

Abstract
Herpes simplex virus type 1 (HSV-1) acquires its mature virus envelope by budding into the lumen of cytoplasmic membranous compartments carrying the viral glycoproteins. In a cellular context, a budding process with identical topology occurs during the formation of intraluminal vesicles in multivesicular bodies. The cellular machinery that mediates this budding process is composed of four protein complexes termed endosomal sorting complexes required for transport (ESCRTs) and several associated proteins, including the ATPase VPS4. We have recently shown that functional VPS4 is specifically required for the cytoplasmic envelopment of HSV-1. We now demonstrate that, consistent with a role of VPS4 in virus envelopment, dominant-negative ESCRT-III proteins potently block HSV-1 production. Retroviruses are known to recruit the ESCRT machinery by small peptide motifs termed late domains. These late domains interact with various ESCRT components and thereby promote ESCRT recruitment. The best-characterized late-domain interacting ESCRT proteins are ALIX and TSG101. The presence of potential ALIX and TSG101 binding sequence motifs in various structural HSV-1 proteins suggested a functional role of these proteins in HSV-1 envelopment. We therefore used a set of dominant-negative proteins, as well as RNA interference, to characterize the contribution of ALIX and TSG101 to HSV-1 production. Interestingly, despite the strict requirement for a functional ESCRT-III complex, our data suggest that HSV-1 production is independent of ALIX and TSG101 expression. In line with these data, we also find that ESCRT-III proteins and VPS4A/B are specifically incorporated into mature HSV-1 virions.
Herpes simplex virus type 1 (HSV-1), a human pathogen, is a member of the large family of Herpesviridae. Herpesviruses are pleiomorphic enveloped particles that contain a double-stranded DNA genome. The viral genome is surrounded by an icosahedral capsid shell that is wrapped in a proteinaceous layer named the tegument. The tegumented capsid is enveloped by a cell-derived lipid bilayer in which numerous virally encoded envelope proteins are embedded.
Herpesviruses acquire their final envelope in the cytoplasm of the host cell in a process that is termed secondary envelopment. During secondary envelopment tegument-coated nucleocapsids bud into the lumen of vesicles that are thought to be derived from the trans-Golgi network. Mature virus particles are then released from the cell in an exocytic manner by fusion of this virus-containing compartment with the plasma membrane (reviewed in reference 26).
Cellular processes that are topologically related to secondary envelopment of herpesviruses are (i) the formation of intraluminal vesicles within multivesicular bodies and (ii) the final abscission step of cytokinesis. Furthermore, the secondary envelopment of herpesviruses is topologically related to the budding process of many enveloped RNA viruses. All of the above-mentioned processes involve membrane deformation and fission events that are mediated by components of up to four protein complexes, termed endosomal sorting complexes required for transport (ESCRT-0, -I, -II, and -III), and their associated proteins (for reviews, refer to references 5, 17, 28, and 35).
In the case of the formation of multivesicular bodies, the ESCRT complexes −0, -I, and -II recognize and sequester ubiquitinated cargo proteins. Concentrated cargo is then assumed to be incorporated into budding vesicles upon assembly of the ESCRT-III complex. In mammalian cells there are at least 11 ESCRT-III proteins, also referred to as charged multivesicular body proteins (CHMPs). It is thought that the otherwise autoinhibited soluble CHMPs are recruited from the cytoplasm to cellular membranes, where they assemble into large complexes that potentially mediate membrane deformation and scission (19, 29, 36, 37). These membrane-associated CHMP complexes are disassembled and thereby recycled by the AAA (named for ATPase associated with various cellular activities) ATPase VPS4 (20, 29, 36).
There is a body of evidence showing that small RNA viruses such as the human immunodeficiency virus type 1 (HIV-1) are able to recruit the ESCRT machinery to viral budding sites by means of conserved sequence motifs (reviewed in reference 1). These peptide motifs are termed late (L) domains since their mutation or deletion leads to a characteristic arrest of virus assembly at late stages. HIV-1 contains two different L domains in its C-terminal p6 domain of the Gag polyprotein. The first L domain contains a PTAP tetrapeptide motif and recruits TSG101 (tumor susceptibility gene 101), a component of the ESCRT-I complex. Disruption of the TSG101-Gag interaction, as well as the depletion of TSG101 by small interfering RNA (siRNA), arrests HIV-1 budding at a late stage (11, 24, 33). The second L domain in HIV-1 p6 is a YPXnL (X = any residues; n = 1 to 3) motif that interacts with the ESCRT-associated protein ALIX (ALG-2-interacting protein X) (30, 34). Although the ALIX binding site in HIV-1 p6 alone is not sufficient for virus release in the absence of a functional PTAP motif, budding of a PTAP deletion mutant HIV-1 can be potently rescued by increasing cellular expression levels of ALIX (10, 31). In addition, the ability of ALIX to rescue release of HIV-1 ΔPTAP depends on its interaction with CHMP4 (ESCRT-III), underlining the importance of the recruitment of ESCRT-III components to the viral budding site (10, 31). Dominant negative ESCRT-III constructs, generated by fusing them to a bulky adduct such as green fluorescent protein (GFP), also potently interfere with retrovirus budding. Although many RNA viruses differ in their use of L domains to recruit ESCRT proteins, the budding of all such viruses that have been tested requires a functional ESCRT-III complex and the activity of VPS4 (16, 23, 34).
Our group and others have recently shown that HSV-1 secondary envelopment is dependent on functional VPS4, suggesting that other ESCRT components may also be required for this process (3, 8). The presence of various L-domain motifs in structural proteins of HSV-1 (Table 1) led us ask whether functional TSG101 (ESCRT-I) and/or ALIX is required for the efficient production of infectious HSV-1. In the present study, we show that HSV-1 production is independent of TSG101 (ESCRT-I) and ALIX by using both dominant-negative proteins and RNA interference. In contrast, expression of any dominant-negative ESCRT-III protein potently blocks production of infectious HSV-1. Together with the identification of ESCRT-III proteins as integral components of HSV-1 virions, our data suggest that secondary envelopment of HSV-1 does not require TSG101 (ESCRT-I) or ALIX but is highly dependent on a functional ESCRT-III complex, in addition to VPS4 activity.
TABLE 1.
Potential late domain motifs in HSV-1 proteinsa
| Late domain motifb | HSV-1 gene | Sequence(s)c | Virion componentd |
|---|---|---|---|
| P[T/S]AP | RL2 (ICP0) | PSAP288, PTAP724 | + |
| UL29 (ICP8) | PTAP438 | − | |
| UL36 (VP1/2) | PSAP390, PTAP2828 | + | |
| UL42 | PTAP420 | − | |
| UL51 | PTAP230 | + | |
| US1 (ICP22) | PSAP166 | − | |
| US8 (gE) | PSAP214*, PTAP513 | + | |
| YPXnL | UL5 | YPYTL167 | − |
| UL8 | YPRPL701 | − | |
| UL15 (terminase) | YPFFLL636 | ? | |
| UL26 | YPGVL386 | +e | |
| UL26.5 | YPGVL80 | − | |
| UL29 (ICP8) | YPLQL201 | − | |
| UL36 (VP1/2) | YPYL221 | + | |
| UL39 (ICP6/10) | YPVPL316 | − | |
| UL41 (vhs) | YPQFL252 | + | |
| UL42 | YPDL120 | − | |
| UL43 | YPLFLL166† | ? | |
| UL53 (gK) | YPLFL251† | ? | |
| US2 | YPVTTL291 | + | |
| US7 (gI) | YPTLEL124* | + | |
| PPXY | RL2 (ICP0) | PPEY720 | + |
| UL19 (VP5) | PPGY11 | + | |
| UL36 (VP1/2) | PPTY2867 | + | |
| UL44 (gC) | PPLY218* | + | |
| UL48 (VP16) | PPLY38 | + | |
| UL52 | PPTY874 | − | |
| UL56 | PPPY26, PPPY52, PPTY147 | + | |
| US8A | PPGY108 | ? |
Individual protein sequences of human herpesvirus 1 complete genome (GenBank accession no. X14112.1) were extracted by using online resources (http://www.bioinformatics.org/sms2/genbank_trans.html), and specific peptide patterns were also identified by using online resources (http://www.bioinformatics.org/sms2/protein_pattern.html).
