

Abstract
The hepatitis B virus (HBV) core protein (CP) forms the shell of an icosahedral nucleocapsid. In a former work, we identified 11 amino acid residues of CP exposed on the capsid surface by an alanine mutation scan as being important for capsid envelopment. We now introduced several other amino acids at six of these positions and found that almost all 27 tested point mutations at S17, K96, and I126 reproduced the phenotype of the alanine mutation (with only two exceptions): the formation of nucleocapsids and of the viral DNA genome was wild type, but capsid envelopment and virion release were strongly inhibited. This indicates that these side chains have a very specific function during nucleocapsid envelopment. We also identified several CP point mutations (e.g., F122V/S/Y and R127D/G) allowing the formation of capsids but preventing the packaging of pregenomic RNA. The envelopment of such mutant capsids was blocked. Apparently, these CP mutations hampered the recognition/packaging of the pregenome-P-protein complex by CP, a process which is still barely understood, and the mutant capsids devoid of HBV-specific nucleic acid did not express the capsid maturation signal required for envelopment.
Hepatitis B virus (HBV), the prototype member of the virus family Hepadnaviridae, is a small, enveloped DNA virus acutely and persistently infecting humans and causing liver diseases in a substantial fraction of infected persons (17). More than 350 million people worldwide are long-term HBV carriers and carry a relatively high risk for the development of liver cirrhosis or hepatocellular carcinomas. Treatment options for HBV carriers are still unsatisfying.
After virus entry into the hepatocyte, the partially double-stranded, circular, 3.2-kb viral DNA genome is translocated into and released in the nucleus and completed to a fully double-stranded circular DNA, which forms the template for transcription by host factors. The formation of progeny virus is initiated in the cytosol by the binding of a viral, 3.5-kb-long, terminally redundant RNA transcript (the pregenome) to the viral reverse transcriptase/DNA polymerase (P protein) (1), together with host factors, and the subsequent packaging of this ribonucleoprotein complex by multiple homodimers of the viral 21-kDa core protein (CP). The CP homodimers form the shell of icosahedral capsids (25), which appear in two forms. One species has a diameter of approximately 32 nm and a T=4 symmetry and consists of 120 CP dimers; the other species is slightly smaller, has a T=3 symmetry, and consists of 90 CP dimers. The T=4 particles prevail, and both forms can also be found in virions (6). The P protein and pregenome recognize each other specifically by an interaction of P with a stem-loop structure in the pregenome (1). How this ribonucleoprotein complex is then identified by CP during nucleocapsid formation and how it is packaged are largely unknown.
The capsid has holes of 1.2 nm to 1.5 nm in diameter allowing the influx of deoxyribonucleotide triphosphates, which are used by the P protein for the synthesis of a DNA minus strand complementary to the pregenome and for subsequent DNA plus-strand synthesis. Prior to completion of plus-strand synthesis, nucleocapsids are enveloped at intracellular membranes containing the viral surface proteins and appear in the luminal compartment of secretory organelles, and the resulting virions are released into the bloodstream.
The envelope proteins are a major player in virion formation, which is underlined by the fact that a viral mutant unable to express all three viral surface proteins, S, M, and L, will not generate lipid-enveloped nucleocapsids (3). The envelope proteins are synthesized as membrane proteins at the endoplasmic reticulum and gain a complex transmembrane topology. It has been demonstrated that the L and S proteins, but not the M protein, are required for virion formation. Mutational analyses revealed that an approximately 22-amino-acid (aa)-long linear stretch in a cytoplasmic domain of L (matrix domain [MD]) is important for capsid envelopment (4, 10, 13), and it has been speculated that this domain functions in contacting the capsid, like a matrix protein.
The second player in hepatitis B virion morphogenesis, the capsid, can be expressed efficiently in Escherichia coli, especially when the arginine-rich C-terminal domain of CP is omitted. If the 185-aa (HBV genotype A; used in this work) or 183-aa (HBV genotypes B, C, D, E, F, and H) CP is truncated at aa 149, it preferentially forms T=4 particles, which have been used for crystal structure determination (25). The CP homodimer in the capsid forms a spike by four alpha-helices protruding from the surface and a rhombus-shaped base (see Fig. 3). Cryo-electron microscopic analyses of nucleocapsids derived from virions showed subtle differences (15). Based on the crystal structure, an alanine mutagenesis scan of aa exposed at the surface of the capsid has been performed, and the mutants were analyzed for the ability to form cytoplasmic capsids competent for DNA genome synthesis and for morphogenesis and release of virions (14). Mutations compatible with nucleocapsid formation but blocking envelopment were clustered at two narrow areas, at the base of the spike and in a lateral region of the base (capsid envelopment determinant [CED]). The function of these determinants is not clear. It is possible that they mediate the contact with the envelope proteins (possibly with the MD in protein L) or with cellular factors required for transport or envelopment or that they are involved in generating the so-called maturation signal of the capsid (15, 20): capsids containing no HBV-related nucleic acid (16) or either pregenomic RNA or single-stranded HBV minus-strand DNA (9, 24) are not competent for envelopment. Rather, the synthesis of the second DNA strand is coupled to a change in the nucleocapsid allowing its incorporation into virions. The nature of the step that is regulated by this change is unclear.
FIG. 3.

