

Abstract
Human T-lymphotropic virus type 1 (HTLV-1) is the etiological agent of adult T-cell leukemia/lymphoma, and it encodes a number of nonstructural proteins that are involved in virus replication and immune evasion. The viral protein p12 previously has been characterized to interfere with major histocompatibility complex class, ICAM-1, and ICAM-2 expression, and it activates STAT5. Using a previously established T-cell line immortalized with an HTLV-1 molecular clone deleted for p12, we assessed the role of p12 in regulating cellular growth and virus transmission. These cells were complemented for p12 expression by the transduction of a lentivirus vector expressing p12. We report that p12 conferred a selective growth advantage in vitro and increased the colony formation of human T cells in soft-agar assays. Consistently with previous studies, p12− and p12+ cell lines produced similar amounts of virus particles released into the supernatant of cultured cells, although we found that p12 expression greatly enhanced virus transmission. Moreover, we found that interleukin-2 (IL-2) stimulation also increased HTLV-1 transmission whether p12 was expressed or not, and inversely, that the inhibition of Jak signaling significantly reduced HTLV-1 transmission. Intriguingly, IL-2/Jak signaling was not associated with changes in viral gene expression, viral RNA encapsidation, the maturation of the virus particle, cell-cell adherence, or Gag polarization and virological synapse formation. We do demonstrate, however, that IL-2 stimulation and p12 expression significantly increased the rate of syncytium formation, revealing a novel role for IL-2 signaling and Jak activation in HTLV-1 virus transmission.
The retrovirus human T-cell leukemia virus type 1 (HTLV-1) is the etiological agent of adult T-cell leukemia/lymphoma (ATLL) (24, 57, 60), a lymphoproliferative disorder with an extremely poor prognosis that develops in approximately 2 to 5% of infected individuals. The viral genome encodes structural and enzymatic proteins, the regulatory proteins Tax and Rex, and additional small regulatory proteins (3, 8, 13, 37, 51). It still is unclear how HTLV-1 transforms human T cells, although HTLV-1-infected cells display alterations in a variety of cellular pathways. The oncoprotein Tax deregulates numerous pathways, such as the cell cycle, NF-κB, p53, and DNA repair pathways (28, 33, 39, 59). The virus also disrupts the proapoptotic network and simultaneously increases cellular proliferation (52). The longevity and replicative potential of HTLV-1-infected T cells also is increased through hTERT promoter expression and telomerase activity (7, 19, 64). Collectively, these events may lead to the accumulation of genetic defects, although direct evidence demonstrating whether these genetic alterations are virus related or are secondary events is lacking.
HTLV-1 infects and immortalizes primary human T cells in vitro, and after several months of culture, these cells become transformed and are able to grow in the absence of interleukin-2 (IL-2). IL-2 independence correlates in most cases with the constitutive activation of the Janus activated kinase/signal transducers and activators of transcription (Jak/STAT) pathway (45, 69) and the decreased expression of src homology 2-containing tyrosine phosphatase 1 protein (44). In early stages of ATLL, the viral proteins Tax and Rex are involved in the upregulation of IL-2 and the IL-2 receptor (IL-2R) (16, 41, 43, 63), possibly leading to the autocrine proliferation of infected cells. Although it is unknown whether these proteins still are expressed at levels sufficient to maintain the activation of the IL-2/IL-2R pathway in later stages of leukemia, ATLL tumor cells always express high levels of the IL-2Rα chain and usually show constitutive Jak/STAT activation (65). One particular HTLV-1 protein, p12, is involved in IL-2/IL-2R signaling and has been shown to interact with the IL-2R beta and gamma chains and increases STAT5-dependent transcription (53). p12 also has been shown to stimulate IL-2 production from infected T cells (23). The expression of p12 in Jurkat T cells elevates basal intracellular calcium and selectively activates the nuclear factor of activated T cells (NFAT), a downstream transcription factor in T lymphocytes (2). As a result, p12 expression was found to enhance IL-2 production in T lymphocytes in a calcium pathway-dependent manner.
The HTLV-1 p12 protein is a small hydrophobic protein that localizes to the endoplasmic reticulum, the Golgi body, and lipid rafts within the plasma membrane (26). The expression of p12 in HTLV-1-infected cells has been demonstrated by the detection of both p12-encoding transcripts in cultured and ex vivo cells from individuals infected with HTLV-1 and antibodies and cytotoxic T lymphocytes to p12 peptides in vivo (20, 56). Importantly, the ablation of the splice acceptor site for the singly spliced p12 mRNA from a molecular clone of HTLV-1 impaired viral infectivity in a rabbit model in vivo (15). This may be related in part to the finding that p12 interferes with major histocompatibility complex class I (MHC-I) heavy-chain trafficking and may facilitate the escape of HTLV-1-infected cells from host immune surveillance mechanisms (34). Recent studies also show the inability of NK cells to eliminate HTLV-1-infected CD4+ T cells despite the downmodulation of MHC-I molecules by HTLV-1 p12 (5). The authors found that the diminished activity of NK cells toward HTLV-1-infected cells was due to the decreased adherence between NK cells and HTLV-1-infected cells, largely through the downregulation of ICAM-1 and ICAM-2 by p12. p12 deletion also reduced virus transmission in quiescent peripheral blood mononuclear cells (PBMC) in the absence of IL-2 and mitogen stimulation (1). The ability of the mutated virus to infect PBMC was restored when mitogen was added to the culture system, indicating a critical role of p12 for viral infection in quiescent T lymphocytes.
Here, we present evidence that p12 provides a proliferative advantage to human T cells infected with HTLV-1 in limiting IL-2 concentrations. We found that p12 increased virus transmission, and this was mediated in part through the stimulation of the IL-2R and the activation of the Jak/STAT pathway in both IL-2-dependent and IL-2-independent HTLV-1-infected cells. While there was no difference in the amount of virus produced, viral genome encapsidation, virion maturation, or Gag polarization, we found that signaling by p12 or IL-2 through the IL-2R increased the rate of membrane fusion between infected cells and target cells. These data suggest that p12 and IL-2R/Jak/STAT signaling affects virus transmission and reveals a new role of the IL-2R/Jak pathway in retrovirus transmission.
MATERIALS AND METHODS
Cell culture, reagents, and cell sorting.
The cell lines A2 and R4 were established using HTLV-1 infectious molecular clone ACH and its syngeneic clone mutated for p12 as previously described (14). Cell lines were maintained in RPMI 1640 medium (Invitrogen Carlsbad, CA) supplemented with 20% fetal bovine serum (FBS), gentamicin, penicillin-streptomycin, and 40 U/ml IL-2. BHK1E6 cells were described previously (4). BHK1E6, ECV, and 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Cellgro) supplemented with 10% FBS, gentamicin, and penicillin-streptomycin. BHK1E6 cells were transfected with 1 μg of pCMV-Tax (61) using Polyfect per the manufacturer's instructions (Qiagen). Pyridone-6 ([P6] Jak inhibitor 1) (1 μM; Calbiochem), AG490 (50 μM; BioMol), and zidovudine (AZT) (30 μM; Sigma) were included where indicated. Complementation for p12 expression in R4 cells was obtained by infection with a lentivirus vector expressing p12IRES-GFP. Cells containing a lentivirus vector expressing only p12 to complement R4 (designated R4+p12 cells) were analyzed and sorted using green fluorescent protein (GFP) as a marker to greater than 90% purity with a FACSAria (BD Biosciences).
p12 lentivirus vector production and purification.
293T cells were transfected with 10 μg pHR′CMVp12IRES-GFP, 5 μg vesicular stomatitis virus G protein, and 5 μg pDNL6 using calcium phosphate (Invitrogen) according to the manufacturer's instructions as previously described (47, 53). The p12 virus was collected in the supernatant for 4 days, concentrated by ultracentrifugation, and resuspended in phosphate-buffered saline (PBS). Virus production was quantified using the RETROtek HTLV p19 antigen enzyme-linked immunosorbent assay (ELISA) (Zepto Metrix Corp) as previously reported (50).