X, any amino acid (n = 1 to 3).
*, Motif present in extracellular domain.; †, motif present in potential transmembrane domain.
Data from Loret et al. (22).
VP24 cleavage product.
MATERIALS AND METHODS
Cell lines and viruses.
COS-7, HaCaT, HeLa, and Vero cells were maintained in Dulbecco modified Eagle medium supplemented with 100 U of penicillin/ml, 100 μg of streptomycin/ml, 2 mM l-glutamine, and 10% (vol/vol) fetal calf serum (complete growth medium). HSV-1 lacking full-length UL36 expression (HSV-1 KΔUL36) and a UL36-complementing cell line (HS30) were provided by P. Desai (9).
Plasmids.
Primers were designed to clone the open reading frames of CHMP1A (NM_002768), CHMP1B (NM_020412), CHMP2A (NM_198426), CHMP3 (NM_016079), CHMP4A (NM_014169) (starting from Met44), CHMP4B (NM_176812), CHMP4C (NM_152284), CHMP5 (NM_016410), CHMP6 (NM_024591), and CHMP7 (NM_152272) in frame into pEYFP-N1 (BD Biosciences Clontech). All PCR products were verified by sequencing. The CHMP2B-GFP expression plasmid was from Chris M. Sanderson (University of Liverpool). pcDNA3-myc-ALIX was provided by Harald Stenmark (University of Oslo). Sequences encoding the ALIX-Bro1 (amino acids [aa] 1 to 358), -Bro1/V (aa 1 to 702), and -V (aa 362 to 702) domains were amplified by PCR and cloned into pEGFP-C3 (BD Biosciences Clontech). All PCR products were verified by sequencing. pCR3.1-YFP-ALIX177-868, pCR3.1-GFP-TSG1011-156, and pCR3.1-YFP-VPS4EQ plasmids were provided by Juan Martin-Serrano (King's College London). The pcDNA3-UL36 plasmid was previously described (8). The plasmid encoding full-length TSG101-CFP was from Walther M. Mothes (Yale University). The plasmids encoding yellow fluorescent protein (YFP)-tagged HIV-1 Gag and Gag-ΔPTAP were constructed by cloning the Gag and GagΔPTAP sequences from GFP-tagged constructs provided by Wesley I. Sundquist (11) into a YFP vector and were provided by Walther M. Mothes (Yale University). Empty pE[G/Y]FP and pcDNA3.1 plasmids were obtained from BD Biosciences Clontech and Invitrogen, respectively.
Antibodies.
For Western blot analysis the following primary antibodies (identifier, dilution) were used: anti-ALIX (UT325, 1:1,000), anti-CHMP1B (UT592, 1:250), anti-VPS4A (UT289, 1:500), and anti-VPS4B (UT292, 1:500) were from Wesley I. Sundquist (University of Utah); anti-gD (LP14, 1:50) and anti-VP16 (LP1, 1:50) were from Tony Minson (University of Cambridge); anti-gB (R69, 1:5,000) was from Gary Cohen (University of Pennsylvania); anti-CHMP6 (1:1,000) was from J. Paul Luzio (Cambridge Institute for Medical Research); anti-actin (AC-40, 1:2,000) was from Sigma; anti-calreticulin (ab22683, 1:2,000), anti-CHMP1A (ab36679, 1:1,000), anti-CHMP2B (ab33174, 1:200), anti-EEA1 (ab2900, 1:1,000), anti-TSG101 (ab83, 1:500), and anti-VP5 (ab6508, 1:500) were from Abcam; anti-GFP (JL-8, 1:5,000) was from Clontech; anti-LAMP1 (H4A3, 1:1,000) from the Developmental Studies Hybridoma Bank, University of Iowa; and anti-tubulin (MCA77G, 1:1,000) was from AbD Serotec.
HIV-1 VLP production assay.
293T cells were seeded at 3 × 105 cells per well in six well dishes. Cells were cotransfected with 500 ng of pGag-YFP or pGagΔPTAP-YFP, together with 500 ng of plasmid DNA encoding either YFP alone, YFP-VPS4EQ, or CHMP-YFP/GFP gene products. Cell culture media were harvested 24 h posttransfection and cleared of cellular debris by two consecutive centrifugation steps at 5,000 × g for 7 min at 4°C. Virus-like particles (VLPs) in these supernatants were pelleted by centrifugation at 30,000 rpm at 4°C using a TLA-55 rotor for 60 min, and VLP pellets were resuspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. The cell samples were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris [pH 7.9], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate) supplemented with protease inhibitor cocktail (Roche) and incubated on ice for 20 min. Lysates were clarified by centrifugation at 17,000 × g for 15 min at 4°C. VLP and cell samples were analyzed by Western blotting with anti-GFP. Western blots were scanned and intensities of protein bands corresponding to released VLPs were quantified by using ImageJ (http://rsb.info.nih.gov/ij/).
Complementation assay.
COS-7 cells were seeded at densities of 2 × 105 cells per well in six-well dishes. Cells were transfected with a total of 1 μg of plasmid DNA per well comprising of 500 ng of pcDNA3-UL36 and various amounts of plasmid encoding the various dominant-negative ESCRT proteins or pEYFP-N1/pcDNA3 as a control. Transfections were performed by using FuGENE 6 (Roche) according to the manufacturer's instructions. At 24 h posttransfection the cells were infected with HSV-1 KΔUL36 at 5 PFU/cell, and unabsorbed virus was inactivated by treatment with acid wash (40 mM citric acid, 135 mM NaCl, 10 mM KCl [pH 3.0]) for 1 min at room temperature. After three washes with phosphate-buffered saline (PBS), the cells were incubated in complete growth medium. Samples were harvested at 16 to 18 h postinfection. Cell-associated virus was liberated by sonication for 20 s at 39% amplitude. Infectious virus titers were determined by plaque assays on HS30 cells. Cell samples were harvested for Western blot analysis as described above.
siRNA treatment.
HaCaT cells were seeded at a density of 2 × 106 cells per 10-cm dish in antibiotic-free media. Cells were transfected twice with 200 nM small interfering RNA (siRNA) in Opti-MEM I (Gibco) using Oligofectamine Reagent (Invitrogen) according to the manufacturer's instructions. TSG101 and ALIX siRNA-treated cells were infected 3 and 4 days, respectively, after the first siRNA transfection.
