

Abstract
We previously demonstrated that the endoplasmic reticulum (ER) chaperone BiP functions in human cytomegalovirus (HCMV) assembly and egress. Here, we show that BiP localizes in two cytoplasmic structures in infected cells. Antibodies to the extreme C terminus, which includes BiP's KDEL ER localization sequence, detect BiP in regions of condensed ER near the periphery of the cell. Antibodies to the full length, N terminus, or larger portion of the C terminus detect BiP in the assembly compartment. This inability of C-terminal antibodies to detect BiP in the assembly compartment suggests that BiP's KDEL sequence is occluded in the assembly compartment. Depletion of BiP causes the condensed ER and assembly compartments to dissociate, indicating that BiP is important for their integrity. BiP and pp28 are in association in the assembly compartment, since antibodies that detect BiP in the assembly compartment coimmunoprecipitate pp28 and vice versa. In addition, BiP and pp28 copurify with other assembly compartment components on sucrose gradients. BiP also coimmunoprecipitates TRS1. Previous data show that cells infected with a TRS1-deficient virus have cytoplasmic and assembly compartment defects like those seen when BiP is depleted. We show that a fraction of TRS1 purifies with the assembly compartment. These findings suggest that BiP and TRS1 share a function in assembly compartment maintenance. In summary, BiP is diverted from the ER to associate with pp28 and TRS1, contributing to the integrity and function of the assembly compartment.
Human cytomegalovirus (HCMV), the largest of the human herpesviruses, is capable of encoding over 200 proteins, which are expressed in temporal fashion as immediate-early, early, delayed-early, and late genes. Despite the extensive coding capacity of HCMV, its replication cycle is slow. During this protracted period, the virus must maintain optimal replication conditions in the host cell. However, the increasing strain of the infection induces cellular stress responses with consequences that may be deleterious to the progress of the infection. We and others have previously shown that HCMV has multiple mechanisms to deal with the deleterious aspects of cellular stress responses while maintaining beneficial ones (2, 8-10, 14, 17, 18, 22-24, 26, 27, 50, 51).
An example of these mechanisms is the viral control of endoplasmic reticulum (ER) stress and the unfolded protein response (UPR). Due to the number of HCMV proteins that are glycosylated, or receive other ER-dependent posttranslational modifications, the load of proteins in the ER can exceed its capacity, resulting in ER stress and the activation of the UPR (18, 47, 51). However, we and others have shown that HCMV controls and modulates the UPR, maintaining aspects that may benefit the viral infection while inhibiting aspects that would be detrimental (18, 51).
The UPR is normally controlled by transmembrane sensors which initiate the complex UPR signaling cascade when activated by ER stress (reviewed in references 20, 35, 38, and 52). The ER molecular chaperone BiP (immunoglobulin heavy chain-binding protein), also called glucose-regulated protein 78 (GRP78), is believed to bind these sensors and keep them inactive during unstressed conditions. However, when unfolded or misfolded proteins accumulate in the ER, BiP leaves these sensors to perform its chaperone function, thus allowing the sensors to activate UPR signaling. We have previously shown that during HCMV infection, BiP is vastly overproduced (8), suggesting that BiP may have other functions in the viral infection. Indeed, it has been shown that BiP binds to the viral proteins US2 and US11; this interaction is necessary for the virus-mediated degradation of major histocompatibility complex class I and II (15, 47). Further, we have shown that depletion of BiP, using either the BiP-specific subtilase cytotoxin SubAB (32) or short hairpin RNAs, caused infectious virion formation in the cytoplasm to cease and nucleocapsids to accumulate just outside the outer nuclear membrane (8). This result suggested that BiP has a significant role in virion formation and cytoplasmic egress.
Although the exact mechanism of virion formation in the cytoplasm is not well understood, studies have identified a perinuclear structure, referred to as the cytoplasmic assembly compartment, that is involved in the process. Several viral proteins, for example, tegument proteins (pp28, pp65) (36) and viral glycoproteins (gB, gH, gL, gO, gp65) (36, 46), have been identified as part of this structure. Defining the exact origin of this compartment has been complicated by the observation of specific organellar markers in and around the compartment, while other markers of the same organelles are not detected. For example, immunofluorescence examination suggests that the early endosomal marker early endosome antigen 1 (EEA1) has been observed in the center of the assembly compartment (12, 13); however, Rab4 and Rab5, other early endosomal markers, were not detected (16). Such observations suggest that the virus directs specific viral and cellular proteins to the assembly compartment as needed for assembly compartment function.
In the present study, we further examine the role of BiP during an HCMV infection, including its localization and interactions with other proteins. We show here that in infected cells, BiP localizes in two distinct structures, regions of condensed ER near the periphery of the cell and the assembly compartment. The data suggest that BiP diversion from the ER to the assembly compartment is due to occlusion of its ER localization signal. Depletion of BiP causes both condensed ER and assembly compartments to disperse, indicating that BiP is important for their formation or maintenance. BiP and pp28 appear to associate in the assembly compartment, since BiP from the assembly compartment coimmunoprecipitates pp28 and vice versa. In addition, both BiP and pp28 copurify with the assembly compartment on sucrose gradients. BiP also coimmunoprecipitates TRS1. Previous studies (1, 4) have shown that cells infected with HCMV with a mutation in the TRS1 gene show cytoplasmic and assembly compartment defects like those seen when BiP is depleted (reference 8 and the studies presented below). We show that a fraction of TRS1 purifies with the assembly compartment, indicating a shared assembly compartment function with BiP. In summary, our data suggest that BiP is diverted from the ER to associate with pp28 and TRS1, contributing to the integrity and function of the assembly compartment.