Proliferation and soft-agar colony formation assays.
Cells were plated in duplicate for each time point at identical concentrations of 5 × 106 cells/ml in 96-well plates with increasing IL-2 concentrations from 0 to 40 U/ml. Cells were stained with trypan blue (Sigma) and then counted on days 0, 2, 4, 8, and 15. For soft-agar colony formation assays, 0.8 million cells were added to semisolid medium containing 0 to 40 U/ml of IL-2 and allowed to grow for 3 weeks. The smallest colony observed at 40 U/ml of IL-2 (approximately 10 cells) was used as a cutoff for all subsequent colony counts at all IL-2 concentrations; groups of cells smaller than this were not counted as a colony. Results represent the average numbers of colonies in 50 randomly chosen fields at ×10 magnification.
Virus transmission and syncytium assays.
BHK1E6 cells harboring a lacZ reporter gene behind a Tax-responsive promoter were cocultured with HTLV-1-infected lymphocytes for 48 h. Monolayers were washed with PBS and fixed with 4% (wt/vol) paraformaldehyde, and cells were stained with 200 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal). β-Galactosidase expression was visualized by bright-field microscopy. To inhibit Jak-STAT signaling, cells were pretreated for 24 h with either P6 (1 μM) or AG490 (50 μM) prior to coculture. To inhibit the HTLV-1 reverse transcriptase, cells were treated with AZT (30 μM) for 1 h at 37°C prior to coculture. To assess syncytium formation, 5 × 105 ECV cells were cocultured with 1 × 106 lymphocytes for 48 h. Following coculture, cells were washed twice with PBS, fixed with 4% (wt/vol) paraformaldehyde, and stained with 0.1% (wt/vol) crystal violet, and syncytia were visualized by bright-field microscopy.
Electron microscopy.
Cell or virus pellets were fixed overnight in 2% glutaraldehyde in 0.1 M cacodylate buffer, rinsed two times with buffer, and then postfixed in 1% osmium tetroxide-0.1 M cacodylate buffer for 1 h. Samples then were dehydrated through a series of graded ethanol solutions (30 to 100%) and placed into half Embed 812 resin/half propylene oxide to infiltrate overnight. Samples then were placed into complete Embed 812 resin (Electron Microscopy Sciences Inc.) for 1 h and placed into the appropriate molds with fresh resin to polymerize at 60°C overnight. Sections were cut at 70 to 80 nm on a Leica UCT ultramictrotome, collected onto 300-mesh Cu thin bar grids, and stained with uranyl acetate and lead citrate. Sections were examined and photographed using a JEOL 100CXII transmission electron microscope at 80 kV.
Western blotting.
Cell lysate or cell supernatant protein concentrations were determined by a modified Bradford assay (Bio-Rad). Proteins were separated by sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis and transferred to a polyvinyl difluoride membrane. Lysates were probed with either anti-p24 or anti-env Sp2,3/4A (NIH Bioreagents Research and Reference Program), followed by the addition of anti-mouse horseradish peroxidase (HRP) or anti-goat HRP (Santa Cruz) and detection by enhanced chemiluminescence (GE Healthcare Life Sciences).
Cell adherence, migration, and Gag polarization.
To assess cell adherence, MT-2 cells were labeled with 5-chloromethylfluorescein diacetate ([CMFDA] 5 μM; Molecular Probes) for 30 min according to the manufacturer's instructions. Following washes, cells were cocultured with BHK1E6 cells for 4 h, and monolayers were washed with three washes of PBS to remove unbound cells. Cells then were fixed with 4% paraformaldehyde and washed with PBS, and CMFDA-labeled cells were visualized using a laser-scanning cytometer (iCys; Compucyte).
Cell migration was assessed essentially as described previously (40, 71). Cells were incubated in the absence of IL-2 for 4 h and then added to the top of a 5-μm polycarbonate membrane (Neuro Probe, Inc.) soaked with 10 μg/ml ICAM (R&D Systems) in a chemotaxis chamber containing various amounts of IL-2 in the lower chamber. Cells were allowed to migrate for 90 min and then fixed to the membrane and stained with acid hematoxylin (Sigma). Membranes were cleared with 100% xylenes (Sigma) and mounted using permount (Sigma). The migration distance through the filter was measured in micrometers using the leading-front technique. Data are representative of 50 fields each of duplicate experiments.
Gag polarization and cell fusion immunofluorescence.
Gag polarization was performed essentially as described previously (31). MT-2 cells or R4 cells were cultured in the presence of IL-2 or P6 (1 μM) for 48 h, washed, and cocultured with CMFDA-labeled Jurkat cells at a 1:1 ratio for 40 min. Cells were centrifuged using a cytospin, fixed with 4% paraformaldehyde, and permeabilized with 0.5% Triton X-100. Cells were stained with anti-p24, followed by costaining with anti-mouse-Texas Red and 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma). Cells were mounted onto glass slides and visualized by laser-scanning confocal microscopy (Zeiss). For fusion analysis, ECV or BHK1E6 cells were plated onto glass coverslips and were transfected with red fluorescent protein (RFP) for 24 h with Lipofectamine 2000 (Invitrogen). R4 cells were stained with CMFDA as described above (Molecular Probes) and were cocultured with either ECV or BHK1E6 cells for 36 h. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, stained with DAPI, mounted, and visualized as described above.
RESULTS
Generation of HTLV-1 p12− and HTLV-1 p12+ cell lines.
Previous studies have established that the small regulatory proteins p12, p13, and p30 are dispensable for virus replication and human T-cell immortalization by HTLV-1 (22). Additional studies also have shown that p12 alone is sufficient to increase the proliferation of human PBMCs after the stimulation of T-cell receptors with low concentrations of anti-CD3 and anti-CD28 antibodies (53). Moreover, the proliferative advantage of p12-transduced PBMCs was evident mainly in limiting concentrations of IL-2. However, these experiments were performed in the absence of any other viral proteins. Here, we used a previously established human T-cell line immortalized with an HTLV-1 molecular clone, ACH, referred to here as A2, and a cell line derived with the corresponding isogenic molecular clone ablated for p12 expression, referred to as R4 (14). To confirm that differences observed between A2 and R4 are due to the absence of p12, we used a lentivirus vector expressing only p12 to complement R4 (R4+p12). The retroviral vector used (Fig. 1A) expresses a polycistronic mRNA encoding the gene of interest and an internal ribosome entry site (IRES) followed by GFP. In this context, GFP-positive transduced cells expressing p12 could be sorted and analyzed independently from nontransduced p12-negative cells (Fig. 1B).
FIG. 1.

Production of p12-expressing lentiviral vector and cells. (A) Schematic representation of the p12 IRES GFP construct. (B) Infection efficiency in 293T cells following overnight infection with a p12-expressing lentiviral vector. Five hundred microliters of concentrated virus yields nearly 100% GFP-positive cells. (C) A2 cells, which naturally express p12, R4 cells, which are devoid of p12, or R4 cells, expressing p12, were cultured for up to 15 days in RPMI medium in the presence of 0, 5, 10, or 40 U/ml of IL-2. Cells were harvested at 2, 4, 8, and 15 days, stained with trypan blue, and counted. Results are representative of two independent experiments done in triplicate, and standard deviations are shown (P < 0.01). Arrows indicate differences in growth at 0 or 5 U of IL-2 in the presence or absence of p12. (D) p12 enhances colony formation at low levels of IL-2. Average numbers of colonies formed by R4 with and without p12 following 3 weeks of growth in semisolid media with 0, 5, 10, 20, and 40 U IL-2/ml are shown. Results represent the average numbers of colonies in 50 randomly chosen fields at ×4 magnification. (E) Representative bright-field images of R4 and R4+p12 colonies grown in 0 U of IL-2, taken at ×10 magnification.
p12 stimulates cell proliferation in limiting concentrations of IL-2.