HeLa cells were seeded at a density of 2.5 × 105 cells per well of a six-well plate in antibiotic-free medium. The cells were transfected twice with 100 nM total siRNA in Opti-MEM I (Gibco) using DharmaFECT 1 (Dharmacon) according to the manufacturer's instructions. Cells were treated only with control or ALIX siRNA for the first transfection and incubated for 48 h. In the second transfection, ALIX siRNA-treated cells were transfected with either ALIX siRNA for the ALIX only depletion or with a 1:1 mixture of ALIX and TSG101 siRNA. For the TSG101-only depletion, control siRNA treated cells were transfected with TSG101 siRNA in the second transfection. Two days after the second siRNA transfection HaCaT and HeLa cells were infected with HSV-1 strain SC16 at 5 PFU/cell at 37°C for 1 h and acid washed as described above. Samples were harvested at various time points after infection, and cell-associated virus was liberated by sonication for 20 s at 39% amplitude. Infectious titers were determined by plaque assay on Vero cells. Cell samples were harvested for Western blot analysis as described above.
The following siRNAs were used and obtained from Dharmacon: the negative control (siNEG; catalog number D-001206-10), the previously published siRNAs targeting ALIX with the sense sequence 5′-GCC GCU GGU GAA GUU CAU CTT (2) and TSG101 with the sense sequence 5′-CCU CCA GUC UUC UCU CGU CTT (11).
Immunofluorescence.
COS-7 cells were seeded on glass coverslips at a density of 2.5 × 105 cells per well per well in six-well dishes. One day later, cells were cotransfected with 500 ng of pcDNA3-UL36, together with 500 ng of plasmid encoding either YFP-tagged CHMP or unfused YFP using FuGENE 6 (Roche) according to the manufacturer's instructions. At 24 h posttransfection, cells were infected with HSV-1 KΔUL36 at 5 PFU/cell and treated as described for the complementation assay above. At 15 h postinfection cells were fixed in 4% paraformaldehyde in PBS and then permeabilized in IF-Wash buffer (PBS containing 0.1% [vol/vol] Triton X-100 and 1% [vol/vol] fetal calf serum). Cells were incubated with the monoclonal anti-VP5 antibody (LP12, from Tony Minson, University of Cambridge) diluted in IF-Wash for 1 h at room temperature. Samples were washed twice with IF-Wash buffer and incubated with Alexa Fluor 568-conjugated goat anti-mouse immunoglobulin G for 1 h (1:1,000; Molecular Probes). The samples were washed twice with IF-Wash, once with PBS, rinsed in water, and mounted on slides in ProLong Gold antifade reagent with DAPI (4′,6′-diamidino-2-phenylindole; Invitrogen). Images were acquired using the 60× oil objective of an Olympus IX81 inverted wide field microscope and processed with Image-Pro 6.3 (MediaCybernetics) and Photoshop (Adobe).
Analysis of released supernatant virus.
COS-7 cells were seeded at 106 cells per 10-cm dish. Cells were either left untreated or transfected with 6 μg of plasmid encoding the UL36 gene product, together with 6 μg of either empty EYFP-N1 or CHMP1A-YFP expression constructs. Transfected and untreated cells were infected with HSV-1 KΔUL36 at 5 PFU/cell as described above. At 24 h postinfection supernatants of two 10-cm dishes per condition were collected and cleared from cellular debris by two consecutive low speed centrifugations (3,500 rpm, 10 min, 4°C; Beckman GH-3.8). Virus particles were pelleted at 20,000 rpm for 60 min at 4°C (Beckman SW 40 Ti) and resuspended in 55 μl of PBS per condition. Then, 20 μl of the concentrated samples was incubated in the presence or absence of 200 μg of trypsin (Worthington Biochemical Corp.)/ml at 37°C for 1 h. The reaction was stopped by adding a 5× final concentration of a protease inhibitor cocktail (Roche) for 10 min at room temperature. One-half of the total sample was loaded per lane for separation by SDS-PAGE and Western blot analysis. Cell samples were harvested for Western blot analysis as described above.
Virus gradient purification.
HaCaT cells growing in 850-cm2 roller bottles were infected either with HSV-1 strain SC16 or StH2 at 0.1 PFU/cell or with medium without virus (mock). Cell culture medium was harvested 48 h after infection, and cellular debris was pelleted by two subsequent low-speed centrifugations (2,900 rpm, 10 min, 4°C; Beckman GH-3.8). Virus particles were pelleted at 18,000 rpm for 90 min at 4°C (Beckman type 19 rotor). Virus and mock pellets were resuspended in 3 ml of PBS containing 1% fetal calf serum and layered onto a 30-ml 5 to 15% Ficoll (in PBS) continuous gradient. After centrifugation at 12,000 rpm for 90 min at 4°C (Beckman SW 32 Ti), a light scattering band of HSV-1 particles of each strain was collected by side puncture and aspiration into a syringe. The same volume of mock sample at the same level in the gradient as the HSV-1 particles was harvested. Samples were diluted in sterile PBS and subsequently pelleted at 20,000 rpm for 90 min at 4°C (Beckman SW 32 Ti). The virus pellets were resuspended in sterile PBS and divided into aliquots. The infectious titer was determined by plaque assay on Vero cells before samples were analyzed by Western blotting. The cell samples were harvested for Western blot analysis as described above.
Trypsin-Triton X-100 treatment.
Gradient-purified StH2 (8 × 109 PFU/ml) was incubated in the presence or absence of 1% Triton X-100 (BDH Chemical, Ltd.) and 200 μg of trypsin/ml at 37°C for 1 h. The reaction was stopped by adding a 4× final concentration of protease inhibitor cocktail (Roche) for 10 min at room temperature. The samples were separated by SDS-PAGE and analyzed by immunoblotting.
RESULTS
Overexpression of dominant-negative constructs of TSG101 and ALIX does not interfere with HSV-1 production.
It has been previously shown that overexpression of a dominant-negative VPS4 construct specifically blocks secondary envelopment of HSV-1, suggesting a role of further ESCRT proteins in HSV-1 assembly (3, 8). Many enveloped RNA viruses recruit the ESCRT machinery through an interaction with TSG101 or ALIX. The TSG101 binding tetrapeptide P[T/S]AP motif and the ALIX binding YPXnL (X = any residues; n = 1 to 3) motif can be found in several structural HSV-1 proteins (Table 1), which led us to ask whether dominant-negative TSG101 or ALIX constructs could interfere with the production of infectious HSV-1.
In order to study virus growth only in cells expressing the dominant-negative protein of interest, a complementation assay was used. This assay is based on the ability to complement the growth defect of a HSV-1 virus lacking the essential UL36 gene (KΔUL36) in trans. COS-7 cells were cotransfected with an UL36 expression construct together with a plasmid encoding the protein of interest. Subsequently, cells were infected with HSV-1 KΔUL36 (9). This resulted in productive virus replication only in cells that provided the UL36 gene product in trans and of which the vast majority would also be expressing the protein of interest. The infectious titers were determined by plaque assay on a complementing cell line (HS30) that stably provides the UL36 gene product (9). The background level of this assay is demonstrated by the mock-transfected sample which did not provide the UL36 gene product (Fig. 1B).
FIG. 1.

