

Abstract
Bcr and Abr are GTPase-activating proteins for the small GTPase Rac. Both proteins are expressed in cells of the innate immune system, including neutrophils and macrophages. The function of Bcr has been linked to the negative regulation of neutrophil reactive oxygen species (ROS) production, but the function of Abr in the innate immune system was unknown. Here, we report that mice lacking both proteins are severely affected in two models of experimental endotoxemia, including exposure to Escherichia coli lipopolysaccharide and polymicrobial sepsis, with extensive microvascular leakage, resulting in severe pulmonary edema and hemorrhage. Additionally, in vivo-activated neutrophils of abr and bcr null mutant mice produced excessive tissue-damaging myeloperoxidase (MPO), elastase, and ROS. Moreover, the secretion of the tissue metalloproteinase MMP9 by monocytes and ROS by elicited macrophages was abnormally high. In comparison, ROS production from bone marrow monocytes was not significantly different from that of controls, and the exocytosis of neutrophil secondary and tertiary granule products, including lactoferrin, was normal. These data show that Abr and Bcr normally curb very specific functions of mature tissue innate immune cells, and that each protein has distinct as well as partly overlapping functions in the downregulation of inflammatory processes.
BCR originally was discovered as a human gene on chromosome 22 that, in chronic myeloid leukemia, becomes fused to the c-ABL tyrosine kinase gene originating from chromosome 9 (18). The normal gene encodes a 160-kDa protein that contains a domain with GTPase-activating (GAP) activity toward Rho family GTPases (7, 11, 12, 32, 36). There is only one other gene in mouse and human, called ABR, that is closely homologous to BCR (17). Abr shares several domains with Bcr, which includes a Dbl homology (DH) domain and a GAP domain. Bcr has an additional N-terminal part consisting of a coiled-coil and a serine/threonine kinase domain that is not present in Abr, suggesting that each GAP has a distinct cellular function.
Rho GTPases, including Rho, Rac, and Cdc42, play important roles in many functions of cells of the innate immune system (16). They cycle between active GTP and inactive GDP-bound conformations. GAP proteins catalyze the conversion of bound GTP to GDP on Rho GTPases and thus act as negative, inactivating regulators.
In previous studies, we showed that both Abr and Bcr specifically act as GAPs for Rac and not for the related Cdc42 (6). To investigate the normal cellular function of these two related GAPs, we generated mice defective in the production of Abr or Bcr through gene targeting. Mice that lack both proteins have defects in the architecture of the inner ear, with the partial absence of otoconia and hair cells. Additionally, postnatal cerebellar development is abnormal, with a persistence of ectopic granule cells at the cerebellar surface. These combined abnormalities cause persistent circling and balance problems (20, 21).
As reported previously, neutrophils from mice lacking Bcr produce increasing amounts of reactive oxygen species (ROS), and bcr−/− mice injected with Escherichia coli lipopolysaccharide (LPS) are much more severely affected than are wild-type mice (39). We further explored the role of Bcr and Abr in the innate immune system with a detailed study of bone marrow-derived macrophages (BMM). Interestingly, macrophages isolated from double-knockout (abr × bcr−/−) mice exhibited multiple defects. These include aberrant actin cytoskeletal organization and the increased colony-stimulating factor 1-stimulated chemotaxis and phagocytosis of opsonized zymosan or E. coli (6).
In the current study, we examined whether the defects observed in vitro result in an observable phenotype in vivo, under inflammatory conditions. Here, we report that Abr plays a distinct role in negatively regulating the innate immune system in vivo, as well as exhibiting overlap with the function of Bcr. Mice lacking both Abr and Bcr have a severely impaired ability to resolve septic shock, showing that the activity of both proteins is required for the appropriate negative control of innate immune responses.
MATERIALS AND METHODS
Mice and LPS injection models.
The studies described here were reviewed and approved by the Childrens Hospital of Los Angeles IACUC. Mice were maintained in accordance with the NIH guide for the care and use of laboratory animals. Bcr knockouts, abr knockouts, wild-type controls, and abr × bcr−/− mice used for some experiments were on a mixed/outbred genetic background obtained by interbreeding mice on 129Sv, Black Swiss, and C57Bl/6J genetic backgrounds (12). Other experiments, as indicated in the text or figure legends, were performed with these same genotypes but on a defined/inbred (FVB/J) genetic background. For all experiments, age-, sex-, and background-matched mice of each genotype were used. Mice were injected intraperitoneally (i.p.) with LPS (E. coli L2630, serotype 0111:B4; Sigma, St. Louis, MO) at the dose indicated for the individual experiments. Animals were monitored by observing their overall condition, as well as by measuring body weight loss and regain and body temperature.
CLP model.
Mice were subjected to cecal ligation/puncture (CLP) in accordance with institutional regulations. We used paired sibling wild-type and double-null mice (8-to 9-month-old outbred females or 2-month-old males on a defined background). Briefly, animals were maintained under anesthesia with an isoflurane-oxygen mixture. A 1- to 2-cm midline incision was made through the skin and abdominal wall, and the cecum was located. One length of suture was applied with light tension, and two to three knots were applied around the cecal-ileal junction. For mice on the inbred background, only 1/3 of the cecum was ligated. The cecum was pierced once with a 22-gauge needle. A forceps was used longitudinally along the cecum to expel a small drop of stool. Before the closure of the abdominal wall and skin, 0.5 ml saline and 2 drops of marcaine were dripped into the peritoneal cavity for fluid resuscitation and pain relief. In addition, buprenorphine (0.1 mg/kg of body weight) was injected subcutaneously before CLP and every 12 h after CLP. Animals were put on a heating pad for recovery and monitored closely thereafter for body temperature and body weight. The body temperature of abr × bcr−/− mice was significantly lower than that of wild-type CLP-treated mice on days 2 and 3 (n = 8 for each genotype, defined genetic background; P < 0.05 by analysis of variance and Student-Newman-Keuls test).
Histology and assessment of liver function.
Tissues were fixed with 10% neutral buffered formalin (Sigma, St. Louis, MO), processed, and embedded in paraffin. For standard histology, sections were cut at 5 μm and stained with hematoxylin and eosin (H&E). b4-isolectin staining was used to identify macrophages. Liver function was assayed by plasma alanine transferase (ALT) and aspartate aminotransferase (AST) tests at the Yale Mouse Phenotyping Center. We scored H&E-stained sections from the ileum of CLP-operated and control mice on the outbred background for villus height and mucosal damage as described previously (3). Ileum villus lengths of sham CLP operation, wild-type CLP, and abr × bcr−/− CLP (n = 103, 155, and 140 villi, respectively) were measured by SPOT software (Diagnostic Instruments, Inc.) on samples of wild-type (n = 5) and abr × bcr−/− (n = 4) mice. For the measurement of the goblet cell numbers and villus length of mice on an inbred background, we used two wild-type mice as sham operation controls and treated three wild-type and three abr × bcr−/− mice with CLP (30 villi from each mouse).