We investigated the proliferation of A2, R4, and R4+p12 in media containing various concentrations of IL-2. Our previous studies showed that, in p12-transduced PBMCs, the increased basal level of STAT5 activation by p12 stimulated cell proliferation at lower concentrations of exogenous IL-2 (53). Experiments examining cell proliferation in the context of other HTLV-1 genes, however, have not been done. To address this, the HTLV-1-infected cell lines A2, R4, and R4+p12 were divided into four aliquots and plated in triplicate in 96-well plates in the presence or absence of 5, 10, or 40 U of IL-2, and cell proliferation was monitored for 15 days. In the absence of p12 expression, the long-term growth of R4 cells was severely impaired in no or low concentrations of IL-2 (<5 U/ml) (Fig. 1C) compared to the growth of p12-expressing A2 cells. Differences in proliferation, however, were not detected when cells were cultured in higher IL-2 concentrations, suggesting that proliferative advantages conferred by p12 are compensated for by IL-2 signaling. Of note, p12 does not appear to offer any short-term proliferative advantage, which agrees with previous observations (14). The long-term differences seen in proliferation were due to the lack of p12 rather than genetic differences between A2 and R4, since the complementation of R4 with p12 (R4+p12) restored proliferation to levels seen with A2 in the presence of 5 U or less of IL-2 (Fig. 1C). Significantly, we observed a twofold increase in the proliferation of R4+p12 or A2 cells above that of R4 cells alone at limiting concentrations of IL-2, suggesting that p12 overcomes a proliferative disadvantage in the absence of IL-2.
Transforming activity of p12 in soft-agar colony formation assay.
It has been shown previously that HTLV-1 Tax exhibits transforming activity and induces fibroblast colony formation in soft agar (42). Although p12 alone cannot induce the formation of colonies, it strongly potentiates the transforming activity of the E5 oncoprotein (25). We sought to measure the ability of p12, in the context of the HTLV-1 genome, to enhance colony formation in different concentrations of IL-2. R4 cells are IL-2 dependent and are not fully transformed, making them suitable for assays in the absence of IL-2. R4 and R4+p12 cells were seeded in soft agar, and colonies were counted after 14 days. In large amounts of IL-2, both R4 and R4+p12 cells formed multiple large colonies in soft agar, and the number and sizes of the colonies were not statistically different (Fig. 1D). The expression of p12, however, facilitated the formation of numerous large colonies when cells were seeded in media containing low levels of IL-2, or even in media without IL-2. In contrast, almost no colonies were observed with R4 cells alone in the absence of IL-2 (Fig. 1D), and any colonies observed were much smaller in size than typical R4+p12 colonies (Fig. 1E). These data suggest that p12 not only stimulates proliferation but also synergizes with other viral genes to increase the colony formation potential of HTLV-1.
p12 expression increases cell-cell virus transmission.
An increase in the proliferation of virus-producing cells often is associated with an increase in viral particle production and the subsequent infection of target cells. We therefore assessed whether p12 expression had an effect on HTLV-1 transmission. We used R4 and R4+p12 as the virus producer cell lines and BHK1E6 as a reporter cell line, which previously has been characterized to measure HTLV-1 transmission (4). BHK1E6 cells are permissive to HTLV-1 and are stably transfected with the HTLV-1 long terminal repeat (LTR) fused to the β-galactosidase gene, and infection can be monitored by X-Gal staining. When p12 was expressed in R4 cells and cocultured with BHK1E6 target cells, the number of infected target cells was increased more that twofold compared to the level for R4 cells lacking p12 (Fig. 2A). The ability of p12 to increase virus transmission also was dose dependent, as an increasing number of virus-producing cells resulted in the increased infection of the reporter BHK1E6 cell line (Fig. 2A). In agreement with previous reports, HTLV virus transmission from T cells required cell-cell contact (35), as fresh cell supernatants did not give rise to LacZ positivity (data not shown). Intriguingly, when R4 cells either expressing or lacking p12 were cocultured with BHK1E6 in the absence of IL-2, virus transmission was reduced nearly threefold compared to that of cells cultured in the presence of IL-2 (Fig. 2B). The removal of IL-2 from the culture media of R4 cells did not affect R4 survival, as assessed by trypan blue exclusion (data not shown). To preclude the possibility that this effect of IL-2 on virus transmission was limited to the R4 cell line, we also used the MT-2 cell line, which is an HTLV-1-transformed cell line that is not dependent on IL-2 for growth but expresses high levels of CD25 (IL-2R alpha chain). Interestingly, virus transmission mediated by MT-2 cells, which occurs in the absence of IL-2, also was increased twofold in the presence of IL-2 (Fig. 2C). Significantly, the inclusion of the reverse transcriptase inhibitor AZT, which prevents the viral reverse transcriptase from generating a provirus, inhibited virus transmission by MT-2 cells, demonstrating that LacZ expression requires virus uncoating and the integration of the HTLV-1 genome (Fig. 2D) and is not due to the transfer of Tax protein to the target BHK1E6 cells.
FIG. 2.
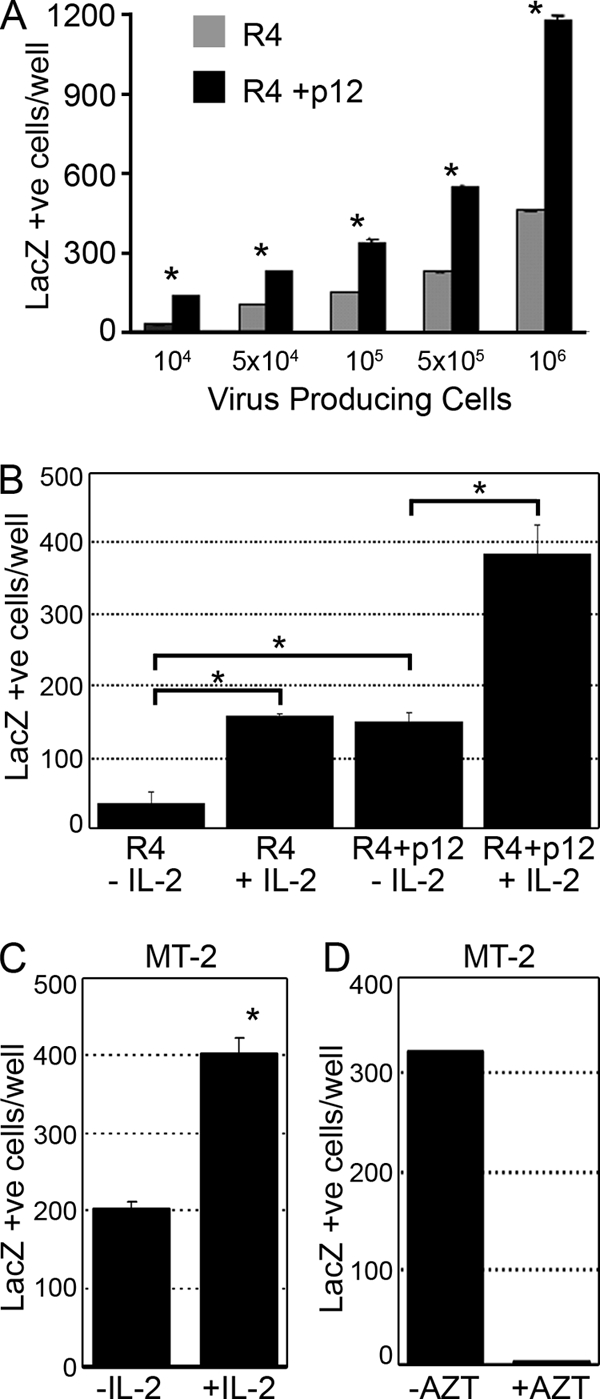
HTLV-1 p12 and IL-2 increase virus transmission in vitro. (A) BHK1E6 cells were cocultured with R4 cells or R4 cells expressing p12 in the presence or absence of IL-2. Cells were fixed and stained with X-Gal, and LacZ-positive cells were counted. Standard deviations are shown. *, P < 0.01. (B) BHK1E6 cells were cocultured with either R4 cells or p12-expressing R4 cells in the absence or presence of IL-2 and were fixed and stained with X-Gal as described for panel A. (C) MT-2 cells were cultured in the presence or absence of IL-2, cocultured with BHK1E6 cells, and stained as described for panel A. (D) MT-2 cells were treated with 10 μM AZT, cocultured with BHK1E6 cells, and stained as described above.
