

Abstract
A structure of adenovirus type 12 (HAdV12) complexed with a soluble form of integrin αvβ5 was determined by cryo-electron microscopy (cryoEM) image reconstruction. Subnanometer resolution (8 Å) was achieved for the icosahedral capsid with moderate resolution (27 Å) for integrin density above each penton base. Modeling with αvβ3 and αIIbβ3 crystal structures indicates that a maximum of four integrins fit over the pentameric penton base. The close spacing (∼60 Å) of the RGD protrusions on penton base precludes integrin binding in the same orientation to neighboring RGD sites. Flexible penton-base RGD loops and incoherent averaging of bound integrin molecules explain the moderate resolution observed for the integrin density. A model with four integrins bound to a penton base suggests that integrin might extend one RGD-loop in the direction that could induce a conformational change in the penton base involving clockwise untwisting of the pentamer. A global conformational change in penton base could be one step on the way to the release of Ad vertex proteins during cell entry. Comparison of the cryoEM structure with bent and extended models for the integrin ectodomain reveals that integrin adopts an extended conformation when bound to the Ad penton base, a multivalent viral ligand. These findings shed further light on the structural basis of integrin binding to biologically relevant ligands, as well as on the molecular events leading to HAdV cell entry.
A growing number of viruses have been identified as using one of the 24 types of integrin heterodimers as a receptor for cell entry (32). Integrins are cell surface molecules involved in the regulation of adhesion, migration, growth, and differentiation (11). The large multidomained extracellular segments of α and β integrin subunits bind a variety of ligands, including viral ligands, while the smaller intracellular domains interact with cytoskeletal proteins (Fig. 1A). These extracellular and intracellular interactions facilitate bidirectional signaling, with the initiating events occurring either outside of the cell (outside-in signaling) or within the cell (inside-out signaling) (24). Integrin clustering has been established as having an important role in outside-in signaling (9, 19, 20, 44). Clustering results in the formation of focal adhesions, which are organized intracellular complexes, that facilitate downstream signaling cascades within the cell (24).
FIG. 1.
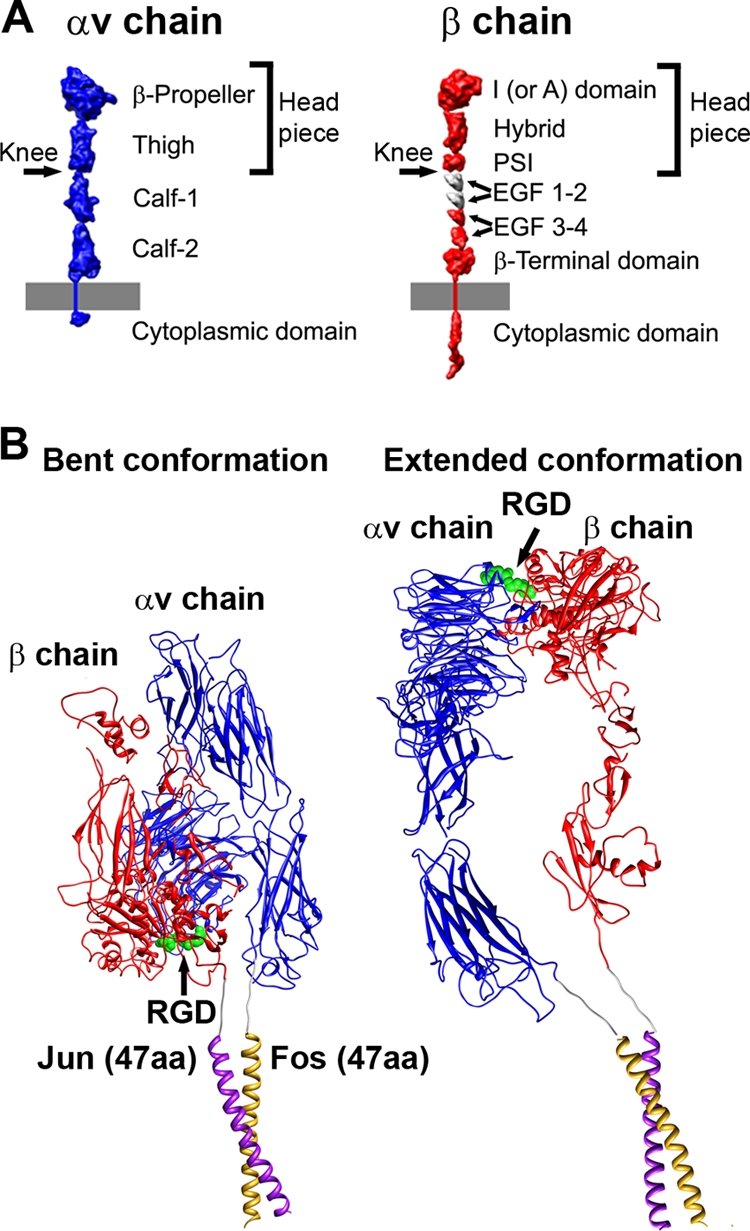
Integrin domains and conformations. (A) Structural domains of integrin αv and β chains, including the extracellular domains, transmembrane-spanning regions, and small cytoplasmic domains, shown in extended schematic forms. The domains are represented as 10Å-resolution density maps based on crystallographic coordinates. The membrane is represented by a gray bar. (Modified from Stewart and Nemerow (32) and reprinted with permission from Elsevier.) (B) Models for soluble αvβ5 integrin with Fos/Jun dimerization domains. Each chain has a six residue glycine-rich linker between the ectodomain and the Fos or Jun dimerization domain. The model of a bent integrin conformation (left) was built as a composite of αvβ3 integrin crystal structures, PDB-IDs 1L5G and 1U8C (42, 43), and the crystal structure of c-Fos/c-Jun bound to DNA, PDB-ID 1FOS (6). The model of an extended integrin conformation (right) is similar to the extended model docked into the HAdV12/αvβ5 cryo structure (Fig. 8B).
Studies of adenovirus (Ad) interactions with αv integrins provided some of the first evidence of the virus-induced signaling events (13, 14). The Ad penton base capsid protein, which sits at the 12 vertices of the icosahedral capsid, has five prominent Arg-Gly-Asp (RGD) containing loops that are flexible and protrude from the viral surface (31, 48). Receptor-mediated endocytosis of Ad is stimulated by interaction of the RGD-containing penton base with αvβ3 and αvβ5 integrins (34). This interaction leads to receptor clustering, followed by tyrosine phosphorylation/activation of focal adhesion kinase, as well as activation of p130CAS, phosphatidylinositol 3-OH-kinase, and the Rho family of small GTPases, and subsequent actin polymerization and Ad internalization (32). Integrin signaling events also lead to production of proinflammatory cytokines (23) and may result in increased survival of certain host cells through subsequent signaling to protein kinase B (AKT) (25).