Positions of aa residues in the core protein homodimer that are relevant for capsid envelopment. A sphere model of the core protein homodimer is shown. Even conservative point mutations at aa residues S17, K96, and I126 and less conservative mutations at L95, exposed at the outer capsid surface, blocked virion formation (Fig. 2A). We assume that they form an area for the interaction with cellular or viral factors (envelope proteins) required for nucleocapsid envelopment.
In the present work, we evaluated how much variability is allowed in CED on the capsid surface without impairing its function in virion formation. We identified four residues in this area which cannot be mutated to almost any other aa without strongly blocking virion formation. In addition, we found several CP point mutations compatible with efficient capsid formation but blocking the packaging of viral RNA.
MATERIALS AND METHODS
Plasmids.
Plasmid pSVHBV1.5 (14) contains a simian virus 40 early promoter followed by a longer-than-full-length terminal redundant copy of a genotype A HBV genome (22) (numbering of the plus strand of the HBV genome starts with the deoxycytidine of the unique EcoRI site) (Fig. 1). This plasmid initiates complete HBV replication after transfection of human hepatoma Huh7 cells. Plasmid pSVHBV1.5core− was derived from pSVHBV1.5 by changing HBV genome nucleotide (nt) 2012 in the 5′ part of the terminally redundant HBV genome from dT to dG, resulting in a stop codon at triplet 38 of the core gene (Fig. 1) (14). Plasmid pSVHBV1.5RT−env− is a derivative of pSVHBV1.5 in which (i) HBV genome nt 740 was changed from dG to dC, destroying the reverse transcriptase activity of the P protein by a missense mutation (RT−); and (ii) HBV genome nt 196 was changed from dT to dG, resulting in a stop codon in the envelope gene blocking expression of all three HBV envelope proteins (env−). Both mutations were transferred from plasmid pRVHBV1.5RT−env− (9) to pSVHBV1.5 by recombining a PstI (nt 26) to HpaI (nt 962) 936-bp-long DNA fragment. The construction of plasmid pSVcore for the expression of HBV core protein (Fig. 1) has been described previously (11).
FIG. 1.

Map of plasmids. (Top) For the expression of the HBV genome, a longer-than-full-length copy of the viral DNA (thick bar) was inserted 3′ of the simian virus 40 (SV40) early promoter (dashed box) in plasmid pSVHBV1.5core−. The open reading frames of the viral C, P, E, and X genes are indicated by open boxes. The position of the single polyadenylation site of the HBV genome is shown by a vertical bar (pA). The C gene carries the point mutation T2012G, introducing a stop codon at triplet 38 (arrow). The positions of restriction sites for BspEI and EcoRI are indicated. Numbers refer to nucleotides of the 3,221-bp-long HBV genome (genotype A), beginning with dC of the unique EcoRI site. (Bottom) For the expression of WT and mutant CPs, plasmid pSVcore was used. In this vector, HBV nt 1816 to nt 1888 at the 5′ end of the HBV insert are missing relative to plasmid pSVHBV1.5core−. As a consequence, the packaging signal of the pregenome is not formed in the 5′ region of the corresponding mRNA. Also, the region from nt 2820 to nt 676 is missing to prevent P and envelope protein expression.
Site-directed mutagenesis.
All point mutations generated within the CP expression vector pSVcore were constructed as described previously (14) by a PCR-based method, with the exception that Power Script DNA Polymerase Long (Pan Biotech, Aidenbach, Germany) was used. All portions of a plasmid generated by PCRs were sequenced after molecular cloning to exclude unintentional mutations. The crystal structure of the capsid (Protein Data Bank accession no. 1QGT.pdb) (25) was visualized using Swiss-PdbViewer v4.0.
Cell culture and transfection.
Human hepatoma Huh7 cells were cultivated in Dulbecco's modified Eagle medium (Invitrogen, Karlsruhe, Germany) containing 10% (vol/vol) fetal bovine serum (Biochrome AG, Berlin, Germany) and antibiotics (100 units/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin [Invitrogen]) at 37°C in a 5% CO2 atmosphere. All transient transfections were performed using the calcium phosphate method as described before (11). For endogenous polymerase reactions (EPR) and reverse transcription-PCR (RT-PCR) experiments, 3 × 105 cells were seeded in six-well plates. After 20 h, 2 μg of plasmid DNA was used for transfection. For Southern and Western blot experiments, 106 cells were cultivated overnight per 10-cm cell culture dish prior to transient transfection with 10 μg of total plasmid DNA. In the case of cotransfections, equal mass amounts of plasmids were mixed.
Preparation of viral particles.
