

Abstract
Osteoblasts and chondrocytes arise from common osteo-chondroprogenitor cells. We show here that inactivation of ERK1 and ERK2 in osteo-chondroprogenitor cells causes a block in osteoblast differentiation and leads to ectopic chondrogenic differentiation in the bone-forming region in the perichondrium. Furthermore, increased mitogen-activated protein kinase signaling in mesenchymal cells enhances osteoblast differentiation and inhibits chondrocyte differentiation. These observations indicate that extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in the lineage specification of mesenchymal cells. The inactivation of ERK1 and ERK2 resulted in reduced beta-catenin expression, suggesting a role for canonical Wnt signaling in ERK1 and ERK2 regulation of skeletal lineage specification. Furthermore, inactivation of ERK1 and ERK2 significantly reduced RANKL expression, accounting for a delay in osteoclast formation. Thus, our results indicate that ERK1 and ERK2 not only play essential roles in the lineage specification of osteo-chondroprogenitor cells but also support osteoclast formation in vivo.
The extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK MAPK) pathway is activated by various stimuli, including a number of growth factors and cytokines. The activation of the Raf members of MAPK kinase kinase leads to the activation of the MAPK kinase, MEK1, and MEK2. MEK1 and MEK2 then phosphorylate and activate MAPK, ERK1, and ERK2. ERK1 and ERK2 phosphorylate various cytoplasmic and nuclear target proteins, ranging from cytoplasmic adaptor proteins and transcription factors to kinases, including ribosomal S6 kinase (RSK) (4, 17, 18, 23, 36, 46). In this pathway, multiple mutations that cause syndromes with various skeletal manifestations have recently been identified. Missense-activating mutations in KRAS, BRAF, MEK1, and MEK2 have been identified in Costello, Noonan, LEOPARD, and Cardio-facio-cutaneous syndromes, while loss-of-function mutations in RSK2, a kinase downstream of ERK1 and ERK2, cause Coffin-Lowry syndrome (2, 42). These observations highlight the importance of the ERK MAPK pathway in human skeletal development.
Both chondrocytes and osteoblasts arise from common osteo-chondro progenitor cells. Bone growth is achieved through two major ossification processes, endochondral ossification and intramembranous ossification, in which chondrocytes and osteoblasts are involved (5, 13, 30, 31, 38). In normal endochondral ossification, the skeletal element is formed as a cartilaginous template that is subsequently replaced by bone. Condensed mesenchymal cells differentiate into chondrocytes. Chondrocytes first proliferate in columnar stacks to form the growth plate and then exit the cell cycle and differentiate into hypertrophic chondrocytes. Hypertrophic chondrocytes are removed by apoptotic cell death, and the cartilaginous matrix is resorbed by chondroclasts/osteoclasts and replaced by trabecular bone. Chondroclast/osteoclast formation is supported by receptor activator of nuclear factor-kappa B ligand (RANKL) secreted from osteoblasts and bone marrow stromal cells (16, 45). In intramembranous ossification, mesenchymal cells directly differentiate into bone-forming osteoblasts; cortical bone is formed by osteoblasts that arise from the osteo-chondroprogenitor cells in the perichondrium. Previous studies with mice have indicated that the ERK MAPK signaling cascade participates in chondrocyte and osteoblast differentiation. Expression of a constitutively active mutant of MEK1 in chondrocytes caused a dwarf phenotype and inhibited hypertrophic chondrocyte differentiation (28). In addition, expression of a constitutively active mutant of MEK1 in mature osteoblasts accelerated bone development, whereas dominant-negative MEK1 was inhibitory at the embryonic stage (11). The roles of ERK/MAPK signaling in mesenchymal cell differentiation require further investigation. In the current study, using the Cre-loxP system, we examined the roles of ERK1 and ERK2 in early steps of chondrocyte and osteoblast differentiation and two major ossification processes, endochondral ossification and intramembranous ossification, by generating loss-of-function models. In addition to the loss-of-function models, we also generated a gain-of-function model (mice that overexpress a constitutively active mutant of MEK1 in undifferentiated mesenchymal cells) and examined the roles of ERK/MAPK signaling in osteo-chondro progenitor cells.
We show here that ERK1 and ERK2 are essential for osteoblast differentiation and that ERK1 and ERK2 inhibit ectopic cartilage formation in the perichondrium. We also show that increased MAPK signaling in osteo-chondroprogenitor cells inhibits cartilage formation and that increased MAPK signaling in the perichondrium stimulates bone formation. Furthermore, inactivation of ERK1 and ERK2 significantly reduced RANKL expression, accounting for a delay in osteoclast formation. Thus, our results indicate that ERK1 and ERK2 not only play essential roles in the lineage specification of osteo-chondroprogenitor cells but also support osteoclast formation in vivo.
MATERIALS AND METHODS
Mutant animals.
The institutional animal care and use committee of Case Western Reserve University approved all animal procedures. All animal care and use were performed in accordance with the institutional animal use methods and policies. ERK1-deficient mice and Prx1-Cre and Col2a1-Cre transgenic mice were described previously (21, 32, 37). The floxed ERK2 allele was created by inserting loxP sites flanking exon2 (35). To generate Prx1-MEK1 transgenic mice, the cDNA for FLAG-tagged MEK1(S218/222E, Δ32-51) and an IRES-LacZ cassette were cloned downstream of a 2.4-kb Prx1 promoter (24, 28). The construct was microinjected into the pronuclei of fertilized C57BL/6-DBA/2 hybrid eggs. ROSA-LacZ reporter mice were purchased from Jackson Laboratories (41).
In situ hybridization, immunohistochemistry, TRAP staining, and BrdU incorporation assay.
In situ hybridization was performed using 35S-labeled riboprobes. Nuclei were visualized with Hoechst 33258. Immunohistochemical staining was performed using a PicTure kit (Invitrogen) or MM biotinylation kit (Biocare). The following antibodies were used: ERK1/2 (Santa Cruz), type X collagen (Quartett), FLAG M5 (Sigma), and β-catenin (BD) antibodies. For immunofluorescent detection of ERK1/2, Alexa Fluor 594-conjugated secondary antibody (Invitrogen) was used. Fluorescence-based tartrate-resistant acid phosphatase (TRAP) staining and alkaline phosphatase staining were performed using ELF97 (Invitrogen) as a phosphatase substrate (6, 10). To reduce background, TRAP staining using ELF97 was performed in the presence of 50 mM tartrate. Naphthol AS-BI phosphoric acid-based TRAP staining was performed using an acid phosphatase leukocyte kit (Sigma). For examining cell proliferation, mice were injected intraperitoneally with BrdU. Embryos were harvested 4 h after injection. BrdU incorporation was detected using a BrdU staining kit (Zymed). All images were acquired with Leica DM 6000B and DM IRB microscopes. Photographs were taken with a digital camera (DC500; Leica) using Leica Application Suite 1.3 software. The signal of in situ hybridization in dark-field images was colored using Photoshop software (Adobe), and brightness and contrast were adjusted by applying brightness/contrast adjustments to the whole image, with the strict intent of not obscuring, eliminating, or misrepresenting any information present in the original, including background. For in situ hybridization, the signal images were overlaid on images of Hoechst 33258.
RNA analysis and semiquantitative and real time PCR.