Multiple studies indicate that after interaction with an RGD-containing ligand a straightening of the integrin extracellular domains occurs, leading to the “extension” or “switchblade” model for integrin activation (16, 45). In the extension model the headpiece domains, which are closest to the RGD interaction site, have a “closed” conformation in the low-affinity, unliganded state. This state is characterized by the close proximity of the α and β subunits at the “knees” or midpoints of the extracellular segments. In contrast, the high-affinity, ligand-bound state in the extension model is distinguished by an “open” headpiece conformation with separation at the knees of the extracellular segments. The location of the RGD binding site between the α-subunit β-propellor and the β-subunit I domain was first visualized in the crystal structure of the αvβ3 extracellular segment with a bound RGD peptide (43). In this structure the RGD site is folded back toward the membrane, and the integrin is in a closed conformation. The closed conformation has also been observed in crystal structures of the αvβ3 ectodomain without an RGD peptide (41) and the αIIbβ3 ectodomain (47).
The open integrin conformation has been characterized as having a large separation of up to ∼70 Å between the knees of α and β subunits (16). Four slightly different open headpiece conformations were observed in crystal structures of the αIIbβ3 headpiece with bound fibrinogen-mimetic therapeutics (38). These structures show that the change from a closed to an open headpiece conformation is accompanied by a piston-like motion of helix α7 in the β-chain I domain and a large swing of the β-chain hybrid domain of up to 69°, as well as extension and separation of the two integrin chains. Comparison of the available αvβ3 and αIIbβ3 crystal structures is providing information on the interdomain angle variation and flexibility between domains (47).
One aspect of the extension model is that separation of the C-terminal, intracellular portions of the α and β subunits leads to inside-out activation. This concept is supported by nuclear magnetic resonance structures of the cytoplasmic tails of αIIbβ3 showing that the membrane-proximal helices engage in a weak interaction that can be disrupted by constitutively activating mutations or by talin, a protein found in high concentrations in focal adhesions (33). The concept that the integrin α and β subunits must also separate during outside-in signaling is supported by a study involving a disulfide-bonded mutant of αIIbβ3 integrin (46). When the α and β subunits are linked in the vicinity of the transmembrane helices the mutant αIIbβ3 is still able to bind ligand, mediate adhesion, and undergo antibody-induced clustering. However, the disulfide-bonded mutant exhibits defects in focal adhesion formation and focal adhesion kinase activation. Reduction of the disulfide bond or single cysteine mutants rescues signaling.
A competing model for integrin activation, called the “deadbolt” model, proposes only small conformational changes in the integrin β-chain I domain upon RGD binding (2). This model is based on crystal structures of the αvβ3 ectodomain with or without an RGD peptide (41, 43). Both of these αvβ3 structures reveal a bent integrin conformation with a closed headpiece conformation. However, the RGD peptide was soaked into a preformed crystal of αvβ3 and crystal contacts may have prevented conformational changes.
There are relatively few and only moderate resolution structures of virus-integrin complexes. A moderate resolution cryoEM structure has been determined for the Picornavirus echovirus 1 (EV1) in complex with the I domain of the α2 integrin subunit (39). Docking of crystal structures of EV1 and the α2 I domain into the cryoEM density indicates that the I domain binds within a canyon on the surface of EV1 and that five integrins could potentially bind at one vertex of the icosahedral capsid. Confocal fluorescence microscopy experiments indicated that EV1 causes integrin clustering on human osteosarcoma cells stably transfected with α2 integrin. However, it could not be determined whether the bound integrins were in the inactive (bent) or active (extended) conformation.
Moderate resolution (∼21 Å) cryoEM structures of Ad type 2 (HAdV2) and HAdV12 in complex with a soluble form of αvβ5 integrin revealed a ring of integrin density over each penton base capsid protein (5). Better-defined integrin density was observed in the HAdV12/integrin complex, supporting the idea suggested from sequence alignments that the RGD loop of the HAdV12 penton base is shorter and less flexible than that of HAdV2. This study also suggested that the precise spatial arrangement of the five RGD protrusions on the penton base might promote integrin clustering, which may lead to the intracellular signaling events required for virus internalization into a host cell. A similar spacing of RGD-containing integrin-binding sites around the fivefold axis of icosahedral virions has been noted for Ad, foot-and-mouth disease virus, and coxsackievirus A9 (32).
We present here a significantly higher-resolution cryoEM structure of HAdV12 complexed with soluble αvβ5 that provides insight into the Ad-integrin interaction. The resolution of the icosahedral capsid portion of the Ad-integrin complex was improved to 8 Å, and the capsid shows clearly resolved α-helices, which allows accurate docking of the penton base crystal structure within the cryoEM density. The resolution of the integrin density is more moderate due to flexibility of the RGD-containing surface loop of penton base and incoherent averaging of integrin heterodimers. Nevertheless, modeling studies with available integrin crystal structures have enabled us to distinguish between a bent or extended conformation (Fig. 1B) when αvβ5 binds to the multivalent ligand presented by the Ad penton base. The cryoEM structural analysis also indicates that integrin induces a conformational change in penton base.
MATERIALS AND METHODS
Preparation and isolation of HAdV12 and soluble αvβ5 integrin.
The integrin used for the cryoEM study was a soluble form of recombinant αvβ5 produced in insect cells as previously described (17). This recombinant form of αvβ5 has an αv chain formed by the complete αv ectodomain followed by a glycine linker and the Fos dimerization domain. The β5 chain is formed by the entire β5 ectodomain, followed by a glycine linker and the Jun dimerization domain.
HAd12V (ATCC) was propagated in 293 cells and purified by two cycles of cesium chloride density gradient ultracentrifugation as previously described (36). The banded virus was then dialyzed against Tris buffer at pH 8.0 (50 mM Tris, 130 mM NaCl, 3 mM KCl).
CryoEM.
The HAdV12 sample (250 μg/ml) and a soluble recombinant form of αvβ5 (17) (309 μg/ml) were incubated for 2 h in a Tris buffer (pH 8.1; 1 mM Ca2+, 1 mM Mg2+). The sample was prepared to contain about five integrin molecules per RGD binding site. CryoEM grids were prepared as described by Saban et al. (28), applying 6 μl of sample to each Quantifoil R2/4 holey carbon grid (Quantifoil Micro Tools GmbH). The Vitrobot cryo-fixation device (FEI Company) was used for flash freezing of the grids in a cryogen. An FEI Polara (300 kV; FEG) transmission cryo-electron microscope operated at liquid nitrogen temperature and 300 kV with a Gatan UltraScan 4kx4k charge-coupled device camera was used for data acquisition with SAM, a semiautomatic data collection routine (30). The absolute magnification for the data was ∼×398,000.
Image processing.
The automatic selection program VIRUS (1) was used to extract particle images from the cryo-electron micrographs. The initial box size of the particle images was chosen to include ∼500 Å beyond the edge of the HAdV12 capsid in order to visualize the bound integrin. An in-house script was used to bin the particle images to three sizes for image processing: 3202 pixels (4.8 Å pixels), 6402 pixels (3.2 Å pixels), and 9602 pixels (1.6 Å pixels). The program CTFFIND3 (18) was used to determine initial estimates for the microscope defocus and astigmatism parameters. The cryoEM structure of Ad35f (29) filtered to 12-Å resolution was used as the starting model for refinement with FREALIGN (8). A total data set of 2,499 particle images was refined with first the coarsest pixel size (4.8 Å), then with the intermediate pixel size (3.2 Å), and lastly with the finest pixel size (1.6 Å). In the initial refinement rounds a modified version of FREALIGN was used to allow the input of externally determined particle centers (29). The orientational parameters (two translational parameters and three Euler angles), as well as the defocus, astigmatism, and magnification parameters, were refined on a per-particle basis. The final reconstruction included 1,141 particle images, selected on the basis of their FREALIGN phase residual parameter. The resolution of the final reconstruction was assessed the Fourier shell correlation (FSC) curve calculated by FREALIGN. Maps were calculated for three radial shells: 300 to 463 Å, corresponding to the icosahedral capsid, 463 to 515 Å for the proximal integrin density, and 515 to 700 Å for the distal integrin density.