Tissue injury study and plasma leakage assay.
LPS at 10, 15, or 20 mg/kg was injected into the peritoneal cavity of conscious mice (n = 6 to 8) to evaluate long-term mortality. For the endotoxin-induced tissue injury, LPS at 15 mg/kg (the 55% lethal dose as determined by the mortality study) was injected into the peritoneal cavity of conscious mice, and 4 h after challenge microvascular permeability was measured using the dye extraction method reported by Udaka et al. (38). Briefly, Evans blue dye (2.5% solution in 0.6% saline; Sigma, St. Louis, MO) was injected intravenously (i.v.) at a dose of 30 mg/kg 30 min prior to sacrifice, and lungs and ileum were removed. Lungs were perfused through the spontaneously beating right ventricle. The extravasated Evans blue was extracted with formamide (Sigma, St. Louis, MO) (3 ml) for 48 to 72 h at 60°C, and the optical density of the extract was measured at 620 nm using a spectrophotometer. Each measured value was converted into an amount of extravasated dye (in micrograms/site).
Lung MPO activity assay.
Total numbers of inflammatory cells were counted in bronchoalveolar lavage fluid (BALF) of matched wild-type (n = 4) and bcr × abr−/− mice (n = 5) on an outbred background 4 h after the i.p. injection of 15 mg/kg LPS. After the harvest of BALF, some of the mice were used to assess neutrophils in the different lung compartments: neutrophils were tracked (untreated wild-type, n = 2; wild-type treated with LPS, n = 2; double-null mutant treated with LPS, n = 4) as described previously (33) after the i.v. injection of 5 μg fluorescein isothiocyanate-Gr-1 antibodies 5 min before sacrifice. Briefly, mice were perfused to remove nonadherent blood vessel neutrophils. Lungs containing interstitial and marginalized intravascular polymorphonuclear leukocytes (PMN) were minced and processed as described previously (33) using fluorescence-activated cell sorting and Gr-1 and CD11b antibodies (BD-Pharmingen). To measure MPO, ice-cold, minced lung tissue was homogenized (10%, wt/vol) in 0.05 M phosphate buffer (pH 6.0) containing 5 mg/ml hexa-decyltrimetylammonium bromide (HTAB; Sigma, St. Louis, MO) using a tissue homogenizer. The tissue homogenates were taken through three freeze/thaw cycles and were centrifuged at 12,500 × g for 15 min. The appearance of a colored product from the MPO-dependent reaction of K-Blue (a commercialized preparation of stabilized hydrogen peroxide, which contains 3,3′,5,5′-tetramethylbenzidine) (Neogen, Lexington, KY) was measured using a spectrophotometer at 620 nm. MPO activity was determined by comparing the absorbance of samples to that of the MPO standard curve (0.016 to 0.5 U/ml in phosphate buffer), and results were expressed as units per gram of tissue.
Isolation of cells and cell culture.
Total bone marrow was obtained from mice by flushing pelvises, femurs, and tibiae with 0.1% bovine serum albumin and Hank's balanced salt solution (HBSS; Invitrogen) with 2 U/ml heparin. BMM were isolated from bone marrow cells according to the protocol of Stanley (35). BMM were cultured as described previously (8). Peritoneally elicited macrophages (PEM) were obtained by the lavage of the peritoneal cavity 5 days after i.p. injection with 3 ml 4% Brewer thioglycolate medium (Difco, Detroit, MI). Neutrophils were isolated from total bone marrow according to Lowell et al. (26), except that erythrocytes were removed by the centrifugation of the collected neutrophils on a cushion of Histopaque 1118 (Sigma, St. Louis, MO).
Measurement of superoxide anion generation.
The cytochrome c reduction assay was used to evaluate extracellular superoxide anion production from PEM and BMM as previously described (12). The superoxide production of abr−/− neutrophils isolated from mice on an outbred background (n = 4) was 180 ± 49 mAU/min; for the wild type it was 115 ± 24 mAU/min (n = 4). The flow-cytometric analysis of isolated PEM with anti-CD11b and anti-Gr-1 demonstrated that 95% of each preparation reacted only with anti-CD11b (macrophages), whereas 1% or less of each population doubly stained with both (neutrophils). No statistically significant difference was observed in the mean fluorescence intensity (MFI) of anti-CD11b-labeled PEM from wild-type mice (MFI = 2,640 ± 240; n = 3) or mice lacking both Abr and Bcr proteins (MFI = 2,264 ± 295; n = 3).
Neutrophil stimulation, measurement of MPO and elastase, and lactoferrin secretion.
Bone marrow-derived neutrophils were resuspended in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Carlsbad, CA) to 1 × 106 cells/ml. Cytochalasin B and N-formyl-methionyl-leucyl-phenylalanine (fMLP) were from Sigma (St. Louis, MO). After stimulation, the neutrophils were pelleted and the supernatants retained. The supernatants were diluted 1:10 in DMEM (without phenol red), and MPO activity was determined using the tetramethylbenzidine liquid substrate system for enzyme-linked immunosorbent assays (ELISA) (Sigma, St. Louis, MO). Elastase activity was determined for a 1:10 dilution of each supernatant after incubation with chromogenic elastase substrate I (Calbiochem, San Diego, CA). The release of lactoferrin was measured by ELISA using an anti-human lactoferrin antibody from Sigma as described previously (30). The reactivity of this antibody with murine lactoferrin was confirmed by Western blotting using supernatants from neutrophils. Only one reactive protein band was observed (data not shown). Supernatants were diluted 1:10 in buffer before addition to the enzyme immunoassay/radioimmunoassay 96-well plates (Corning, Corning NY).
Gelatin zymography.
Conditioned medium from BMM treated for 20 h with 100 ng/ml LPS in 0.5% fetal calf serum (FCS) (Invitrogen, Carlsbad, CA) or supernatants from neutrophils were incubated at room temperature in an equal volume of 2× nonreducing sodium dodecyl sulfate (SDS) sample buffer. The samples were resolved on SDS-7.5% PAGE containing 1 mg/ml gelatin (Type A; Sigma, St. Louis, MO). After being washed in 2.5% Triton, the gel was developed by incubation at 37°C in 50 mM Tris-HCl, pH 7.6, 10 mM CaCl2, 50 mM NaCl, and 0.05% Brij 35 for 16 h. Bands of activity were detected by Coomassie blue staining. Un-Scan-It software (Silk Scientific) was used to quantify the resulting bands.