MATERIALS AND METHODS
Tissue culture, reagents, and primary antibodies.
Low passage life-extended human foreskin fibroblasts (5) were cultured in Dulbecco's modified Eagle's medium (supplemented with 10% fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM GlutaMax) at 37°C in 5% CO2. SubAB toxin and its nontoxic derivative SubAA272B (32) were purified by nickel-nitrilotriacetic acid chromatography as described previously (33, 45) and added at 100 ng/ml to tissue culture 12 h before harvesting. The TRS1 antibody was a gift from T. Shenk (34). All other antibodies used are commercially available and are described in Table 1.
TABLE 1.
Commercial antibodies useda
| Name | Type | Source (catalog no.) | Epitope (amino acids) | Use in this paper |
|---|---|---|---|---|
| BiPA | Rabbit poly | Abgent (AP1335a) | 1-654 | IF |
| BiPB | Mouse mono | BD Bioscience (610978) | 525-628 | W |
| BiPC | Goat poly | Santa Cruz (C-20, sc-1051) | 20 amino acids within 600-654 | IF, W |
| BiPD | Mouse mono | R+D Systems (MAb 4846) | 1-654 | IF |
| BiPH | Rabbit poly | Santa Cruz (H-129, sc-13968) | 525-654 | IF |
| BiPR | Rabbit poly | Sigma (GL-19, G8918) | 636-654 | IF |
| BiPS | Rabbit poly | Sigma (ET-21, G9043) | 71-91 | IF |
| BiPY | Rabbit poly | Abcam (ab21685) | 600-654 | IF, IP |
| KDEL BiP K | Mouse mono | MBL (SR-827F) | KDEL (649-654) | IF, IP |
| gB | Mouse mono | EastCoast Bio (CA005) | IF, W | |
| pp28 | Mouse mono | Abcam (ab6502) | IF, W, IP | |
| PFK-1 | Goat poly | Santa Cruz (K-15, sc-31712) | W |
mono, monoclonal; poly, polyclonal; IF, immunofluorescence; W, Western blotting; IP, immunoprecipitation.
Virus preparation, titration, and infections.
Virus stocks (Towne strain) were prepared as previously described (22), and titers were determined by using the 50% tissue culture infective dose method. All experiments were performed using a multiplicity of infection of 3.
Indirect immunofluorescence.
Coverslips containing either mock or HCMV-infected human foreskin fibroblasts were washed in phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde at room temperature. Cells were permeabilized in PBS containing 0.5% Triton X-100 and blocked in PBS containing 5% bovine serum albumin (BSA) and 0.5% Tween 20; alternatively, BSA was replaced with 10% human or goat serum and 5% glycine (blocking buffer) as indicated in the text. Primary and secondary antibodies (Alexa Fluor 594 and 647; Invitrogen) were diluted in the respective blocking buffers. Coverslips were washed in PBS. Before mounting, coverslips were rinsed in H2O and mounted on slides using Vectashield mounting medium containing 4′6′-diamidino-2-phenylindole (DAPI) (Vector Laboratories). Slides were examined using a Nikon Eclipse E600 microscope. Micrographs were acquired, processed by deconvolution, and analyzed using Image-Pro 6.3 and Autoquant X2 (MediaCybernetics) software.
Assembly compartment purification.
Assembly compartments were isolated on sucrose gradients using extracts from mock-infected or infected fibroblasts which had been treated with SubAB toxin, or the inactive mutant toxin SubAA272B, for 12 h prior to harvest at 72 h postinfection (hpi). Extraction protocols and sucrose gradient fractionation conditions have been previously described (36). Fractions were collected from the top of the gradient, and aliquots of the fractions were analyzed by Western blotting.
Western analysis.
Western blot samples were added to 3× loading solution (187.5 mM Tris-HCl [pH 6.8], 6% sodium dodecyl sulfate, 30% glycerol, 0.3% bromophenol blue, 467 mM β-mercaptoethanol) and boiled for 5 min. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% gels). Following electrophoresis, the gel was transferred to nitrocellulose in transfer buffer (33.3 mM Tris, 256 mM glycine, 20% methanol) under a constant current. Membranes were blocked in Tris-buffered saline containing 1% Tween 20 (TBST) plus 5% nonfat milk. Primary antibodies were diluted in TBST containing 2% BSA; horseradish peroxidase-conjugated secondary antibodies (Thermo) were diluted in TBST containing 5% milk. Membranes were washed in TBST and developed with Lumi-Light Western blotting substrate (Roche).
Immunoprecipitation.
Mock- and HCMV-infected cell lysates were prepared at 96 hpi by harvesting in 1% Triton buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, 1% Triton X-100, 10% glycerol, 1 mM EDTA, 10 mM tetrasodium pyrophosphate, 100 mM sodium fluoride, 17.5 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, 1.25 μg/ml pepstatin, 8.5 μg/ml aprotinin). Immunoprecipitation reactions were performed using Dynabeads Protein G (Invitrogen) according to the manufacturer's instructions. One hundred micrograms of either mock- or HCMV-infected lysates and 20 μl of Dynabeads were used for each reaction. Antibody amounts varied according to the manufacturer's recommendations. After immunoprecipitation, protein was removed from the beads using 20 μl of 1× loading dye containing 2× β-mercaptoethanol. Samples were then prepared for Western analysis as described above.
RESULTS
CMV infection localizes BiP to two distinct cellular structures.