A temperature factor (B = −300Å2 or B = −500Å2 as specified) and cosine edge filtering was applied to the final density maps with the BFACTOR program (http://emlab.rose2.brandeis.edu/grigorieff/download_b.html). The graphics figures were produced with USCF Chimera (22).
The CoLoRes routine from SITUS (4) was used to refine the absolute pixel size of the cryoEM density map to within 0.05 Å. The absolute pixel size was taken as the value that gave the maximal cross correlation coefficient between the X-ray crystal structure of penton base (PDB-ID 1X9T) (48) and a selected portion of the cryoEM density map, including one penton base pentamer.
De novo model building for penton base RGD loop.
Alignment of penton base sequences from multiple human Ad serotypes and comparison with the HAdV2 penton base crystal structure (48) indicates that the HAdV12 penton base is likely to have a flexible and extended RGD-containing loop of 15 amino acids. Within this loop there are 6 amino acids on either side of the RGD residues. The crystal structure of HAdV2 penton base (PDB-ID 1X9T) (48) does not contain coordinates for the RGD loop. A Rosetta loop building protocol was used (27) to build models for the RGD loop in HAdV12 (residues 296 to 312). A total of 25 loop models were built. Ten of the twenty-five models were selected for Fig. 3. This subset is representative of the maximum variation within the set of 25 models.
FIG. 3.

Penton base in the HAdV12/αvβ5 integrin complex and models for the integrin-binding RGD loop. (A) Top view of the HAdV12 penton base cryoEM density (mesh) with the docked crystal structure of the HAdV2 penton base pentamer and fiber peptide (PDB-ID 1X9T) (48). (B) Side view of the same but with the cryoEM density shown at a higher isosurface contour level to reveal α-helical density rods. The arrows indicate the longest α-helix in the penton base monomer. The rectangle indicates the α-helical density below the penton base assigned to protein IIIa (29). Each penton base monomer is in a different color with the fiber peptide at the top of the penton base in green. (C and D) Top and side views of the HAdV2 penton base crystal structure (gray) with 10 different models for the HAdV12 RGD loop (amino acids 296 to 312) built with Rosetta (27).
Integrin docking within the HAdV12/αvβ5 integrin cryoEM density.
Given the high homology between the β3 and β5 integrin chains (identity, 55%; similarity, 70%; gaps, 4%), the crystal structures of αvβ3 (41, 42) and the αvβ3/RGD peptide complex (43) are reasonable for comparison with the integrin density in the HAdV12/αvβ5 complex. There is also significant homology between the αv and αIIb integrin chains (identity, 30%; similarity, 41%; gaps, 4%). Therefore, we have also compared the cryoEM density with the platelet integrin αIIbβ3 crystal structures complexed with fibrinogen-mimetic therapeutics, which display open headpiece conformations (38).
A density section encompassing the vertex region of the HAdV12/αvβ5 integrin cryoEM structure was extracted for docking with atomic resolution integrin structures. Two integrin domains (the β-propeller of the αv chain, and the I domain of the β3 chain) together with RGD peptide were selected from the crystal structure of the extracellular segment of integrin αvβ3 in complex with an RGD ligand (PDB-ID 1L5G) (43) for initial docking experiments. Interactive docking was performed with UCSF Chimera (22). The two αvβ3 integrin domains and RGD peptide were docked as a rigid body into the cryoEM integrin density.
In a second phase of docking experiments four conformations of the β3 integrin chain in crystal structures of αIIbβ3 complexes with fibrinogen-mimetic therapeutics (PDB-IDs 2VDK and 1TYE) (38) were evaluated in the context of the HAdV12/αvβ5 cryoEM integrin density. Each of the four β3 conformations has a different angle (57°, 59°, 61°, or 69°) between the β chain I and hybrid domains. The I domain of each of these conformers was aligned with the previously docked I domain from the αvβ3/RGD crystal structure by using the UCSF Chimera Matchmaker tool.
A model for the complete αvβ5 integrin ectodomain in a bent conformation was built as a composite of crystal structures of αvβ3 (PDB-ID 18UC) (42) and the αvβ3/RGD peptide complex (PDB-ID 1L5G) (43). A model for the complete integrin ectodomain in an extended conformation was built as a composite of crystal structures of the αvβ3/RGD peptide complex and an αIIbβ3/ligand mimetic complex (PDB-ID 2VDK) (38). The α-chain domains calf-1 and calf-2 and the β-chain domains EGF1-EGF4 and terminal domain in the extended model were taken from an αvβ3 crystal structure (PDB-ID 1U8C) and roughly positioned into the cryoEM density.
RESULTS
CryoEM structure of HAdV12/αvβ5 complex indicates disorder for the bound integrin.
HAdV12/αvβ5 integrin complexes were formed with a soluble, recombinant form of αvβ5 (17). This form of αvβ5 retains the ability to recognize the Ad penton base, as well as vitronectin, an RGD-containing extracellular matrix protein. A kinetic analysis of the interaction between HAdV2 virions with this soluble form of αvβ5 indicated ∼4.2 integrin molecules bound per penton base at close to saturation (5). The first cryoEM study of the HAdV12/αvβ5 complex reached only moderate (∼21-Å) resolution (5), and at that time there were no atomic resolution structures of αv integrins available for docking into the cryoEM density. We have performed additional cryoEM analyses of HAdV12/αvβ5, pushed the resolution beyond 10 Å for the icosahedral capsid, and analyzed the resulting cryoEM density with the available αvβ3 and αIIbβ3 crystal structures.
A cryoEM data set with 2,499 particle images of HAdV12/αvβ5 complexes was acquired on an FEI Polara (300 kV; FEG) microscope, which allowed significant improvement in the resolution. The final cryoEM structure presented in Fig. 2 is based on a subset of ∼45% of the data set (1,141 particle images) selected to yield the highest resolution for the icosahedral capsid. The resolution was assessed in three radial shells corresponding to the icosahedral capsid (radii of 300 to 463 Å), the proximal integrin density (radii of 443 to 515 Å), and the more distal diffuse integrin density extending away from the virion (radii of 515 to 600 Å). Overlapping radial shells were selected for the capsid and proximal integrin density, since the integrin extends over the RGD containing protrusions of the penton base.
FIG. 2.