Production of infectious HSV-1 is not inhibited by dominant-negative ALIX or TSG101 fragments. (A) Schematic representation of TSG101 and ALIX constructs used. ALIX-V or ALIX(177-868) and TSG101(1-156) have been previously shown to block YPXnL- and PTAP-dependent retrovirus budding, respectively. (B) COS-7 cells were cotransfected with a plasmid expressing the essential HSV-1 UL36 gene product, together with a plasmid encoding the protein of interest, an empty pcDNA3, or a concentration range of a YFP-encoding plasmid. Mock-treated cells (mock) did not receive any plasmid DNA. Transfected cells were infected with HSV-1 lacking the UL36 gene (KΔUL36), which will only produce infectious virions in UL36-expressing cells. Infected cells were scraped and sonicated to release infectious progeny virus and HSV-1 titers were determined by plaque assay on a UL36 complementing cell line (HS30). Bars represent the mean PFU per ml, and error bars indicate the standard errors of the mean of triplicate samples. (C) Protein samples collected from each condition were analyzed by Western blotting to demonstrate the expression levels of GFP, YFP, and myc-tagged proteins, as well as the viral tegument protein VP16.
An empty pcDNA3 plasmid or a GFP-encoding plasmid cotransfected with the UL36 expression plasmid served as negative controls indicating conditions of no inhibition of virus growth. Although the expression of unfused GFP served as a negative control, a small reduction in HSV-1 titers in a dose-dependent manner was noted. The striking reduction in virus titers caused by the overexpression of dominant-negative VPS4EQ compared to the GFP control served as a positive control for a block of HSV-1 production (8).
The overexpression of full-length myc-tagged ALIX did not affect virus titers compared to the pcDNA3 control. To further investigate of the role of ALIX in HSV-1 production we sought to determine whether the overexpression of various GFP-tagged ALIX fragments affects HSV-1 titers. The ALIX protein can be subdivided into three domains: the Bro1 domain (aa 1 to 358) that interacts with CHMP4, the V domain (aa 362 to 702) that interacts with YPXnL viral late-domain motifs, and the proline-rich region (PRR; aa 703 to 868) (10, 31) (Fig. 1A). Previous studies have demonstrated that expression of the ALIX V domain or expression of a N-terminally truncated ALIX lacking the first half of the Bro1 domain (ALIX177-868) acts in a dominant-negative fashion and blocks ALIX-dependent virus release (23, 27). The overexpression of the dominant-negative V domain or the N-terminally truncated ALIX177-868 showed little effect on HSV-1 titers. In addition, overexpression of the ALIX-Bro1 domain or a construct consisting of both the Bro1 and V domains (ALIX-Bro1/V) exhibited little effect on production of infectious HSV-1 (Fig. 1B). HIV-1 budding is sensitive to the overexpression of either full-length TSG101 or an N-terminal PTAP binding fragment of TSG101 (TSG1011-156) (12, 13, 25). We therefore sought to determine whether the overexpression of these two TSG101 constructs could interfere with HSV-1 production. As shown in Fig. 1B, neither overexpression of full-length TSG101 nor of its N-terminal PTAP binding fragment affected HSV-1 titers compared to unfused GFP. All of the ALIX constructs and the dominant-negative GFP-TSG1011-156 were expressed at levels similar to those of GFP and YFP-VPS4EQ, and all of the samples demonstrated a comparable level of expression of the late HSV-1 protein, VP16, suggesting an equivalent level of infection by HSV-1 in all conditions (Fig. 1C).
Depletion of endogenous TSG101 and ALIX has no effect on HSV-1 growth.
Although overexpression of certain TSG101 and ALIX subdomains act in a dominant-negative fashion to block retroviral budding, presumably by masking viral late-domain motifs from endogenous ESCRT proteins, it is possible that expression levels might not be high enough to saturate potential HSV-1 L domains. Therefore, as an alternative approach siRNAs were used to deplete endogenous TSG101 and ALIX expression levels.
Cells were transfected with siRNAs targeting either TSG101 or ALIX. Subsequently, the siRNA and control treated cells were infected with HSV-1, and virus growth was monitored over time (Fig. 2A and B). Although the protein expression of TSG101 and ALIX was significantly reduced compared to control lysates, virus growth remained unaffected.
FIG. 2.

ALIX and TSG101 depletion does not affect growth of HSV-1. HaCaT (A and B) or HeLa (C) cells were transfected with specific siRNA in order to deplete the endogenous ALIX and TSG101 proteins. The siRNA- and mock-treated cells were subsequently infected with HSV-1 strain SC16 at 5 PFU/cell. Infected cells were scraped and sonicated at various time points (A and B) or 16 h postinfection (C) to release infectious progeny virus. Infectious HSV-1 titers were determined for each sample by plaque assay on Vero cells. The data represent mean PFU per ml, and error bars indicate the standard errors of the mean of triplicate samples. Protein samples from each siRNA-treated and untreated (mock) condition were analyzed by Western blotting to demonstrate the expression levels of endogenous TSG101 and ALIX at the time of infection for panels A and C or at 25 h postinfection for panel B (right-hand panels).
Furthermore, we addressed a potential redundancy of TSG101 and ALIX in mediating HSV-1 release by simultaneously depleting the expression of both proteins. Although protein levels of TSG101 and ALIX were reduced by >90% in case of the simultaneous depletion of both proteins [si(TSG101+ALIX), Fig. 2C], neither the simultaneous knockdown nor the individual depletion of those proteins had an effect on the production of infectious HSV-1 (Fig. 2C).
Taken together, these data demonstrate that the production of HSV-1 is independent from TSG101 and ALIX expression.
Dominant-negative ESCRT-III proteins block production of infectious HSV-1.
The ESCRT-III complex, consisting of at least 11 individual CHMPs in humans, plays a central role in ESCRT-mediated budding events. To address whether a functional ESCRT-III complex mediates the production of infectious HSV-1, we generated a set of dominant-negative CHMPs by fusing a YFP tag to their C termini (16). These CHMP-YFP constructs blocked the release of HIV-1 VLPs, in agreement with previous reports (15, 23, 34), although CHMP6 and CHMP7 showed a weaker inhibition of HIV-1 particle release than the other dominant-negative CHMPs (Fig. 3A). The expression level of HIV-1 Gag was equivalent in the cells under all conditions examined (data not shown).
FIG. 3.

Dominant-negative ESCRT-III proteins potently inhibit the production of infectious HSV-1. (A) HIV-1 Gag VLP release assays were performed to test the ability of dominant-negative CHMPs to block an ESCRT-mediated virus budding process. 293T cells were cotransfected with a plasmid expressing YFP-tagged HIV-1 Gag together with a plasmid encoding YFP/GFP-tagged CHMPs, YFP-VPS4EQ or YFP. Cells cotransfected with YFP-Gag-ΔPTAP- and YFP-expressing plasmids served as a negative control since little VLP release is expected in the absence of the PTAP late-domain motif. Cell culture medium samples from three parallel samples of each condition were harvested 24 h posttransfection and clarified by low-speed centrifugation, and then VLPs were harvested by high-speed centrifugation (30,000 rpm, 4°C, 60 min; TLA-55 rotor). Samples were analyzed by Western blotting, and the amount of HIV-1 Gag present in each sample was quantified by densitometry analysis of scanned blots using ImageJ. The data were normalized to the mean level of YFP-tagged HIV-1 Gag released in the presence of YFP. Bars represent the mean VLP release, and error bars indicate the standard errors of the mean of triplicate samples. (B) COS-7 cells were cotransfected with a plasmid expressing the essential HSV-1 UL36 gene product, together with a plasmid encoding YFP/GFP-tagged CHMPs, YFP-VPS4EQ, or YFP. Mock-treated cells (mock) did not receive any plasmid DNA. Transfected cells were infected with HSV-1 lacking the UL36 gene (KΔUL36), which will only produce infectious virions in UL36 expressing cells. Infected cells were scraped and sonicated to release infectious progeny virus and HSV-1 titers were determined by plaque assay on a UL36 complementing cell line (HS30). Bars represent mean PFU per ml, and error bars indicate the standard errors of the mean of triplicate samples. (C) Protein samples collected from each condition were analyzed by Western blotting to demonstrate the expression levels of the YFP-tagged proteins, as well as the viral tegument protein VP16.