The intracellular localization of BiP was examined by immunofluorescence microscopy using a panel of commercially available anti-BiP antibodies made using different regions and epitopes of the protein (see Table 1). As shown in Table 1, for several antibodies, a rather large region of the protein is listed because the manufacturers considered the exact epitope to be proprietary. All of these antibodies can specifically detect BiP in Western analyses of mock- and HCMV-infected cell extracts (data not shown). In mock-infected cells, each of the antibodies visualized the known cytoplasmic ER localization of BiP (Fig. 1), with the exception of antibody BiPD, which will be discussed below (Fig. 2). It is important to note that all of the micrographs of BiP in mock-infected cells were taken with longer exposure times than those for the infected cells. This is because BiP levels in uninfected cells are significantly lower than in infected cells.
FIG. 1.

Immunofluorescence analysis of BiP shows that it is localized in two distinct locations during HCMV infection. Mock- or HCMV-infected cells were prepared for immunofluorescence microscopy at 96 hpi. BiP (red) was detected using the following antibodies described in Materials and Methods and Table 1: BiPA (A), BiPH (H), BiPR (R), BiPS (S), BiPY (Y), BiPC (C), and KDEL (K). Normal rabbit IgG (IgG) was used as a control. Nuclei were stained with DAPI (blue).
FIG. 2.

BiP colocalizes with assembly compartment markers. (A) Mock- and HCMV-infected cells were prepared for immunofluorescence at 96 hpi and stained for BiPD (red), a mouse monoclonal antibody. (B) HCMV-infected cells were prepared at 96 hpi and stained for BiP (red) using BiPS and BiPR antibodies (Table 1) and either pp28 or gB (green). Colocalization of BiP and pp28 is indicated in yellow. Nuclei shown in panels A and B were stained with DAPI (blue). Comp., composite.
In cells infected for 96 h, antibodies made against the full-length protein (BiPA), the N terminus (BiPS), or a larger portion of the C terminus (BiPH and BiPY) detect BiP in a round, perinuclear structure resembling the cytoplasmic assembly compartment. A clue that these are assembly compartments is the observation that the nuclei next to the perinuclear structures is often the familiar kidney shape known to appear with assembly compartments in HCMV-infected cells.
In addition to this perinuclear structure, BiPH and BiPY also detected BiP in cytoplasmic regions often seen at the periphery of the infected cells. These regions looked condensed or clumped compared to the cytoplasmic appearance of BiP in mock-infected cells. Three antibodies detected the condensed cytoplasmic regions but did not distinctly stain the perinuclear structure. The epitopes used to make each of these contained the extreme C terminus of BiP. These were BiPC and BiPR, as well as BiPK, which is made against the KDEL ER localization signal at the extreme C-terminal end of BiP. Since these antibodies failed to detect BiP in the perinuclear structure, we feel that the C-terminal region, including the KDEL sequence, may be occluded by interactions with other proteins and that this may account for BiP's relocation from the ER to the perinuclear region.
We note that the antibodies that detect BiP in the perinuclear structure are rabbit polyclonal antibodies (BiPA, -S, -H, and -Y). This raises some concern that the rabbit antisera may be interacting, at least in part, with HCMV-encoded Fc receptors which have an affinity for rabbit immunoglobulin G (IgG) (28) and may cause spurious localization of IgG to the assembly compartment. However, one rabbit polyclonal antibody, BiPR, localizes BiP in the condensed cytoplasmic regions, not the perinuclear region. In addition, control rabbit IgG used under the same staining and exposure conditions as those for BiPA, -S, -H, and -Y does not stain the perinuclear region (Fig. 1). If the exposure times and gain are increased, one can detect spurious IgG staining in the assembly compartment.
However, these controls may not be sufficient. Previous studies have suggested that the HCMV-induced Fc receptors have much greater affinities for some IgG subclasses than for others (28). Thus, if some rabbit antibody preparations were predominantly high affinity Fc binding IgGs, and others were not, then this could account for the variable rabbit IgG results in Fig. 1. To test this possibility, we used BiPD, a mouse monoclonal antibody that is not recommended by its manufacturer for immunofluorescence studies. Indeed, on mock-infected cells, BiPD does not provide a normal ER staining (Fig. 2A). However, in infected cells, BiPD stains a perinuclear region, but the staining is different from the perinuclear staining with rabbit antibodies. BiPD visualized a small dot of BiP in the center of the perinuclear region as well as a ring around the periphery (Fig. 2A).
This result supports the conclusion that BiP is associated with the perinuclear region; however, since BiPD is not a recommended antibody for immunofluorescence, we wanted to return to the rabbit antibodies and use techniques that would block the Fc receptors prior to the addition of the specific rabbit IgG. The experiments in Fig. 1 were blocked with BSA, which would not be sufficient to block the Fc receptors. The use of goat serum for blocking has been used with HCMV-infected samples (13). However, it has been shown that goat IgG has very little affinity for the HCMV Fc receptors (3), and our utilization of it for blocking made very little difference in the perinuclear staining using the rabbit antibodies (not shown). Thus, we blocked with human serum, which binds well to the Fc receptors (3).
BiP is contained in the assembly compartment, where it colocalizes with pp28.
After being blocked with human serum, the samples were stained for BiP and then costained for either pp28 or gB, which are known components of the assembly compartment. The rabbit anti-BiP antibodies used for the human serum blocking experiment were BiPS and BiPR, which come from the same source (Sigma) and recognize BiP in either the assembly compartment (BiPS) or the condensed cytoplasmic regions (BiPR). Figure 2B shows that after Fc receptor blocking with human serum, BiPS detected the perinuclear region, showing a spot in the middle and a ring around the periphery, similar to what was detected with the mouse monoclonal BiPD. The peripheral circle of BiP colocalized with pp28, whereas the space between the peripheral circle and the central dot stained for gB (Fig. 2B). The agreement between the BiPD and BiPS staining suggests that blocking the Fc receptors with human serum allows the rabbit anti-BiP IgG to accurately locate BiP in the assembly compartment.