Inhibition of IL-2R/Jak signaling decreases virus transmission.
Since virus transmission mediated by both R4 and MT-2 cells was significantly increased in the presence of IL-2 (Fig. 2C) and both IL-2 and p12 induce Jak/STAT activation, we hypothesized that this increase in virus transmission is the result of Jak/STAT activation through the IL-2R. We therefore tested the ability of two known antagonists of Jak/STAT signaling, P6 and AG490, to modulate HTLV-1 virus transmission. Interestingly, the inclusion of either P6 or AG490 inhibited HTLV-1 transmission by both R4 cells and MT-2 cells (Fig. 3A and B), suggesting that Jak/STAT signaling is involved in virus transmission. The inclusion of these inhibitors did not affect cell viability (data not shown) and did not inhibit the Tax-mediated transactivation of the HTLV-1 LTR and the activation of the LacZ reporter gene in BHK1E6 cells following the transient transfection of Tax (Fig. 3C).
FIG. 3.
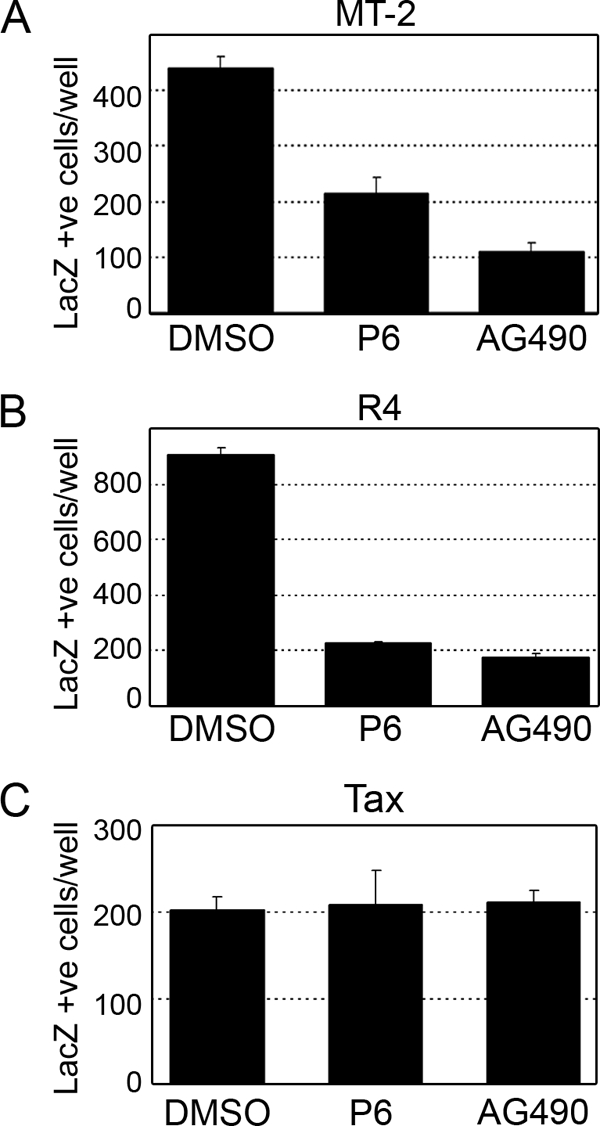
Inhibition of Jak/STAT signaling decreases HTLV-1 infectivity. (A and B) MT-2 cells (A) or R4 cells (B) were treated with P6 or AG490 for 24 h and were cocultured with BHK16 cells for an additional 48 h. Cells were fixed and stained with X-Gal, and LacZ-positive cells were counted. (C) Jak/STAT signaling is not required for the Tax-mediated expression of LacZ in BHK1E6 cells. Cells were transfected with 1 μg of pCMV-Tax in the presence of dimethylsulfoxide (DMSO), AG490, or P6 for 24 h, fixed, and stained with X-Gal.
p12 increases chemotaxis but not cell adherence or virion morphology.
The increase in virus transmission due to p12 expression or IL-2 stimulation was independent of an increase in virus production, as an ELISA to detect the viral p19 Gag protein showed similar amounts of p19 present in the supernatant of R4 and R4+p12 cells (Fig. 4A). Viral Gag p24 expression in cell pellets or in the supernatants from infected cells also was unchanged in the presence or absence of either IL-2 or Jak/STAT inhibitors (Fig. 4B). We also tested the encapsidation of the viral genome by performing quantitative real-time RT-PCR to detect the HTLV-1 genome from purified viral particles, but we did not observe any significant difference between controls and IL-2-stimulated samples, suggesting that variations in genome encapsidation are not involved (data not shown).
FIG. 4.

Neither p12 nor IL-2 signaling alters Gag expression, virus production, or cell adherence. (A) Supernatants from R4 or R4 cells expressing p12 were analyzed by ELISA to detect Gag p19 expression. (B) Extracellular virus and cell-associated (intracellular) protein preparations from R4 cells treated with IL-2, P6, or AG490 were analyzed by Western blotting (WB) for Gag p24 expression. (C and D) IL-2 has no effect on the adherence of HTLV-1-infected cells to target cells. MT-2 cells (1 × 106) grown in the presence or absence of IL-2 were stained with CMFDA, cocultured with a BHK1E6 monolayer for 4 h, analyzed by laser-scanning cytometry (C), and quantified (D). (E) Electron microscopy of virus particles released from MT-2 cells grown in the presence or absence of IL-2 (scale bar = 100 nm); 1 representative image of 20 is shown. (F) Chemotaxis of R4 or R4+p12 cells toward increasing amounts of IL-2 was performed as described in Materials and Methods. The distance migrated is indicated in micrometers, and standard deviations are shown. *, P < 0.001.
One defining characteristic of HTLV-1-infected T cells is that cell-to-cell contact is required for the efficient transmission of virus particles to target cells. We addressed the ability of infected cells to adhere to target cells by labeling HTLV-1-infected producer cells with a green fluorescent dye, coculturing them with BHK1E6 cells, and quantifying the number of adherent producer cells using a fluorescent scanning cytometer. The culturing of cells in the presence or absence of IL-2 had no effect on the ability of HTLV-1-infected cells to adhere to BHK1E6 monolayers (Fig. 4C and D), suggesting that adherence is unaffected by IL-2 signaling. The electron microscopy of virus particles released from either MT-2 or R4 cells in the presence or absence of IL-2 also showed no significant difference in virion morphology, as the majority of particles possessed an electron-dense core, which contains the viral RNA (Fig. 4E) (67).