As described above, the UL36 complementation assay was used to assess the role of CHMPs in HSV-1 production. The expression of all dominant-negative CHMPs caused a strong inhibition of viral growth, with the majority of CHMP-YFP fusion proteins reducing HSV-1 titers to almost background levels (Fig. 3B). Both CHMP6-YFP and CHMP7-YFP showed a smaller decrease in HSV-1 titer, in agreement with the weaker inhibition of these two dominant-negative CHMPs observed for HIV-1 VLP release (Fig. 3A). Interestingly, CHMP4A-YFP and CHMP4C-YFP also showed a smaller decrease in HSV-1 titer. Recently, careful analysis of the CHMP4 isoforms demonstrated that dominant-negative CHMP4B had a greater effect on HIV-1 release than CHMP4A or CHMP4C (4). Our data suggest that the same may be true for HSV-1 assembly. The expression of the YFP-tagged proteins was confirmed by Western blot analysis (Fig. 3C). We also monitored expression levels of the HSV-1 tegument protein VP16 expression as an indication for virus replication (Fig. 3B). Overall, these data suggest that a functional ESCRT-III complex is required for productions of infectious HSV-1.
Entry is not affected by the overexpression of dominant-negative ESCRT-III proteins.
In order to determine whether the expression of dominant-negative ESCRT-III proteins blocked HSV-1 production by affecting virus entry, an immunofluorescence-based entry assay was performed. Transfected COS-7 cells expressing the UL36 gene product and either CHMP-YFP or YFP were infected with HSV-1 KΔUL36 at 3 PFU/cell and fixed 15 h posttransfection. Virus infection was visualized by immunofluorescence staining with a monoclonal antibody recognizing the HSV-1 major capsid protein (VP5). Fluorescence microscopy was used to score for infection. We scored cells that simultaneously showed a fluorescent signal in the YFP channel and clear intranuclear assembly sites as infected (Fig. 4A). The data are presented as the proportion of transfected cells that are infected (Fig. 4B). A total of 71% of YFP-expressing cells were infected, whereas 75% of CHMP1A-expressing cells and 83% of CHMP6-expressing cells were infected. Therefore, these data suggest that overexpression of CHMP-YFP fusion proteins did not inhibit HSV-1 entry or expression of viral late genes, such as UL19 (VP5).
FIG. 4.

Overexpression of dominant-negative ESCRT-III proteins does not affect entry of HSV-1. COS-7 cells were either left untreated or cotransfected with a plasmid expressing UL36 together with a plasmid expressing CHMP1A-YFP, CHMP6-YFP, or YFP. Cells were infected at 24 h posttransfection with HSV-1 KΔUL36 at 5 PFU/cell. Cells were fixed 15 h postinfection, permeabilized and incubated with a monoclonal antibody specific for the HSV-1 major capsid protein (VP5), followed by an Alexa Fluor 568-conjugated anti-mouse secondary antibody. (A) Epifluorescence image of an infected CHMP1A-YFP-expressing cell. CHMP1A-YFP-positive cytoplasmic compartments can be observed (top left-hand panel) in a cell with high levels of VP5 in the nucleus (bottom left-hand panel), indicating that HSV-1 can enter cells and initiate viral gene expression in the presence of a dominant-negative CHMP. Right-hand panel shows merged image. Scale bar, 20 μm. (B) Cells were counted and scored for the simultaneous expression of YFP-tagged proteins and a characteristic nuclear staining of the HSV-1 major capsid protein (VP5), as an indication of virus entry and subsequent viral gene expression. Bars indicate the percentage of YFP or CHMP-YFP-expressing cells that were also VP5 positive from a total of 200 cells or the percentage of all cells that were VP5 positive from a total of 400 cells in the untransfected sample.
HSV-1 particle release is blocked by dominant-negative CHMP expression.
We have previously demonstrated that the inhibition of ESCRT function through expression of dominant-negative VPS4 blocks the final envelopment of HSV-1 in the cytoplasm, leading to an almost complete loss of mature enveloped virus particles (8). However, we cannot formally rule out that the reduction in HSV-1 titer observed in the presence of dominant-negative CHMPs is due to the release of noninfectious particles rather than a block in virus assembly. To address this question, COS-7 cells were transfected with control plasmids or with the UL36 expression plasmid together with either CHMP1A-YFP dominant-negative or YFP control, infected with HSV-1 KΔUL36 at 5 PFU/cell, and the tissue culture medium containing released virus particles was collected. HSV-1 particles were concentrated by ultracentrifugation, and the infectious titer present in the samples was determined by plaque assay on the UL36 complementing cell line, HS30 (Fig. 5, top panel). In agreement with the observed reduction in total HSV-1 titer in the presence of dominant-negative CHMPs (Fig. 3), the HSV-1 titer present in the supernatant fraction was reduced by ∼50-fold in the presence of CHMP1A-YFP. The concentrated virus samples were left untreated or treated with trypsin in order to distinguish between enveloped and nonenveloped capsids. These samples were analyzed by Western blotting to determine the presence of viral capsid (VP5), tegument (VP16), and envelope (gB) proteins. The pelleted supernatant fraction from HSV-1-infected cells would be expected to contain a mixture of mature virions and capsidless light-particles, as well as potential contamination of free capsids from lysed cells. Tegument proteins, such as VP16, that are packaged into virions and light particles, or capsid proteins, such as VP5, contained within mature virions would be protected from protease digestion by a lipid bilayer. Envelope glycoproteins such as gB in either virions or light particles would have their extracellular domains exposed and would therefore be sensitive to protease digestion. Any free capsids would also be sensitive to protease treatment. As can be seen in Fig. 5, the untreated released virus fraction from the CHMP1A-YFP sample showed a marked reduction in the level of tegument (VP16) and envelope (gB) compared to the YFP control, suggesting the reduction in HSV-1 titer is indeed due to a defect in virion formation rather than the formation of noninfectious virions. Surprisingly, we observed a similar level of capsid (VP5) in all samples in the absence of trypsin. However, the only sample exhibiting VP5 that was resistant to trypsin treatment, and thus membrane enclosed, was in the UL36+YFP control. These data suggest that in this assay there is a large amount of free capsids present in the supernatant, but enveloped, capsid-containing virions are only produced in the presence of UL36 and that this virion formation is strongly inhibited by dominant-negative CHMP1A. Interestingly, these data also suggest that the formation of light particles is independent of the UL36 gene product (VP1/2) due to the presence of trypsin protected VP16 in the absence of UL36 and that this light particle formation is also inhibited by dominant-negative CHMP1A; if light particle formation was not effected by CHMP1A-YFP, then an equivalent trypsin-insensitive VP16 signal would have been expected in this sample. Overall, these data strongly support the proposal that the reduction in HSV-1 titer observed when ESCRT-III function is inhibited is due to a block of HSV-1 assembly.
FIG. 5.