Total RNA was isolated using an RNeasy kit (Qiagen). RNA was reverse transcribed to cDNA with a high-capacity cDNA archive kit (Applied Biosystems). Real-time PCR was performed with the Applied Biosystems 7500 real-time PCR detection system. TaqMan probe sets were designed and synthesized by Applied Biosystems (Erk2, Mm00442479_m1; Rankl, Mm00441908_m1; Opg, Mm00435452_m1; Dkk1, Mm00438422_m1; Tcf1, Mm03053891_s1; Cbfb, Mm00491551_m1; c-Fos, Mm00487425_m1; Fra1, Mm00487429_m1; Fra2, Mm00484442_m1; JunB, Mm00492781_s1; Krox20, Mm00456650_m1; Col2a1, Mm01309562_g1; Col10a1, Mm00487041_m1; Gapdh, 4352932E). To compare gene expression levels, the comparative cycle threshold method was used. Gapdh was used as an endogenous control to correct for potential variation in RNA loading or in efficiency of amplification. Semiquantitative PCR was performed with the Applied Biosystems 9700 GeneAmp PCR system, using the following primer sets: for Runx2, 5′-GAACCAAGAAGGCACAGACA-3′ (forward) and 5′-AACTGCCTGGGGTCTGAAAA-3′ (reverse) (PCR product, 452 bp); for Osteocalcin, 5′-CTCTGTCTCTCTGACCTCACAG-3′ (forward) and 5′-CAGGTCCTAAATAGTGATACCG-3′ (reverse) (PCR product, 252 bp); for BSP, 5′-GAAACGGTTTCCAGTCCAG-3′ (forward) and 5′-CTGAAACCCGTTCAGAAGG-3′ (reverse) (PCR product, 568 bp); for Col1a2, 5′-TGAAGTGGGTCTTCCAGGTC-3′ (forward) and 5′-GACCAGGCTCACCAACAAGT-3′ (reverse) (PCR product, 200 bp); for Alkaline phosphatase, 5′-AATGCCCTGAAACTCCAAAAGC-3′ (forward) and 5′-CCTCTGGTGGCATCTCGTTATC-3′ (reverse) (PCR product, 472 bp); and for Gapdh, 5′-ACCACAGTCCATGCCATCAC-3′ (forward) and 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse) (PCR product, 452 bp). The band intensities were compared, while the band intensity and cycle numbers are linear.
Cell culture.
Primary rib chondrocytes and calvarium osteoblasts were isolated as described previously (12, 28). Cells were infected with adenovirus (Ad) expressing Cre, green fluorescent protein (GFP) (Gene Transfer Vector Core, University of Iowa), or constitutively active MEK1 (Vector Biolabs) at a multiplicity of infection of 150 to 200. Osteoblasts were grown in differentiation medium (alpha minimum essential medium, 10% fetal calf serum, 5 mM β-glycerophosphate, 100 μg/ml ascorbic acid). Alizarin red and von Kossa staining were done by following standard protocols. For in vitro osteoclast differentiation, spleen cells were isolated and cultured in alpha minimal essential medium supplemented with 10% fetal calf serum, 10 ng/ml RANKL (R&D), and L929 cell-conditioned medium containing macrophage colony-stimulating factor (M-CSF).
Western blot analysis.
Total cellular protein was prepared by lysing cells in 62.5 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate, 10% glycerol, 1 mM sodium orthovanadate, 1 mM sodium fluoride, 1 mM β-glycerophosphate, supplemented with proteinase inhibitor cocktail Complete Mini (Roche). Protein concentrations were determined by a bicinchoninic acid protein assay kit (Pierce). Twenty to thirty micrograms of protein was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene difluoride filters (Millipore). The filters were blocked in 5% nonfat dry milk in Tris-buffered saline (pH 7.5) containing 0.1% Tween 20 and then incubated with the following antibodies: ERK1 and ERK2 (Santa Cruz), Osterix (Santa Cruz), ATF4 (Santa Cruz), RSK2 (Cell Signaling Technology), and β-actin (Cell Signaling Technology) antibodies. Filters were then incubated with the second antibody (horseradish peroxidase-conjugated anti-rabbit or anti-goat immunoglobulin G), and the signal was detected by enhanced chemiluminescence (Pierce). Images were captured using a KODAK Image Station 4000MM device.
RESULTS
Inactivation of ERK2 in mesenchymal cells of ERK1-null mice causes severe limb deformity and bone defects.
We first analyzed ERK1-null mice and ERK2flox/flox; Prx1-Cre mice, in which ERK2 was inactivated in the limb and head mesenchyme by using the Prx1-Cre transgene. These mice did not show obvious skeletal abnormalities (data not shown). To totally inactivate ERK1 and ERK2, we further inactivated ERK2 in the ERK1-null background. ERK1−/−; ERK2flox/flox; Prx1-Cre mice were born at the expected Mendelian ratio. Skeletal preparation of ERK1−/−; ERK2flox/flox; Prx1-Cre mice by use of alcian blue and alizarin red staining showed severe limb deformity at postnatal day 0 (P0) (Fig. 1A; see also Fig. S1 in the supplemental material). In addition, ERK1−/−; ERK2flox/flox; Prx1-Cre mice showed bone defects in the calvaria. The lambdoid suture did not close at least up to P12 (Fig. 1B). Histological examination showed no distinct cortical bone formation in the humeri of ERK1−/−; ERK2flox/flox; Prx1-Cre mice at P5 (Fig. 1C). ERK2 inactivation was confirmed to occur in the tibiae, femurora, and humeri of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos at embryonic day 16.5 (E16.5) by real-time PCR (Fig. 1D and data not shown). ERK2 mRNA was decreased about 90% compared with the level in littermate ERK1−/−; ERK2flox/flox embryos. The inactivation of ERK1 and ERK2 was also confirmed by immunohistochemistry using an antibody that recognizes both ERK1 and ERK2 (Fig. 1E). Immunoreactivity of chondrocytes and cells in the bone-forming area was significantly reduced in ERK1−/−; ERK2flox/flox; Prx1-Cre mice. These observations indicate that ERK1 and ERK2 are essential for bone formation.
FIG. 1.

ERK1−/−; ERK2flox/flox; Prx1-Cre embryos and mice. (A) Skeletal preparation after alizarin red and alcian blue staining at P1. Limbs were severely deformed in ERK1−/−; ERK2flox/flox; Prx1-Cre mice. (B) Skeletal preparation of the cranium after alizarin red staining. ERK1−/−; ERK2flox/flox; Prx1-Cre mice showed bone defects in the calvaria at P12. (C) Hematoxylin, eosin, and alcian blue staining of the humerus showing an absence of the primary ossification center and cortical bone formation in ERK1−/−; ERK2flox/flox; Prx1-Cre mice at P5. (D) Real-time PCR showed ERK2 inactivation in the tibiae and humeri of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos at E16.5. (E) Immunohistochemistry using anti-ERK1 and ERK2 antibody showed reduced immunoreactivity in chondrocytes and cells in the bone-forming region of tibiae of ERK1−/−; ERK2flox/flox; Prx1-Cre mice at P5. (a) ERK1−/+; ERK2flox/flox mice. The boxed area in the upper panel is magnified in the lower panel. (b) ERK1−/−; ERK2flox/flox; Prx1-Cre mice. The upper left panel shows immunostaining for ERK1 and ERK2. Boxed areas 1 and 2 are magnified in the corresponding right panels. While chondrocytes showed reduced immunoreactivity (boxed area 1), endothelial cells show positive staining (boxed area 2). The lower left panel shows alcian blue, hematoxylin, and eosin staining of a neighboring section. Bars indicate 100 μm.
ERK1 and ERK2 are essential for osteoblast differentiation.
To examine osteoblast differentiation, we performed in situ hybridization analyses. Early osteoblast marker Col1a1 and master transcription factors for osteoblast differentiation Runx2, Osterix, and Atf4 were normally expressed in the long bones and calvaria of ERK1−/−; ERK2flox/flox; Prx1-Cre mice, indicating that ERK1 and ERK2 are not required for their expression (Fig. 2A and C and data not shown). However, the expression of Osteocalcin, a marker of mature osteoblasts, was undetectable in the long bones and calvaria of ERK1−/−; ERK2flox/flox; Prx1-Cre mice (Fig. 2B and C). Because Osterix regulates Osteocalcin expression downstream of Runx2, these observations indicate that osteoblast differentiation was arrested after Osterix expression and before differentiation into fully mature osteoblasts.
FIG. 2.

In situ hybridization analyses of the femur (A, B) and calvaria (C), showing normal levels of Col1a1, Runx2, and Osterix (Osx) expression and markedly decreased Osteocalcin (OCN) expression in ERK1−/−; ERK2flox/flox; Prx1-Cre embryos and mice. (A) E15.5; (B) E16.5 and P5; (C) P1.
To examine whether the block in osteoblast differentiation is due to the cell autonomous effects of ERK1 and ERK2 inactivation, we isolated calvarium osteoblasts from ERK1−/−; ERK2flox/flox embryos. The cells were infected with Ad expressing Cre recombinase (Ad-Cre) or a control virus expressing GFP (Ad-GFP). Reverse transcription-PCR showed marked reduction of Osteocalcin and Bsp expression in cells infected with Ad-Cre, while Alkaline phosphatase expression was slightly reduced and Col1a2 expression remained unaltered (Fig. 3A). These observations indicate that the block in osteoblast differentiation is cell autonomous, and ERK1 and ERK2 are required for osteoblast differentiation into mature osteoblasts. Consistent with in vivo observations, the inactivation of ERK1 and ERK2 did not abolish Runx2 mRNA. In addition, ERK1 and ERK2 inactivation did not affect Osterix, ATF4, and RSK2 protein expression (Fig. 3B). The block in osteoblast differentiation was also confirmed by von Kossa staining (Fig. 3C).
