884. doi: 10.1016/s0022-3476(69)80317-3. [DOI] [PubMed] [Google Scholar]
- 47.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 48.Soffer D, Grotsky HW, Rapin I, Suzuki K. Cockayne syndrome: Unusual pathological findings and review of the literature. Ann Neurol. 1979;6:340–348. doi: 10.1002/ana.410060407. [DOI] [PubMed] [Google Scholar]
- 49.Lahiri S, Davies N. Cockayne’s syndrome: Case report of a successful pregnancy. Br J Obstet Gynaecol. 2003;110:871–872. [PubMed] [Google Scholar]
- 50.Cleaver JE, Thompson IH, Richardson AS, States JC. A summary of mutations in the UV-sensitive disorders: Xeroderma pigmento-sum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat. 1999;14:9–22. doi: 10.1002/(SICI)1098-1004(1999)14:1<9::AID-HUMU2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 51.Barrett SF, Robbins JH, Tarone RE, Kraemer KH. Evidence for defective repair of cyclobutane pyrimidine dimers with normal repair of other DNA photoproducts in a transcriptionally active gene transfected into Cockayne syndrome cells. Mutat Res DNA Repair. 1991;255:281–291. doi: 10.1016/0921-8777(91)90032-k. [DOI] [PubMed] [Google Scholar]
- 52.Parris CN, Kraemer KH. Ultraviolet-induced mutations in Cockayne syndrome cells are primarily caused by cyclobutane dimer photoproducts while repair of other photoproducts is normal. Proc Natl Acad Sci U S A. 1993;90:7260–7264. doi: 10.1073/pnas.90.15.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lanning M, Similä S. Cockayne’s syndrome: Report of a case with normal intelligence. Z Kinderheilk. 1970;109:70–75. [PubMed] [Google Scholar]
- 54.Czeizel AE, Marchalko M. Cockayne syndrome type III with high intelligence. Clin Genet. 1995;48:331–333. doi: 10.1111/j.1399-0004.1995.tb04121.x. [DOI] [PubMed] [Google Scholar]
- 55.Rowlatt U. Cockayne’s syndrome: Report of case with necropsy findings. Acta Neuropathol (Berl) 1969;14:52–61. doi: 10.1007/BF00687702. [DOI] [PubMed] [Google Scholar]
- 56.Leech RW, Brumback RA, Miller RH, et al. Cockayne syndrome: Clinicopathologic and tissue culture studies of affected siblings. J Neuropathol Exp Neurol. 1985;44:507–519. [PubMed] [Google Scholar]
- 57.Levin PS, Green R, Victor DI, MacLean AL. Histopathology of the eye in Cockayne’s syndrome. Arch Ophthalmol. 1983;101:1093–1097. doi: 10.1001/archopht.1983.01040020095016. [DOI] [PubMed] [Google Scholar]
- 58.Lindenbaum Y, Dickson DW, Rosenbaum PS, et al. Xeroderma pigmentosum/Cockayne syndrome complex: First neuropathological study and review of eight other cases. Eur J Paediatr Neurol. 2001;5:225–242. doi: 10.1053/ejpn.2001.0523. [DOI] [PubMed] [Google Scholar]
- 59.Gandolfi A, Houroupian D, Rapin I, et al. Deafness in Cockayne’s syndrome: Morphological, morphometric, and quantitative study of the auditory pathway. Ann Neurol. 1984;15:135–143. doi: 10.1002/ana.410150205. [DOI] [PubMed] [Google Scholar]
- 60.Kohji T, Hayashi M, Shioda K, et al. Cerebellar neurodegeneration in human hereditary DNA repair disorders. Neurosci Lett. 1998;243:133–136. doi: 10.1016/s0304-3940(98)00109-8. [DOI] [PubMed] [Google Scholar]
- 61.Challa VR, Thore CR, Moody DM, et al. Increase of white matter string vessels in Alzheimer’s disease. J Alzheimers Dis. 2004;6:379–383. doi: 10.3233/jad-2004-6404. [DOI] [PubMed] [Google Scholar]
- 62.Dickson DW. Structural changes in the aged brain. In: Mattson MP, Geddes JW, editors. Advances in Cell Aging and Gerontology, Volume 2, The Aging Brain. New York: Elsevier; 1997. pp. 51–76. [Google Scholar]
- 63.Kipling D, Davis T, Ostler EL, Faragher RGA. What can progeroid syndromes tell us about human aging? Science. 2004;305:1426–1431. doi: 10.1126/science.1102587. [DOI] [PubMed] [Google Scholar]
- 64.Hannawalt PC. The bases for Cockayne syndrome. Nature. 2000;405:415–416. doi: 10.1038/35013197. [DOI] [PubMed] [Google Scholar]
- 65.Egly J-M. TFIIH: From transcription to clinic. FEBS Lett. 2001;498:124–128. doi: 10.1016/s0014-5793(01)02458-9. [DOI] [PubMed] [Google Scholar]
- 66.Hasty P, Campisi J, Hoeijmakers J, et al. Aging and genome maintenance: Lessons from the mouse. Science. 2003;299:1355–1359. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- 67.Itoh M, Hayashi M, Shioda K, et al. Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev. 1999;21:326–333. doi: 10.1016/s0387-7604(99)00033-9. [DOI] [PubMed] [Google Scholar]
- 68.Kuraoka I, Bender C, Romieu A, et al. Removal of oxygen free-radical-induced 5′,8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc Natl Acad Sci U S A. 2000;97:3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le Page F, Kwoh EE, Avrutskaya A, et al. Transcription-coupled repair of 8-oxoguanine: Requirements for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell. 2000;101:159–171. doi: 10.1016/s0092-8674(00)80827-2. [DOI] [PubMed] [Google Scholar]
- 70.Hayashi M, Itoh M, Araki S, et al. Oxidative stress and disturbed glutamate transport in hereditary nucleotide repair disorders. J Neuropathol Exp Neurol. 2001;60:350–356. doi: 10.1093/jnen/60.4.350. [DOI] [PubMed] [Google Scholar]
- 71.Beal MF. Mitochondria, NO and neurodegeneration. Biochem Soc Symp. 1999;66:43–54. doi: 10.1042/bss0660043. [DOI] [PubMed] [Google Scholar]
- 72.Rosenblum JS, Gilula NB, Lerner RA. On signal sequence polymorphisms and diseases of distribution. Proc Natl Acad Sci U S A. 1996;93:4471–4473. doi: 10.1073/pnas.93.9.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]