Figure 2B also shows that BiPR recognized the cytoplasmic clumps in samples blocked with human serum just as it had with BSA blocking in Fig. 1; thus, human serum blocking did not alter its staining pattern, consistent with the idea that the BiPR IgG preparation does not contain IgG subclasses which bind the Fc receptors.
To support the microscopic evidence that BiP was contained in the assembly compartment, we used a nonmicroscopic approach. Previously described procedures (36) provided a means to purify the assembly compartment on sucrose gradients. Western analyses of gradient fractions (Fig. 3A) showed that a large amount of BiP from infected cells sediments well into the gradient and cosediments with gB and pp28 in the assembly compartment region. A control protein, the glycolytic enzyme phosphofructokinase-1 (PFK-1), does not sediment with the assembly compartment.
FIG. 3.

BiP cosediments with assembly compartment markers in a sucrose gradient. Lysates were prepared from infected cells treated with SubAA272B (mutant toxin) (A) or SubAB (WT toxin) (B) and sedimented on a discontinuous sucrose gradient as previously described (36). The gradient was fractionated from the top (300 μl/fraction) and analyzed by Western analysis. AC, assembly compartment.
We conclude from these data and the data in Fig. 2 and 3A that BiP is a stable component of the assembly compartment.
Depletion of BiP using the SubAB subtilase cytotoxin disrupts the assembly compartment in infected cells.
Previously, we presented an electron microscope (EM) analysis showing that depletion of BiP by cleavage using the BiP-specific SubAB subtilase cytotoxin (32) caused a cessation of cytoplasmic viral activity in infected cells, with nucleocapsids accumulating in the nucleus and just outside the outer nuclear membrane (8). Using cells infected for 72 h, the assembly compartment was localized using anti-pp28 or anti-gB (Fig. 4). The specific cells used for staining were treated for 12 h prior to harvesting with the SubAB subtilase cytotoxin or its defective mutant form, which has no effect on infected cells (8), or were left untreated. Figure 4 shows that the assembly compartments, visualized by either anti-gB or anti-pp28, are intact in untreated cells and in cells treated with the mutant toxin. However, the integrity of the assembly compartments is disrupted after BiP cleavage using the toxin. These data suggest that BiP is important for maintaining the integrity of the assembly compartment. To further prove that BiP is important for maintaining the integrity of the assembly compartment, we analyzed sucrose gradient-purified assembly compartments from infected cells treated with the SubAB toxin. The assembly compartments analyzed in Fig. 3A (discussed above) were derived from infected cells treated with the mutant SubAB cytotoxin, which has no effect on assembly compartment integrity. In Fig. 3B, the cells had been treated with the wild-type toxin 12 h prior to assembly compartment isolation. Under these conditions, there is a significant loss of BiP, gB, and pp28 migrating in the assembly compartment region. The combined data from Fig. 3 and 4 strongly indicate that BiP is essential for maintaining the integrity of the assembly compartment.
FIG. 4.
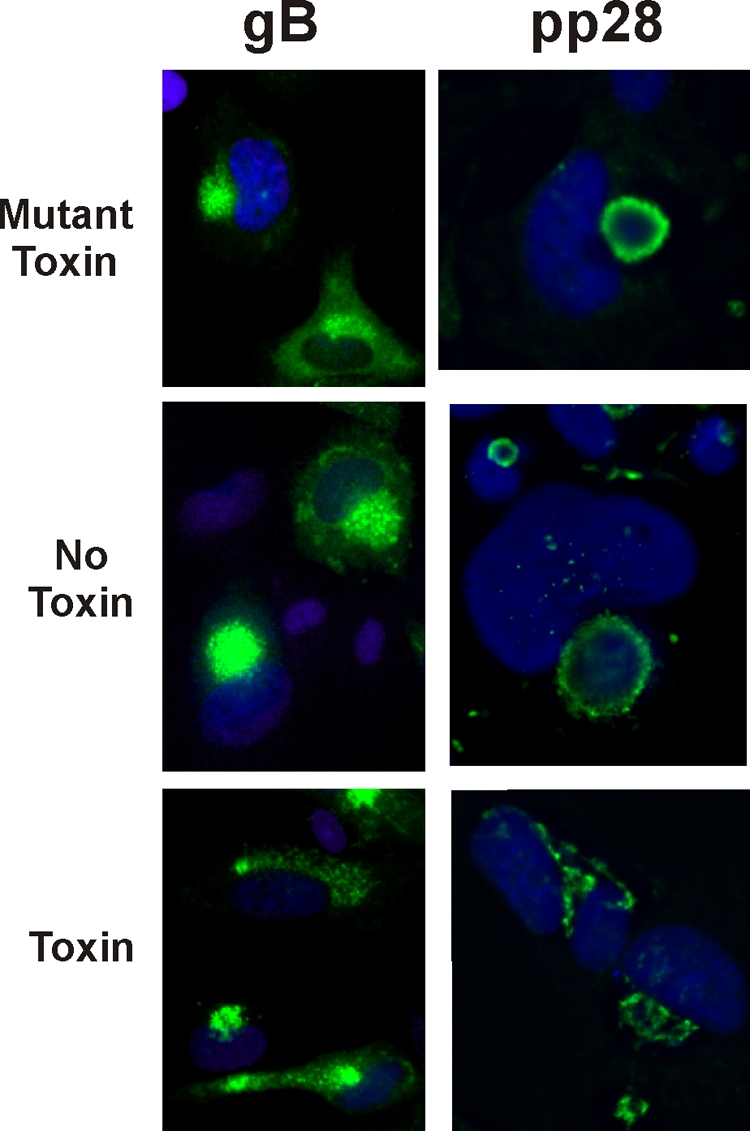
Cleavage of BiP disrupts the assembly compartment. At 60 hpi, HCMV-infected cells were treated with SubAB (Toxin) or SubAA272B (Mutant Toxin) or left untreated (No Toxin). At 72 hpi, the cells were prepared for immunofluorescence microscopy, and assembly compartment integrity was examined by staining for gB and pp28. Nuclei were stained with DAPI (blue).