Rac activation assay.
Murine bone marrow-derived neutrophils were isolated as described previously (5). A total of 5 × 106 cells were suspended in 300 μl of HBSS containing glucose and were incubated at 37°C for 5 min. Dimethylsulfoxide (DMSO) or 1 to 5 μM fMLP was added. In two experiments, cells were preincubated for 5 min at 37°C with 1 μg/ml cytochalasin B. Incubation at 37°C was continued for the times indicated in the figure legends. Cells were lysed with ice-cold 2× MLB lysis buffer (50 mM Tris, pH 7.5, 300 mM NaCl, 2% Igepal CA-630, 20 mM MgCl2, 2 mM EDTA, 20% glycerol, 20 μg/ml leupeptin, 20 μg/ml aprotinin, 2 mM sodium pervanadate, 2 mM phenylmethylsulfonyl fluoride). Rac activation assays were performed as described previously (6). Affinity-precipitated proteins as well as 20 μl of the supernatants of each sample were separated by SDS-PAGE, followed by immunoblotting with anti-Rac2 (Millipore) or anti-Rac1 (Cytoskeleton) antibody. The resulting blots were scanned and analyzed with Un-Scan-It software (Silk Scientific, Orem, UT). Differences (n-fold) were calculated based on the signal of GTP-bound Rac and of total Rac for each sample. Mice within individual experiments all were on an outbred or on a defined genetic background. We used three to four mice of each genotype per experiment.
Statistical analysis.
Results for the functional studies are shown mainly as means ± standard errors of the means (SEM). Statistical analyses were performed on original data using either a paired Student's t test or one-way analysis of variance, followed by a Dunnett's multiple comparison test. P < 0.05 was considered significant, and n represents the number of animals.
RESULTS
Mice lacking Abr and Bcr exhibit increased responses to E. coli LPS in vivo.
To examine if Abr also regulates inflammation in vivo, we compared the reactions of wild-type, abr−/−, and bcr−/− mice to endotoxin challenge. Within the first hours after LPS administration, all animals became lethargic and showed reduced activity, and all animals rapidly lost weight during the first 24 h (Fig. 1). Whereas in wild-type mice the body weight gradually returned to normal by day 4, abr−/− and bcr−/− mice were much slower to recover (Fig. 1). Interestingly, abr × bcr−/− mice showed no signs of recovery and died within 2 days (data not shown). Similar results were obtained with mice on an outbred background (data not shown). This indicates that Bcr and Abr independently affect recovery from septicemia.
FIG. 1.
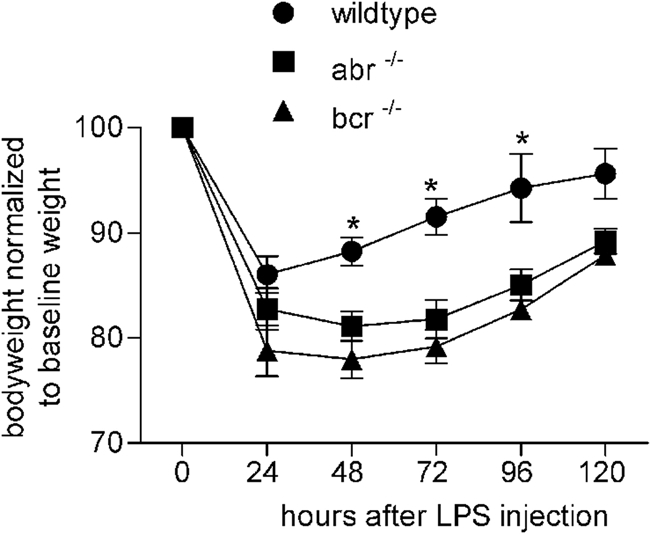
Increased sensitivity of mice lacking Abr or Bcr to LPS-induced endotoxemia. The body weight loss and regain of the indicated genotypes given LPS (15 mg/kg given i.p.) is shown. Genotypes include abr−/− (squares; n = 4), bcr−/− (triangles; n = 4), and wild type (circles; n = 6). Body weights are normalized to baseline weight in each animal. *, P < 0.01 by paired Student's t test compared to results for abr−/− and bcr−/− mice. Only weights of surviving mice are included in the graphs, and all genotypes were on the FVB/J background.
Death due to septic shock frequently is caused by severe damage to target organs. One hallmark of such injury is acute pulmonary edema caused by endothelial cell barrier dysfunction. To examine if the lack of Abr or Bcr was associated with increased endotoxin-induced microvascular lung injury, LPS was injected in all four genotypes, and plasma leakage was determined using the Evans blue assay. As expected, LPS clearly caused lung edema in all mice, which did not occur with saline-injected controls. In mice lacking Abr, Bcr, or both, a severe dysfunction of the microvasculature was observed, resulting in extensive edema (Fig. 2A).
FIG. 2.

Lack of Abr and Bcr is associated with increased acute microvascular lung injury upon endotoxin challenge. (A) Plasma leakage and (C) myeloperoxidase levels in lungs 4 h after the administration of LPS (15 mg/kg, i.p.). Plasma leakage was measured using the Evans blue assay, and all data are expressed as means ± SEM (n = 4 to 6) of the concentration of Evans blue dye (in micrograms per gram of tissue). P < 0.05 (*) and P < 0.001 (***) compared to results for wild-type (wt) mice injected with LPS. MPO activity was expressed as units per gram of tissue, and all data are expressed as means ± SEM (n = 4 to 6). P < 0.01 (**) and P < 0.001 (***) compared to results for treated wild-type mice. All genotypes were on the FVB/J background. (B) BALF total cell count 4 h after i.p. injection with 15 mg/kg LPS. (D to G) Histology of H&E-stained sections of lungs from wild-type controls (D and F) and double-null mutants (E and G) 24 h after i.v. LPS injection. Representative samples are shown from two wild-type and three abr × bcr−/− animals on an outbred background. The superior lung lobe at the same location is shown for both the wild-type (D) and abr × bcr−/− lung (E). Note the reduced airspace and severe interstitial thickening in the double-null mutant lungs, whereas wild-type mice exhibit localized areas of reduced airspace resulting mainly from edema. The image in panel F shows larger areas of lightly stained plasma between cells of the interstitium, whereas panel G shows high cellularity. Arrow heads in panel G and the inset identify highly vacuolated macrophages. Bars represent 200 μm (D and E) and 20 μm (F and G).