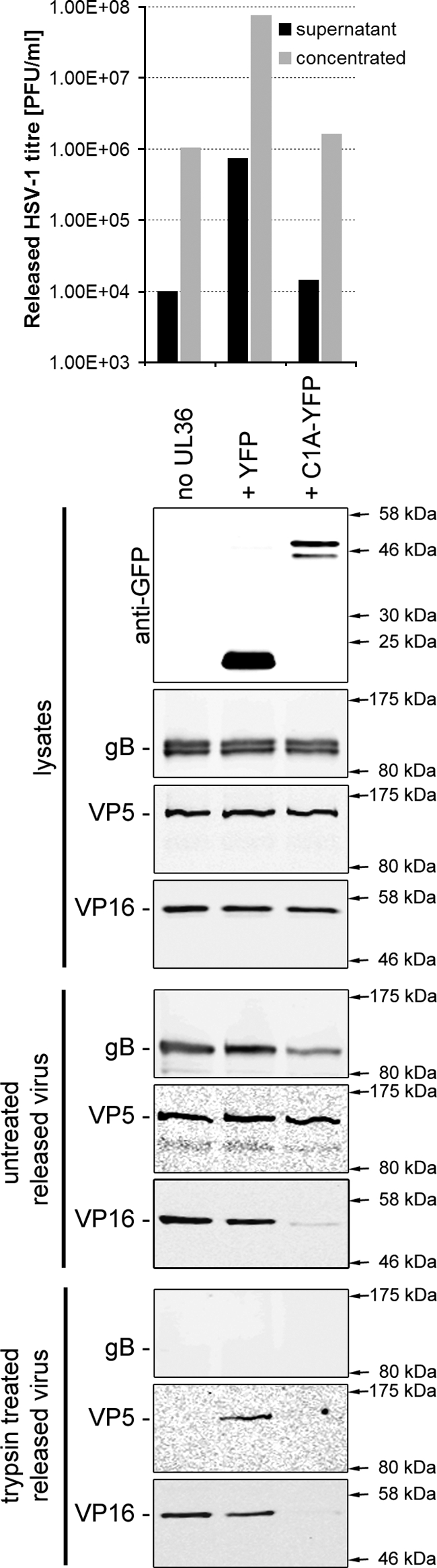
Dominant negative CHMP1A blocks the release of HSV-1 particles from infected cells. COS-7 cells were left untreated or cotransfected with a plasmid expressing UL36, together with a plasmid expressing YFP or CHMP1A-YFP. Cells were infected 24 h posttransfection with HSV-1 KΔUL36 at 5 PFU/cell. At 24 h postinfection cell culture medium samples were collected and clarified by low-speed centrifugation. Released (supernatant) HSV-1 particles were harvested by high-speed centrifugation (20,000 rpm, 4°C, 60 min; SW 40Ti rotor) and resuspended in a small volume of PBS to concentrate the released HSV-1 fraction. The infectious HSV-1 titers in the superna-tant and concentrated HSV-1 samples were determined by plaque assay on a UL36 complementing cell line (HS30; upper panel). Concentrated HSV-1 particles were treated with PBS (untreated) or 200 μg of trypsin/ml at 37°C for 1 h. The reactions were stopped by the addition of protease inhibitor cocktail (Roche). Protein samples were harvested from infected cells and samples analyzed by Western blotting for the presence of YFP-tagged proteins in cell lysates and HSV-1 envelope (gB), capsid (VP5), and tegument (VP16) proteins in all samples (lower panels).
ESCRT proteins are present in purified virus preparations.
It has previously been reported that certain ESCRT components, such as TSG101, VPS28, ALIX, and VPS4B, are specifically packaged into HIV-1 virions (6, 34). In order to test whether ESCRT proteins are also incorporated into HSV-1 particles, virions were purified and analyzed by Western blotting. HaCaT cells were either infected with two different HSV-1 strains, SC16 and StH2, or mock infected as a control for potential contaminants such as exosomes and cellular debris. Extracellular growth media were collected 48 h postinfection and clarified by two subsequent low-speed centrifugations. The virus was then concentrated by centrifugation and purified on a 5 to 15% Ficoll gradient. A clearly visible light scattering band of virus particles was collected by side puncture. A fraction of the same volume from the same sedimentation level was harvested from the mock-infected sample as a control. After a further concentration step by centrifugation, titers of the samples were determined. The same number of virus particles as estimated by the PFU, or the same volume for the mock control sample, was separated by SDS-PAGE and subjected to Western blot analysis. The absence of a signal for the viral proteins VP16 and gD for the samples from mock-infected cells reflected their lack of any infectivity as expected (Fig. 6). We analyzed our virus preparations for various cellular markers to assess the quality of our purifications. Infected and uninfected cellular lysates were used to test for potential cross-reactivity of the antibodies with viral proteins. Neither the early endosomal marker EEA1, the late endosomal/lysosomal marker LAMP1, the cytoskeletal proteins actin/tubulin, nor the endoplasmic reticulum marker calreticulin gave significant signals for the gradient purified samples, whereas they were readily detectable in the lysates. After we assessed the quality of our virus preparations, we sought to determine whether ESCRT proteins can be detected in these gradient-purified virus samples.
FIG. 6.

ESCRT proteins are associated with purified HSV-1 virions. HaCaT cells were grown in roller bottles until confluent and then infected either with the HSV-1 strains SC16 or StH2 at 0.1 PFU/cell or with medium without virus (mock). Cell culture medium was harvested at 48 h postinfection and clarified of cell debris by two subsequent low-speed centrifugation steps (2,900 rpm, 10 min, 4°C; Beckman GH-3.8). After concentration by high-speed centrifugation (18,000 rpm, 4°C, 90 min; type 19 rotor), virus particles were purified on a 5 to 15% Ficoll continuous gradient (12,000 rpm, 4°C. 90 min; SW 32Ti rotor). The appropriate density light scattering band corresponding to mature, infectious HSV-1 particles was collected by side puncture. The same volume of mock sample at the same level in the gradient as the light scattering band was harvested. After determination of the infectious virus titers on Vero cells, 1.5 × 108 PFU and 5 × 107 PFU of each virus preparation and an identical volume of the mock sample were analyzed by Western blotting with specific antibodies to markers of cellular compartments and ESCRT proteins. Then, 1.5 × 107 PFU and 5 × 106 PFU of each virus preparation and an identical volume of the mock sample were analyzed by Western blotting with specific antibodies to the viral proteins VP16 and gD.
Although TSG101 and ALIX could be detected in virus preparations, their signals were either very faint or absent in different independent experiments. In contrast, we repeatedly detected the presence of all ESCRT-III proteins that could be tested, such as CHMP1A, CHMP1B, and CHMP2B, as well as the ATPases VPS4A and VPS4B (Fig. 6).
The ESCRT proteins are protected from trypsin digestion by a lipid envelope.
To determine whether the ESCRT-III proteins that copurified with HSV-1 were physically incorporated into the virus particle, we assessed whether these proteins were resistant to trypsin treatment. Treatment of purified HSV-1 with trypsin resulted in a complete degradation of the exposed HSV-1 membrane protein gD, whereas the internal tegument protein VP16 was not affected (Fig. 7). A simultaneous treatment with trypsin in the presence of the detergent TritonX-100 resulted in a complete degradation of VP16 due to the disruption of the virus envelope. VP16 and gD levels were not affected in untreated virions or TritonX-100-treated purified virions. The same samples were analyzed for the ESCRT-III proteins CHMP2B and CHMP6 that showed a similar pattern as the internal virus component VP16 (Fig. 7). In contrast to the virus preparations analyzed in Fig. 6, ALIX could be detected as a faint signal but TSG101 was not detectable in purified virus from this preparation. Finally, the ATPase VPS4A showed the same signal pattern as the analyzed ESCRT-III components and the internal tegument protein VP16. These data suggest that the tested ESCRT-III components and the ATPase VPS4 are internal components of purified HSV-1 particles.