FIG. 3.

(A) Semiquantitative reverse transcription-PCR showing reduced Osteocalcin (OCN) and Bone sialoprotein (BSP) expression in primary ERK1−/−; ERK2flox/flox calvarium mesenchymal cells that were infected with Ad expressing Cre recombinase (Ad-Cre). Primary calvarium mesenchymal cells were isolated from E15.5 ERK1−/−; ERK2flox/flox embryos and infected with Ad-Cre or Ad expressing GFP (Ad-GFP). RNA was extracted 20 days after infection. (B) Western blot analysis showing Osterix (OSX), ATF4, and RSK2 expression in primary ERK1−/−; ERK2flox/flox calvarium cells that were infected with Ad-Cre or Ad-GFP. ERK2 expression was inhibited 80% by Ad-Cre infection, while Osterix, ATF4, and RSK2 expression remained largely unaffected. Total cell lysates were prepared 10 days after infection. (C) von Kossa staining of ERK1−/−; ERK2flox/flox calvarium cell cultures 20 days after infection with Ad-Cre or Ad-GFP. Ad-Cre infection inhibited mineralization. (D) Real-time PCR analysis showed reduced Krox20, c-Fos, Fra1, Fra2, and Cbfb expression in the humeri of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos at E16.5, while JunB was not affected. Real-time PCR analysis of the tibia and femur showed similar results. (E) Real time PCR analysis showed reduced Erk2, Krox20, and c-Fos expression in ERK1−/−; ERK2flox/flox calvarium cells infected with Ad-Cre. ERK1−/−; ERK2flox/flox calvarium cells were infected with Ad-Cre or Ad-GFP, and RNA was extracted 9 days after infection.
We also isolated the tibiae, femurora, and humeri of E16.5 embryos and examined expression of transcription factors implicated in osteoblast differentiation by real-time PCR. We found that a number of transcription factors were downregulated in the skeletal elements of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos. Krox20 was strongly inhibited in bones lacking ERK1 and ERK2 (Fig. 3D). In addition, AP-1 family members Fra1, Fra2, and c-Fos were all downregulated in bones lacking ERK1 and ERK2. In contrast, JunB expression remained unchanged, indicating that the downregulation of Krox20, Fra1, Fra2, and c-Fos is not a consequence of general suppression of transcription. Furthermore, Cbfb, which encodes a coregulator of runt domain transcriptional factors, was also downregulated in bones lacking ERK1 and ERK2 (27, 47). Therefore, the severe bone phenotype of ERK1−/−; ERK2flox/flox; Prx1-Cre mice likely involves multiple regulatory factors of osteoblast differentiation. Consistent with these observations, at least Krox20 and c-Fos were strongly inhibited in calvarium cells infected with Ad-Cre (Fig. 3E).
Inactivation of ERK1 and ERK2 in mesenchymal cells causes ectopic cartilage formation in the perichondrium.
Interestingly, we observed ectopic cartilage formation in the perichondria of ERK1−/−; ERK2flox/flox; Prx1-Cre mice (Fig. 4A). The ectopic cartilage formation was observed as early as E13.5 in the mutant humerus (see Fig. S2B in the supplemental material). Cells in the ectopic cartilage expressed a master transcription factor for chondrocyte differentiation, Sox9, and a cartilage-specific marker, Col2a1, indicating chondrogenic differentiation. These observations suggest that osteo-chondroprogenitor cells in the perichondrium were blocked in their differentiation into osteoblasts and instead differentiated into chondrocytes. Interestingly, the cells in the ectopic cartilage also expressed markers for prehypertrophic chondrocytes (Indian hedgehog [Ihh] and Parathyroid hormone/Parathyroid hormone-related peptide receptor) as well as markers for hypertrophic chondrocytes (Col10a1 and Mmp13) (Fig. 4A and data not shown; see also Fig. S2G and H in the supplemental material). It is possible that chondrocytes in the ectopic cartilage accelerated differentiation into hypertrophic chondrocytes in the absence of ERK1 and ERK2.
FIG. 4.

Ectopic cartilage formation in the perichondria of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos. (A) Alcian blue staining and in situ hybridization of the femur at E15.5. The ectopic cartilage (arrowheads) in the perichondrium expresses Sox9, Col2a1, and Indian hedgehog (Ihh). (B) Alcian blue staining (top panel) and immunohistochemical staining of the radius for β-catenin (middle panel). ERK1−/−; ERK2flox/flox; Prx1-Cre embryos showed reduced β-catenin protein levels in the perichondrium at E16.5 (arrows). The boxed area is magnified in the bottom panel. (C) Tcf1 and Dkk1 expression quantitated by real-time PCR. ERK1−/−; ERK2flox/flox; Prx1-Cre embryos showed reduced Tcf1 and Dkk1 expression in the tibia and humerus at E16.5.
Since the inactivation of β-catenin in mesenchymal cells causes similar ectopic cartilage formation in the perichondrium (7, 12, 34), we examined β-catenin expression by immunohistochemistry. Strong β-catenin expression was observed in the bone-forming region of the periostea and perichondria of control embryos at E15.5 (Fig. 4B). In contrast, β-catenin staining was decreased in the periostea and perichondria of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos. We further examined Tcf1 and Dkk1 expression by real-time PCR, because both Tcf1 and Dkk1 expression depends on intact β-catenin signaling (12, 34). We observed strong inhibition of Tcf1 and Dkk1 expression, supporting the notion that β-catenin signaling is reduced in the skeletal elements (Fig. 4C).
Ectopic chondrocyte differentiation in the perichondrium in association with a block in osteoblast differentiation has been also reported to occur in Osterix-null mice and in mice in which Ihh signaling is disrupted (22, 29). Osterix, Ihh, and its downstream target Patched were normally expressed in the skeletal elements of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos (Fig. 2A and 4A; see also Fig. S2G in the supplemental material). In addition, coexpression of a constitutively active mutant of MEK1 did not affect the transcriptional activity of Osterix in transient-transfection experiments in vitro (data not shown). These observations suggest that Osterix and Ihh signaling are not affected by MAPK signaling.
Inactivation of ERK1 and ERK2 caused an expansion of terminally differentiated chondrocytes in the growth plates and decreased osteoclasts.
The inactivation of ERK1 and ERK2 resulted in a remarkable widening of the zone of hypertrophic chondrocytes in the growth plate (Fig. 5A). Some of the epiphyseal chondrocytes closer to the articular surface stained positive for type X collagen, suggesting accelerated hypertrophy. This notion is further supported by an increase in Col10a1 expression in ERK1−/−; ERK2flox/flox primary chondrocytes that were infected with Ad-Cre (see Fig. 7C). In addition, in situ hybridization indicated that the widening of the zone of hypertrophic chondrocytes is associated with a remarkable expansion of terminally differentiated hypertrophic chondrocytes (Fig. 5B). Although the matrix showed intense staining with anti-type X collagen antibody, these cells expressed Col10a1 at a reduced level and instead expressed markers for terminally differentiated hypertrophic chondrocytes (Vegf, Mmp13, and Osteopontin). These observations suggest that the expansion of the hypertrophic zone is caused by impaired removal of terminally differentiated hypertrophic chondrocytes. Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling staining did not show significant difference between control and mutant mice, suggesting that chondrocyte apoptosis is not affected (data not shown).
FIG. 5.