CryoEM structure of the HAdV12/αvβ5 integrin complex. (A) Full structure viewed along a twofold icosahedral axis and shown as three radial shells: icosahedral capsid (300 to 463 Å, blue), integrin ring and fiber (443 to 515 Å, gold), and diffuse integrin density (515 to 600 Å, red). The icosahedral capsid shell is shown filtered to 8 Å with an applied B-factor of −300 Å2, the integrin ring and fiber filtered to 19 Å with an applied B-factor of −500 Å2, and the diffuse integrin density filtered to 33 Å with an applied B-factor of −500 Å2. (B) Enlarged top and side views of the vertex region colored as in panel A with the fiber shaft in green. The diffuse integrin density (red) is shown with a lower isosurface contour level in panel B (1.2σ versus 1.7σ in panel A) to display a fuller extent of this density. Scale bars, 100 Å. (C) An FSC plot indicating a resolution range for the HAdV12/αvβ5 icosahedral capsid (radial shell, 300 to 463 Å) of 8.3 to 6.9Å (8.3 Å, FSC = 0.5; 7.5 Å, FSC = 0.3; 6.9 Å, FSC = 0.143) (blue); the HAdV12/αvβ5 radial shell (443-515Å) including the integrin ring and fiber above the penton base of 27Å to 19Å (27 Å, FSC = 0.5; 24 Å, FSC = 0.3; 19 Å, FSC = 0.143) (gold); and the HAdV12/αvβ5 radial shell (515-600Å) with diffuse integrin density of 85Å to 33Å (85 Å, FSC = 0.5; 50 Å, FSC = 0.3; 33 Å, FSC = 0.143) (red).
The resolution of the icosahedral HAdV12 capsid is 8.3 Å (FSC 0.5 threshold) and, as expected for this resolution, α-helices within the capsid proteins, including penton base and protein llla, are clearly resolved as density rods (Fig. 3B). A ring of integrin density is observed over each penton base with well-defined connector regions stretching away from the capsid surface and surrounding the fiber shaft (Fig. 2B, gold density). The resolution of the radial shell with the integrin ring is 27 Å (FSC = 0.5), which is significantly worse than that of the capsid, indicating static or dynamic disorder for the bound integrin. Although the integrin ring displays fivefold symmetry and gives the impression that there are five copies of integrin bound to the penton base, icosahedral symmetry has been imposed during the reconstruction process. In the outermost radial shell the resolution is 85 Å (FSC = 0.5). This shell has weak and diffuse density over the vertices (Fig. 2B, red density), indicating that the disorder of the bound integrin increases with greater distance from the capsid.
When the resolution is assessed over all radial shells (radii of 0 to 700 Å), the overall resolution of the HAdV12/αvβ5 structure is 10.9 Å (FSC = 0.5). This is significantly better than the resolution previously reported for the same complex (5).
Steric hindrance limits the number of integrins bound per penton base.
The crystal structure the HAdV2 penton base (48) fits well within the cryoEM density for the HAdV12 penton base in the HAdV12/αvβ5 complex (Fig. 3A and B). The HAdV2 penton base has a high degree of homology (65% identity, 71% similarity) with the HAdV12 penton base and thus serves as a reasonable model for analysis of the cryoEM density. The biggest difference between the two sequences is that the HAdV12 RGD loop is much shorter, with just 15 amino acids in the HAdV12 loop corresponding to 78 amino acids in the HAdV2 penton base RGD loop. The longer HAdV2 RGD loop is missing from the crystal structure due to disorder. Presumably, the HAdV12 RGD loop is also flexible. In the absence of a crystal structure for the HAdV12 penton base, we used the Rosetta loop building protocol to model the HAdV12 RGD loop. Rosetta was first developed for de novo protein structure prediction and has been extended to model loops and protein segments. In a benchmark study, it was shown that segments of 13 to 18 amino acids can be modeled with accuracies of 1 to 7 Å root mean square deviation (27). Rosetta loop building produced 25 models for the HAdV12 RGD loop all with approximately the same score and with various conformations (Fig. 3C and D). We interpret this result to indicate that it is possible for the HAdV12 RGD loop to extend in a variety of directions from the top of the penton base.
The crystal structure of the αvβ3 extracellular segment with a bound RGD peptide reveals the RGD binding site between the αv-chain β-propeller domain and the β-chain I domain (43). Manual docking of these two integrin domains within the integrin ring above the penton base shows a basic level of agreement between the size of these domains and the dimensions of the ring in the HAdV12/αvβ5 structure (Fig. 2B and 4). A comparison of the interdomain angles in the various integrin crystal structures indicates little variability (∼3°) between the α-chain β-propeller and β-chain I domain (47). Therefore, this two-domain integrin unit (β-propeller/I domain/RGD peptide) from the αvβ3/RGD crystal structure (43) was docked as a rigid body within the cryoEM density. The HAdV2 penton base crystal structure was simultaneously docked in the capsid density. During the integrin docking performed with UCSF Chimera (22), distances were monitored between the residues on either side of the RGD-loop gap in the penton base crystal structure with the Arg and Asp residues of the RGD peptide within the αvβ3/RGD crystal structure. There are seven residues on either side of the central G residue in the RGD sequence of the HAdV12 loop. Our goal was to keep the monitored distances at <25 Å, corresponding to the maximum span of seven fully extended amino acid residues (26).
FIG. 4.

The RGD-binding integrin domains form the ring of density over the penton base in the HAdV12/αvβ5 structure. (A) The two RGD-binding integrin domains, αv chain β-propeller (blue) and β chain I domain (red), together with the RGD residues (green) from the αvβ3/RGD crystal structure (PDB-ID 1L5G), are shown modeled over the HAdV2 penton base crystal structure (PDB-ID 1X9T). One monomer of penton base is shown in gold, the rest in gray, and the fiber peptides are depicted as wide black ribbons. The missing residues in the penton base RGD loop are represented by dashed lines. (B) The penton base is shown with four docked copies of the two-domain integrin unit. (C) The penton base is shown with simulated density (light blue) generated from the integrin models in panel B with fivefold averaging and filtered to 27-Å resolution.
Although previous cryoEM analyses of Ad-integrin complexes at lower resolution suggested the possibility that up to five integrins could bind to each penton base, our present high-resolution analysis reveals that only four β-propeller/I domain/RGD peptide units can be docked without clashes between neighboring integrins (Fig. 4B). Three of the four integrin units can be docked without exceeding the RGD-loop distance constraint, and the fourth integrin unit only seems to fit with an apparent violation in the RGD-loop constraint (with distances of ∼30 Å). If one assumes that small conformational changes at the top of the penton base monomer are possible, a fourth integrin could be bound at the top of the penton base. After docking four integrin β-propeller/I domain/RGD peptide units over the penton base, the fifth RGD protrusion is effectively shielded by the bound integrin.
The cryoEM density for the integrin ring appears to have fivefold symmetry, but this may only be due to the fact that icosahedral symmetry has been imposed on the cryoEM structure. We tried to saturate the RGD binding sites by adding a fivefold excess of integrin molecules when preparing the HAdV12/αvβ5 complex, but there may be penton bases with fewer than the maximum number of integrin molecules bound. The cryoEM structure represents an icosahedrally averaged representation of the particle images included in the final structure. Therefore, the cryoEM density alone does not indicate the number of integrin molecules bound per penton base. However, docking of integrin crystal structures into the cryoEM density indicates that a maximum of four integrins can bind to one HAdV12 penton base. This is consistent with the previous Biacore measurement of ∼4.2 integrin molecules bound per HAdV2 penton base at close to saturation (5).
Integrin molecules bind in various orientations with respect to the penton base.