The condensed BiP-containing cytoplasmic structures contain ER markers.
We used cells infected for 72 h to localize BiP using the BiPC antibody, which detects BiP only in the condensed cytoplasmic regions (Fig. 5). In addition, antibodies were used to visualize another ER marker, protein disulfide isomerase (PDI). As in the previous experiment, the specific cells used for staining were treated with the SubAB subtilase cytotoxin or its defective mutant form for 12 h prior to harvesting. The mutant toxin-treated cells show the condensed BiP-containing cytoplasmic clumps similar to those seen in untreated infected cells (Fig. 1, BiPC). In addition, the mutant toxin-treated sample (Fig. 5) shows that BiP colocalizes with PDI, suggesting that the condensed regions are of ER origin. Such condensed regions of organelles at the periphery of the infected cell have been previously described (29). Cleavage of BiP using the toxin returned both BiP and PDI staining results to a diffuse cytoplasmic localization (Fig. 5). It should be noted that cleavage of BiP using the toxin does not destroy the epitope for BiPC, and so it can still be visualized. These data suggest that BiP is in condensed ER and that depletion of BiP relieves these condensed regions. The disappearance of the clumped ER correlates with our previous EM data, which showed that upon BiP depletion, all obvious cytoplasmic viral activity disappeared and the cytoplasm regained a relatively normal appearance (8).
FIG. 5.

The condensed cytoplasmic BiP-containing regions colocalize with ER markers. Samples were prepared as described for Fig. 4 except that they were examined for the ER marker PDI (green) and BiP (red) using the BiPC antibody, which detects BiP in the condensed regions and not in the assembly compartment. Mut Toxin, mutant toxin.
BiP associates with pp28 and TRS1.
In Fig. 6, we used immunoprecipitation to assess the possibility of interactions between BiP and specific viral proteins. The first was pp28, since the data above suggest a close association between the two. The second was TRS1, because it has been shown by immunofluorescence (4) and electron microscopy (1) that the intracellular phenotype of cells infected with TRS1-mutant HCMV is very similar to the phenotype we have reported for infected cells depleted of BiP (see above and reference 8). Specifically, an immunofluorescence analysis shows that the loss of BiP or TRS1 disrupts the integrity of the assembly compartment; an EM analysis shows that the loss of BiP or TRS1 causes the disappearance of the cytopathic effects of viral infection seen in the cytoplasm. The similarity of these phenotypes suggests a shared function.
FIG. 6.

BiP associates with pp28 and TRS1 during infection. (A) Lysates from mock (M)- and HCMV (V)-infected HFs were prepared at 96 hpi for immunoprecipitation with BiPY, an antibody that detects BiP in the assembly compartment, and the KDEL antibody, which does not detect BiP in the assembly compartment. Following immunoprecipitation, reactions were analyzed by Western blotting using antibodies against BiP, TRS1, and pp28. Western analysis of 20% of the input is also shown (Input). (B and C) Similar lysates from mock- and HCMV-infected cells were immunoprecipitated with anti-pp28, and Western blots were probed with anti-BiP (B) or anti-TRS1 (C). A cross-reaction with light chains by the pp28 antibody is shown (LC) in panels B and C.
Immunoprecipitation using BiPY, which recognizes BiP in the assembly compartment, showed that BiP coprecipitated pp28 and TRS1 (Fig. 6A). An association between BiP and pp28 has also been detected by others (N. Moorman and T. Shenk, personal communication). Interestingly, pp28 and TRS1 were not immunoprecipitated using BiPK, the KDEL antibody which does not detect BiP in the assembly compartment. This finding provides further support that the KDEL epitope is blocked when BiP is in the assembly compartment.
The interaction with TRS1 suggests that TRS1 is in the assembly compartment; this idea is supported by the analysis of purified assembly compartments (Fig. 3A), which shows that a fraction of the total TRS1 sediments with BiP and other assembly compartment components. The amount of TRS1 sedimenting with the assembly compartment appears to be lower than that expected from the amount in the immunoprecipitation. However, the various components of the assembly compartment may have different affinities; therefore, some may be dissociated during purification and sedimentation.
In Fig. 6B, the reverse experiment shows that immunoprecipitating pp28 coimmunoprecipitates BiP. We do not have a TRS1 antibody that performs well in immunoprecipitation; however, Fig. 6C shows that immunoprecipitation of pp28 coprecipitates TRS1. Cross-reaction of the pp28 antibody with the light chains is noted in Fig. 6B and C. The pp28 protein is an abundant component of the assembly compartment, and it also multimerizes in the assembly compartment (40). Thus, immunoprecipitation of pp28 would be expected to bring down large multimers of the protein; we feel that this accounts for the large amount of pp28 precipitated by anti-pp28 relative to results for BiP and TRS1. Overall, the precipitation data suggest that complexes containing BiP, TRS1, and multimerized pp28 exist in the assembly compartment.