Delayed formation of primary ossification centers in ERK1−/−; ERK2flox/flox; Prx1-Cre mice. (A) Immunohistochemistry for type X collagen showed a widening of the zone of hypertrophic chondrocytes in the tibiae of ERK1−/−; ERK2flox/flox; Prx1-Cre mice at P0. (B) In situ hybridization for Col10a1, Vegf, Mmp-13, and Osteopontin showed an expansion of terminally differentiated chondrocytes in the hypertrophic zone of the tibia at P0. (C) TRAP staining showed an absence of TRAP-positive cells in the tibiae of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos at E16.5. Arrows indicate TRAP-positive cells. The upper images in panels A, B, and C show ERK1−/−, and the lower images show ERK1−/−; ERK2flox/flox; Prx1-Cre mice. (D) Real-time PCR showed reduced RANKL expression in the tibiae and humeri of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos at E16.5. (E, F) ERK2, RANKL, and Osteoprotegerin (OPG) expression examined by real-time PCR. Inactivation of ERK2 strongly inhibited RANKL expression in ERK1−/−; ERK2flox/flox rib chondrocytes (E) and calvarium osteoblasts (F) in vitro. RNA was extracted from ERK1−/−; ERK2flox/flox chondrocytes and osteoblasts 5 days and 8 days after infection with Ad expressing Cre recombinase or GFP. (G) ELF97-based fluorescent TRAP staining (green fluorescence) in combination with immunofluorescence for ERK protein (red fluorescence), showing the presence of ERK protein in TRAP-positive osteoclasts (arrows) in the femoral metaphysis of an ERK1−/−; ERK2flox/flox; Prx1-Cre mouse at P0. The immunofluorescent signal for ERK protein was indistinguishable from that of ERK1−/−; ERK2flox/flox mice (data not shown). The lower panels show a magnification of an osteoclast indicated by arrowheads in the upper panels. Nuclei were visualized by DAPI (4′,6-diamidino-2-phenylindole). (H) X-Gal staining followed by TRAP staining showing no β-galactosidase activity in TRAP-positive osteoclast-like cells derived from spleen cells of Prx1-Cre mice harboring the ROSA-LacZ reporter allele (right panel). The staining results were indistinguishable from those observed for TRAP-positive osteoclast-like cells derived from control ROSA-LacZ reporter mice (left panel). (I) Spleen cells from ERK1−/−; ERK2flox/flox; Prx1-Cre mice formed TRAP-positive multinucleated osteoclast-like cells in the presence of M-CSF and RANKL (left panel). Nuclei were visualized by DAPI. These osteoclast-like cells express ERK protein (right panel), and the staining intensity was indistinguishable from that of osteoclast-like cells generated from spleen cells of ERK1−/−; ERK2flox/flox mice (data not shown).
FIG. 7.

(A) Hematoxylin, eosin, and alcian blue staining of the femur showing a dosage-dependent widening of the zone of hypertrophic chondrocytes at E16.5. Bars indicate the width of the zone of hypertrophic chondrocytes. (B) In situ hybridization of the tibia showing an expansion of Col10a1-expressing domains in ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos at E18.5. (C) Primary ERK1−/−; ERK2flox/flox chondrocytes were infected with Ad expressing Cre recombinase or GFP at a multiplicity of infection of 200. Col10a1 expression was examined by real-time PCR at 5 days after Ad infection. ERK2 inactivation in ERK1−/−; ERK2flox/flox chondrocytes increased Col10a1 expression. (D) Disorganization of columnar structures in the tibial growth plates of ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos at E18.5. (E) BrdU incorporation of the distal femoral growth plate. ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos showed reduced chondrocyte proliferation at E18.5. Data represent means ± standard deviations. N.S., not significant; wt, wild type. Analysis of variance was used to detect significant difference. *, P < 0.01.
Inactivation of ERK1 and ERK2 caused decreases in osteoclast formation and RANKL expression.
We also examined osteoclasts by TRAP staining. TRAP-positive cells were decreased in the long bones of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos at E16.5 (Fig. 5C), suggesting that the decreased osteoclasts account at least in part for the delayed cartilage removal in ERK1−/−; ERK2flox/flox; Prx1-Cre embryos. Consistent with the reduced number of osteoclasts, immunohistochemistry for MMP9 showed reduced staining in ERK1−/−; ERK2flox/flox; Prx1-Cre embryos (see Fig. S3A in the supplemental material).
Since osteoclastogenesis is regulated by RANKL and osteoprotegerin (OPG) produced by mesenchymal cells, we examined RANKL and OPG expression in the long bones of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos. RANKL expression was strongly inhibited in the long bones of ERK1−/−; ERK2flox/flox; Prx1-Cre embryos, while OPG expression remained unaltered (Fig. 5D and data not shown). Because the Prx1-Cre transgene inactivates ERK2 both in chondrocytes and in osteoblasts, we examined RANKL and OPG regulation in each cell lineage in vitro. We harvested calvarial osteoblasts and rib chondrocytes from ERK1−/−; ERK2flox/flox mice and inactivated ERK2 by infecting Ad expressing Cre recombinase. In both cell types, ERK2 inactivation strongly inhibited RANKL expression, while OPG expression was not affected (Fig. 5E and F). Consistent with these observations, treatment with MEK inhibitor U0126 strongly inhibited RANKL expression in both cell types (see Fig. S3B and C in the supplemental material). These observations indicate that RANKL expression depends on MAPK signaling both in the osteoblast and in the chondrocyte lineages.
Since reduced osteoclastogenesis could be due to unspecific deletion of ERK1 and ERK2 in the osteoclast lineage, we examined ERK expression in osteoclasts of ERK1−/−; ERK2flox/flox; Prx1-Cre mice by double staining for TRAP activity and ERK protein. Fluorescence-based TRAP staining using ELF97 showed green fluorescence in osteoclasts of ERK1−/−; ERK2flox/flox; Prx1-Cre mice at P0 (Fig. 5G). The staining intensity for ERK is similar to that of osteoclasts of littermate ERK1−/−; ERK2flox/flox mice (data not shown). To check the activity of the Prx1-Cre transgene in the monocyte/macrophage lineage, we further isolated spleen cells from ROSA-LacZ reporter mice harboring the Prx1-Cre transgene and control ROSA-LacZ reporter mice without the Prx1-Cre transgene. These cells were induced to differentiate into osteoclast-like cells in the presence of M-CSF and RANKL. X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining followed by TRAP staining showed no X-Gal staining in TRAP-positive osteoclast-like cells, indicating that the Prx1-Cre transgene is not active during in vitro osteoclast differentiation (Fig. 5H).
To further exclude the possibility that the reduced osteoclast formation in ERK1−/−; ERK2flox/flox; Prx1-Cre mice is due to defects in osteoclast precursor cells, we isolated spleen cells from ERK1−/−; ERK2flox/flox; Prx1-Cre and littermate ERK1−/−; ERK2flox/flox mice at P0. Spleen cells from ERK1−/−; ERK2flox/flox; Prx1-Cre mice formed osteoclast-like multinucleated giant cells in the presence of M-CSF and RANKL, similar to ERK1−/−; ERK2flox/flox cells (Fig. 5I and data not shown). These giant cells displayed TRAP activity and expressed ERK, similar to ERK1−/−; ERK2flox/flox cells. These observations further support the notion that reduced osteoclastogenesis in ERK1−/−; ERK2flox/flox; Prx1-Cre embryos is due to reduced support from mesenchymal cells.
ERK1 and ERK2 inactivation in chondrocytes causes severe chondrodysplasia.
To further examine the roles of ERK1 and ERK2 in differentiated chondrocytes, we inactivated ERK2 in chondrocytes of ERK1-null mice by using the Col2a1-Cre transgene. ERK1−/−; ERK2flox/flox; Col2a1-Cre mice died immediately after birth, presumably due to respiratory insufficiency caused by the defective rib cage. We obtained ERK1−/−; ERK2flox/flox; Col2a1-Cre mice at a Mendelian ratio at E18.5. The inactivation of ERK1 and ERK2 in chondrocytes was confirmed by immunofluorescence using the antibody that recognizes both ERK1 and ERK2 (Fig. 6A). We further confirmed ERK expression in osteoblasts and osteoclasts of ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos by performing ELF97-based fluorescent alkaline phosphatase staining and TRAP staining along with immunofluorescence for ERK (Fig. 6B and C). The fluorescent signal for ERK was indistinguishable from that of control ERK1−/−; ERK2flox/flox embryos (data not shown). Consistent with the normal expression of ERK in osteoblasts, calvarium osteoblasts isolated from ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos showed mineralization, similar to ERK1+/−; ERK2flox/flox cells, in vitro (see Fig. S4 in the supplemental material).
FIG. 6.

ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos. (A) Immunofluorescence using anti-ERK1 and ERK2 antibody showed reduced immunoreactivity in chondrocytes in the tibiae of ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos at E18.5. (B) ELF97-based fluorescent TRAP staining in combination with immunofluorescence for ERK protein, showing the presence of ERK protein in TRAP-positive osteoclasts (arrows) of ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos. (C) ELF97-based fluorescent alkaline phosphatase staining in combination with immunofluorescence for ERK protein, showing the presence of ERK protein in alkaline phosphatase-positive osteoblasts (arrows) of ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos. (D) Skeletal preparation after alizarin red and alcian blue staining. ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos showed severe kyphotic deformity in the thoracic spine at E18.5. (E, F) Hematoxylin, eosin, and alcian blue staining of the spine (E) and cranial base (F) showing an absence of ossification centers in ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos at E18.5. * indicates the ossification center in the vertebral body, and arrowheads indicate the corresponding areas in ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos. VB, vertebral body; D, intervertebral disc; SC, spinal cord; Pi, pituitary gland; wt, wild type.
ERK1−/−; ERK2flox/flox; Col2a1-Cre mice showed a strong cartilage phenotype both in the axial and in the appendicular skeletons. Skeletal preparation with alcian blue and alizarin red staining showed kyphotic deformity in the spines of ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos (Fig. 6D). Histological examinations showed an absence of primary ossification centers in the spine and cranial base at E18.5 (Fig. 6E and F). In long bones, there was a gene dosage-dependent widening of the zone of hypertrophic chondrocytes (Fig. 7A). ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos showed an expansion of the Col10a1 expression domain (Fig. 7B). Similar to that in ERK1−/−; ERK2flox/flox; Prx1-Cre embryos, Col10a1 expression was also observed in chondrocytes that were closer to the articular surface in ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos, suggesting premature hypertrophy. To further confirm the role of ERK1 and ERK2 in hypertrophic differentiation of chondrocytes, we performed in vitro experiments. ERK2 inactivation in primary ERK1−/−; ERK2flox/flox chondrocytes by Ad-mediated expression of Cre recombinase resulted in increased Col10a1 expression, further supporting the notion that ERK1 and ERK2 inhibit hypertrophic differentiation of chondrocytes (Fig. 7C). Loss of ERK1 and ERK2 also resulted in severe disorganization of the epiphyseal cartilage. Chondrocytes failed to form columnar structures in the growth plates (Fig. 7D). BrdU incorporation experiments indicated significant reduction in chondrocyte proliferation in ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos at E18.5 (Fig. 7E). These observations indicate that ERK1 and ERK2 are required for proper formation of columnar structures and chondrocyte proliferation.
Similar to those in ERK1−/−; ERK2flox/flox; Prx1-Cre embryos, TRAP-positive osteoclasts were decreased in ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos at E16.5, suggesting that chondrocytes support osteoclastogenesis through ERK1 and ERK2 at this stage (Fig. 8A). The reduced osteoclastogenesis is unlikely to be due to unspecific inactivation of ERK in the osteoclast lineage, since TRAP-positive osteoclasts of ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos express ERK (Fig. 6B). In addition, when osteoclast-like cells were generated in vitro from spleen cells of Col2a1-Cre transgenic mice harboring the ROSA-LacZ reporter allele, TRAP-positive osteoclast-like cells did not stain positive for X-Gal (data not shown). Furthermore, spleen cells isolated from E18.5 ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos formed TRAP-positive osteoclast-like cells in the presence of M-CSF and RANKL, and these cells express ERK, similar to ERK1−/−; ERK2flox/flox cells (Fig. 8B).
FIG. 8.

(A) TRAP staining of the femur showing decreased TRAP-positive cells in ERK1−/−; ERK2flox/flox; Col2a1-Cre embryos at E16.5. (B) ELF97-based fluorescent TRAP staining in combination with immunofluorescence for ERK protein. Spleen cells from ERK1−/−; ERK2flox/flox (a) and ERK1−/−; ERK2flox/flox; Col2a1-Cre (b,c) embryos formed TRAP-positive, multinucleated osteoclast-like cells in the presence of M-CSF and RANKL. Nuclei were visualized by DAPI (4′,6-diamidino-2-phenylindole). Lower panels show immunofluorescence using anti-ERK antibody (a′ and b′) or nonimmune immunoglobulin G (IgG) (c′) in corresponding cells.
Increased bone formation in mice that express a constitutively active mutant of MEK1(S218/222E, Δ32-51) under the control of a 2.4-kb prx1 promoter.
To further examine the roles of MAPK in mesenchymal cells, we generated Prx1-MEK1 transgenic mice that express a constitutively active mutant of MEK1(S218/222E, Δ32-51) and LacZ under the control of a 2.4-kb prx1 promoter (Fig. 9A). X-Gal staining showed the transgene expression in the developing limb bud, the perichondrium and periosteum of the long bones, some of the periarticular chondrocytes, and the lambdoid suture in the cranium (Fig. 9B, C, and F). Protein expression of the FLAG-tagged MEK1 mutant was also confirmed by immunohistochemistry using anti-FLAG antibody (Fig. 9D). In remarkable contrast to the defective bone formation in ERK1−/−; ERK2flox/flox; Prx1-Cre mice, Prx1-MEK1 transgenic mice showed a dramatic increase in cortical bone formation, fusion of long bones (Fig. 10A) and carpal and tarsal bones (Fig. 9E), and accelerated closure of the lambdoid suture (Fig. 9F). We observed relatively normal expression of Gdf5 and Wnt 14 at the presumptive joint region (data not shown), suggesting that the bone fusions are not due to the altered joint formation but due to increased bone formation. The increase in cortical bone thickness was preceded by a thickening of the Runx2-, Osterix-, and Bsp-expressing perichondrium, suggesting that MAPK signaling recruits and directs osteo-chondral progenitor cells toward the osteoblastic lineage (Fig. 10B). We also observed accelerated osteocalcin expression in the perichondria of Prx1-MEK1 transgenic mice in comparison with that in wild-type mice (see Fig. S5A in the supplemental material), suggesting an accelerated osteoblast differentiation by the increased MAPK signaling.
FIG. 9.

(A) Schematic representation of the construct that drives the expression of a constitutively active mutant of MEK1 and LacZ under the control of a 2.4-kb prx1 promoter. (B) X-Gal staining of an E15.5 embryo showing transgene expression in the limb and cranium. (C) X-Gal staining of the distal ulna of an E15.5 embryo showing transgene expression in the periarticular chondrocytes, periosteum, and perichondrium. (D) Immunostaining of the FLAG-tagged MEK1(S218/222E, Δ32-51) using anti-M5 FLAG antibody showing transgene expression in the periarticular chondrocytes (arrows), periosteum (arrowheads), and perichondrium of the distal radius of a Prx1-MEK1 transgenic embryo at E15.5 (middle panel). No immunoreactivity was observed in a wild-type (Wt) littermate embryo (left panel). The immunostained section was further stained with alcian blue and eosin (right panel). The cartilaginous matrix of transgene-expressing periarticular chondrocytes (arrows) shows reduced alcian blue staining. (E) Skeletal preparation of the forelimbs after alizarin red and alcian blue staining. Transgenic mice showed a thickening and shorting of long bones at P8. Wt, wild type; Tg, transgenic. (F) Skeletal preparation of the cranium after X-Gal and alizarin red staining showing transgene expression in the mesenchyme of the lambdoid suture. Transgenic mice showed an accelerated closure of the lambdoid suture at E17.5.
FIG. 10.

(A) Cross section of the forelimb stained with hematoxylin and eosin. Prx1-MEK1 transgenic mice showed increased bone formation and fusion of long bones (arrowhead) at P10. Wt, wild type. (B) Hematoxylin, eosin, and alcian blue staining and in situ hybridization of the tibia at E15.5. Prx1-MEK1 transgenic embryos showed a thickening of the perichondrium, which expresses Runx2, Osterix (Osx), and Bone sialoprotein (BSP) (arrowheads). (C) Hematoxylin, eosin, and alcian blue staining of the foot. Prx1-MEK1 transgenic embryos showed a delay in the formation of cartilage anlage at E15.5. Ti, tibia; Ta, talus; Ca, calcaneus. (D) In situ hybridization of the carpal bones. Prx1-MEK1 transgenic embryos showed reduced Col2a1 expression at E15.5. (E) Primary wild-type chondrocytes were infected with Ad expressing a constitutively active mutant of MEK1 (Ad-MEK1) or empty virus (Ad-Null). Col2a1 expression was examined by real-time PCR at 48 h after Ad infection. Expression of a constitutively active mutant of MEK1 strongly inhibited Col2a1 expression. (F) FGF18 treatment (20 ng/ml) downregulated Col2a1 expression in wild-type primary chondrocytes at 24 h. The downregulation was inhibited by U0126. C, control; F, FGF18; U, U0126; F+U, FGF18 plus U0126.