During the docking procedure it became apparent that the close spacing of the RGD protrusions on the penton base relative to the size of the two-domain integrin unit precludes the possibility of integrin binding in the same orientation to each of the neighboring RGD sites. All four docked integrin units are approximately contained within the cryoEM envelope for the integrin ring, but they are each in a different orientation relative to the underlying penton base monomer. Incoherent averaging of different integrin orientations relative to the icosahedral HAdV12 capsid, would explain the markedly different resolutions (8.3 Å versus 27 Å) between the capsid and the radial shell with the integrin ring. The integrin ring is only ∼40 Å above the capsid surface at its midpoint, and thus it must be bound in an asymmetric or highly flexible manner in order to explain the dramatic fall off in resolution from the icosahedral capsid. To test the effect of incoherent averaging, coordinates were saved for the four docked 2-domain integrin units and converted to a fivefold averaged density map (Fig. 4C). This resulted in a density ring that strongly resembles that observed above the penton base in the cryoEM structure (Fig. 2B).
The adjacent integrin domains, αv thigh and β-chain hybrid and PSI, were included in the analysis in an attempt to model the connector regions extending radially outward above the integrin ring in the cryoEM structure (Fig. 5). There is much greater variability observed in crystal structures for the interdomain angle between the β-chain I and hybrid domains (0° to 70°) than between the α-chain β-propeller and thigh domains (up to 10°) (47). For the docking analysis we built composite integrin models from the αvβ3/RGD crystal structure (43) and the αIIbβ3/ligand crystal structures with various swing angles for the β-chain hybrid domain (38). Although the individual integrin domains are not resolved in the cryoEM HAdV12/αvβ5 structure, composite models with an angle between the β-chain I and hybrid domains of 57° to 69° fit the best. With the adjacent integrin domains included with a 69° I/hybrid domain angle the fivefold averaged density map shows connectors similar to those in the cryoEM structure (Fig. 2B and 5C).
FIG. 5.

The integrin headpiece forms the ring and connector regions stretching away from the penton base in the HAdV12/αvβ5 structure. (A) The additional integrin headpiece domains are labeled. The RGD-binding integrins domains are modeled above the penton base as in Fig. 4. This is a composite integrin headpiece model built from PDB-IDs 1L5G and chain B of 2VDK with a 69° angle between the β-chain I and hybrid domains (38). (B) The penton base is shown with four docked copies of the integrin headpiece. (C) The penton base is shown with simulated density (light blue) generated from the integrin models in panel B with fivefold averaging and filtered to 27-Å resolution.
In order to estimate the degree of incoherent averaging that would be caused by four integrins bound as in our integrin/penton base model; we aligned the penton base monomers and measured the distances between all pairs RGD peptides (Fig. 6B). These distances vary from 10 to 29 Å. This large variation would cause a significant reduction, presumably on the order of 10 Å, in the resolution of the integrin ring compared to the icosahedral capsid.
FIG. 6.

Natural twist of penton base and possible untwisting by integrin. (A) Space-filling representation of the penton base pentamer (PDB-ID 1X9T) with each subunit in a different color. The natural twist of one subunit from the bottom of the pentamer to the top solvent accessible surface in the virion is represented by the arrow. (B) Side view of four superimposed penton base monomers with four copies of the RGD residues (magenta, cyan, blue, and green) as modeled in Fig. 4 and 5. Note that the magenta copy of the RGD residues extends the RGD loop counter to the natural twist of the penton base monomer. (C) Side view of the penton base pentamer shown with one monomer in magenta, fiber peptides in green, and the modeled RGD-binding integrin domains (blue and red) with the magenta copy of the RGD residues. (D) The integrin domains are removed, and an arrow indicates the direction that an integrin bound in the manner of panel C would tend to extend the RGD loop of penton base and lead to untwisting. (E) Top view of the penton base pentamer with a curved arrow indicating clockwise untwisting of the penton base.
Evidence of conformational change in penton base.
One of the docked integrin positions effectively pulls the RGD loop in a direction that is counter to the natural twist of the pentamer (Fig. 6). The HAdV2 penton base crystal structure shows that each monomer has two domains, with a right-handed twist between the lower and upper domains (Fig. 6A) (48). When viewed from the top this appears as a counterclockwise rotation from the lower to the upper domain. We propose, based on our model of the penton base/integrin interaction, that when four integrins are bound a clockwise rotation is induced in one penton base monomer (Fig. 6D and E). This rotation direction is counter to the natural twist of the pentamer. A conformational change of this sort could lead to a reduced number of intermolecular contacts between the penton base and the neighboring hexons in the capsid and to release of the penton base from the capsid. The penton base is known to be released along with fiber and protein IIIa in heat denaturation assays designed to mimic cell entry (35).
Close examination of the capsid density in the cryoEM HAdV12/αvβ5 structure and comparison with the cryoEM structure of the Ad35f vector (29) suggests that a small percentage of the penton bases and the associated copies of fiber and protein IIIa have been lost in the HAdV12/αvβ5 sample. When the Ad35f cryoEM structure is displayed with an isocontour level of 1.7σ, α-helices within both hexon and penton base are observed as density rods (Fig. 7A). When the HAdV12/αvβ5 structure is isocontoured to display the hexon α-helices as rods (at 2.8σ), the penton base density is weak (Fig. 7B). Alternatively, when the HAdV12/αvβ5 structure is isocontoured to display penton base α-helices as rods (at 2.3σ), the hexon density includes significantly more than just α-helical density rods. These results indicate that some percentage of the penton bases are missing in the HAdV12/αvβ5 particle images that contribute to the cryoEM structure. The hexon density in the HAdV12/αvβ5 structure also appears stronger than the protein IIIa density underneath the penton base (data not shown). We speculate that integrin binding to Ad might predispose the penton base and protein IIIa to release from the capsid, even in the absence of the host cell membrane and the low pH environment of the endosome. The loss of penton base and protein llla at the vertex region is consistent with our model of induced conformational changes by integrin engagement.
FIG. 7.

The penton base has low partial occupancy in the HAdV12/αvβ5 cryoEM structure. (A) Thin (∼20-Å) slab of the Ad35f cryoEM structure (29) with docked crystal structures of hexon (cyan) (PDB-ID 1P30) and penton base (gold) with the N-terminal fiber peptide (green). The cryoEM density (mesh) is isocontoured at 1.7σ and shows rods for α-helices in both hexon and penton base. (B) Thin slab of the HAdV12/αvβ5 cryoEM structure with the same docked atomic resolution structures. The cryoEM density is isocontoured at 2.8σ and shows rods for α-helices in hexon and almost no density for the penton base. (C) Same as panel B but with the HAdV12/αvβ5 cryoEM density isocontoured at 2.3σ showing rods for α-helices in penton base similar to those shown for Ad35f in panel A, but with significantly more hexon density than in panel A. Both maps are shown filtered to 7.5 Å with the same applied B factor (−300 Å2).
CryoEM density is consistent with an extended integrin conformation.