Acute inflammation in the lung is characterized by a substantial influx of immune cells from the circulation. We measured both BALF cells and interstitial/intravascular lung neutrophils shortly after LPS injection. Compared to those of wild-type controls, abr × bcr−/− BALF contained elevated numbers of cells 4 h after LPS injection (Fig. 2B). There was a reduced number of intravascular CD11b+ Gr-1+ neutrophils and increased numbers of CD11b+ Gr-1− interstitial neutrophils (as defined in reference 33) in the lungs of LPS-injected mice compared to those of saline-injected controls, but no significant difference between samples of abr × bcr−/− and wild-type mice was found (not shown). This suggests that in the double-null mutants, a more substantial diapedesis of neutrophils into the airspace had occurred within this short time frame.
Activated neutrophils that accumulate in injured tissue can produce substantial amounts of MPO. Therefore, MPO activity was assessed in LPS-treated lungs. As shown in Fig. 2C, both abr−/− and bcr−/− lungs contained significantly higher levels of MPO than those of wild-type mice. Interestingly, lungs of mice lacking both proteins contained the highest levels of MPO (Fig. 2C).
We next examined the lungs of abr × bcr−/− mice on a histological level at a later time point, 24 h after endotoxin challenge. Compared to the lungs from wild-type mice, which exhibited only localized areas of interstitial thickening (Fig. 2D and F), lungs from abr × bcr−/− mice at this point displayed a diffuse, prominently enhanced thickening of the interstitium, resulting in significantly reduced alveolar space, severe edema, and hemorrhaging (Fig. 2E and G). Presumed resident macrophages (identified morphologically and with b4-isolectin staining; data not shown) in the lungs from mice lacking both Abr and Bcr were abnormally and heavily vacuolated (Fig. 2G). Similar results were obtained with mice on an inbred background (data not shown). In single-null mutant mice, no clear differences were observable between lung tissue sections taken 24 h after LPS injection compared to that of wild-type mice (not shown).
Mice lacking Abr and Bcr in a model of polymicrobial sepsis.
To investigate the combined impact of the loss of bcr and abr in a model of polymicrobial sepsis, we performed CLP on matched sets of wild-type and double-null mutant (abr × bcr−/−) mice. Animals were supported by fluid resuscitation. In this model, a nidus of infection and/or ischemia causes persistent intestinal inflammation (reviewed in references 10 and 31). To limit damage, we punctured the cecum only once using a thin (22-gauge) needle, but all animals became ill. However, mice lacking abr and bcr lost significantly more weight, at both the 24- and 48-h time points (Fig. 3A), and at the 48-h time point, 56% of abr × bcr−/− and 30% of control wild-type mice had died (Fig. 3B). To investigate intestinal damage, sections were prepared from the surviving animals sacrificed 48 h after CLP. As illustrated in Fig. 3C, increased damage in the ileum of double-null mutants, such as edema, villi with flattened or distorted tips, and exposed lamina propria, were observed. To quantify the degree of mucosal damage, H&E-stained sections of the ileum were analyzed as described previously (3). This yielded a significantly higher score for the abr × bcr−/− null mutant samples than that of the control wild-type mice (mucosal damage score of 3.5 for the mutant and 1.0 for the wild-type mice; n = 4 in each group; P = 0.03). The mean length of double-null mutant ileum villi was 266 ± 95 μm, significantly shorter (P < 0.0001 by paired t test) than that of wild-type CLP-operated (344 ± 59 μm) or sham CLP-operated mice (301 ± 63 μm). The analysis of null mutant mice on an inbred background showed a similar degree of damage, as determined by an increase of the goblet cell number per villus and a decrease in villus length (Fig. 3D).
FIG. 3.

Increased sensitivity of mice lacking abr and bcr to CLP. (A) Seven to eight sets of 8- to 10-month-old sibling female wild-type (wt) or abr × bcr−/− mice were subjected to CLP. Weight loss is indicated as the percentage of original body weight at 24 and 48 h after CLP. *, P < 0.01 by paired Student's t test. (B) Percentage of mice surviving at the indicated times (n = 9 wild-type mice and 10 abr × bcr−/− mice). (C) Increased intestinal damage to the ileum of abr × bcr−/− mice 48 h after CLP. Magnifications and genotypes are indicated. Scale bars, 100 μm. (D) Decreased villus length and increased goblet cell number in mice lacking Abr and Bcr. #, P < 0.05 compared to results for sham operation; *, P < 0.05 compared to results for wild-type CLP. For sham-operated mice, n = 2; for wild-type CLP and abr × bcr−/− CLP mice, n = 3 in each group (30 villi from each mouse). (E) Elevated ALT and AST levels in plasma of abr × bcr−/− mice. P < 0.005 (*) and P < 0.001 (**) compared to results for sham operation and PBS. ⋄, P < 0.05 compared to results for abr × bcr−/− by paired t test. (A, B, C, and E) Mice on outbred background; (D) mice on inbred background.
At 48 h after CLP, AST and ALT levels were assessed in the plasma to determine liver and/or heart dysfunction, respectively, and thus provide indications of organ failure. As shown in Fig. 3E, double-null mutant mice demonstrated significantly higher levels of plasma AST and ALT than their wild-type siblings.
Elevated levels of ROS from abr × bcr−/− phagocytic cells.
Since the lack of Abr and Bcr leads to an increase in inflammatory reactions, we next investigated whether this could be ascribed to intrinsic abnormalities in their phagocytic cells.
LPS-induced lung injury is characterized by an initial influx of neutrophils into the lungs, later followed by macrophages (28). To examine if both Bcr and Abr are expressed in the two cell types, we performed Western blot analysis. As shown in Fig. 4A, Bcr and Abr both normally are present in these innate immune cells.
FIG. 4.

Increased ROS production in myeloid cells lacking Bcr and Abr. (A) SDS lysates of BMM and neutrophils from wild-type and null mutant mice were analyzed using Abr antiserum (10) or anti-Bcr N20 antibodies by Western blotting (top and bottom, respectively). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (B and C) Elevated superoxide production in PEM from null mutant mice. Thioglycolate-elicited macrophages from wild-type and single-null mutant mice (B) or wild-type and double-null mutants (C) were stimulated with 150 nM PMA in the presence of cytochrome c. Results were calculated as nanomoles of reduced cytochrome c/milligram of protein/time and then converted to differences (n-fold) for comparison. Assays were performed in triplicate. For panel B, four wild-type mice and five abr−/− and bcr−/− mice from two independent, representative experiments were used. For panel C, 3 mice each for both wild-type and abr × bcr−/− mice from one representative experiment are shown (*, P = 4 × 10−9). Error bars indicate the standard deviations for each mean.