Cell-free infection with purified HTLV-1 particles from T cells is extremely inefficient compared to virus transmission through cell-cell contact. p12 increases cell-to-cell spread, and such a mechanism has been shown to be relevant to de novo T-cell infection following p12-induced LFA-1 clustering on T cells via calcium-dependent signaling (36). HTLV-1-infected lymphocytes also have been shown to produce cell surface protrusions (70) that are similar to those seen during cell spreading. As a result, we hypothesized that p12 increases chemotaxis as a method to facilitate virus transmission. Cell migration was analyzed using a polycarbonate membrane that served as a barrier to discriminate migratory cells from nonmigratory cells (71). Migrating cells are able to extend protrusions in the presence of a chemoattractant and pass through the pores of the polycarbonate membrane. Finally, the cells are removed from the top of the membrane, and the migratory cells are stained and quantified. The migration distance through the filter was measured in micrometers using the leading-front technique as previously described (40). Our data demonstrate that p12 expression increased the ability of cells to migrate through filters (Fig. 4F) both in the presence and absence of IL-2.
IL-2- and Jak-dependent virus transmission is independent of Gag polarization but increases the rate of membrane fusion.
Recent studies have shown that the cell-cell transmission of HTLV can occur through a virological synapse, where viral core proteins such as Gag are polarized at the site of cell-cell contact (31). We tested the ability of Gag to cluster at the virological synapse in the presence or absence of IL-2. Jurkat cells were labeled with the green fluorescent dye CMFDA and were cocultured with R4 or MT-2 cells for 40 min. Cells were fixed and stained with anti-p24 to label Gag, and synapse formation was evaluated by immunofluorescence. As seen in Fig. 5, treatment with IL-2 or the Jak/STAT inhibitor P6 had no observable effect on Gag polarization. At least 100 conjugates from each sample were analyzed, with approximately 60% of all conjugates from each sample displaying the polarization of Gag (data not shown).
FIG. 5.

IL-2 expression does not affect Gag polarization and synapse formation. R4 or MT-2 cells were cultured in the presence or absence of IL-2 or P6 for 48 h and were cocultured with CMFDA-labeled Jurkat cells for 40 min. Cells were fixed and stained with Gag-specific antisera (α-Gag) and were visualized by confocal microscopy. DIC, differential interference contrast.
Since free HTLV virions are essentially noninfectious and virus transmission can occur through cell-cell contact and fusion (21, 30, 46), we addressed the ability of IL-2 and p12 to regulate membrane fusion induced by R4 cells using a syncytium assay. R4 cells grown in the absence of IL-2 and cocultured with ECV cells induced the formation of small multinuclear syncytia. R4 cells cultured in the presence of IL-2 and cocultured with ECV cells, however, induced the formation of nearly three times the number of syncytia, suggesting that signaling through the IL-2 pathway increases the ability of infected cells to undergo membrane fusion (Fig. 6A and C). To verify that membrane fusion occurs between R4 and ECV cells, we examined the transfer of soluble RFP from transiently transfected ECV cells to CMFDA-labeled green R4 cells. As seen in Fig. 6B, RFP was readily transferred to R4 cells within syncytia. This was in stark contrast to the complete lack of transfer of RFP from BHK1E6 cells to R4 cells, as BHK cells do not exhibit membrane fusion or syncytium formation (Fig. 6B) (4). In addition to increasing the number of syncytia, treatment with IL-2 also increased the average number of nuclei seen in each syncytium (data not shown). Similar results were shown in R4 cells expressing p12, as these cells also displayed a nearly threefold increase in syncytium formation (Fig. 6D). To demonstrate the involvement of the Jak/STAT pathway during IL-2 stimulation in syncytium formation, the inclusion of Jak/STAT inhibitors inhibited the formation of syncytia by upwards of threefold (Fig. 6E), indicating that Jak/STAT activation plays a significant role in syncytium formation. While previous work has shown that the envelope protein from HTLV-1 is responsible for cell fusion, flow cytometry and Western blot analysis failed to detect any significant change in Env expression or cleavage in the presence or absence of IL-2 (Fig. 6F and G), suggesting that IL-2 and Jak/STAT signaling enhance virus transmission and syncytium formation without altering envelope protein expression.
FIG. 6.

Membrane fusion and syncytium formation is enhanced by p12 expression and IL-2. (A) ECV cells were cocultured with R4 cells in the presence or absence of IL-2 for 48 h, fixed, and stained with crystal violet, and cells were visualized by bright-field microscopy. (B) R4 cells were stained with green CMFDA and cocultured with either ECV or BHK1E6 cells transfected with red fluorescent protein (RFP). Cells were fixed, stained with DAPI, and visualized by immunofluorescence. (C) Multinucleated syncytia depicted in panel A were counted from 30 fields of view from experiments performed in triplicate, and data are presented as the average number of syncytia per field of view (f.o.v.). Standard deviations are shown, and asterisks indicate P < 0.01. (D) R4 cells or R4 cells expressing p12 were cocultured with ECV cells for 48 h, fixed, stained, and visualized as described for panel C. (E) Inhibition of Jak/STAT signaling inhibits syncytium formation. R4 cells were treated with P6 or AG490, cocultured with ECV cells, fixed, stained, and counted as described for panel C. (F) R4 cells cultured for 48 h in the presence or absence of IL-2 were fixed, stained with anti-Env, and analyzed by flow cytometry. (G) Cell lysates from cells cultured as described for panel F were analyzed by Western blotting (WB) with anti-Env antibody.
DISCUSSION
The importance of p12 in HTLV-1 transmission and persistence was demonstrated first by the selective ablation of p12 from an HTLV-1 proviral clone that prevented virus transmission in vivo and in quiescent PBMCs in the absence of IL-2 (1, 15). Our results support the observations that p12 also is important for virus transmission in vitro, and this likely occurs through the induction of the IL-2/Jak/STAT signaling pathway. p12 expression also enhanced cell proliferation in the absence of IL-2, suggesting that Jak/STAT activation is important for both HTLV-1-mediated cell growth and virus transmission. Although p12 expression is dispensable for HTLV-1 T-cell immortalization and growth in vitro, our results indicate that p12 increases the proliferation of HTLV-1-infected cells at low IL-2 concentrations, particularly between 8 and 15 days posttreatment (Fig. 1C).
Despite the fact that no significant difference in the amount of virus produced between HTLV-1 p12+ and p12− cells was seen (Fig. 4A), we found that p12 expression and IL-2/Jak signaling significantly increased virus transmission in coculture assays (Fig. 2), suggesting that IL-2 signaling through the Jak/STAT pathway plays a role in the transmission of HTLV-1 to uninfected target cells. While our investigation demonstrated that p12 increases the chemotaxis of HTLV-1-infected cells, we did not see a difference in cell adherence, Gag polarization, synapse formation, viral RNA encapsidation, or virion structure. We did, however, observe an increase in syncytium formation between IL-2-stimulated or p12-expressing HTLV-1-infected cells and ECV cells. These data suggest that IL-2 and p12 affect the maturation of the viral envelope. Cell-cell contact and fusion events are complex and are not well understood. Certain cell types readily undergo fusion, other cell types are nonfusiogenic, and still other cell types remarkably have been shown to undergo cell fusion without any apparent increase in virus transmission or the resulting trans-activation of reporter constructs (17, 18, 29, 32). It has been hypothesized that membrane fusion between producer and target cells occurs through a process of at least two steps following binding (17), so it will be of interest to see whether p12 expression and IL-2 signaling affect any of the various steps involving fusion and virus transmission. Syncytium formation and virus transmission also are regulated by multiple cell surface proteins, further complicating these events (11, 17, 18, 27, 29, 32).
Several other possibilities explain the effect of p12/IL-2/Jak signaling on the increase in HTLV-1 transmission. Recent studies have shown that p12 localizes to lipid rafts at the plasma membrane, and that the disruption of lipid rafts alters virus transmission (12, 54, 58, 68). Since the IL-2R is a known component of lipid rafts, lipid raft formation induced by IL-2/Jak/STAT signaling may be required for the efficient transmission of HTLV. Alternatively, Jak/STAT activation induced by p12 and IL-2 may regulate host or viral proteins involved in virus transmission. Indeed, we observed an almost threefold increase in syncytium formation in the presence of p12 or IL-2, but no change was seen in the expression of Env, which is intimately involved in syncytium formation. The human disc large protein (Dlg1) has been shown to interact with the envelope protein from HTLV-1, and this interaction is required for syncytium formation (9, 62). Env also is known to undergo a number of other changes, including disulfide isomerization and a series of glycosylation events, that are required for syncytium formation (38, 55). Whether such posttranslational modifications rely on the activity of nonstructural viral proteins such as p12 or are regulated by IL-2 and Jak/STAT signaling remains to be investigated.