DISCUSSION
BiP is an important ER chaperone which also functions to maintain calcium homeostasis and monitor ER stress. Previous data have shown that these functions are important for an HCMV infection and that each function is manipulated by the virus during infection (2, 17, 18, 42, 51). Previous data have also suggested that in infected cells, BiP has additional roles in the suppression of immune surveillance (15, 47) and virion formation and egress (8). We show here that during infection, BiP is relocalized to two distinct cytoplasmic localizations. One pool is located in condensed structures in the periphery of the cytoplasm which also contain other ER markers, thus appearing to originate from the ER. It is likely that this pool of BiP is performing its known ER functions as well as its virus-induced ER function of interacting with HCMV proteins US2 and US11, which mediates the degradation of major histocompatibility complex classes I and II (15, 47).
The second structure containing BiP is the assembly compartment, which is a complex perinuclear structure. The assembly compartment has been reported to be a modified compartment made of many vesicles derived from the cellular secretory system, early endosomes, or the trans-Golgi network. It is not believed to be derived from the ER, ER-Golgi intermediate compartment, cis- or medial Golgi, or lysosomes. Components of the secretory apparatus become reoriented in the assembly compartment in conjunction with virion maturation (11, 36, 39, 48). In agreement with its role in virion maturation, the assembly compartment also contains many different virally encoded tegument, envelope, and nonstructural proteins (12, 16, 37, 39, 46). Microscopically, the assembly compartment has a cylindrical appearance with a height that is similar to that of the nucleus (13); its perinuclear location and the adjacent enlarged, kidney-shaped nucleus gives HCMV-infected cells a distinct morphology (41). It has been suggested that the many vesicles that make up the assembly compartment each hold tegument proteins which can be deposited in virions as they move through the space between the vesicles (13). Our data suggest two locations of BiP in the assembly compartment, namely, (i) a small space in the center of the assembly compartment and (ii) a ring at the outer edge of the cylindrical assembly compartment where BiP colocalizes with pp28. In between these two locations is a BiP-free region; our data show that one occupant of this region is gB. The outer ring of BiP fits well with the Pellett model (13) that suggests that the assembly compartment is made up of concentric rings of vesicles, each with specific tegument proteins that are sequentially picked up by nucleocapsids as they move from the outside to the center of the assembly compartment. In this scenario, BiP's protein binding and chaperone functions could aid in depositing tegument proteins on nucleocapsids, a mechanism of action that is supported by the finding that BiP is in the tegument layer of purified virions (49).
It has been suggested that the formation of the assembly compartment causes the condensation of other cytoplasmic structures (13, 29). These studies noted that the ER becomes located toward the periphery of the cell relative to the assembly compartment. This is similar to what we report by examining BiP's ER localization in infected cells. Thus, there is a connection between the formation of the assembly compartment and the BiP-containing condensed ER structures.
Although the mechanisms of assembly compartment formation and cytoplasmic ER condensation are unclear, our data suggest that the integrity of these structures is dependent on BiP, since BiP depletion causes both structures to dissociate. This agrees with our previously reported EM data, which showed that viral cytopathic effect in the cytoplasm of infected cells disappeared after depletion of BiP using either the SubAB toxin or short hairpin RNAs (8). A similar situation occurs in infections with HCMV with a mutation in the TRS1 gene (1, 4), suggesting that BiP and TRS1 may have shared or coordinate functions. The TRS1 gene product has been reported to have several functions during HCMV infection. Transient expression analyses suggest that it is a transcriptional activator, and other studies have suggested that it promotes oriLyt-dependent DNA replication (30, 31, 34, 44). Additionally, both TRS1 and its homolog IRS1 prevent the phosphorylation of eIF2α and the activation of RNase L (10, 14). More importantly for our study is the suggestion that TRS1 has a role in viral capsid assembly (1, 4). Our observations that BiP and TRS1 associate in the assembly compartment, and that a fraction of TRS1 copurifies with the assembly compartment, suggest that there is a functional relationship between BiP and TRS1 which is needed for assembly compartment integrity.
We have also characterized the association between BiP and pp28 in the assembly compartment. The pp28 protein (UL99 open reading frame) is a very abundant constituent of the tegument layer (6, 25, 49); it is a true late protein that is myristoylated and phosphorylated (19, 21, 37, 43). It is essential for the production of infectious virions, and mutation of the UL99 open reading frame resulted in nonenveloped and noninfectious cytoplasmic particles (7, 19, 43). This phenotype is similar to what we have reported for cells depleted of BiP (8). Thus, the association of BiP and pp28 appears to contribute to the same steps in virion maturation. Studies have demonstrated higher-molecular-weight forms of pp28 in infected cells, and both FRET assays and subcellular fractionation of infected cells suggested that pp28 multimerization occurs in the assembly compartment (40). Given BiP's chaperone function, it is possible that the close association between BiP and pp28 contributes to the multimerization of pp28.
The pattern of detection of BiP in the two cytoplasmic compartments by different antibodies suggests that antibodies to the C-terminal end of BiP cannot detect or immunoprecipitate BiP localized in the assembly compartment. This is especially true for antibodies to the KDEL sequence at the very C terminus of the protein, suggesting that C-terminal epitopes are blocked when BiP is in the assembly compartment. Such blockage of the KDEL ER localization signal by a viral protein would explain how BiP can be diverted from its normal ER localization to the assembly compartment.