Inhibition of cartilage formation in Prx1-MEK1 transgenic mice.
In contrast to the increased bone formation, we observed inhibition of cartilage formation in the transgenic mice. Skeletal preparation of E14.5 embryos showed a delay in the formation of cartilage anlagen, and alcian blue staining of histological sections showed smaller cartilage anlagen in the transgenic mice at E15.5 (Fig. 10C; see also Fig. S5C in the supplemental material). Immunohistochemistry for the constitutively active MEK1 indicated that cartilage develops within the transgene-expressing mesenchymal condensation (see Fig. S5B in the supplemental material). In addition, alcian blue staining was reduced in the periarticular cartilage corresponding to the expression domain of the constitutively active MEK1 (Fig. 9D). Furthermore, Col2a1 expression was decreased in the developing cartilage primordia (Fig. 10D). Consistent with these observations in vivo, Ad-mediated expression of a constitutively active mutant of MEK1 in primary chondrocytes strongly inhibited Col2a1 expression (Fig. 10E). In addition, FGF18, a potent activator of the MAPK pathway, also downregulated Col2a1 expression, and the downregulation was inhibited by U0126 (Fig. 10F). Collectively, these observations indicate that increased MAPK signaling inhibits cartilage formation.
DISCUSSION
In this study, we showed that ERK1 and ERK2 are essential for osteoblast differentiation and bone formation. Osteocalcin-expressing mature osteoblasts did not develop in the absence of ERK1 and ERK2, indicating that ERK1 and ERK2 are essential for differentiation into mature osteoblasts. Despite severe impairment in osteoblast differentiation, master transcription factors for osteoblast differentiation (Runx2, Osterix, and Atf4) are normally expressed, indicating that ERK1 and ERK2 are not required for their expression. In addition to transcriptional regulation, ERK1 and ERK2 can regulate the activity of transcription factors posttranslationally. Indeed, Runx2 has been shown to be phosphorylated and activated by ERK MAPK (11, 43). In addition, Atf4 is phosphorylated and activated by RSK2, which is a cytoplasmic substrate of ERK1 and ERK2 (44). Phosphorylation by ERK1 and ERK2 is essential for the complete activation of RSK2 (40, 50). Therefore, reduced activity of Runx2 and Atf4 may have a role in the severe defects in osteoblast differentiation. However, downstream targets of Runx2, such as Osterix, Atf4, Vegf, Ihh, and Col10a1, are normally expressed in the absence of ERK1 and ERK2, suggesting that Runx2 is not totally inactive in the absence of ERK1 and ERK2 (29, 44, 48, 49, 51). In addition, the bone phenotype of ERK1−/−; ERK2flox/flox; Prx1-Cre mice is apparently more severe than that of Atf4 and Rsk2-null mice, suggesting additional mechanisms for regulating osteoblast differentiation.
Consistent with this notion, we found that a number of transcription factors implicated in bone formation were downregulated in ERK1−/−; ERK2flox/flox; Prx1-Cre embryos. Notably, Krox20, a zinc finger transcription factor expressed in endosteal and periosteal osteoblasts, was strongly downregulated in the absence of ERK1 and ERK2. Krox20 enhances the Osteocalcin promoter activity (19), and Krox20-null mice show a strong decrease in trabecular bone formation and Osteocalcin-positive cells (20). In addition, AP-1 family members Fra1, Fra2, c-Fos, and Cbfb (which encodes a coregulator of runt-domain transcriptional factors) were all downregulated in the skeletal elements lacking ERK1 and ERK2. Since these transcriptional regulators are all implicated in bone formation (1, 9, 15, 26, 27, 47), these observations strongly suggest that ERK1 and ERK2 regulate osteoblast differentiation and bone formation through multiple regulators.
In addition to the block in osteoblast differentiation, we observed ectopic cartilage formation in the perichondria of ERK1−/−; ERK2flox/flox; Prx1-Cre mice (Fig. 4A; see also Fig. S2 in the supplemental material). These observations suggest that osteochondral progenitor cells were blocked in their differentiation into osteoblasts and differentiated into chondrocytes. A similar phenotype has been reported to occur in β-catenin conditional knockout mice (7, 12). We observed decreased β-catenin protein levels in the perichondria of ERK1−/−; ERK2flox/flox; Prx1-Cre mice. Consistent with this observation, expression of Tcf1 and Dkk1, which is dependent on β-catenin signaling, was strongly inhibited in these mice. These results suggest a role for reduced β-catenin signaling in ectopic cartilage formation in the perichondrium. Loss of ERK1 and ERK2 may result in low β-catenin protein levels through intracellular cross talk between ERK and canonical Wnt signaling. Such interaction has been reported to occur in hepatocellular carcinoma, in which ERK inactivates GSK-3β, leading to the stabilization of β-catenin (8). Alternatively, the effect of ERK inactivation on β-catenin expression may be indirect, and loss of ERK1 and ERK2 results in a block in osteoblast differentiation before differentiating osteoblasts express β-catenin at an increased level. The inactivation of β-catenin in calvarial mesenchymal cells by use of the Prx1-Cre or Dermo1-Cre transgene caused chondrogenic differentiation (7, 12). In contrast, loss of ERK1 and ERK2 did not cause ectopic cartilage formation in the calvaria, indicating distinct roles in the lineage specification of cranial mesenchyme. Further investigation is necessary to elucidate the interaction between MAPK and β-catenin in the lineage specification of mesenchymal cells.
In remarkable contrast to the defective bone formation in ERK1−/−; ERK2flox/flox; Prx1-Cre mice, Prx1-MEK1 transgenic mice that express a constitutively active mutant of MEK1 in the perichondrium/periosteum showed a dramatic increase in cortical bone formation. The increase in cortical bone thickness was preceded by a thickening of the Runx2-, Osterix-, and Bsp-expressing perichondrium, suggesting that MAPK signaling recruits and directs osteo-chondral progenitor cells toward the osteoblastic lineage. In addition, Prx1-MEK1 transgenic mice showed a delay in the formation of cartilage anlage. This is consistent with the notion that MAPK signaling inhibits chondrogenic differentiation of mesenchymal cells. Interestingly, the accelerated cranial suture closure and bone fusions in Prx1-MEK1 transgenic mice were similar to those of human skeletal syndromes caused by activating mutations in fibroblast growth factor receptor 2 (14). These include Apert, Pfeiffer, Jackson-Weiss, and Crouzon syndromes. These syndromes show craniosynostosis and variable degrees of limb abnormalities, including cutaneous and osseous syndactyly and fusion of various bones. Since the MAPK pathway is one of the major downstream pathways of FGF signaling, these observations suggest that the MAPK pathway is responsible for some of the clinical features of fibroblast growth factor receptor 2-related skeletal syndromes. This notion is further supported by the recent observation in mice that express an Apert syndrome mutation, S252W, in which MEK inhibitor U0126 treatment rescued craniosynostosis (39).
In contrast to osteoblast differentiation, loss of ERK1 and ERK2 did not affect the formation of cartilage anlage, indicating that ERK1 and ERK2 are dispensable for cartilage formation. However, loss of ERK1 and ERK2 caused severe disorganization of the epiphyseal cartilage and significant reduction in chondrocyte proliferation. These observations indicate that ERK1 and ERK2 are required for proper organization of the epiphyseal cartilage and chondrocyte proliferation. The cartilage phenotype of ERK1, ERK2 conditional knockout mice was substantially alleviated by one functional allele of either ERK1 or ERK2, indicating that one allele of either ERK1 or ERK2 is sufficient for restoring the growth plate architecture and chondrocyte proliferation. We have previously shown that chondrocyte proliferation is not affected in mice that express a constitutively active mutant of MEK1 in chondrocytes (28). Apparently, basal MAPK signaling is sufficient for chondrocyte proliferation, and further increasing MAPK signaling does not stimulate proliferation. Recently, B-raf was conditionally inactivated in chondrocytes in the A-raf-null background (33). Surprisingly, these mice did not show obvious cartilage abnormalities. Our results support the notion that intact C-raf signaling activates ERK1 and ERK2 to a level sufficient for normal cartilage development.