Virus transmission from HTLV-1-infected T cells occurs through a virological synapse, whereby the microtubule-organizing center (MTOC) and other cytoskeletal components of the cell are reorganized and polarized following contact with an uninfected cell (31). Our observation that the stimulation of the IL-2R (CD25) has an effect on HTLV-1 transmission is intriguing. Recently, to identify the cell surface signal that triggers MTOC polarization during virological synapse formation, Barnard et al. coated latex beads with monoclonal antibodies against a variety of T-cell surface molecules (6). They found ICAM-1 to be one such surface molecule that could be stimulated to induce MTOC rearrangement (6). Tax expression increases the expression of ICAM-1, and the cross-linking of cell surface ICAM-1 leads to MTOC polarization (48, 49, 66), while p12 appears to induce LFA-1 clustering on the cell surface (36). It has been postulated that Tax and p12 play different regulatory roles in regulating the expression of cell surface markers, which may control synapse formation and subsequent virus transmission (49). Since the IL-2R physically associates with ICAM-1 in the plasma membrane (10), it is possible that the activation and cross-linking of the IL-2R by IL-2 or p12 expression activates microtubule polarization indirectly via its association with ICAM-1. Previous studies also showed that HTLV-1 Gag protein appears to be transported to the MTOC by a microtubule-dependent process (48), which is similar to observations involving a number of other viruses. The further analysis of the ability of p12- and IL-2-dependent signaling to induce changes in HTLV-1 infectivity may lead to the development of future antiretroviral therapeutic strategies to limit virus replication. Overall, our results show that signaling through the IL-2R/Jak pathway increases the transmission potential and syncytium formation ability of HTLV-1-infected cells and demonstrate a novel role for IL-2/Jak signaling in retrovirus transmission.
Acknowledgments
We thank Joyce Slusser at the KU Medical Center Flow Cytometry Core and Barbara Fegley at the KU Medical Center Electron Microscopy Research Laboratory for their technical assistance. We also thank Charles Bangham for helpful discussions.
L. Ratner was supported by grants CA63417 and CA10521. M. Larimore was supported by grants CA100730 and CA70529. This work was supported by grants CA115398 and CA106258 from the National Cancer Institute to C. Nicot.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Published ahead of print on 2 September 2009.
REFERENCES
- 1.Albrecht, B., N. D. Collins, M. T. Burniston, J. W. Nisbet, L. Ratner, P. L. Green, and M. D. Lairmore. 2000. Human T-lymphotropic virus type 1 open reading frame I p12I is required for efficient viral infectivity in primary lymphocytes. J. Virol. 74:9828-9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albrecht, B., C. D. D'Souza, W. Ding, S. Tridandapani, K. M. Coggeshall, and M. D. Lairmore. 2002. Activation of nuclear factor of activated T cells by human T-lymphotropic virus type 1 accessory protein p12I. J. Virol. 76:3493-3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albrecht, B., and M. D. Lairmore. 2002. Critical role of human T-lymphotropic virus type 1 accessory proteins in viral replication and pathogenesis. Microbiol. Mol. Biol. Rev. 66:396-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Astier-Gin, T., J. P. Portail, F. Lafond, and B. Guillemain. 1995. Identification of HTLV-1- or HTLV-1I-producing cells by cocultivation with BHK-21 cells stably transfected with a LTR-lacZ gene construct. J. Virol. Methods 51:19-29. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee, P., G. Feuer, and E. Barker. 2007. Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and -2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J. Virol. 81:9707-9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnard, A. L., T. Igakura, Y. Tanaka, G. P. Taylor, and C. R. Bangham. 2005. Engagement of specific T-cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood 106:988-995. [DOI] [PubMed] [Google Scholar]
- 7.Bellon, M., and C. Nicot. 2008. Regulation of telomerase and telomeres: human tumor viruses take control. J. Natl. Cancer Inst. 100:98-108. [DOI] [PubMed] [Google Scholar]
- 8.Berneman, Z. N., R. B. Gartenhaus, M. S. Reitz, Jr., W. A. Blattner, A. Manns, B. Hanchard, O. Ikehara, R. C. Gallo, and M. E. Klotman. 1992. Expression of alternatively spliced human T-lymphotropic virus type I pX mRNA in infected cell lines and in primary uncultured cells from patients with adult T-cell leukemia/lymphoma and healthy carriers. Proc. Natl. Acad. Sci. USA 89:3005-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blot, V., L. Delamarre, F. Perugi, D. Pham, S. Benichou, R. Benarous, T. Hanada, A. H. Chishti, M. C. Dokhelar, and C. Pique. 2004. Human Dlg protein binds to the envelope glycoproteins of human T-cell leukemia virus type 1 and regulates envelope mediated cell-cell fusion in T lymphocytes. J. Cell Sci. 117:3983-3993. [DOI] [PubMed] [Google Scholar]
- 10.Burton, J., C. K. Goldman, P. Rao, M. Moos, and T. A. Waldmann. 1990. Association of intercellular adhesion molecule 1 with the multichain high-affinity interleukin 2 receptor. Proc. Natl. Acad. Sci. USA 87:7329-7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceccaldi, P. E., F. Delebecque, M. C. Prevost, A. Moris, J. P. Abastado, A. Gessain, O. Schwartz, and S. Ozden. 2006. DC-SIGN facilitates fusion of dendritic cells with human T-cell leukemia virus type 1-infected cells. J. Virol. 80:4771-4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chazal, N., and D. Gerlier. 2003. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 67:226-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciminale, V., G. N. Pavlakis, D. Derse, C. P. Cunningham, and B. K. Felber. 1992. Complex splicing in the human T-cell leukemia virus (HTLV) family of retroviruses: novel mRNAs and proteins produced by HTLV type I. J. Virol. 66:1737-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins, N. D., C. D'Souza, B. Albrecht, M. D. Robek, L. Ratner, W. Ding, P. L. Green, and M. D. Lairmore. 1999. Proliferation response to interleukin-2 and Jak/Stat activation of T cells immortalized by human T-cell lymphotropic virus type 1 is independent of open reading frame I expression. J. Virol. 73:9642-9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins, N. D., G. C. Newbound, B. Albrecht, J. L. Beard, L. Ratner, and M. D. Lairmore. 1998. Selective ablation of human T-cell lymphotropic virus type 1 p12I reduces viral infectivity in vivo. Blood 91:4701-4707. [PubMed] [Google Scholar]
- 16.Cross, S. L., M. B. Feinberg, J. B. Wolf, N. J. Holbrook, F. Wong-Staal, and W. J. Leonard. 1987. Regulation of the human interleukin-2 receptor alpha chain promoter: activation of a nonfunctional promoter by the transactivator gene of HTLV-1. Cell 49:47-56. [DOI] [PubMed] [Google Scholar]
- 17.Daenke, S., and S. Booth. 2000. HTLV-1-induced cell fusion is limited at two distinct steps in the fusion pathway after receptor binding. J. Cell Sci. 113:37-44. [DOI] [PubMed] [Google Scholar]