ROS produced by phagocytic cells can contribute significantly to tissue damage during septic shock (9, 13). Therefore, we investigated if there are differences in ROS production in BMM from the different genotypes. Cells were primed in vitro with LPS, tumor necrosis factor α, or granulocyte-macrophage colony-stimulating factor and subsequently stimulated with phorbol myristate acetate (PMA) or zymosan to induce superoxide anion production. However, despite exhaustive experimentation, no significant differences in the levels of ROS were measured between the four genotypes of BMM (not shown).
We next examined ROS production in PEM, which are representative of macrophages that are activated in vivo. PEM were generated by inducing aseptic peritonitis with thioglycolate and were stimulated with PMA. As shown in Fig. 4B, PEM from mice deficient in Abr generated superoxide anion to the same extent as those from wild-type mice. Interestingly, PEM from bcr−/− mice produced more superoxide anion than those from wild-type and abr−/− mice (Fig. 4B). In addition, PEM derived from abr × bcr−/− mice showed the highest level of superoxide anion production (Fig. 4C), and bone marrow-derived neutrophils from double-null mutant mice also generated more ROS than those of wild-type mice (n = 4 for each genotype; wild type, 115 ± 23.7 mAU/min; abr × bcr−/−, 167 ± 36.6 mAU/min).
Increased secretion of specific products from double-null mutant phagocytic cells.
Neutrophils contain primary, secondary, and tertiary granules as well as secretory vesicles, which are exocytosed in response to stimulation with inflammatory molecules and cytokines (13). They are released in a hierarchical order (24), and the spatial or temporal deregulation of this process may result in excessive tissue damage (2). To assess possible differences in the rate of degranulation in neutrophils from abr−/−, bcr−/−, and abr × bcr−/− mice, we isolated bone marrow-derived neutrophils and stimulated them with fMLP or cytochalasin B plus fMLP as described previously (14). MPO and elastase were used as markers for primary granules and lactoferrin as a secondary granule marker. MMP9, found in tertiary granules, was used as a marker for the export of these granules (4). The cell surface upregulation of CD11b was used as a marker for the release of secretory vesicles (24).
Compared to wild-type controls, BMM neutrophils isolated from abr × bcr−/− mice demonstrated a statistically significant 1.5-fold-enhanced release of MPO in response to costimulation by cytochalasin B and fMLP (Fig. 5A). A similar pattern of release of elastase (Fig. 5B) also was observed. The release of MPO and elastase by bone marrow-derived neutrophils from single-null mutant mice (abr−/− or bcr−/−) was not significantly different from that of wild-type controls (not shown). Also, the release of lactoferrin (Fig. 5C) or of MMP9 (Fig. 5D) or CD11b cell surface expression (Fig. 5E) was similar for wild-type and abr × bcr−/− neutrophils.
FIG. 5.

Abnormal release of tissue-damaging products in phagocytic cells from mice deficient in Abr and Bcr. (A to E) Neutrophils (5 × 105) isolated from the bone marrow of wild-type (W; open bars) and double-null mutant mice (D; shaded bars) either were kept on ice, incubated at 37°C without further stimulus for 20 min, incubated at 37°C with 1 μM fMLP for 15 min, or incubated with 1 μg/ml cytochalasin B (CB) for 5 min, followed by incubation with 1 μM fMLP for 15 min. Supernatants were assayed for MPO activity (A), elastase activity (B), or lactoferrin release (C). Activity in panels A and B is depicted as the difference (n-fold) normalized to the activity obtained from 1% Triton lysates of nonstimulated PMN. Shown are the means ± standard deviations from two independent, representative experiments. (A) *, P = 0.02 (n = 4); (B) *, P = 0.03 (n = 4). For lactoferrin release, absorbance from the ELISA was converted to levels (n-fold) of differences. (D). MMP9 activity in the resulting supernatant was analyzed by gelatin zymography (W, wild type; D, abr × bcr−/−). (E) To quantify CD11b upregulation, flow cytometry was performed on the stimulated neutrophils using an antibody specific for CD11b. The MFI of each is compared graphically. (F and G) Increased secretion of MMP9 in BMM from mice lacking Abr and Bcr in response to stimulation with LPS. BMM were incubated for 20 h in 0.5% FCS containing 100 ng/ml LPS. Conditioned media (26 μl) were analyzed by gelatin zymography. (G) Representative gel showing bands of MMP9 activity (upper) and MMP2 (lower; from the FCS) and the location of molecular size standards to the right. (F) Graphical representation of MMP9 activity in conditioned media from BMM stimulated as described for panel G. Activity is expressed as the difference (n-fold) normalized to the protein content of each monolayer. Means ± standard deviations are shown. *, P < 0.05 (n = 5).
Macrophages also secrete gelatinases (14). Therefore, we also assessed the release of MMP9 from BMM stimulated with LPS. Interestingly, BMM from abr−/− and abr × bcr−/− mice exhibited a statistically significant increased release of MMP9 (Fig. 5G). The quantification of the bands of MMP9 activity (Fig. 5F) indicated that BMM from abr−/− and abr × bcr−/− mice had an approximately 1.8- to 2.3-fold increase, respectively, compared to levels for wild-type BMM.
Increased levels of activated Rac in neutrophils stimulated with cytochalasin B and fMLP.
Mitchell et al. (29) reported that the exocytosis of primary granules in human neutrophils is regulated by Rac-dependent actin remodeling. We therefore compared Rac activation in abr × bcr−/− and wild-type neutrophils stimulated with fMLP and cytochalasin B or fMLP alone. As illustrated in Fig. 6, at t = 0, nonstimulated abr × bcr−/− neutrophils had a higher baseline level of GTP-bound Rac1 and of GTP-bound Rac2 than wild-type neutrophils. The stimulation of the wild-type and null mutant cells for 15 and 30 s with 5 μM fMLP increased levels of both GTP-Rac1 and GTP-Rac2. Interestingly, higher levels of GTP-Rac1 and GTP-Rac2 were present after 15 s of stimulation in abr × bcr−/− neutrophils compared to that of wild-type neutrophils (2.82 versus 1.97 and 2.08 versus 1.45, respectively).
FIG. 6.