The cryoEM structure of the HAdV12/αvβ5 integrin complex also provides information on the overall conformation of the extracellular portion of αvβ5 integrin in complex with a multivalent ligand, the Ad penton base. Keeping the RGD-binding domains as modeled in Fig. 4, we added the remaining αv (blue) and β (red) extracellular domains to form either a bent conformation or an extended integrin conformation (Fig. 8). The αvβ3/RGD crystal structure represents a bent conformation for an almost complete ectodomain just lacking the β-chain PSI domain (43). The PSI domain was added from another αvβ3 crystal structure of the complete ectodomain in a bent conformation (42). There is currently no available atomic resolution structure for a complete ectodomain in the extended conformation. Therefore, we built a composite model starting with the headpiece domains as modeled in Fig. 5 and adding the remaining αv and β chain domains to roughly approximate the diffuse integrin density in the outer radial shell (515 to 600 Å) of the cryoEM structure.
FIG. 8.

Comparison of HAdV12/αvβ5 integrin density with bent and extended integrin models. (A) The cryoEM integrin density (mesh) is shown with four copies of the integrin ectodomain in a bent conformation. This is a composite integrin model built from PDB-IDs 1L5G (43) and 18UC (42). (B) The penton base is shown with four copies of the integrin ectodomain in an extended conformation. This is a composite integrin model built from the domains shown in Fig. 5 with the remaining domains modeled to approximate the cryoEM density in the outermost radial shell (515 to 600 Å) of the HAdV12/αvβ5 structure. In both panels the αv chains are blue, the β chains are red, the bound RGD peptides are green, and the penton base is gold. Note that the gap between the upper and lower cryoEM integrin density regions is an artifact from calculating the density in separate radial shells.
Comparison of the bent integrin model with the cryoEM density shows that a significant portion of the integrin model folds back toward the penton base rather than filling the diffuse integrin density in the outer radial shell (Fig. 8A). Overall, the bent integrin conformation is inconsistent with the shape of the cryoEM density. Instead, we found that the integrin density in the HAdV12/αvαβ5 structure is more consistent with an extended conformation with the integrin α and β chains extending away from the penton base (Fig. 8B). In our cryoEM-based model for extended integrins bound to penton base, the C-terminal ends of the integrin ectodomains are all clustered around the Ad fiber and are within ∼100 Å of one another.
The soluble form of αvβ5 integrin used in this cryoEM study has the C-terminal ends of the αv and β5 chains tethered together with 7-amino-acid Gly-rich linkers and Fos/Jun dimerization domains (17). The distance between the ends of the Fos/Jun domains when they are dimerized is 12 Å, as measured in a crystal structure of c-Fos/c-Jun bound to DNA (PDB-ID 1FOS) (6). The distance between the C-terminal ends of the αv and β3 integrin chains in the bent conformation in the αvβ3/RGD crystal structure is surprisingly similar at 13 Å (Fig. 1B). Therefore, the Fos/Jun dimerization domains plus Gly-rich linkers should not impede the formation of a bent integrin conformation as observed in the αvB3/RGD crystal structure. The soluble αvβ5 integrin has one additional residue at the C-terminal end of both the αv and β5 chains compared to the αvβ3/RGD crystal structure. So, we have modeled the effect of the linkers and Fos/Jun dimerization domains on the extended integrin conformation assuming 8-amino-acid linkers in each integrin chain. Our calculations indicate that the Fos/Jun dimerization domains plus two 8-amino-acid linkers would allow the C-terminal ends of the αv and β5 chains to spread apart by up to 70 Å (assuming an extended length of 3.6 Å per residue in the two linkers [26]). This maximum distance is consistent with our cryoEM-based model for four extended integrin ectodomains bound to penton base (Fig. 8B). Although our model has the αv and β5 C-terminal pairs within ∼15 Å of each other (Fig. 1B), the diffuse nature and low resolution (85 Å) of the integrin density in the outermost radial shell (Fig. 2B) suggest that the integrin chains in this region are highly disordered and may well spread farther apart.
DISCUSSION
Symmetry or asymmetry of receptor binding.
CryoEM structures of various picornavirus/receptor complexes, including rhinovirus and coxsackievirus with ICAM-1 (12, 37) and poliovirus with the poliovirus receptor CD155 (3, 10, 40), have revealed one receptor per asymmetric unit or 60 receptor molecules per virion. This is in contrast to what we have observed for the interaction of adenovirus with its integrin receptor. In the case of both ICAM-1 and the poliovirus receptor, elongated density was observed for 60 discrete receptor binding sites on the viral capsid after imposed icosahedral symmetry. The highest resolution of these cryoEM studies is an 8.0 Å (FSC = 0.5) resolution structure of coxsackievirus A21 complexed with an ICAM-1 variant (37). This resolution assessment was made for the radial shells corresponding to the viral protein shell and the first domain (D1) of ICAM-1. CryoEM density was also observed for four additional ICAM-1 domains (D2 to D5) extending outward from the viral surface. However, the resolution was worse, and the density strength was progressively lower for each successive domain, suggesting flexibility at each elbow between domains. Although the strength of the cryoEM density for the D1 domain of ICAM-1 suggested only 80% occupancy of ICAM-1 binding sites, docking with crystal structures for both coxsackievirus A21 and ICAM-1 indicated no reason why 60 receptors molecules might not bind to one virion. Our cryoEM structure of HAdV12/αvβ5 is similar to CAV21/ICAM-1 in that more moderate resolution and weaker density is observed at progressively greater distances from the viral capsid. However, the HAdV12/αvβ5 structure differs in that discrete density is not observed at 60 receptor binding sites. In fact, docking with crystal structures of integrin indicates that 60 integrin molecules are unlikely to bind to one HAdV12 virion simultaneously.
The HAdV12/αvβ5 cryoEM structure provides a model for the penton base/integrin interaction.
HAdV12 was chosen for this cryoEM structural study since its penton base has the shortest integrin-binding RGD loop. We reasoned that the short HAdV12 RGD-loop would be the least flexible among the various Ad types and thus the HAdV12/αvβ5 complex would yield the highest resolution for the bound integrin. We present here a cryoEM structure of the HAdV12/αvβ5 complex with a resolution of 8.3 Å (FSC = 0.5) for the icosahedral capsid. This allowed visualization of α-helices in the capsid and facilitated accurate docking of the penton base crystal structure within the cryoEM density. The Rosetta de novo protein modeling software was used to build atomic models for the RGD-containing loop of the HAdV12 penton base. These models suggest that the RGD loop is quite flexible and might extend in various directions from the top of the penton base.
The HAdV12/αvβ5 cryoEM structure displayed significantly lower resolution for the integrin density than that observed for the capsid. The structure showed a ring of integrin density on top of the penton base RGD-protrusions and surrounding the Ad fibers. The resolution of this radial shell of integrin density is 27 Å (FSC = 0.5). Beyond this integrin ring, the cryoEM structure showed additional diffuse integrin density with an even lower resolution of 85 Å (FSC = 0.5) extending out ∼160 Å from the top of the penton base. Rigid body modeling with the RGD-binding domains of the homologous αvβ3 integrin indicates that four integrin molecules can be modeled above the HAdV12 penton base. However, because of the close spacing of the penton base RGD protrusions (∼60 Å) our model indicates that each of the four bound integrins must adopt a different orientation relative to the underlying penton base monomer. The significant variability in the modeled integrin positions with respect to the penton base RGD protrusions is estimated to be in the range of 10 to 29 Å after alignment of the penton base monomers. This positional variability for bound integrin may explain the lower resolution observed for the integrin density compared to the icosahedral capsid in the cryoEM structure of the complex.