- 18.Daenke, S., S. A. McCracken, and S. Booth. 1999. Human T-cell leukaemia/lymphoma virus type 1 syncytium formation is regulated in a cell-specific manner by ICAM-1, ICAM-3 and VCAM-1 and can be inhibited by antibodies to integrin beta2 or beta7. J. Gen. Virol. 80:1429-1436. [DOI] [PubMed] [Google Scholar]
- 19.Datta, A., M. Bellon, U. Sinha-Datta, A. Bazarbachi, Y. Lepelletier, D. Canioni, T. A. Waldmann, O. Hermine, and C. Nicot. 2006. Persistent inhibition of telomerase reprograms adult T-cell leukemia to p53-dependent senescence. Blood 108:1021-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dekaban, G. A., A. A. Peters, J. C. Mulloy, J. M. Johnson, R. Trovato, E. Rivadeneira, and G. Franchini. 2000. The HTLV-1 orfI protein is recognized by serum antibodies from naturally infected humans and experimentally infected rabbits. Virology 274:86-93. [DOI] [PubMed] [Google Scholar]
- 21.Delamarre, L., A. R. Rosenberg, C. Pique, D. Pham, and M. C. Dokhelar. 1997. A novel human T-leukemia virus type 1 cell-to-cell transmission assay permits definition of SU glycoprotein amino acids important for infectivity. J. Virol. 71:259-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derse, D., J. Mikovits, and F. Ruscetti. 1997. X-I and X-II open reading frames of HTLV-1 are not required for virus replication or for immortalization of primary T-cells in vitro. Virology 237:123-128. [DOI] [PubMed] [Google Scholar]
- 23.Ding, W., S. J. Kim, A. M. Nair, B. Michael, K. Boris-Lawrie, A. Tripp, G. Feuer, and M. D. Lairmore. 2003. Human T-cell lymphotropic virus type 1 p12I enhances interleukin-2 production during T-cell activation. J. Virol. 77:11027-11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franchini, G. 1995. Molecular mechanisms of human T-cell leukemia/lymphotropic virus type I infection. Blood 86:3619-3639. [PubMed] [Google Scholar]
- 25.Franchini, G., J. C. Mulloy, I. J. Koralnik, A. Lo Monico, J. J. Sparkowski, T. Andresson, D. J. Goldstein, and R. Schlegel. 1993. The human T-cell leukemia/lymphotropic virus type I p12I protein cooperates with the E5 oncoprotein of bovine papillomavirus in cell transformation and binds the 16-kilodalton subunit of the vacuolar H+ ATPase. J. Virol. 67:7701-7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukumoto, R., M. Dundr, C. Nicot, A. Adams, V. W. Valeri, L. E. Samelson, and G. Franchini. 2007. Inhibition of T-cell receptor signal transduction and viral expression by the linker for activation of T cells-interacting p12I protein of human T-cell leukemia/lymphoma virus type 1. J. Virol. 81:9088-9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gauthier, S., I. Pelletier, M. Ouellet, A. Vargas, M. J. Tremblay, S. Sato, and B. Barbeau. 2008. Induction of galectin-1 expression by HTLV-1 Tax and its impact on HTLV-1 infectivity. Retrovirology 5:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grassmann, R., M. Aboud, and K. T. Jeang. 2005. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene 24:5976-5985. [DOI] [PubMed] [Google Scholar]
- 29.Hildreth, J. E., A. Subramanium, and R. A. Hampton. 1997. Human T-cell lymphotropic virus type 1 (HTLV-1)-induced syncytium formation mediated by vascular cell adhesion molecule-1: evidence for involvement of cell adhesion molecules in HTLV-1 biology. J. Virol. 71:1173-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoshino, H., M. Shimoyama, M. Miwa, and T. Sugimura. 1983. Detection of lymphocytes producing a human retrovirus associated with adult T-cell leukemia by syncytia induction assay. Proc. Natl. Acad. Sci. USA 80:7337-7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Igakura, T., J. C. Stinchcombe, P. K. Goon, G. P. Taylor, J. N. Weber, G. M. Griffiths, Y. Tanaka, M. Osame, and C. R. Bangham. 2003. Spread of HTLV-1 between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713-1716. [DOI] [PubMed] [Google Scholar]
- 32.Imai, T., K. Fukudome, S. Takagi, M. Nagira, M. Furuse, N. Fukuhara, M. Nishimura, Y. Hinuma, and O. Yoshie. 1992. C33 antigen recognized by monoclonal antibodies inhibitory to human T cell leukemia virus type 1-induced syncytium formation is a member of a new family of transmembrane proteins including CD9, CD37, CD53, and CD63. J. Immunol. 149:2879-2886. [PubMed] [Google Scholar]
- 33.Jeang, K. T., C. Z. Giam, F. Majone, and M. Aboud. 2004. Life, death, and tax: role of HTLV-1 oncoprotein in genetic instability and cellular transformation. J. Biol. Chem. 279:31991-31994. [DOI] [PubMed] [Google Scholar]
- 34.Johnson, J. M., J. C. Mulloy, V. Ciminale, J. Fullen, C. Nicot, and G. Franchini. 2000. The MHC class I heavy chain is a common target of the small proteins encoded by the 3′ end of HTLV type 1 and HTLV type 2. AIDS Res. Hum. Retrovir. 16:1777-1781. [DOI] [PubMed] [Google Scholar]
- 35.Jones, K. S., C. Petrow-Sadowski, Y. K. Huang, D. C. Bertolette, and F. W. Ruscetti. 2008. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4+ T cells. Nat. Med. 14:429-436. [DOI] [PubMed] [Google Scholar]
- 36.Kim, S. J., A. M. Nair, S. Fernandez, L. Mathes, and M. D. Lairmore. 2006. Enhancement of LFA-1-mediated T cell adhesion by human T lymphotropic virus type 1 p12I1. J. Immunol. 176:5463-5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koralnik, I. J., A. Gessain, M. E. Klotman, A. Lo Monico, Z. N. Berneman, and G. Franchini. 1992. Protein isoforms encoded by the pX region of human T-cell leukemia/lymphotropic virus type I. Proc. Natl. Acad. Sci. USA 89:8813-8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li, K., S. Zhang, M. Kronqvist, M. Wallin, M. Ekstrom, D. Derse, and H. Garoff. 2008. Intersubunit disulfide isomerization controls membrane fusion of human T-cell leukemia virus Env. J. Virol. 82:7135-7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maggirwar, S. B., E. Harhaj, and S. C. Sun. 1995. Activation of NF-kappa B/Rel by Tax involves degradation of I kappa B alpha and is blocked by a proteasome inhibitor. Oncogene 11:993-998. [PubMed] [Google Scholar]
- 40.Makishima, H., T. Ito, N. Asano, H. Nakazawa, S. Shimodaira, Y. Kamijo, Y. Nakazawa, T. Suzuki, H. Kobayashi, K. Kiyosawa, and F. Ishida. 2005. Significance of chemokine receptor expression in aggressive NK cell leukemia. Leukemia 19:1169-1174. [DOI] [PubMed] [Google Scholar]
- 41.Maruyama, M., H. Shibuya, H. Harada, M. Hatakeyama, M. Seiki, T. Fujita, J. Inoue, M. Yoshida, and T. Taniguchi. 1987. Evidence for aberrant activation of the interleukin-2 autocrine loop by HTLV-1-encoded p40x and T3/Ti complex triggering. Cell 48:343-350. [DOI] [PubMed] [Google Scholar]