At 5 days posttransfection, the cell culture supernatant was harvested, and cells were washed twice with phosphate-buffered saline (PBS) and treated for 10 min with 1 ml of lysis buffer (150 mM NaCl, 50 mM Tris-Cl [pH 7.5], 5 mM MgCl2, 0.2% [vol/vol] Nonidet P-40 [NP-40]). The medium and cell lysate were cleared by centrifugation (14,000 rpm, 4°C, 10 min). The supernatants of the cell lysate and medium fractions were used for immunoprecipitation of intracellular capsids with polyclonal rabbit anti-HBc (Dako, Hamburg, Germany) and of secreted virions with polyclonal goat anti-HBs (Dako), respectively, bound to swollen protein A-Sepharose beads (Sigma-Aldrich, Hamburg, Germany). After overnight incubation at 4°C, bound viral particles were pelleted and used for EPR, Southern blotting, or RT-PCR. For polyethylene glycol (PEG) precipitation of intracellular capsids, dry PEG-8000 and NaCl were dissolved in cleared cellular lysates (final concentrations, 10% [wt/vol] and 2% [wt/vol], respectively), incubated overnight at 4°C, and spun for 1 h at 4,000 rpm at room temperature. The pellet was dissolved with 20 μl of 100 mM NaCl, 0.1 mM EDTA, and 10 mM Tris-Cl (pH 8.0) and loaded into a 1% (wt/vol) agarose-Tris-acetate-EDTA (TAE) gel for separation. For isopycnic CsCl gradient centrifugation, 10 ml of cleared medium was used to dissolve 3.84 g of solid CsCl and spun for 48 h at 48,000 rpm and 20°C in a Beckman 70.1Ti rotor, allowing virions to reach their buoyant densities between 1.23 g/ml and 1.27 g/ml. After opening the tube by slicing it at the top, 10 1-ml fractions were taken from the top, and their buoyant densities were determined by measuring the refractive index. Subsequently, the fractions were diluted with 1 volume of 1% (vol/vol) NP-40-0.2% (wt/vol) dithiothreitol and precipitated with PEG as described above.
Detection of viral particles by EPR.
Radioactive labeling of the encapsidated viral DNA by the endogenous polymerase, isolation of the DNA, and its separation in agarose gels were done as described previously (11). The radioactive signals were visualized by a phosphorimager (Molecular Imager FX; Bio-Rad, Munich, Germany).
Southern blotting.
After immunoprecipitation of intracellular capsids, exogenous DNA was digested by DNase treatment (end concentration, 0.07 mg/ml; 30 min at 37°C). To isolate the encapsidated viral DNA and to inactivate the DNase, proteinase K was added (end concentration, 0.3 mg/ml) and incubated for 30 min at 37°C in 20 mM Tris-Cl, pH 7.5, 20 mM EDTA, 2% (wt/vol) sodium dodecyl sulfate. After extraction with 1 volume of phenol-chloroform (1:1), the nucleic acid was pelleted by two successive ethanol precipitations. Resolved DNA was loaded and separated in a 1% (wt/vol) agarose-TAE gel. All following steps were done according to the protocol for an AlkPhos direct labeling kit (Amersham Bioscience, Freiburg, Germany). Blotting was performed by capillary transfer, using a positively charged nylon membrane (Hybond-N+; Amersham Bioscience). After neutralization, cross-linking, hybridization, and washing, the signals were detected by chemiluminescence and Kodak Biomax MR films (Sigma-Aldrich, Hamburg, Germany).
Western blotting of cytosolic nucleocapsids and secreted virions.
After PEG precipitation of capsids from cellular lysates or CsCl gradient fractions, the samples were separated by native 1% (wt/vol) agarose-TAE gel electrophoresis. The gel was blotted overnight by capillary transfer, using a nitrocellulose transfer membrane (Protran BA 85 [pore size, 0.45 μm]; Schleicher & Schuell, Dassel, Germany) and 10× SSC buffer (1.5 M NaCl, 150 mM sodium citrate [pH 7.0]). The membrane was blocked by incubation for 1 h at room temperature in 50 ml of PBS supplemented with 10% (wt/vol) skim milk powder and 0.1% (vol/vol) Tween 20 (blocking buffer), followed by washing three times for 15 min each with PBS-0.1% (vol/vol) Tween 20 and incubation with polyclonal rabbit anti-HBc (dilution, 1:2,000; Dako) in 50 μl blocking buffer per cm2 of membrane, with shaking, for 2 h at room temperature. The membrane was washed as described above, and horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (heavy plus light chains) F(ab′)2 fragments from donkey (Dianova, Hamburg, Germany) (dilution, 1:10,000) were added in blocking buffer to the membrane for 1.5 h at room temperature, with agitation. Finally, the membrane was washed as described above an additional two times for 10 min each with PBS. Detection of HBcAg was achieved by the addition of 50 μl/cm2 membrane of equally mixed ECL Luminol and Enhancer detection solutions (Amersham Bioscience) and exposure of a Kodak BioMax MR film for 1 min to 2 h.
Detection of HBV pregenomes by RT-PCR.
Cytosolic nucleocapsids were enriched from cell lysates in one 10-cm dish by immunoprecipitation with 10 μl protein A bead slurry/1 μl anti-HBc. After pelleting of the beads, exogenous DNA and RNA were destroyed by a combined incubation with DNase I and RNase A (end concentration, 0.07 mg/ml [each]) in 70 μl of 50 mM Tris-Cl (pH 7.5), 75 mM NH4Cl, 1 mM EDTA, 20 mM MnCl2, 0.07% (vol/vol) beta-mercaptoethanol, and 0.03% (vol/vol) NP-40 for 1 h at 37°C. RNase activity was stopped by the addition of 1 U RNasin Plus RNase inhibitor (Promega, Mannheim, Germany) per μl of digestion mix and incubation for 1 h at 37°C. To isolate encapsidated nucleic acids, 70 μl of 20 mM Tris-Cl (pH 7.5), 20 mM EDTA, 2% (wt/vol) sodium dodecyl sulfate, 0.6 mg/ml proteinase K, and 0.6 mg/ml tRNA was added, and the mixture was incubated for 1 h at 50°C. Proteins were removed by extraction with 1 volume of phenol-chloroform-isoamyl alcohol (25:24:1), and nucleic acids were precipitated by ethanol. Resolved RNA was treated for 20 min at room temperature with 2 U of AMP-D1′ DNase I (Sigma-Aldrich) as described by the manufacturer in a 20-μl volume to remove DNA and to keep the RNA. The reaction was stopped by the addition of 2 μl of 50 mM EDTA and incubation at 70°C for 10 min.