In conclusion, our studies have shown that BiP, an important ER chaperone and mediator of the UPR, is diverted from the ER to the assembly compartment, where it associates with pp28 and TRS1, contributing to the formation, integrity, and function of this important virion maturation structure.
Acknowledgments
We thank all the members of the Alwine lab for critical evaluations of the data and encouraging discussions. In addition, we thank Bill Britt, Tom Shenk, and Nat Moorman for helpful discussions, reagents, and data sharing.
N.J.B. was supported by training grant T32 CA115299-02 awarded to Erle S. Robertson. The work was supported by the Abramson Family Cancer Research Institute and Public Health Service grant R01 CA028379-29, awarded to J.C.A. by the National Cancer Institute.
Footnotes
Published ahead of print on 9 September 2009.
REFERENCES
- 1.Adamo, J. E., J. Schroer, and T. Shenk. 2004. Human cytomegalovirus TRS1 protein is required for efficient assembly of DNA-containing capsids. J. Virol. 78:10221-10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alwine, J. C. 2008. Modulation of host cell stress responses by human cytomegalovirus, p. 263-279. In T. E. Shenk and M. F. Stinski (ed.), Current topics in microbiology and immunology, vol. 325. Human cytomegalovirus. Springer, New York, NY. [DOI] [PubMed] [Google Scholar]
- 3.Antonsson, A., and P. J. H. Johansson. 2001. Binding of human and animal immunoglobulins to the IgG Fc receptor induced by human cytomegalovirus. J. Gen. Virol. 82:1137-1145. [DOI] [PubMed] [Google Scholar]
- 4.Blankenship, C. A., and T. E. Shenk. 2002. Mutant human cytomegalovirus lacking the immediate-early TRS1 coding region exhibits a late defect. J. Virol. 76:12290-12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bresnahan, W. A., G. E. Hultman, and T. Shenk. 2000. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 74:10816-10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Britt, W. J., and S. Boppana. 2004. Human cytomegalovirus virion proteins. Hum. Immunol. 65:395-402. [DOI] [PubMed] [Google Scholar]
- 7.Britt, W. J., M. Jarvis, J. Y. Seo, D. Drummond, and J. Nelson. 2004. Rapid genetic engineering of human cytomegalovirus by using a lambda phage linear recombination system: demonstration that pp28 (UL99) is essential for production of infectious virus. J. Virol. 78:539-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchkovich, N. J., T. G. Maguire, A. W. Paton, J. C. Paton, and J. C. Alwine. 2008. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J. Virol. 82:31-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchkovich, N. J., Y. Yu, C. A. Zampieri, and J. C. Alwine. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signaling pathway. Nat. Rev. Microbiol. 6:265-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Child, S. J., M. Hakki, K. L. De Niro, and A. P. Geballe. 2004. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 78:197-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crump, C. M., C. H. Hung, L. Thomas, L. Wan, and G. Thomas. 2003. Role of PACS-1 in trafficking of human cytomegalovirus glycoprotein B and virus production. J. Virol. 77:11105-11113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das, S., and P. E. Pellett. 2007. Members of the HCMV US12 family of predicted heptaspanning membrane proteins have unique intracellular distributions, including association with the cytoplasmic virion assembly complex. Virology 361:263-273. [DOI] [PubMed] [Google Scholar]
- 13.Das, S., A. Vasanji, and P. E. Pellett. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 81:11861-11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hakki, M., E. E. Marshall, K. L. De Niro, and A. P. Geballe. 2006. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 80:11817-11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hegde, N. R., M. S. Chevalier, T. W. Wisner, M. C. Denton, K. Shire, L. Frappier, and D. C. Johnson. 2006. The role of BiP in endoplasmic reticulum-associated degradation of major histocompatibility complex class I heavy chain induced by cytomegalovirus proteins. J. Biol. Chem. 281:20910-20919. [DOI] [PubMed] [Google Scholar]
- 16.Homman-Loudiyi, M., K. Hultenby, W. Britt, and C. Söderberg-Nauclér. 2003. Envelopment of human cytomegalovirus occurs by budding into Golgi-derived vacuole compartments positive for gB, Rab 3, trans-Golgi network 46, and mannosidase II. J. Virol. 77:3191-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isler, J. A., T. G. Maguire, and J. C. Alwine. 2005. Production of infectious HCMV virions is inhibited by drugs that disrupt calcium homeostasis in the endoplasmic reticulum. J. Virol. 79:15338-15397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isler, J. A., A. H. Skalet, and J. C. Alwine. 2005. Human cytomegalovirus infection activates and regulates the unfolded protein response. J. Virol. 79:6890-6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones, T. R., and S. W. Lee. 2004. An acidic cluster of human cytomegalovirus UL99 tegument protein is required for trafficking and function. J. Virol. 78:1488-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaufman, R. J., D. Scheuner, M. Schroder, X. Shen, K. Lee, C. Y. Liu, and A. S. M. 2002. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 3:411-421. [DOI] [PubMed] [Google Scholar]
- 21.Kerry, J. A., M. A. Priddy, C. P. Kohler, T. L. Staley, D. Weber, T. R. Jones, and R. M. Stenberg. 1997. Translational regulation of the human cytomegalovirus pp28 (UL99) late gene. J. Virol. 71:981-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kudchodkar, S., Y. Yu, T. Maguire, and J. C. Alwine. 2004. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J. Virol. 78:11030-11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kudchodkar, S. B., G. Q. Del Prete, T. G. Maguire, and J. C. Alwine. 2007. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate-early times of human cytomegalovirus infection. J. Virol. 81:3649-3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kudchodkar, S. B., Y. Yu, T. G. Maguire, and J. C. Alwine. 2006. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc. Natl. Acad. Sci. USA 103:14182-14187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer, H., A. T. Bankier, M. P. Landini, C. M. Brown, B. G. Barrell, B. Ruger, and M. Mach. 1988. Identification and procaryotic expression of the gene coding for the highly immunogenic 28-kilodalton structural phosphoprotein (pp28) of human cytomegalovirus. J. Virol. 62:2243-2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohr, I. 2006. Phosphorylation and dephosphorylation events that regulate viral mRNA translation. Virus Res. 119:89-99. [DOI] [PubMed] [Google Scholar]