- 42.Matsumoto, K., H. Shibata, J. I. Fujisawa, H. Inoue, A. Hakura, T. Tsukahara, and M. Fujii. 1997. Human T-cell leukemia virus type 1 Tax protein transforms rat fibroblasts via two distinct pathways. J. Virol. 71:4445-4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGuire, K. L., V. E. Curtiss, E. L. Larson, and W. A. Haseltine. 1993. Influence of human T-cell leukemia virus type I tax and rex on interleukin-2 gene expression. J. Virol. 67:1590-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Migone, T. S., N. A. Cacalano, N. Taylor, T. Yi, T. A. Waldmann, and J. A. Johnston. 1998. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc. Natl. Acad. Sci. USA 95:3845-3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Migone, T. S., J. X. Lin, A. Cereseto, J. C. Mulloy, J. J. O'Shea, G. Franchini, and W. J. Leonard. 1995. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-1. Science 269:79-81. [DOI] [PubMed] [Google Scholar]
- 46.Nagy, K., P. Clapham, R. Cheingsong-Popov, and R. A. Weiss. 1983. Human T-cell leukemia virus type I: induction of syncytia and inhibition by patients’ sera. Int. J. Cancer 32:321-328. [DOI] [PubMed] [Google Scholar]
- 47.Naldini, L., U. Blomer, P. Gallay, D. Ory, R. Mulligan, F. H. Gage, I. M. Verma, and D. Trono. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263-267. [DOI] [PubMed] [Google Scholar]
- 48.Nejmeddine, M., A. L. Barnard, Y. Tanaka, G. P. Taylor, and C. R. Bangham. 2005. Human T-lymphotropic virus, type 1, tax protein triggers microtubule reorientation in the virological synapse. J. Biol. Chem. 280:29653-29660. [DOI] [PubMed] [Google Scholar]
- 49.Nejmeddine, M., V. S. Negi, S. Mukherjee, Y. Tanaka, K. Orth, G. P. Taylor, and C. R. Bangham. 2009. HTLV-1-Tax and ICAM-1 act on T-cell signal pathways to polarize the microtubule-organizing center at the virological synapse. Blood 114:1016-1025. [DOI] [PubMed] [Google Scholar]
- 50.Nicot, C., M. Dundr, J. M. Johnson, J. R. Fullen, N. Alonzo, R. Fukumoto, G. L. Princler, D. Derse, T. Misteli, and G. Franchini. 2004. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat. Med. 10:197-201. [DOI] [PubMed] [Google Scholar]
- 51.Nicot, C., R. L. Harrod, V. Ciminale, and G. Franchini. 2005. Human T-cell leukemia/lymphoma virus type 1 nonstructural genes and their functions. Oncogene 24:6026-6034. [DOI] [PubMed] [Google Scholar]
- 52.Nicot, C., R. Mahieux, S. Takemoto, and G. Franchini. 2000. Bcl-XL is up-regulated by HTLV-1 and HTLV-1I in vitro and in ex vivo ATLL samples. Blood 96:275-281. [PubMed] [Google Scholar]
- 53.Nicot, C., J. C. Mulloy, M. G. Ferrari, J. M. Johnson, K. Fu, R. Fukumoto, R. Trovato, J. Fullen, W. J. Leonard, and G. Franchini. 2001. HTLV-1 p12I protein enhances STAT5 activation and decreases the interleukin-2 requirement for proliferation of primary human peripheral blood mononuclear cells. Blood 98:823-829. [DOI] [PubMed] [Google Scholar]
- 54.Niyogi, K., and J. E. Hildreth. 2001. Characterization of new syncytium-inhibiting monoclonal antibodies implicates lipid rafts in human T-cell leukemia virus type 1 syncytium formation. J. Virol. 75:7351-7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pique, C., D. Pham, T. Tursz, and M. C. Dokhelar. 1992. Human T-cell leukemia virus type I envelope protein maturation process: requirements for syncytium formation. J. Virol. 66:906-913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pique, C., A. Ureta-Vidal, A. Gessain, B. Chancerel, O. Gout, R. Tamouza, F. Agis, and M. C. Dokhelar. 2000. Evidence for the chronic in vivo production of human T cell leukemia virus type I Rof and Tof proteins from cytotoxic T lymphocytes directed against viral peptides. J. Exp. Med. 191:567-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Poiesz, B. J., F. W. Ruscetti, M. S. Reitz, V. S. Kalyanaraman, and R. C. Gallo. 1981. Isolation of a new type C retrovirus (HTLV) in primary uncultured cells of a patient with Sezary T-cell leukaemia. Nature 294:268-271. [DOI] [PubMed] [Google Scholar]
- 58.Popik, W., T. M. Alce, and W. C. Au. 2002. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4+ T cells. J. Virol. 76:4709-4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Portis, T., J. C. Harding, and L. Ratner. 2001. The contribution of NF-kappa B activity to spontaneous proliferation and resistance to apoptosis in human T-cell leukemia virus type 1 Tax-induced tumors. Blood 98:1200-1208. [DOI] [PubMed] [Google Scholar]
- 60.Ratner, L. 2004. Adult T cell leukemia lymphoma. Front. Biosci. 9:2852-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rimsky, L., J. Hauber, M. Dukovich, M. H. Malim, A. Langlois, B. R. Cullen, and W. C. Greene. 1988. Functional replacement of the HIV-1 rev protein by the HTLV-1 rex protein. Nature 335:738-740. [DOI] [PubMed] [Google Scholar]
- 62.Round, J. L., T. Tomassian, M. Zhang, V. Patel, S. P. Schoenberger, and M. C. Miceli. 2005. Dlgh1 coordinates actin polymerization, synaptic T cell receptor and lipid raft aggregation, and effector function in T cells. J. Exp. Med. 201:419-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Siekevitz, M., M. B. Feinberg, N. Holbrook, F. Wong-Staal, and W. C. Greene. 1987. Activation of interleukin 2 and interleukin 2 receptor (Tac) promoter expression by the trans-activator (tat) gene product of human T-cell leukemia virus, type I. Proc. Natl. Acad. Sci. USA 84:5389-5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sinha-Datta, U., I. Horikawa, E. Michishita, A. Datta, J. C. Sigler-Nicot, M. Brown, M. Kazanji, J. C. Barrett, and C. Nicot. 2004. Transcriptional activation of hTERT through the NF-kappaB pathway in HTLV-1-transformed cells. Blood 104:2523-2531. [DOI] [PubMed] [Google Scholar]
- 65.Takemoto, S., J. C. Mulloy, A. Cereseto, T. S. Migone, B. K. Patel, M. Matsuoka, K. Yamaguchi, K. Takatsuki, S. Kamihira, J. D. White, W. J. Leonard, T. Waldmann, and G. Franchini. 1997. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc. Natl. Acad. Sci. USA 94:13897-13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanaka, Y., K. Fukudome, M. Hayashi, S. Takagi, and O. Yoshie. 1995. Induction of ICAM-1 and LFA-3 by Tax1 of human T-cell leukemia virus type 1 and mechanism of down-regulation of ICAM-1 or LFA-1 in adult-T-cell-leukemia cell lines. Int. J. Cancer 60:554-561. [DOI] [PubMed] [Google Scholar]
- 67.Wang, H., K. M. Norris, and L. M. Mansky. 2002. Analysis of bovine leukemia virus gag membrane targeting and late domain function. J. Virol. 76:8485-8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wielgosz, M. M., D. A. Rauch, K. S. Jones, F. W. Ruscetti, and L. Ratner. 2005. Cholesterol dependence of HTLV-1 infection. AIDS Res. Hum. Retrovir. 21:43-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu, X., S. H. Kang, O. Heidenreich, M. Okerholm, J. J. O'Shea, and M. I. Nerenberg. 1995. Constitutive activation of different Jak tyrosine kinases in human T cell leukemia virus type 1 (HTLV-1) tax protein or virus-transformed cells. J. Clin. Investig. 96:1548-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zacharopoulos, V. R., M. E. Perotti, and D. M. Phillips. 1992. Lymphocyte-facilitated infection of epithelia by human T-cell lymphotropic virus type I. J. Virol. 66:4601-4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zigmond, S. H., and J. G. Hirsch. 1973. Leukocyte locomotion and chemotaxis. New methods for evaluation, and demonstration of a cell-derived chemotactic factor. J. Exp. Med. 137:387-410. [DOI] [PMC free article] [PubMed] [Google Scholar]