FIG. 7.

ESCRT proteins are present in enveloped HSV-1 virions. HSV-1 stain StH2 that had been purified on a 5 to 15% Ficoll continuous gradient was incubated in the presence or absence of 1% Triton X-100, together with the presence or absence of trypsin at 37°C for 1 h. The reaction was stopped by adding protease inhibitor cocktail (Roche). Any proteins incorporated into the virus particle would be protected from protease treatment by the virus envelope in the absence of detergent, and proteins that were not incorporated into the virus particle and so present outside the virus envelope would be sensitive to protease treatment irrespective of the presence of detergent. All samples were analyzed by Western blotting for the presence of the envelope protein gD, the tegument protein VP16, and cellular ESCRT proteins. Totals of 4 × 107 PFU and 4 × 106 PFU per lane, respectively, were loaded for the detection of cellular proteins and viral proteins.
It is interesting that all of the ESCRT proteins showing a reproducible incorporation into HSV-1 particles, strongly inhibit HSV-1 production when expressed as dominant-negative proteins.
Overall, our data show that efficient HSV-1 production requires a functional ESCRT-III complex in addition to enzymatically active VPS4. In contrast to small RNA viruses such as HIV-1, HSV-1 can efficiently produce infectious particles in the absence of TSG101 and/or ALIX. The specific packaging of ESCRT-III components and VPS4A/B into HSV-1 virions further suggests their involvement in the final envelopment step of HSV-1.
DISCUSSION
In the present study, we have shown that a functional ESCRT-III complex is required for the efficient production of infectious HSV-1. The involvement of the ESCRT-III complex was suggested by previous reports that demonstrated the inhibitory effects of ATPase-deficient VPS4 and dominant-negative CHMP3 (VPS24) on HSV-1 release (3, 8). Here, we have utilized a complete set of human CHMPs to demonstrate that overexpression of any dominant-negative ESCRT-III protein strongly inhibits HSV-1 replication. Some interesting differences were observed in the ability of the different dominant-negative CHMPs to inhibit HSV-1 replication, with the weakest inhibition observed with dominant-negative CHMP4A, CHMP4C, CHMP6, and CHMP7. The weaker inhibition in the presence of CHMP6 and CHMP7 dominant negatives parallels our observations that both of these dominant-negative ESCRT-III proteins have a weaker effect on the formation of HIV-1 VLPs. Furthermore, the weaker inhibition of HSV-1 replication by CHMP4A and CHMP4C correlates well with a recent study of the roles of the CHMP4 isoforms (4). In that study, the authors demonstrated that dominant-negative CHMP4B had a greater effect on HIV-1 budding than CHMP4A or C, whereas dominant-negative CHMP4C showed a greater inhibition of cytokinesis than CHMP4A or CHMP4B (4). Overall, these data suggest that HSV-1 and HIV-1 share similarities in their requirements for functional ESCRT-III proteins.
The reduction in infectious HSV-1 in the presence of dominant-negative CHMPs appears to be due to a specific block in the formation of enveloped virions, as shown by a complete absence of detectable enveloped HSV-1 capsids in the culture media of infected cells expressing a dominant-negative CHMP. These data demonstrate that inhibiting the function of the ESCRT-III complex is likely to be causing a block in the final cytoplasmic envelopment of HSV-1 rather than causing the formation of noninfectious virions. Interestingly, our data also suggest that the formation of HSV-1 light particles requires a functional ESCRT-III complex and that light particles can form in the absence of UL36 expression. This second observation may have implications for the mechanism of ESCRT recruitment by HSV-1; if the gene product of UL36, VP1/2, is not necessary for light particle budding, it seems unlikely that VP1/2 would be the major route of recruiting ESCRT proteins to HSV-1 assembly sites, despite the presence of several late-domain motifs in VP1/2 (Table 1). Which structural proteins of HSV-1 are directly responsible for recruiting ESCRT complexes is likely to be complicated due to the presence of numerous viral proteins in a mature HSV-1 particle and potential redundancy between them for recruiting ESCRT complexes. Such potential for a number of parallel mechanisms by which HSV-1 could recruit ESCRTs is highlighted by the fact that there are many potential late-domain motifs present in different HSV-1 proteins.
Interestingly, despite the presence of TSG101 and ALIX binding sites in several structural HSV-1 proteins (Table 1), both proteins are dispensable for HSV-1 release. Therefore, our data suggest that HSV-1 must use an alternative route to recruit the ESCRT machinery. A potential candidate would be via the recruitment of HECT ubiquitin ligases by a PPXY motif. In fact, the UL56 protein of HSV-2 has been recently shown to interact with the HECT ubiquitin ligase Nedd4. However, a deletion of UL56 had no effect on viral growth, indicating that the pUL56-Nedd4 interaction is not essential for HSV-2 assembly (32). Several other HSV-1 structural proteins contain PPXY motifs, although whether any of these motifs or any HECT ubiquitin ligases play a role in ESCRT recruitment to HSV-1 assembly sites is currently under investigation.
The ESCRT-III complex together with VPS4 are thought to be the driving force for membrane deformation and ultimately membrane fission. C-terminally truncated mutants of CHMP4A have been previously shown to assemble into filaments that form circular arrays associated with membrane deformation (14). Recent in vitro observations have shown that assembly of purified yeast ESCRT-III subunits induces inward invaginations and even membrane scission further supports the hypothesis that the ESCRT-III complex plays a central role in ESCRT-mediated budding processes (29, 36). It seems likely that HSV-1 would not absolutely rely on the ESCRT-III complex to deform the membrane during virus budding, since the network of interactions between the cytoplasmic tails of viral glycoproteins, tegument, and capsid proteins could support membrane deformation during secondary envelopment (7, 18, 21). It is more likely that ESCRT-III assembly, together with the ATPase VPS4, is required for the final membrane scission step of secondary envelopment. This notion is supported by the observation that HSV-1 assembly is inhibited at a very late stage in cells expressing dominant-negative VPS4, where particles can appear almost fully formed except for a small gap in the envelope (8).
In the present study, we also showed that ESCRT components are packaged into purified HSV-1 virions. In contrast to HIV-1, TSG101 and ALIX were only detectable at low levels and not consistently between different virus preparations. The presence of these proteins in viral particles, despite being dispensable for HSV-1 production, could be explained by an interaction with other ESCRT components that are specifically recruited to virus buds. In every virus preparation, we could easily detect all ESCRT-III components that could be analyzed, and both isoforms of the cellular ATPase VPS4 in purified virions. Considering that VPS4B has been shown to be packaged into HIV-1 particles, it is not surprising that ESCRT-III proteins that are not fully disassembled by VPS4 can be trapped in a budding virus particle (6, 34). A recently published proteomic characterization of purified extracellular HSV-1 virions indicated the presence of up to 49 distinct potential cellular proteins in mature virions (22). Among these cellular proteins was actin, which we did not detect under our experimental conditions but, to our surprise, no ESCRT proteins. It is unclear whether the membrane-associated ESCRT proteins were lost during sample preparation for the published proteomic characterization of HSV-1, since the authors did not detect certain HSV-1 glycoproteins that have previously been confirmed to be present in the viral envelope (22). It will be interesting to ascertain whether a complete set or just a subset of the ESCRT-III proteins are incorporated into mature HSV-1 particles as more reagents become available for specific detection of endogenous CHMPs. Such information will give us clues as to the core ESCRT machinery that is necessary and sufficient for the final membrane scission event during HSV-1 assembly.