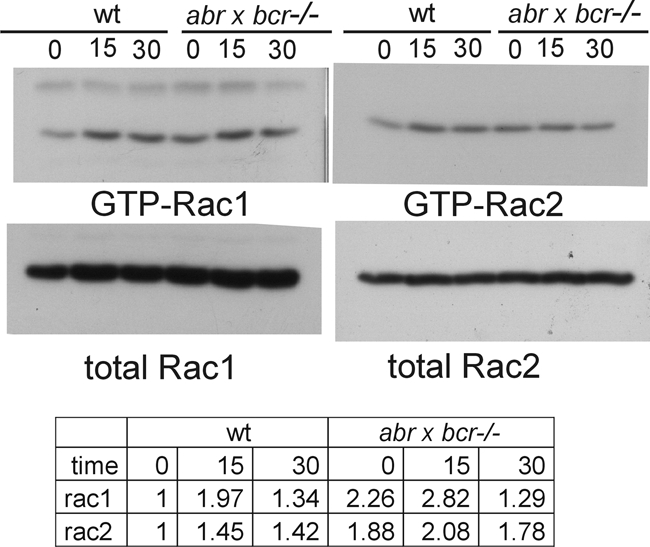
Increased activation of Rac1 and Rac2 in abr × bcr−/− neutrophils. Bone marrow-derived neutrophils were left at 37°C or were stimulated for 15s or 30s with 5 μM fMLP. Assays for activated Rac were performed using a glutathione S-transferase-Pak RBD pulldown assay. Samples represent the pooled neutrophils of three to four mice of each genotype. The Western blot of one experiment is shown; experimental results of fMLP alone and cytochalasin B plus fMLP were combined for the table, which shows the average increase (n-fold) for all experiments (n = 2 for Rac1; n = 3 for Rac2). wt, wild type.
DISCUSSION
To date, the normal cellular function of the Abr gene product has not been investigated. In this report, we show that mice lacking abr gene function exhibit increased susceptibility to endotoxemia compared to that of control wild-type mice, demonstrating that this gene product functions to negatively regulate inflammation. The finding that Abr single knockouts also develop septic shock indicates that Bcr cannot compensate for its function. In concordance with this, mice deficient for Abr as well as Bcr were more severely affected than single-null mutants and died after injection with endotoxin. Similarly, in a distinct model of sepsis caused by the puncture of the cecum, mice lacking both Bcr and Abr were the most severely affected.
The lungs from abr × bcr−/− mice 24 h after injection with LPS demonstrated the development of a severe pathology, including diffuse and severe edema and the hemorrhaging of the interstitium, resulting in reduced alveolar space. Wild-type mice mainly experienced interstitial edema, with a much less severe pathology and a reduced response to LPS injection. The more severe damage in the lungs of the abr × bcr−/− mice likely was initiated by an early phase of the increased migration of leukocytes into the alveolar airspace, as measured in the BALF at the 4-h time point. Our previous studies showed that monocytes lacking both Abr and Bcr have enhanced motility. Here, we show that in addition, there also is the enhanced generation of superoxide anion, degranulation, activation, and the secretion of MMP9 by isolated phagocytic leukocytes derived from abr × bcr−/− mice.
Many factors contribute to the development of sepsis, among which superoxide anion and derived ROS are thought to play an important role in its pathology. ROS increase the permeability of the vascular endothelium and cause vascular damage (15, 25, 34). Mutant mice missing components of the NADPH complex and thus having defective ROS production have reduced susceptibility to endotoxin-induced sepsis (19). It therefore is highly significant that in comparison to wild-type mice, PEM derived from double-null mutant mice produce nearly three times the amount of superoxide anion in response to PMA. Thus, the severity of the septic condition in the abr × bcr−/− mice can be attributed partly to the enhanced superoxide anion generation by phagocytic leukocytes.
We also observed an enhanced level of the degranulation of specific products in neutrophils from mice lacking both Abr and Bcr function. MPO and elastase, contained within primary granules, showed enhanced release in double-null mutant neutrophils compared to that of wild-type neutrophils. Several studies have demonstrated the importance of primary granule enzymes, including elastase and cathepsin G, in the development of sepsis. For example, mice deficient for elastase and cathepsin G are more resistant to LPS-induced septic shock and exhibit little alveolar damage compared to that of wild-type mice (37). Therefore, the enhanced or deregulated exocytosis of the contents of these neutrophil granules very likely significantly contributed to the severity of sepsis in the double-null mutant mice.
In addition, enhanced secretion of MMP9 was measured in LPS-stimulated BMM from abr × bcr−/− mice. Since MMP9 also processes plasma membrane-bound cytokines and growth factors that participate in the inflammatory response (40), the uncontrolled secretion of this serine protease from macrophages in the lung may have contributed to the fatal outcome of LPS exposure in the double-null mutants. Overall, these combined abnormalities and the importance of each in contributing to tissue damage can explain the development of terminal septic shock in abr × bcr−/− mice.
There was no difference between double-null mutant and wild-type neutrophils in secreted lactoferrin levels, MMP9 activity, or the upregulation of CD11b. These are present in distinct compartments within the neutrophil: the secondary and tertiary granules and the secretory vesicles, respectively. Of the three Rac proteins present in human and mouse, Rac2 is restricted to hematopoietic cells (4). The lack of Rac2 was found to have a significant negative impact on the degranulation of primary granules but not on secondary granule, tertiary granule, or secretory vesicle release (1). Since Bcr and Abr are negative regulators of Rac function in vivo (1, 39), we conclude that in mice, Abr and Bcr are specific negative regulators of primary granule release in neutrophils promoted by Rac2. A role of these GAPs in regulating Rac2 is consistent with our previous results showing that the lack of Abr and Bcr caused enhanced colony-stimulating factor 1-directed chemotaxis and the phagocytosis of opsonized E. coli in BMM (6). Null mutants lacking Rac2 also show defects in these specific functions (22, 41).
However, it is clear from these results, as well as from our previous studies, that Abr and Bcr do not act exclusively on Rac2. Other phenotypic abnormalities in the double-null mutant mice are found associated with cell types that are not known to express Rac2, which is hematopoietic specific. Mice lacking Abr and Bcr exhibit vestibular defects in the inner ear, including the partial or complete loss of otoconia, and also have abnormalities in the extension of Bergman glial cells in the cerebellum (20, 21). We show here, using Rac1- or Rac2-specific antibodies, that neutrophils from abr × bcr−/− mice have elevated baseline levels of both activated Rac1 and activated Rac2, indicating that both Rac1 and Rac2 are in vivo substrates for Bcr and Abr.
After a 15-s stimulation with fMLP or cytochalasin B plus fMLP, we detected increased levels of GTP-Rac1 and GTP-Rac2 in abr × bcr−/− neutrophils. Interestingly, Mitchell et al. (29), using human neutrophils, showed that both Rac1 and Rac2 become activated in human neutrophils stimulated with cytochalasin B and fMLP, and they further suggested that the two Racs participate in different actin reorganization processes needed for the exocytosis of granule content. Therefore, it will be of interest to investigate if Abr or Bcr individually regulates an isoform-specific function of Rac in granule exocytosis.