Eight- and 2-μl aliquots of the sample were used separately for RT-PCR (Qiagen, Hilden, Germany), with an initial 35-min incubation for cDNA synthesis at 50°C and heat inactivation of the reverse transcriptase for 15 min at 95°C, followed by a PCR as described in the manufacturer's protocol (sense primer, 5′CCTGAATGGCAAACTCCTTCC [nt 2524 to 2544 of the HBV DNA sequence]; antisense primer, 5′GAATGCAGGGTCCAACTGATGATCG [nt 2953 to 2929 of the HBV DNA sequence]). Treatment of another 8 μl of the RNA preparation from capsids started directly with the heat inactivation step, followed by the PCR, and was used as a control for DNA contaminations. All samples were separated by agarose gel electrophoresis and depicted by ethidium bromide staining and UV light transillumination.
RESULTS
Identification of aa side chains on the capsid surface strictly required for virion formation.
An extensive alanine mutation scan of the capsid surface identified 11 aa as important for capsid envelopment (14). We selected six of these positions (Fig. 2A), including three from the cluster at the base of the spike (S17, L95, and K96) and three from the lateral cluster (F122, I126, and R127), and changed them individually to a number of different aa in order to challenge the model that these residues are functionally relevant for virion formation. As a control, 3 aa (S21, N90, and R98) also exposed on the capsid surface but tolerating alanine mutations were also included (Fig. 2B). Huh7 human hepatoma cells were transiently cotransfected with expression plasmids for the core variants (pSVcore derivatives) (Fig. 1) and the core-negative but otherwise replication-competent genomic HBV construct pSVHBV1.5core− (Fig. 1). Capsids and virions were harvested from cell lysates and culture media by immunoprecipitation with anti-capsid and anti-envelope antisera, respectively, and detected by radioactive labeling of the viral genome, using the endogenous viral DNA polymerase.
FIG. 2.

Influence of C mutations on virion formation. Several point mutations, indicated in single-letter code, were introduced at selected positions in CP. After cotransfection of Huh7 cells with a core-negative HBV genome and the expression vector for the core derivative (Fig. 1), cytoplasmic nucleocapsids (bottom) and virions from the culture medium (top) were immunoprecipitated with anti-HBc and anti-HBs, respectively. The viral genome was radioactively labeled by EPR, isolated, and visualized by agarose gel electrophoresis and autoradiography. (A) Mutations at positions where alanine mutations allowed capsid formation and HBV genome synthesis but blocked virion formation. Almost all mutations at S17, K96, and I126 reproduced this phenotype. (B) Mutations at positions where alanine mutations were WT. Almost all mutations at these positions were also WT. Controls included cotransfection with the WT core gene (WT), transfection with the genomic core-negative HBV construct alone (C−), and 1 μl of a highly viremic serum from a human HBV carrier used for EPR (s).
For S17 and K96, all five and six point mutants, respectively, reproduced the phenotype of the alanine mutation: cytoplasmic capsids were well detectable (c+), but there were no secreted virions (v−) (Fig. 2A). Even conservative mutations like S17T or K96R behaved like this. In the case of I126, 11 mutations, including the conservative changes to valine and leucine, reproduced the phenotype of the I126A mutant (c+ v−), but only methionine showed a wild-type (WT) phenotype and proline blocked the detection of capsids in this assay. This result demonstrates that the molecular structures of residues S17, K96, and I126 play a pivotal role in the envelopment of capsids. For L95, only the conservative mutations to isoleucine and valine were tolerated, but a change to glycine or serine blocked nucleocapsid envelopment, like the alanine mutation. Apparently, small, hydrophobic, branched residues are required at this site.
At position R127, the result was less clear-cut: a conservative change, R127K, was WT, a change to G or D blocked capsid detection (c− v−), whereas mutation to H or S showed c+ v− behavior, and a mutation to L allowed low-level virion secretion. For F122, all five mutations but W blocked capsid detection (c− v−). The F122W change resulted in a low level of cytoplasmic capsids, like in the case of the F122A mutation. The mutations blocking intracellular capsid detection (c− v−) were analyzed in more detail and are described below.
In summary, almost any change of residues S17, K96, and I126 and nonconservative changes of L95 were tolerated with respect to capsid formation and synthesis of the viral DNA genome but specifically and strongly blocked the appearance of secreted virions. These residues form the bottom of a furrow reaching from the bottom of the spike to the lateral end of the rhombus-like base (Fig. 3).
To support the specificity of this result, we also mutated three positions where alanine mutations produced a WT phenotype. All eight additional point mutations at either S21 or N90 were also WT (Fig. 2B). At R98, three mutations reproduced the WT behavior of the alanine mutation and two mutations blocked capsid detection (see below). In summary, none of these 13 new mutations had a specific influence on capsid envelopment because they were either compatible with this step or blocked nucleocapsid formation at a prior step. This result supports the relevance of residues S17, K96, I126, and L95 for capsid envelopment and the specificity of the result.
C mutations allowing capsid formation but blocking pregenome packaging.