This penton base/integrin modeling analysis also showed that with four integrins bound to the HAdV12 penton base the fifth RGD protrusion is effectively shielded from binding integrin. An earlier Biacore study with HAdV2 virions indirectly attached to a sensor chip via an anti-fiber monoclonal antibody, indicated that at close to saturation ∼4.2 soluble αvβ5 integrin molecules could bind to each penton base (5). The HAdV2 penton base has a significantly longer RGD-loop than HAdV12 (78 versus 15 amino acids), and thus the HAdV2 RGD-loop may provide enough additional flexibility that either four or five integrin molecules may bind to one penton base.
The HAdV12/αvβ5 cryoEM structure is consistent with the extension model for integrin activation.
A variety of experiments including the addition of disulfide bonds to lock integrins in the bent conformation have shown that integrin extension is required for ligand binding during integrin inside-out signaling (45). In the extension model, the open headpiece conformation, observed in a crystal structure of αIIbβ3 and a therapeutic antagonist (38), represents the high-affinity, ligand-bound state. The open headpiece conformation has the β-chain hybrid domain swung by up to 70° from its position in the bent integrin conformation (47). This large swing of the hybrid domain presumably leads to separation of the two integrin cytoplasmic tails. Separation of integrin transmembrane domains has also been shown to be an important component of integrin outside-in signal transduction (15). Unfortunately, there is no atomic resolution structure of a complete integrin ectodomain with a bound RGD ligand, except for the αvβ3/RGD crystal structure, which involved soaking an RGD peptide into a preformed αvβ3 crystal. Given the flexible nature of the multidomained integrin heterodimer and the flexibility of integrin-binding RGD loops, it may be difficult to obtain an atomic resolution structure of an integrin/RGD ligand complex with a complete integrin ectodomain in an extended conformation. Our cryoEM structure of HAdV12 complexed with a soluble form of αvβ5 complex, albeit at a moderate resolution for the bound integrin, offers support for the idea that αv integrins adopt an extended conformation upon binding to a multivalent RGD ligand.
Integrin binding may induce a conformational change in penton base and initiate the process of vertex protein release.
The cryoEM structure of the HAdV12/αvβ5 complex also indicates that integrin binding may extend the penton base RGD loop in a direction counter to the natural twist of the pentamer and thus induce an untwisting of penton base. This is supported by a finding of significantly weaker penton base density in the HAdV12/αvβ5 complex than in the Ad35f vector. Since the N-terminal fiber peptide binds at the top of the penton base at the interface between monomers (48), an untwisting of the penton base multimer would likely disrupt the fiber binding site. This would offer a possible explanation for the finding that fiber is released at the cell surface early in the Ad cell entry process, perhaps after interaction with integrin (21).
In conclusion, this considerably improved cryoEM structure of the HAdV12/αvβ5 complex together with crystal structures of penton base, αvβ3, and αIIbβ3 have enabled a more accurate and revealing molecular analysis of the adenovirus integrin interaction. The cryoEM structure indicates that αvβ5 integrin adopts an extended conformation when bound αvβ5 to the multivalent viral ligand and that the viral penton base may be conformationally affected by the binding of multiple integrin molecules to one vertex. The close spacing (∼60 Å) of the integrin-binding RGD sites on the penton base may promote integrin clustering, lead to the intracellular signaling events required for virus internalization, and prime the adenovirus capsid for programmed disassembly (7). The similar spacing of integrin-binding RGD loops noted for foot-and-mouth disease virus and coxsackievirus A9 (32) suggests that these viruses might undergo analogous conformational changes after interaction with integrin at the host cell membrane.
Acknowledgments
Sources of funding for this study include NIH AI42929 (P.L.S.), NIH EY011431 (G.R.N.), and NIH HL054352 (G.R.N.). M.S. acknowledges support from the NIH Molecular Biophysics Training Grant at Vanderbilt (T32-GM008320).
We also gratefully acknowledge computer support from the Advanced Computing Center for Research and Education at Vanderbilt.
Footnotes
Published ahead of print on 2 September 2009.
REFERENCES
- 1.Adiga, U., W. T. Baxter, R. J. Hall, B. Rockel, B. K. Rath, J. Frank, and R. Glaeser. 2005. Particle picking by segmentation: a comparative study with SPIDER-based manual particle picking. J. Struct. Biol. 152:211-220. [DOI] [PubMed] [Google Scholar]
- 2.Arnaout, M. A., B. Mahalingam, and J. P. Xiong. 2005. Integrin structure, allostery, and bidirectional signaling. Annu. Rev. Cell Dev. Biol. 21:381-410. [DOI] [PubMed] [Google Scholar]
- 3.Belnap, D. M., B. M. McDermott, Jr., D. J. Filman, N. Cheng, B. L. Trus, H. J. Zuccola, V. R. Racaniello, J. M. Hogle, and A. C. Steven. 2000. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. USA 97:73-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chacon, P., and W. Wriggers. 2002. Multi-resolution contour-based fitting of macromolecular structures. J. Mol. Biol. 317:375-384. [DOI] [PubMed] [Google Scholar]
- 5.Chiu, C. Y., P. Mathias, G. R. Nemerow, and P. L. Stewart. 1999. Structure of adenovirus complexed with its internalization receptor, αvβ5 integrin. J. Virol. 73:6759-6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glover, J. N., and S. C. Harrison. 1995. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 373:257-261. [DOI] [PubMed] [Google Scholar]
- 7.Greber, U. F., M. Willetts, P. Webster, and A. Helenius. 1993. Stepwise dismantling of adenovirus 2 during entry into cells. Cell 75:477-486. [DOI] [PubMed] [Google Scholar]
- 8.Grigorieff, N. 2007. FREALIGN: high-resolution refinement of single particle structures. J. Struct. Biol. 157:117-125. [DOI] [PubMed] [Google Scholar]
- 9.Hato, T., N. Pampori, and S. J. Shattil. 1998. Complementary roles for receptor clustering and conformational change in the adhesive and signaling functions of integrin αIIb β3. J. Cell Biol. 141:1685-1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He, Y., V. D. Bowman, S. Mueller, C. M. Bator, J. Bella, X. Peng, T. S. Baker, E. Wimmer, R. J. Kuhn, and M. G. Rossmann. 2000. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. USA 97:79-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hynes, R. O. 1987. Integrins: a family of cell surface receptors. Cell 48:549-554. [DOI] [PubMed] [Google Scholar]