Clearly, neutrophils derived from the in vivo inflammatory environment have different characteristics and responses than cells activated in culture or derived from a healthy mouse. When we measured MPO release from neutrophils isolated from nonstimulated bone marrow, only those isolated from mice lacking both Bcr and Abr showed significantly increased secretion. However, in vivo, upon the development of septic shock, the lungs of both single-null mutants as well as double-null mutants contained elevated levels of MPO compared to those of wild-type controls. We found a similar trend using macrophages: whereas activated macrophages isolated from the peritoneum of double-null mutants did show increased ROS production, macrophage precursors isolated from the bone marrow that were differentiated and activated in vitro did not. Therefore, the effect of the loss of Bcr and Abr on various responses in macrophages and neutrophils differs depending on if the cell was matured, activated, or stimulated; in vivo, the inflammatory environment consists of multiple proinflammatory signals that may be impossible to reproduce in vitro.
We conclude that Bcr and Abr have many overlapping functions that are uncovered only when both proteins are ablated. This overlap may be mediated through the shared domains that are 68% identical in amino acid residues and include the DH-, PH-, GAP-, and PDZ-binding sequence. Indeed, proteins that were found to bind to these domains in Bcr, such as RhoGDIα and Mint3, also bind to Abr (23, 27, and unpublished data). However, Bcr also contains an N-terminal domain that is very different than the N termini of Abr and that, in Bcr, contains binding sites for a number of signal transduction proteins, including, among others, tyrosine kinases, serine/threonine kinases, and scaffolding proteins. Therefore, the regulation of functions of which the single-null mutants displayed a measurable phenotype, such as in vivo endothelial permeability upon exposure to LPS, may be mediated molecularly through interactions with the N-terminal domains.
The innate immune system is receiving increased attention, as more pathological states are associated with chronic inflammation. This includes, for example, arteriosclerosis and arthritis, as well as some forms of cancer. Compared to what is know about the initiation of inflammation, much less is known about the negative regulation of myeloid cell functions. Since Bcr and Abr are the only known proteins to date that have very specific activity in downregulating in vivo-activated macrophages and neutrophils, it will be of interest to examine if they play a role in chronic inflammatory conditions as well as septic shock.
Acknowledgments
We are indebted to Martine Torres for helpful discussions and suggestions and Louis Dubeau for comments on the lung pathology. Bin Zhang and Niklas Feldhahn are acknowledged for performing experiments related to this study. We thank Donna Foster for the excellent care of the mice. Pavinder Kaur is acknowledged for help with the CLP experiments.
This work was supported by PHS grants HL071945 (J.G.) and HL060231 (D.W., P.M., J.G., and N.H.). Parviz Minoo is a Hastings Foundation Professor of Pediatrics.
Footnotes
Published ahead of print on 24 August 2009.
REFERENCES
- 1.Abdel-Latif, D., M. Steward, D. L. Macdonald, G. A. Francis, M. C. Dinauer, and P. Lacy. 2004. Rac2 is critical for neutrophil primary granule exocytosis. Blood 104:832-839. [DOI] [PubMed] [Google Scholar]
- 2.Aldridge, A. J. 2002. Role of the neutrophil in septic shock and the adult respiratory distress syndrome. Eur. J. Surg. 168:204-214. [DOI] [PubMed] [Google Scholar]
- 3.Atkinson, C., H. Song, B. Lu, F. Qiao, T. A. Burns, V. M. Holers, G. C. Tsokos, and S. Tomlinson. 2005. Targeted complement inhibition by C3d recognition ameliorates tissue injury without apparent increase in susceptibility to infection. J. Clin. Investig. 115:2444-2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borregaard, N., and J. B. Cowland. 1997. Granules of the human neutrophil polymorphonuclear leukocyte. Blood 89:3503-3521. [PubMed] [Google Scholar]
- 5.Boxio, R., C. Bossenmeyer-Pourié, N. Steinckwich, C. Dournon, and O. Nüsse. 2004. Mouse bone marrow contains large numbers of functionally competent neutrophils. J. Leukoc. Biol. 75:604-611. [DOI] [PubMed] [Google Scholar]
- 6.Cho, Y., J. M. Cunnick, S-J. Yi, V. Kaartinen, J. Groffen, and N. Heisterkamp. 2007. Abr and Bcr, two homologous RacGTPase-activating proteins, control multiple cellular functions of murine macrophages. Mol. Cell. Biol. 27:899-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chuang, T. H., X. Xu, V. Kaartinen, N. Heisterkamp, J. Groffen, and G. M. Bokoch. 1995. Abr and Bcr are multifunctional regulators of the Rho GTP-binding protein family. Proc. Natl. Acad. Sci. USA 92:10282-10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunnick, J., P. Kaur, Y. Cho, J. Groffen, and N. Heisterkamp. 2006. Use of bone marrow-derived macrophages to model murine innate immune responses. J. Immunol. Methods 311:96-105. [DOI] [PubMed] [Google Scholar]
- 9.Dallegri, F., and L. Ottonello. 1997. Tissue injury in neutrophilic inflammation. Inflamm. Res. 46:382-391. [DOI] [PubMed] [Google Scholar]
- 10.De Maio, A., M. B. Torres, and R. H. Reeves. 2005. Genetic determinants influencing the response to injury, inflammation, and sepsis. Shock 23:11-17. [DOI] [PubMed] [Google Scholar]
- 11.Didsbury, J., R. F. Weber, G. M. Bokoch, T. Evans, and R. Snyderman. 1989. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J. Biol. Chem. 264:16378-16382. [PubMed] [Google Scholar]
- 12.Diekmann, D., S. Brill, M. D. Garrett, N. Totty, J. Hsuan, C. Monfries, C. Hall, L. Lim, and A. Hall. 1991. Bcr encodes a GTPase-activating protein for p21rac. Nature 351:400-402. [DOI] [PubMed] [Google Scholar]
- 13.Faurschou, M., and N. Borregaard. 2003. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 5:1317-1327. [DOI] [PubMed] [Google Scholar]