Ten of 53 new (non-alanine) core gene point mutations generated in this work prevented the detection of cytoplasmic capsids by EPR labeling of the viral genome. To characterize the reason for this deficiency, we first assayed whether these mutant CPs were able to assemble into capsids. For this purpose, cell lysates of cells cotransfected with the core-negative HBV genome and the CP expression vectors were loaded on a native agarose gel and blotted onto a membrane after electrophoresis. Capsids on the membrane were then detected as in a Western blot with anti-HBc antibodies (Fig. 4A). The major epitope of the capsid antigen is formed by the tip of the spike (2), which was not changed directly in any of the mutants used in this study, and therefore all mutants should be detectable with similar sensitivities if they form capsids. WT capsids formed a band running at a position similar to that for a 3-kb plasmid in the 1% (wt/vol) agarose gel, indicative of the particulate nature and uniform size/charge ratios of the antigen. When the CP expression vector was omitted, no signal appeared, as expected (Fig. 4A, lane C−). Recombinant capsids from E. coli consisting of the full-length CP packaging bacterial RNA behaved identically to WT capsids from Huh7 cells (Fig. 4A, lane WT). The mutants showed differences in this assay, as follows. The F122V, F122Y, and F122S variants clearly formed capsids, the F122Y variant apparently even in WT amounts. The I126P, F122G, F122K, and R98D mutants generated a smear or no signal, demonstrating that no authentic capsids were formed. The R98H mutant yielded two bands, below and above the position for WT capsids. The R127D and R127G mutants probably formed capsids running slightly faster than authentic particles, compatible with the elimination of a positively charged side chain. In summary, at least the F122V, F122Y, F122S, R127D, and R127G mutants, and probably also the R98H mutant, did form capsids, although the viral genome was not detectable in the corresponding particles by EPR (Fig. 2).
FIG. 4.

Assay of EPR-negative core mutants for capsid formation. (A) Cytoplasmic capsids of cotransfected cells were concentrated and separated in a native agarose gel, blotted, and detected with anti-HBc. Some EPR-negative mutants, e.g., the F122V mutant, produced well-detectable amounts of capsids. Controls included 1 ng and 10 ng of capsids expressed in E. coli (lanes st) (21), WT capsids from cotransfected cells (lane WT), and material from cells transfected with the core-negative HBV genomic construct alone (lane C−). (B) DNAs from cytoplasmic capsids were isolated and analyzed by Southern blotting. No viral DNA was detectable in mutant capsids. Controls included 100 pg and 50 pg of linearized full-length HBV DNA from a plasmid (lanes st) and the WT and C− lanes, as in panel A.
We next checked whether the mutant capsids contained viral DNA detectable by Southern blotting (Fig. 4B). Viral DNA isolated from cytoplasmic WT capsids was clearly visible. However, all 10 mutants analyzed showed no detectable viral DNA packaged within capsids.
Six mutants (F122V, F122Y, F122S, R127D, R127G, and R98H) showing particulate core antigen in the native agarose gel/Western blot assay but no encapsidated viral DNA by EPR or Southern blotting were tested for the presence of viral RNA within capsids. After immunoprecipitation of cytoplasmic capsids with anti-HBc antibodies, external DNA and RNA were digested with DNase and RNase, the enzymes were inactivated, and the nucleic acids were isolated by proteinase K treatment. Eight microliters of a total volume of 22 μl of each sample was used directly for an HBV DNA-specific PCR (Fig. 5B). No signal appeared, demonstrating that the samples were not contaminated with detectable amounts of viral DNA. Another 8 μl (and, separately, 2 μl of the samples from the mutants, as well as 4, 0.8, 0.08, and 0.008 μl of the WT sample) was first treated with reverse transcriptase, and then HBV cDNA was detected by PCR (Fig. 5A). An HBV genome with a missense mutation in the active center of the viral reverse transcriptase blocking the reverse transcriptase activity of the enzyme produced a well-detectable signal of viral pregenomic RNA isolated from cytoplasmic capsids (Fig. 5A, lane RT− env−). When the WT core expression vector (lane core) or the core-negative HBV genome (lane C−) was transfected separately, no signal was generated, as expected. Cotransfection of these plasmids produced a strong signal (lanes WT). The RNA sample from the WT capsids was diluted 2-, 10-, 100-, and 1,000-fold prior to the RT-PCR assay. Even after 100-fold dilution, a clear signal was obtained. The samples from the six core mutants contained small amounts of detectable viral RNA. A fourfold dilution of the samples reduced the RT-PCR signal, showing that the samples from the mutants contained only 1/100 or less viral RNA than the WT samples.
FIG. 5.

Assay of encapsidated pregenomic RNA. (A) RNAs from cytoplasmic capsids were isolated and used for RT-PCR. Samples shown in panel B were heat treated prior to the RT reaction to destroy the reverse transcriptase; the negative result excluded DNA contamination of the samples. The WT sample was diluted 2-, 10-, 100, and 1,000-fold (triangle). HBV RNA was detectable in mutant capsids, but in amounts at least 100-fold less than those of the WT. Controls included RNA from capsids of an HBV mutant carrying a missense mutation in P blocking viral reverse transcriptase activity (RT− env−), encapsidated RNA from cells transfected with the core expression vector alone (core), material from cells transfected with the core-negative HBV genome alone (C−), 10 pg HBV DNA from a plasmid (PCR+), water (PCR−), and a 100-bp ladder (st).
In summary, we showed that F122V, F122Y, F122S, R127D, R127G, and possibly R98H mutations allowed the formation of intracellular capsids, with the F122V level even close to WT levels, but strongly reduced the encapsidation of pregenomic RNA. This was apparently the cause for the negative EPR results (Fig. 2) and the lack of a Southern blot signal (Fig. 4B).