In both ERK1−/−; ERK2flox/flox; Prx1-Cre mice and ERK1−/−; ERK2flox/flox; Co2a1-Cre embryos, we observed premature Col10a1 expression in chondrocytes in the epiphysis, suggesting accelerated hypertrophic differentiation. Interestingly, Col10a1-positive chondrocytes were observed mainly at the lateral edges of the epiphysis in ERK1−/−; ERK2flox/flox; Prx1-Cre embryos, while chondrocytes in the center expressed Col10a1 at lower levels. This might be related to the timing of ERK inactivation using the Prx1-Cre transgene. The inactivation of ERK1 and ERK2 upregulated Col10a1 expression in chondrocytes in vitro, further supporting the notion that ERK1 and ERK2 inhibit hypertrophic differentiation. These observations are consistent with our previous observations that increased MEK1 signaling in chondrocytes inhibits hypertrophic differentiation in transgenic mice (28).
Another important function of ERK1 and ERK2 revealed in this study is the role of ERK1 and ERK2 in supporting osteoclastogenesis. Osteoclast formation is stimulated by RANKL that is secreted from osteoblasts, osteoblast precursor cells, and bone marrow stromal cells, and its action is counterbalanced by its decoy receptor OPG (3, 16, 45). Our in vitro analyses indicated that ERK1 and ERK2 are required for RANKL expression both in the osteoblast and in the chondrocyte lineages. Consistent with this observation, loss of ERK1 and ERK2 in the developing skeleton caused a strong decrease in RANKL expression and reduced formation of osteoclasts. Most interestingly, chondrocyte-specific inactivation of ERK1 and ERK2 resulted in reduced osteoclastogenesis. Similar decreases in RANKL expression and osteoclast formation have been shown to occur in mice in which VDR is conditionally inactivated in chondrocytes (25). These observations indicate that chondrocytes regulate osteoclast formation at the developing primary ossification center. Our results indicate that ERK1 and ERK2 play essential roles in supporting osteoclastogenesis through RANKL expression.
In summary, our results demonstrate that ERK1 and ERK2 are essential for osteoblast differentiation, and ERK1 and ERK2 inhibit chondrogenic differentiation in the perichondrium (Fig. 11). Increased MAPK signaling promotes differentiation of mesenchymal cells into osteoblasts and inhibits chondrogenic differentiation. On the basis of these observations, we propose that ERK1 and ERK2 play essential roles in the lineage specification of mesenchymal cells. Furthermore, ERK1 and ERK2 regulate RANKL expression both in the osteoblast and in the chondrocyte lineages, which in turn regulates osteoclast formation. Further analyses will provide novel insights into the roles of MAPK in mesenchymal cells and skeletal development.
FIG. 11.
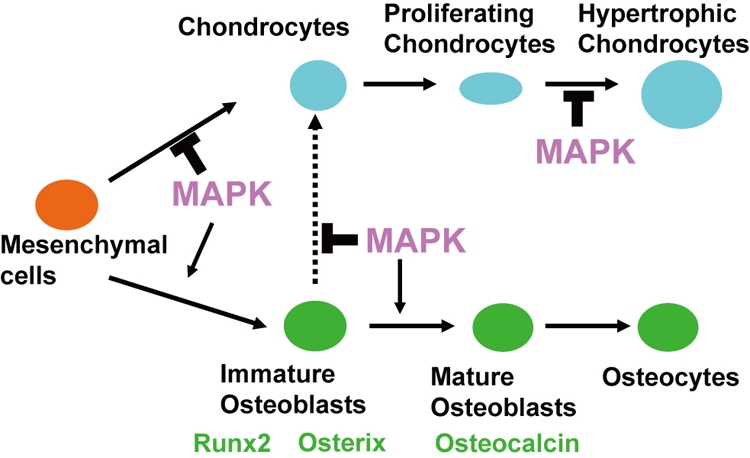
Model for the roles of MAPK in chondrocyte and osteoblast differentiation. While MAPK inhibits chondrogenic differentiation of osteo-chondroprogenitor cells, MAPK enhances osteoblast differentiation. MAPK is essential for osteoblast differentiation into Osteocalcin-expressing mature osteoblasts. MAPK also inhibits hypertrophic chondrocyte differentiation.
Supplementary Material
Acknowledgments
We thank J. Martin for Prx1-Cre mice and the 2.4-kb Prx1 promoter. We are grateful to M. Kurosaka for his continuous support for this work. We also thank Edward Greenfield for reagents and technical suggestions, Yoichi Ezura and Guang Zhou for critical reading of the manuscript, and Valerie Schmedlen for editorial assistance.
This work was supported by research grant no. 6-FY06-341 from the March of Dimes Birth Defects Foundation and NIH grants R21DE017406 and R01AR055556 to S.M.
Footnotes
Published ahead of print on 8 September 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Banerjee, C., J. L. Stein, A. J. Van Wijnen, B. Frenkel, J. B. Lian, and G. S. Stein. 1996. Transforming growth factor-beta 1 responsiveness of the rat osteocalcin gene is mediated by an activator protein-1 binding site. Endocrinology 137:1991-2000. [DOI] [PubMed] [Google Scholar]
- 2.Bentires-Alj, M., M. I. Kontaridis, and B. G. Neel. 2006. Stops along the RAS pathway in human genetic disease. Nat. Med. 12:283-285. [DOI] [PubMed] [Google Scholar]
- 3.Boyce, B. F., and L. Xing. 2008. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 473:139-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chuderland, D., and R. Seger. 2005. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol. Biotechnol. 29:57-74. [DOI] [PubMed] [Google Scholar]
- 5.Colnot, C. 2005. Cellular and molecular interactions regulating skeletogenesis. J. Cell. Biochem. 95:688-697. [DOI] [PubMed] [Google Scholar]
- 6.Cox, W. G., and V. L. Singer. 1999. A high-resolution, fluorescence-based method for localization of endogenous alkaline phosphatase activity. J. Histochem. Cytochem. 47:1443-1456. [DOI] [PubMed] [Google Scholar]
- 7.Day, T. F., X. Guo, L. Garrett-Beal, and Y. Yang. 2005. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 8:739-750. [DOI] [PubMed] [Google Scholar]
- 8.Ding, Q., W. Xia, J. C. Liu, J. Y. Yang, D. F. Lee, J. Xia, G. Bartholomeusz, Y. Li, Y. Pan, Z. Li, R. C. Bargou, J. Qin, C. C. Lai, F. J. Tsai, C. H. Tsai, and M. C. Hung. 2005. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 19:159-170. [DOI] [PubMed] [Google Scholar]
- 9.Eferl, R., A. Hoebertz, A. F. Schilling, M. Rath, F. Karreth, L. Kenner, M. Amling, and E. F. Wagner. 2004. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J. 23:2789-2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filgueira, L. 2004. Fluorescence-based staining for tartrate-resistant acidic phosphatase (TRAP) in osteoclasts. J. Histochem. Cytochem. 52:411-414. [DOI] [PubMed] [Google Scholar]
- 11.Ge, C., G. Xiao, D. Jiang, and R. T. Franceschi. 2007. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J. Cell Biol. 176:709-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill, T. P., D. Spater, M. M. Taketo, W. Birchmeier, and C. Hartmann. 2005. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 8:727-738. [DOI] [PubMed] [Google Scholar]
- 13.Hunziker, E. B. 1994. Mechanism of longitudinal bone growth and its regulation by growth plate chondrocytes. Microsc. Res. Tech. 28:505-519. [DOI] [PubMed] [Google Scholar]
- 14.Ibrahimi, O. A., E. S. Chiu, J. G. McCarthy, and M. Mohammadi. 2005. Understanding the molecular basis of Apert syndrome. Plast. Reconstr. Surg. 115:264-270. [PubMed] [Google Scholar]
- 15.Jochum, W., J. P. David, C. Elliott, A. Wutz, H. Plenk, Jr., K. Matsuo, and E. F. Wagner. 2000. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat. Med. 6:980-984. [DOI] [PubMed] [Google Scholar]
- 16.Kim, N., P. R. Odgren, D. K. Kim, S. C. Marks, Jr., and Y. Choi. 2000. Diverse roles of the tumor necrosis factor family member TRANCE in skeletal physiology revealed by TRANCE deficiency and partial rescue by a lymphocyte-expressed TRANCE transgene. Proc. Natl. Acad. Sci. USA 97:10905-10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondoh, K., S. Torii, and E. Nishida. 2005. Control of MAP kinase signaling to the nucleus. Chromosoma 114:86-91. [DOI] [PubMed] [Google Scholar]