- 14.Goetzl, E. J., M. J. Banda, and D. Leppert. 1996. Matrix metalloproteinases in immunity. J. Immunol. 156:1-4. [PubMed] [Google Scholar]
- 15.Hassan, F., S. Islam, N. Koide, M. M. Mu, H. Ito, I. Mori, T. Yoshida, and T. Yokochi. 2004. Role of p38 mitogen-activated protein kinase (MAPK) for vacuole formation in lipopolysaccharide (LPS)-stimulated macrophages. Microbiol. Immunol. 48:807-815. [DOI] [PubMed] [Google Scholar]
- 16.Heasman, S. J., and A. J. Ridley. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9:690-701. [DOI] [PubMed] [Google Scholar]
- 17.Heisterkamp, N., V. Kaartinen, S. van Soest, G. M. Bokoch, and J. Groffen. 1993. Human ABR encodes a protein with GAPrac activity and homology to the DBL nucleotide exchange factor domain. J. Biol. Chem. 268:16903-16906. [PubMed] [Google Scholar]
- 18.Heisterkamp, N., K. Stam, J. Groffen, A. de Klein, and G. Grosveld. 1985. Structural organization of the bcr gene and its role in the Ph′ translocation. Nature 315:758-761. [DOI] [PubMed] [Google Scholar]
- 19.Jagels, M. A., and T. E. Hugli. 1994. Mechanisms and mediators of neutrophilic leukocytosis. Immunopharmacology 28:1-18. [DOI] [PubMed] [Google Scholar]
- 20.Kaartinen, V., I. Gonzalez-Gomez, J. W. Voncken, L. Haataja, E. Faure, A. Nagy, J. Groffen, and N. Heisterkamp. 2001. Abnormal function of astroglia lacking Abr and Bcr RacGAPs. Development 128:4217-4227. [DOI] [PubMed] [Google Scholar]
- 21.Kaartinen, V., A. Nagy, I. Gonzalez-Gomez, J. Groffen, and N. Heisterkamp. 2002. Vestibular dysgenesis in mice lacking Abr and Bcr Cdc42/RacGAPs. Dev. Dyn. 223:517-525. [DOI] [PubMed] [Google Scholar]
- 22.Koh, A. L., C. X. Sun, F. Zhu, and M. Glogauer. 2005. The role of Rac1 and Rac2 in bacterial killing. Cell Immunol. 235:92-97. [DOI] [PubMed] [Google Scholar]
- 23.Kweon, S. M., Y. J. Cho, P. Minoo, J. Groffen, and N. Heisterkamp. 2008. Activity of the Bcr GTPase-activating domain is regulated through direct protein/protein interaction with the Rho guanine nucleotide dissociation inhibitor. J. Biol. Chem. 283:3023-3030. [DOI] [PubMed] [Google Scholar]
- 24.Lacy, P. 2005. The role of Rho GTPases and SNAREs in mediator release from granulocytes. Pharmacol. Ther. 107:358-376. [DOI] [PubMed] [Google Scholar]
- 25.Lagasse, E., and I. L. Weissman. 1996. Flow cytometric identification of murine neutrophils and monocytes. J. Immunol. Methods 197:139-150. [DOI] [PubMed] [Google Scholar]
- 26.Lowell, C. A., L. Fumagalli, and G. Berton. 1996. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J. Cell Biol. 133:895-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malmberg, E. K., C. X. Andersson, M. Gentzsch, J. H. Chen, A. Mengos, L. Cui, G. C. Hansson, and J. R. Riordan. 2004. Bcr (breakpoint cluster region) protein binds to PDZ-domains of scaffold protein PDZK1 and vesicle coat protein Mint3. J. Cell Sci. 117:5535-5541. [DOI] [PubMed] [Google Scholar]
- 28.Maus, U. A., K. Waelsch, W. A. Kuziel, T. Delbeck, M. Mack, T. S. Blackwell, J. W. Christman, D. Schlondorff, W. Seeger, and J. Lohmeyer. 2003. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J. Immunol. 170:3273-3278. [DOI] [PubMed] [Google Scholar]
- 29.Mitchell, T., A. Lo, M. R. Logan, P. Lacy, and G. Eitzen. 2008. Primary granule exocytosis in human neutrophils is regulated by Rac-dependent actin remodeling. Am. J. Physiol. Cell Physiol. 295:C1354-C1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mócsai, A., E. Ligeti, C. A. Lowell, and G. Berton. 1999. Adhesion-dependent degranulation of neutrophils requires the Src family kinases Fgr and Hck. J. Immunol. 162:1120-1126. [PubMed] [Google Scholar]
- 31.Parker, S. J., and P. E. Watkins. 2001. Experimental models of gram-negative sepsis. Br. J. Surg. 88:22-30. [DOI] [PubMed] [Google Scholar]
- 32.Peck, J., G. Douglas, C. H. Wu, and P. D. Burbelo. 2002. Human RhoGAP domain-containing proteins: structure, function and evolutionary relationships. FEBS Lett. 528:27-34. [DOI] [PubMed] [Google Scholar]
- 33.Reutershan, J., M. A. Morris, T. L. Burcin, D. F. Smith, D. Chang, M. S. Saprito, and K. Ley. 2006. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J. Clin. Investig. 116:695-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Springer, T. A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76:301-314. [DOI] [PubMed] [Google Scholar]
- 35.Stanley, E. R. 1997. Murine bone marrow-derived macrophages. Methods Mol. Biol. 75:301-304. [DOI] [PubMed] [Google Scholar]
- 36.Tan, E. C., T. Leung, E. Manser, and L. Lim. 1993. The human active breakpoint cluster region-related gene encodes a brain protein with homology to guanine nucleotide exchange proteins and GTPase-activating proteins. J. Biol. Chem. 268:27291-27298. [PubMed] [Google Scholar]
- 37.Tkalcevic, J., M. Novelli, M. Phylactides, J. P. Iredale, A. W. Segal, and J. Roes. 2000. Impaired immunity and enhanced resistance to endotoxin in the absence of neutrophil elastase and cathepsin G. Immunity 12:201-210. [DOI] [PubMed] [Google Scholar]
- 38.Udaka, K., Y. Takeuchi, and H. Z. Movat. 1970. Simple method for quantitation of enhanced vascular permeability. Proc. Soc. Exp. Biol. Med. 133:1384-1387. [DOI] [PubMed] [Google Scholar]
- 39.Voncken, J. W., H. van Schaick, V. Kaartinen, K. Deemer, T. Coates, B. Landing, P. Pattengale, O. Dorseuil, G. M. Bokoch, J. Groffen, et al. 1995. Increased neutrophil respiratory burst in bcr-null mutants. Cell 80:719-728. [DOI] [PubMed] [Google Scholar]
- 40.Wiedow, O., and U. Meyer-Hoffert. 2005. Neutrophil serine proteases: potential key regulators of cell signalling during inflammation. J. Intern. Med. 257:319-328. [DOI] [PubMed] [Google Scholar]
- 41.Yamauchi, A., C. Kim, S. Li, C. C. Marchal, J. Towe, S. J. Atkinson, and M. C. Dinauer. 2004. Rac2-deficient murine macrophages have selective defects in superoxide production and phagocytosis of opsonized particles. J. Immunol. 173:5971-5979. [DOI] [PubMed] [Google Scholar]