No envelopment of capsids lacking viral DNA.
Previous work showed that early nucleocapsids containing pregenomic RNA (9, 24) or capsids containing no HBV-related nucleic acid (16) are not competent for envelopment. We therefore asked whether the mutant capsids deficient in pregenome packaging are incorporated into virion-like particles and released from the transfected cells. To answer this question, media from cotransfected cells were separated in an isopycnic CsCl gradient. The gradient was harvested from the top, yielding 10 fractions, and capsids were concentrated from each fraction and detected by native agarose gel electrophoresis and Western blotting (Fig. 6). Capsids could be detected in fractions 3 and 4, at a density of around 1.24 g/ml in the case of the WT, indicative of virions, and in fractions 6 to 9, at densities of around 1.34 g/ml, characteristic of naked capsids. Apparently, naked capsids appeared in the medium in much higher quantities than did virions. The reason for this is unknown. As a negative control, the sample from cells transfected with the core-negative HBV genome alone gave no signal. All six mutants tested, except for the R127D mutant, produced naked capsids in the medium. It is possible that the R127D mutant formed capsids which were unstable in the CsCl solution. Again, the F122V mutant showed the strongest HBcAg signal, but virion-like particles could not be detected for any of the mutants. Apparently, these variants did not express the “mature” phenotype of the capsid coupled to the formation of viral DNA and prerequisite for its envelopment.
FIG. 6.
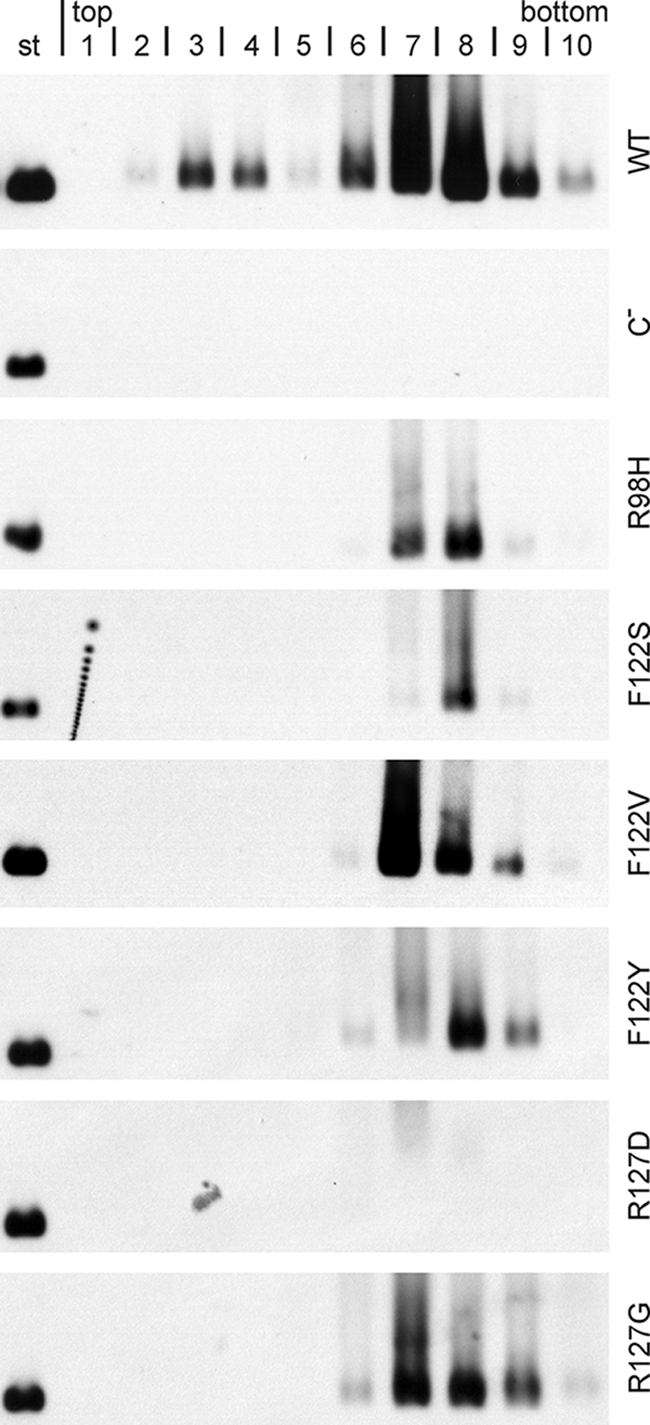
Mutant capsids devoid of HBV-specific nucleic acid produced no virion-like particles. Culture media were separated by isopycnic CsCl gradients. Fractions were taken from the top, mild detergent was added, and capsids were concentrated and detected, as in Fig. 4A, by native agarose gel electrophoresis and Western blotting. The signal around fraction 4 (density, approximately 1.24 g/ml) is indicative of virions, and the signal around fractions 7 and 8 (density, approximately 1.35 g/ml) represents naked capsids. None of the mutants produced virion-like particles. Controls included WT capsids and virions (WT), material from cell cultures transfected with the core-negative HBV genome alone (C−), and 10 ng of capsids from E. coli (st).