- 18.Kyriakis, J. M., H. App, X. F. Zhang, P. Banerjee, D. L. Brautigan, U. R. Rapp, and J. Avruch. 1992. Raf-1 activates MAP kinase-kinase. Nature 358:417-421. [DOI] [PubMed] [Google Scholar]
- 19.Leclerc, N., T. Noh, A. Khokhar, E. Smith, and B. Frenkel. 2005. Glucocorticoids inhibit osteocalcin transcription in osteoblasts by suppressing Egr2/Krox20-binding enhancer. Arthritis Rheum. 52:929-939. [DOI] [PubMed] [Google Scholar]
- 20.Levi, G., P. Topilko, S. Schneider-Maunoury, M. Lasagna, S. Mantero, R. Cancedda, and P. Charnay. 1996. Defective bone formation in Krox-20 mutant mice. Development 122:113-120. [DOI] [PubMed] [Google Scholar]
- 21.Logan, M., J. F. Martin, A. Nagy, C. Lobe, E. N. Olson, and C. J. Tabin. 2002. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33:77-80. [DOI] [PubMed] [Google Scholar]
- 22.Long, F., U. I. Chung, S. Ohba, J. McMahon, H. M. Kronenberg, and A. P. McMahon. 2004. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 131:1309-1318. [DOI] [PubMed] [Google Scholar]
- 23.Marshall, C. J. 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80:179-185. [DOI] [PubMed] [Google Scholar]
- 24.Martin, J. F., and E. N. Olson. 2000. Identification of a prx1 limb enhancer. Genesis 26:225-229. [PubMed] [Google Scholar]
- 25.Masuyama, R., I. Stockmans, S. Torrekens, R. Van Looveren, C. Maes, P. Carmeliet, R. Bouillon, and G. Carmeliet. 2006. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J. Clin. Investig. 116:3150-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCabe, L. R., M. Kockx, J. Lian, J. Stein, and G. Stein. 1995. Selective expression of fos- and jun-related genes during osteoblast proliferation and differentiation. Exp. Cell Res. 218:255-262. [DOI] [PubMed] [Google Scholar]
- 27.Miller, J., A. Horner, T. Stacy, C. Lowrey, J. B. Lian, G. Stein, G. H. Nuckolls, and N. A. Speck. 2002. The core-binding factor beta subunit is required for bone formation and hematopoietic maturation. Nat. Genet. 32:645-649. [DOI] [PubMed] [Google Scholar]
- 28.Murakami, S., G. Balmes, S. McKinney, Z. Zhang, D. Givol, and B. de Crombrugghe. 2004. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 18:290-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakashima, K., X. Zhou, G. Kunkel, Z. Zhang, J. M. Deng, R. R. Behringer, and B. de Crombrugghe. 2002. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108:17-29. [DOI] [PubMed] [Google Scholar]
- 30.Opperman, L. A. 2000. Cranial sutures as intramembranous bone growth sites. Dev. Dyn. 219:472-485. [DOI] [PubMed] [Google Scholar]
- 31.Ornitz, D. M., and P. J. Marie. 2002. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 16:1446-1465. [DOI] [PubMed] [Google Scholar]
- 32.Ovchinnikov, D. A., J. M. Deng, G. Ogunrinu, and R. R. Behringer. 2000. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 26:145-146. [PubMed] [Google Scholar]
- 33.Provot, S., G. Nachtrab, J. Paruch, A. P. Chen, A. Silva, and H. M. Kronenberg. 2008. A-Raf and B-Raf are dispensable for normal endochondral bone development, and parathyroid hormone-related peptide suppresses extracellular signal-regulated kinase activation in hypertrophic chondrocytes. Mol. Cell. Biol. 28:344-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodda, S. J., and A. P. McMahon. 2006. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133:3231-3244. [DOI] [PubMed] [Google Scholar]
- 35.Samuels, I. S., J. C. Karlo, A. N. Faruzzi, K. Pickering, K. Herrup, J. D. Sweatt, S. C. Saitta, and G. E. Landreth. 2008. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. 28:6983-6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seger, R., and E. G. Krebs. 1995. The MAPK signaling cascade. FASEB J. 9:726-735. [PubMed] [Google Scholar]
- 37.Selcher, J. C., T. Nekrasova, R. Paylor, G. E. Landreth, and J. D. Sweatt. 2001. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn. Mem. 8:11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shapiro, I. M., C. S. Adams, T. Freeman, and V. Srinivas. 2005. Fate of the hypertrophic chondrocyte: microenvironmental perspectives on apoptosis and survival in the epiphyseal growth plate. Birth Defects Res. C 75:330-339. [DOI] [PubMed] [Google Scholar]
- 39.Shukla, V., X. Coumoul, R. H. Wang, H. S. Kim, and C. X. Deng. 2007. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 39:1145-1150. [DOI] [PubMed] [Google Scholar]
- 40.Smith, J. A., C. E. Poteet-Smith, K. Malarkey, and T. W. Sturgill. 1999. Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo. J. Biol. Chem. 274:2893-2898. [DOI] [PubMed] [Google Scholar]
- 41.Soriano, P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21:70-71. [DOI] [PubMed] [Google Scholar]
- 42.Trivier, E., D. De Cesare, S. Jacquot, S. Pannetier, E. Zackai, I. Young, J. L. Mandel, P. Sassone-Corsi, and A. Hanauer. 1996. Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome. Nature 384:567-570. [DOI] [PubMed] [Google Scholar]
- 43.Xiao, G., D. Jiang, P. Thomas, M. D. Benson, K. Guan, G. Karsenty, and R. T. Franceschi. 2000. MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J. Biol. Chem. 275:4453-4459. [DOI] [PubMed] [Google Scholar]
- 44.Yang, X., K. Matsuda, P. Bialek, S. Jacquot, H. C. Masuoka, T. Schinke, L. Li, S. Brancorsini, P. Sassone-Corsi, T. M. Townes, A. Hanauer, and G. Karsenty. 2004. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell 117:387-398. [DOI] [PubMed] [Google Scholar]
- 45.Yasuda, H., N. Shima, N. Nakagawa, K. Yamaguchi, M. Kinosaki, S. Mochizuki, A. Tomoyasu, K. Yano, M. Goto, A. Murakami, E. Tsuda, T. Morinaga, K. Higashio, N. Udagawa, N. Takahashi, and T. Suda. 1998. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 95:3597-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoon, S., and R. Seger. 2006. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21-44. [DOI] [PubMed] [Google Scholar]
- 47.Yoshida, C. A., T. Furuichi, T. Fujita, R. Fukuyama, N. Kanatani, S. Kobayashi, M. Satake, K. Takada, and T. Komori. 2002. Core-binding factor beta interacts with Runx2 and is required for skeletal development. Nat. Genet. 32:633-638. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida, C. A., H. Yamamoto, T. Fujita, T. Furuichi, K. Ito, K. Inoue, K. Yamana, A. Zanma, K. Takada, Y. Ito, and T. Komori. 2004. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18:952-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zelzer, E., D. J. Glotzer, C. Hartmann, D. Thomas, N. Fukai, S. Soker, and B. R. Olsen. 2001. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech. Dev. 106:97-106. [DOI] [PubMed] [Google Scholar]
- 50.Zhao, Y., C. Bjorbaek, and D. E. Moller. 1996. Regulation and interaction of pp90(rsk) isoforms with mitogen-activated protein kinases. J. Biol. Chem. 271:29773-29779. [DOI] [PubMed] [Google Scholar]
- 51.Zheng, Q., G. Zhou, R. Morello, Y. Chen, X. Garcia-Rojas, and B. Lee. 2003. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell Biol. 162:833-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.