- 12.Kolatkar, P. R., J. Bella, N. H. Olson, C. M. Bator, T. S. Baker, and M. G. Rossmann. 1999. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 18:6249-6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li, E., D. Stupack, G. M. Bokoch, and G. R. Nemerow. 1998. Adenovirus endocytosis requires actin cytoskeleton reorganization mediated by Rho family GTPases. J. Virol. 72:8806-8812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li, E., D. Stupack, R. Klemke, D. A. Cheresh, and G. R. Nemerow. 1998. Adenovirus endocytosis via α(v) integrins requires phosphoinositide-3-OH kinase. J. Virol. 72:2055-2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo, B. H., C. V. Carman, and T. A. Springer. 2007. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 25:619-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo, B. H., and T. A. Springer. 2006. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 18:579-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathias, P., M. Galleno, and G. R. Nemerow. 1998. Interactions of soluble recombinant integrin αvβ5 with human adenoviruses. J. Virol. 72:8669-8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mindell, J. A., and N. Grigorieff. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142:334-347. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto, S., S. K. Akiyama, and K. M. Yamada. 1995. Synergistic roles for receptor occupancy and aggregation in integrin transmembrane function. Science 267:883-885. [DOI] [PubMed] [Google Scholar]
- 20.Miyamoto, S., H. Teramoto, O. A. Coso, J. S. Gutkind, P. D. Burbelo, S. K. Akiyama, and K. M. Yamada. 1995. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J. Cell Biol. 131:791-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakano, M. Y., K. Boucke, M. Suomalainen, R. P. Stidwill, and U. F. Greber. 2000. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J. Virol. 74:7085-7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersen, E. F., T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng, and T. E. Ferrin. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605-1612. [DOI] [PubMed] [Google Scholar]
- 23.Philpott, N. J., M. Nociari, K. B. Elkon, and E. Falck-Pedersen. 2004. Adenovirus-induced maturation of dendritic cells through a PI3 kinase-mediated TNF-α induction pathway. Proc. Natl. Acad. Sci. USA 101:6200-6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin, J., O. Vinogradova, and E. F. Plow. 2004. Integrin bidirectional signaling: a molecular view. PLoS Biol. 2:e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rajala, M. S., R. V. Rajala, R. A. Astley, A. L. Butt, and J. Chodosh. 2005. Corneal cell survival in adenovirus type 19 infection requires phosphoinositide 3-kinase/Akt activation. J. Virol. 79:12332-12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramachandran, G. N., A. S. Kolaskar, C. Ramakrishnan, and V. Sasisekharan. 1974. The mean geometry of the peptide unit from crystal structure data. Biochim. Biophys. Acta 359:298-302. [DOI] [PubMed] [Google Scholar]
- 27.Rohl, C. A., C. E. Strauss, D. Chivian, and D. Baker. 2004. Modeling structurally variable regions in homologous proteins with Rosetta. Proteins 55:656-677. [DOI] [PubMed] [Google Scholar]
- 28.Saban, S. D., R. R. Nepomuceno, L. D. Gritton, G. R. Nemerow, and P. L. Stewart. 2005. CryoEM structure at 9 Å resolution of an adenovirus vector targeted to hematopoietic cells. J. Mol. Biol. 349:526-537. [DOI] [PubMed] [Google Scholar]
- 29.Saban, S. D., M. Silvestry, G. R. Nemerow, and P. L. Stewart. 2006. Visualization of alpha-helices in a 6-Å resolution cryoelectron microscopy structure of adenovirus allows refinement of capsid protein assignments. J. Virol. 80:12049-12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi, J., D. R. Williams, and P. L. Stewart. 2008. A Script-Assisted Microscopy (SAM) package to improve data acquisition rates on FEI Tecnai electron microscopes equipped with Gatan CCD cameras. J. Struct. Biol. 164:166-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart, P. L., C. Y. Chiu, S. Huang, T. Muir, Y. Zhao, B. Chait, P. Mathias, and G. R. Nemerow. 1997. Cryo-EM visualization of an exposed RGD epitope on adenovirus that escapes antibody neutralization. EMBO J. 16:1189-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stewart, P. L., and G. R. Nemerow. 2007. Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol. 15:500-507. [DOI] [PubMed] [Google Scholar]
- 33.Vinogradova, O., A. Velyvis, A. Velyviene, B. Hu, T. Haas, E. Plow, and J. Qin. 2002. A structural mechanism of integrin αIIbβ3 “inside-out” activation as regulated by its cytoplasmic face. Cell 110:587-597. [DOI] [PubMed] [Google Scholar]
- 34.Wickham, T. J., P. Mathias, D. A. Cheresh, and G. R. Nemerow. 1993. Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell 73:309-319. [DOI] [PubMed] [Google Scholar]
- 35.Wiethoff, C. M., H. Wodrich, L. Gerace, and G. R. Nemerow. 2005. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 79:1992-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu, E., S. A. Trauger, L. Pache, T. M. Mullen, D. J. von Seggern, G. Siuzdak, and G. R. Nemerow. 2004. Membrane cofactor protein is a receptor for adenoviruses associated with epidemic keratoconjunctivitis. J. Virol. 78:3897-3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao, C., C. M. Bator-Kelly, E. Rieder, P. R. Chipman, A. Craig, R. J. Kuhn, E. Wimmer, and M. G. Rossmann. 2005. The crystal structure of coxsackievirus A21 and its interaction with ICAM-1. Structure 13:1019-1033. [DOI] [PubMed] [Google Scholar]
- 38.Xiao, T., J. Takagi, B. S. Coller, J. H. Wang, and T. A. Springer. 2004. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432:59-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xing, L., M. Huhtala, V. Pietiainen, J. Kapyla, K. Vuorinen, V. Marjomaki, J. Heino, M. S. Johnson, T. Hyypia, and R. H. Cheng. 2004. Structural and functional analysis of integrin α2I domain interaction with echovirus 1. J. Biol. Chem. 279:11632-11638. [DOI] [PubMed] [Google Scholar]
- 40.Xing, L., K. Tjarnlund, B. Lindqvist, G. G. Kaplan, D. Feigelstock, R. H. Cheng, and J. M. Casasnovas. 2000. Distinct cellular receptor interactions in poliovirus and rhinoviruses. EMBO J. 19:1207-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiong, J. P., T. Stehle, B. Diefenbach, R. Zhang, R. Dunker, D. L. Scott, A. Joachimiak, S. L. Goodman, and M. A. Arnaout. 2001. Crystal structure of the extracellular segment of integrin αVβ3. Science 294:339-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong, J. P., T. Stehle, S. L. Goodman, and M. A. Arnaout. 2004. A novel adaptation of the integrin PSI domain revealed from its crystal structure. J. Biol. Chem. 279:40252-40254. [DOI] [PubMed] [Google Scholar]
- 43.Xiong, J. P., T. Stehle, R. Zhang, A. Joachimiak, M. Frech, S. L. Goodman, and M. A. Arnaout. 2002. Crystal structure of the extracellular segment of integrin αVβ3 in complex with an Arg-Gly-Asp ligand. Science 296:151-155. [DOI] [PubMed] [Google Scholar]
- 44.Yamada, K. M., and S. Miyamoto. 1995. Integrin transmembrane signaling and cytoskeletal control. Curr. Opin. Cell Biol. 7:681-689. [DOI] [PubMed] [Google Scholar]
- 45.Zhu, J., B. Boylan, B. H. Luo, P. J. Newman, and T. A. Springer. 2007. Tests of the extension and deadbolt models of integrin activation. J. Biol. Chem. 282:11914-11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu, J., C. V. Carman, M. Kim, M. Shimaoka, T. A. Springer, and B. H. Luo. 2007. Requirement of alpha and beta subunit transmembrane helix separation for integrin outside-in signaling. Blood 110:2475-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu, J., B. H. Luo, T. Xiao, C. Zhang, N. Nishida, and T. A. Springer. 2008. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol. Cell 32:849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zubieta, C., G. Schoehn, J. Chroboczek, and S. Cusack. 2005. The structure of the human adenovirus 2 penton. Mol. Cell 17:121-135. [DOI] [PubMed] [Google Scholar]