DISCUSSION
Enveloped viruses can gain their outer coat by a variety of mechanisms (8). The Gag protein of retroviruses, for example, forms the viral capsid and, in addition, causes capsid envelopment by directly interacting with cellular membranes and fusion factors independent of viral surface proteins (23). Other viruses, such as paramyxoviruses, have specialized matrix proteins bridging the nucleocapsid and envelope. For HBV, the viral membrane proteins S and L are essential for enclosing the cytoplasmic nucleocapsid with a lipid-containing envelope. It was therefore proposed that in analogy to togaviruses, cytosolic portions of the transmembrane HBV envelope proteins bind to the cytosolic HBV capsid, and that this interaction drives virion budding. Cryo-electron microscopy of virions revealed that the tips of the core's spikes were close to the viral envelope (6, 19), but the resolution was not sufficient to describe a potential envelope-core interaction on a molecular level. Point mutations in the HBV envelope and core protein have been identified which specifically and strictly block virion formation without influencing other detectable features of these proteins (4, 10, 13, 14). It is possible that the corresponding regions defined by these mutations (MD in the L protein and CED in CP) either bind directly to each other or bind to a cellular factor bridging the capsid and envelope. One candidate for such a factor could be γ-2 adaptin (12). In any case, the molecular interactions by which MD and CED are involved in virion formation require a very precise and narrow interplay, since single point mutations efficiently block the whole process.
In the present work, we showed that the exchange of the WT aa with almost any other residue at four positions of the CED strongly impaired nucleocapsid envelopment. Even conservative changes, such as S17T, K96R, or I126A/V, were not tolerated, demonstrating that not only single aa exchanges abrogated virion formation but even very subtle alterations in certain side chains were similarly disruptive. These residues are also much conserved between different HBV isolates: a comparison of 1,165 HBV core protein sequences revealed that I126 was present in 1,157 isolates. Residue S17 could be found in 1,159 sequences, and K96 was present in 1,156 sequences, with 3 sequences carrying R at position 96. In the background of the HBV strain used in this work, a K96R mutation was not compatible with virus formation; however, this might be different for other HBV strains. Indeed, Garcia and colleagues found that the K96R mutation in CP allowed virion formation by an ayw serotype HBV strain (7).
However, the conservation of S17, K96, and I126 among HBV isolates has only limited significance because the whole HBV core gene is relatively conserved (http://www.helmholtz-muenchen.de/uploads/media/Pairan_Bruss_Tab_S1.xls) (5). This is also the case for residues N90 and R98, which are located at the capsid surface. In these cases, 1,156 and 1,155 of 1,165 sequences carried the WT aa, respectively, although most mutations at these spots had no apparent phenotype in our assay for capsid and virion formation. In particular, none of the 16 point mutations introduced at these sites specifically blocked capsid envelopment, emphasizing the local restriction of CED. Overall, the extensive mutagenesis done in this work impressively confirmed the role of individual aa residues, as mapped by a former alanine scan (14).
In summary, the results suggest a pivotal role, especially for CP aa S17, K96, and I126, in late steps of virion formation. Which step is blocked by these mutations is unclear. It might be capsid interaction with envelope proteins but also, e.g., transport of the capsid to budding sites, interactions with cellular factors, or emergence of the obscure maturation signal.
During our screen, we found that several CP point mutations prevented the detection of cytoplasmic nucleocapsids by use of the activity of the viral DNA polymerase. A closer analysis revealed, as expected, that the reasons for this phenotype were diverse. Some mutations, like R98D, apparently just impeded capsid formation (Fig. 4A). The most trivial explanation for this phenotype would be that the folding or stability of the mutant CP was aberrant. However, some variants, like the F122V mutant, efficiently generated capsids with WT behavior in nondenaturing agarose gel electrophoresis and were expressed in close-to-WT quantities. Why did these mutants not support EPR? One possible reason was that the viral DNA plus strand was almost complete, such that no radioactive nucleotides could be incorporated. However, this explanation was excluded because no viral DNA could be detected in these capsids by Southern blotting (Fig. 4A). Another possibility was that RT of the packaged pregenome was not supported by the mutant capsids. However, this possibility could also be excluded because only very small amounts of RNA pregenome could be found within the capsid lumen. Therefore, the most likely explanation for this phenotype is that the mutations inhibited the packaging of a functional pregenome-P-protein complex. One caveat of this interpretation is that the mutant capsids might be less stable than WT capsids and may have allowed the RNase used in the packaging assay to access and destroy the pregenome in the capsid lumen. This may indeed be the case, e.g., for mutants F122Y and R127D producing reduced signals in Western blots after CsCl gradient centrifugation (compare corresponding lanes in Fig. 4A and 6), indicating a denaturing effect of the CsCl solution for these mutants and a lower stability of the particles. However, at least the R96H, F122V, F122S, and R127G mutants were relatively stable in the CsCl solution, arguing against capsid instability in these cases.
A molecular mechanism for a block of pregenome-P-protein packaging by the core protein mutations is not obvious. The mutations are located at the outer surface of the capsids, and the pregenome-P-protein complex is expected to interact with the interior side of the capsid. Possibly, the packaging process requires a complex interplay of CP domains which is disturbed in the case of the mutants. Very little is known about the selective packaging of the pregenome-P-protein complex into capsids. Capsid assembly by CP occurs at lower CP dimer concentrations when a pregenome-P-protein complex is participating (18). However, if such a complex is missing, then capsid assembly still occurs, although it requires higher concentrations of the capsid building blocks.