- 27.Moorman, N. J., I. M. Cristea, S. S. Terhune, M. P. Rout, B. T. Chait, and T. Shenk. 2008. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3:253-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murayama, T., S. Natsuume-Sakai, K. Shimokawa, and T. Furukawa. 1986. Fc receptor(s) induced by human cytomegalovirus bind differentially with human immunoglobulin G subclasses. J. Gen. Virol. 67:1475-1478. [DOI] [PubMed] [Google Scholar]
- 29.Ogawa-Goto, K., S. Irie, A. Omori, Y. Miura, H. Katano, H. Hasegawa, T. Kurata, T. Sata, and Y. Arao. 2002. An endoplasmic reticulum protein, p180, is highly expressed in human cytomegalovirus-permissive cells and interacts with the tegument protein encoded by UL48. J. Virol. 76:2350-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pari, G. S., and D. G. Anders. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979-6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pari, G. S., M. A. Kacica, and D. G. Anders. 1993. Open reading frames UL44, IRS1/TRS1, and UL36-38 are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA synthesis. J. Virol. 67:2575-2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paton, A. W., T. Beddoe, C. M. Thorpe, J. C. Whisstock, M. C. Wilce, J. Rossjohn, U. M. Talbot, and J. C. Paton. 2006. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443:548-552. [DOI] [PubMed] [Google Scholar]
- 33.Paton, A. W., P. Srimanote, U. M. Talbot, H. Wang, and J. C. Paton. 2004. A new family of potent AB5 cytotoxins produced by Shiga toxigenic Escherichia coli. J. Exp. Med. 200:35-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romanowski, M. J., E. Garrido-Guerrero, and T. E. Shenk. 1997. pIRS1 and pTRS1 are present in human cytomegalovirus virions. J. Virol. 71:5703-5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutkowski, D. R., and R. J. Kaufman. 2004. A trip to the ER: coping with stress. Trends Cell Biol. 14:20-28. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez, V., K. D. Greis, E. Sztul, and W. J. Britt. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez, V., E. Sztul, and W. J. Britt. 2000. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-Golgi-intermediate compartment. J. Virol. 74:3842-3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schroder, M., and R. J. Kaufman. 2006. Divergent roles of IRE1alpha and PERK in the unfolded protein response. Curr. Mol. Med. 6:5-36. [DOI] [PubMed] [Google Scholar]
- 39.Seo, J., and W. Britt. 2006. Sequence requirements for localization of human cytomegalovirus tegument protein pp28 to the virus assembly compartment and for assembly of infectious virus. J. Virol. 80:5611-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seo, J. Y., and W. J. Britt. 2008. Multimerization of tegument protein pp28 within the assembly compartment is required for cytoplasmic envelopment of human cytomegalovirus. J. Virol. 82:6272-6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Severi, B., M. P. Landini, and E. Govoni. 1988. Human cytomegalovirus morphogenesis: an ultrastructural study of the late cytoplasmic phases. Arch. Virol. 98:51-64. [DOI] [PubMed] [Google Scholar]
- 42.Sharon-Friling, R., J. Goodhouse, A. M. Colberg-Poley, and T. Shenk. 2006. Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc. Natl. Acad. Sci. USA 130:19117-19122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silva, M. C., Q. C. Yu, L. Enquist, and T. Shenk. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77:10594-10605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stasiak, P. C., and E. S. Mocarski. 1992. Transactivation of the cytomegalovirus ICP36 gene promoter requires the α gene product TRS1 in addition to IE1 and IE2. J. Virol. 66:1050-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talbot, U. M., J. C. Paton, and A. W. Paton. 2005. Protective immunization of mice with an active-site mutant of subtilase cytotoxin of Shiga toxin-producing Escherichia coli. Infect. Immun. 73:4432-4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Theiler, R. N., and T. Compton. 2002. Distinct glycoprotein O complexes arise in a post-Golgi compartment of cytomegalovirus-infected cells. J. Virol. 76:2890-2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tirosh, B., N. N. Iwakoshi, B. N. Lilley, A. Lee, L. H. Glimcher, and H. L. Ploegh. 2005. Human cytomegalovirus protein US11 provokes an unfolded protein response that may facilitate the degradation of class I major histocompatibility complex products. J. Virol. 79:2768-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tooze, J., M. Hollinshead, B. Reis, K. Radsak, and H. Kern. 1993. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 601:163-178. [PubMed] [Google Scholar]
- 49.Varnum, S. M., D. N. Streblow, M. E. Monroe, P. Smith, K. J. Auberry, L. Paša-tolić, D. Wang, D. G. Camp II, K. Rodland, S. Wiley, W. Britt, T. Shenk, R. D. Smith, and J. A. Nelson. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960-10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walsh, D., C. Perez, J. Notary, and I. Mohr. 2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J. Virol. 79:8057-8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xuan, B., Z. Qian, E. Torigoi, and D. Yu. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 83:3463-3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang, K., and R. J. Kaufman. 2006. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology 66:S102-S109. [DOI] [PubMed] [Google Scholar]