The mutant capsids devoid of viral nucleic acid were not competent for envelopment (Fig. 6). This reproduces the behavior of capsids consisting of WT CP but lacking viral nucleic acid (16). Apparently, none of the CP mutations analyzed in Fig. 6 induced a “mature” phenotype of the capsids independent of viral genome synthesis.
The capsid surface formed by residues S17, K96, and I126 might be a suitable target for an antiviral therapy, e.g., with a low-molecular-weight substance binding tightly to these residues. Apparently, escape mutations abolishing binding of the substance would not be viable, constraining the emergence of resistant strains.
Acknowledgments
We thank Andrea Koch and Udo Goldmann for excellent technical support.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG), SFB 402, Teilprojekt C2, and Graduiertenkolleg 521.
Footnotes
Published ahead of print on 26 August 2009.
REFERENCES
- 1.Beck, J., and M. Nassal. 2007. Hepatitis B virus replication. World J. Gastroenterol. 13:48-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belnap, D. M., N. R. Watts, J. F. Conway, N. Cheng, S. J. Stahl, P. T. Wingfield, and A. C. Steven. 2003. Diversity of core antigen epitopes of hepatitis B virus. Proc. Natl. Acad. Sci. USA 100:10884-10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruss, V. 2007. Hepatitis B virus morphogenesis. World J. Gastroenterol. 13:65-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruss, V. 1997. A short linear sequence in the pre-S domain of the large hepatitis B virus envelope protein required for virion formation. J. Virol. 71:9350-9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chain, B. M., and R. Myers. 2005. Variability and conservation in hepatitis B virus core protein. BMC Microbiol. 5:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dryden, K. A., S. F. Wieland, C. Whitten-Bauer, J. L. Gerin, F. V. Chisari, and M. Yeager. 2006. Native hepatitis B virions and capsids visualized by electron cryomicroscopy. Mol. Cell 22:843-850. [DOI] [PubMed] [Google Scholar]
- 7.Garcia, M. L., R. Byfield, and M. D. Robek. 2009. Hepatitis B virus replication and release are independent of core lysine ubiquitination. J. Virol. 83:4923-4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garoff, H., R. Hewson, and D. J. Opstelten. 1998. Virus maturation by budding. Microbiol. Mol. Biol. Rev. 62:1171-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerelsaikhan, T., J. E. Tavis, and V. Bruss. 1996. Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J. Virol. 70:4269-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kluge, B., M. Schlager, A. Pairan, and V. Bruss. 2005. Determination of the minimal distance between the matrix and transmembrane domains of the large hepatitis B virus envelope protein. J. Virol. 79:7918-7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koschel, M., D. Oed, T. Gerelsaikhan, R. Thomssen, and V. Bruss. 2000. Hepatitis B virus core gene mutations which block nucleocapsid envelopment. J. Virol. 74:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert, C., T. Doring, and R. Prange. 2007. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and gamma 2-adaptin. J. Virol. 81:9050-9060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Seyec, J., P. Chouteau, I. Cannie, C. Guguen-Guillouzo, and P. Gripon. 1999. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J. Virol. 73:2052-2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponsel, D., and V. Bruss. 2003. Mapping of amino acid side chains on the surface of hepatitis B virus capsids required for envelopment and virion formation. J. Virol. 77:416-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roseman, A. M., J. A. Berriman, S. A. Wynne, P. J. Butler, and R. A. Crowther. 2005. A structural model for maturation of the hepatitis B virus core. Proc. Natl. Acad. Sci. USA 102:15821-15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schormann, W., A. Kraft, D. Ponsel, and V. Bruss. 2006. Hepatitis B virus particle formation in the absence of pregenomic RNA and reverse transcriptase. J. Virol. 80:4187-4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seeger, C., and W. S. Mason. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seifer, M., and D. N. Standring. 1995. Assembly and antigenicity of hepatitis B virus core particles. Intervirology 38:47-62. [DOI] [PubMed] [Google Scholar]
- 19.Seitz, S., S. Urban, C. Antoni, and B. Bottcher. 2007. Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J. 26:4160-4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Summers, J., and W. S. Mason. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-415. [DOI] [PubMed] [Google Scholar]
- 21.Uy, A., V. Bruss, W. H. Gerlich, H. G. Kochel, and R. Thomssen. 1986. Precore sequence of hepatitis B virus inducing e antigen and membrane association of the viral core protein. Virology 155:89-96. [DOI] [PubMed] [Google Scholar]
- 22.Valenzuela, P., M. Quiroga, J. Zaldivar, R. Gray, and W. Rutter. 1980. The nucleotide sequence of the hepatitis B viral genome and the identification of the major viral genes. UCLA Symp. Mol. Cell. Biol. 18:57-70. [Google Scholar]
- 23.von Schwedler, U. K., M. Stuchell, B. Muller, D. M. Ward, H. Y. Chung, E. Morita, H. E. Wang, T. Davis, G. P. He, D. M. Cimbora, A. Scott, H. G. Krausslich, J. Kaplan, S. G. Morham, and W. I. Sundquist. 2003. The protein network of HIV budding. Cell 114:701-713. [DOI] [PubMed] [Google Scholar]
- 24.Wei, Y., J. E. Tavis, and D. Ganem. 1996. Relationship between viral DNA synthesis and virion envelopment in hepatitis B viruses. J. Virol. 70:6455-6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wynne, S. A., R. A. Crowther, and A. G. Leslie. 1999. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 3:771-780. [DOI] [PubMed] [Google Scholar]