In conclusion, our data provide evidence for a functional role of the ESCRT-III complex and the ATPase VPS4 in the production of infectious HSV-1 in a TSG101/ALIX-independent manner. It will be interesting to establish how HSV-1 recruits the cellular ESCRT machinery to the budding site, whereas the complexity of HSV-1 virions, together with a potential functional redundancy among viral structural proteins are challenges that have to be overcome.
Acknowledgments
We thank W. I. Sunquist (University of Utah), J. Martin-Serrano (King's College, London, United Kingdom), H. Stenmark, (University of Oslo), C. M. Sanderson (University of Liverpool), W. M. Mothes and N. Sherer (Yale University), J. P. Luzio (Cambridge Institute for Medical Research), P. Desai (Johns Hopkins University), and T. Minson (University of Cambridge) for their generous gifts of reagents. We also thank S. Bell for excellent technical assistance and H. Zenner for helpful discussions and critical reading of the manuscript.
This study was funded by the Royal Society (University Research Fellowship to CMC) and the Medical Research Council (grant no. G0700129). T.P. was supported by the Peter Wildy studentship of Gonville and Caius College, Cambridge, England.
Footnotes
Published ahead of print on 19 August 2009.
REFERENCES
- 1.Bieniasz, P. D. 2006. Late budding domains and host proteins in enveloped virus release. Virology 344:55-63. [DOI] [PubMed] [Google Scholar]
- 2.Cabezas, A., K. G. Bache, A. Brech, and H. Stenmark. 2005. Alix regulates cortical actin and the spatial distribution of endosomes. J. Cell Sci. 118:2625-2635. [DOI] [PubMed] [Google Scholar]
- 3.Calistri, A., P. Sette, C. Salata, E. Cancellotti, C. Forghieri, A. Comin, H. Gottlinger, G. Campadelli-Fiume, G. Palu, and C. Parolin. 2007. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 81:11468-11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlton, J. G., M. Agromayor, and J. Martin-Serrano. 2008. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc. Natl. Acad. Sci. USA 105:10541-10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlton, J. G., and J. Martin-Serrano. 2009. The ESCRT machinery: new functions in viral and cellular biology. Biochem. Soc. Trans. 37:195-199. [DOI] [PubMed] [Google Scholar]
- 6.Chertova, E., O. Chertov, L. V. Coren, J. D. Roser, C. M. Trubey, J. W. Bess, Jr., R. C. Sowder II, E. Barsov, B. L. Hood, R. J. Fisher, K. Nagashima, T. P. Conrads, T. D. Veenstra, J. D. Lifson, and D. E. Ott. 2006. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 80:9039-9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chi, J. H., C. A. Harley, A. Mukhopadhyay, and D. W. Wilson. 2005. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 86:253-261. [DOI] [PubMed] [Google Scholar]
- 8.Crump, C. M., C. Yates, and T. Minson. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 81:7380-7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Desai, P. J. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608-11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher, R. D., H. Y. Chung, Q. Zhai, H. Robinson, W. I. Sundquist, and C. P. Hill. 2007. Structural and biochemical studies of ALIX/AIP1 and its role in retrovirus budding. Cell 128:841-852. [DOI] [PubMed] [Google Scholar]
- 11.Garrus, J. E., U. K. von Schwedler, O. W. Pornillos, S. G. Morham, K. H. Zavitz, H. E. Wang, D. A. Wettstein, K. M. Stray, M. Cote, R. L. Rich, D. G. Myszka, and W. I. Sundquist. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55-65. [DOI] [PubMed] [Google Scholar]
- 12.Goff, A., L. S. Ehrlich, S. N. Cohen, and C. A. Carter. 2003. Tsg101 control of human immunodeficiency virus type 1 Gag trafficking and release. J. Virol. 77:9173-9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goila-Gaur, R., D. G. Demirov, J. M. Orenstein, A. Ono, and E. O. Freed. 2003. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J. Virol. 77:6507-6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanson, P. I., R. Roth, Y. Lin, and J. E. Heuser. 2008. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 180:389-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horii, M., H. Shibata, R. Kobayashi, K. Katoh, C. Yorikawa, J. Yasuda, and M. Maki. 2006. CHMP7, a novel ESCRT-III-related protein, associates with CHMP4b and functions in the endosomal sorting pathway. Biochem. J. 400:23-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howard, T. L., D. R. Stauffer, C. R. Degnin, and S. M. Hollenberg. 2001. CHMP1 functions as a member of a newly defined family of vesicle trafficking proteins. J. Cell Sci. 114:2395-2404. [DOI] [PubMed] [Google Scholar]
- 17.Hurley, J. H. 2008. ESCRT complexes and the biogenesis of multivesicular bodies. Curr. Opin. Cell Biol. 20:4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamen, D. E., S. T. Gross, M. E. Girvin, and D. W. Wilson. 2005. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 79:6134-6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lata, S., M. Roessle, J. Solomons, M. Jamin, H. G. Gottlinger, D. I. Svergun, and W. Weissenhorn. 2008. Structural basis for autoinhibition of ESCRT-III CHMP3. J. Mol. Biol. 378:818-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lata, S., G. Schoehn, A. Jain, R. Pires, J. Piehler, H. G. Gottlinger, and W. Weissenhorn. 2008. Helical structures of ESCRT-III are disassembled by VPS4. Science 321:1354-1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, J. H., V. Vittone, E. Diefenbach, A. L. Cunningham, and R. J. Diefenbach. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347-354. [DOI] [PubMed] [Google Scholar]
- 22.Loret, S., G. Guay, and R. Lippe. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605-8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-Serrano, J., A. Yarovoy, D. Perez-Caballero, and P. D. Bieniasz. 2003. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. USA 100:12414-12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin-Serrano, J., T. Zang, and P. D. Bieniasz. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313-1319. [DOI] [PubMed] [Google Scholar]
- 25.Martin-Serrano, J., T. Zang, and P. D. Bieniasz. 2003. Role of ESCRT-I in retroviral budding. J. Virol. 77:4794-4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mettenleiter, T. C., B. G. Klupp, and H. Granzow. 2006. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 9:423-429. [DOI] [PubMed] [Google Scholar]
- 27.Munshi, U. M., J. Kim, K. Nagashima, J. H. Hurley, and E. O. Freed. 2007. An Alix fragment potently inhibits HIV-1 budding: characterization of binding to retroviral YPXL late domains. J. Biol. Chem. 282:3847-3855. [DOI] [PubMed] [Google Scholar]
- 28.Piper, R. C., and D. J. Katzmann. 2007. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 23:519-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saksena, S., J. Wahlman, D. Teis, A. E. Johnson, and S. D. Emr. 2009. Functional reconstitution of ESCRT-III assembly and disassembly. Cell 136:97-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strack, B., A. Calistri, S. Craig, E. Popova, and H. G. Gottlinger. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689-699. [DOI] [PubMed] [Google Scholar]
- 31.Usami, Y., S. Popov, and H. G. Gottlinger. 2007. Potent rescue of human immunodeficiency virus type 1 late domain mutants by ALIX/AIP1 depends on its CHMP4 binding site. J. Virol. 81:6614-6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ushijima, Y., T. Koshizuka, F. Goshima, H. Kimura, and Y. Nishiyama. 2008. Herpes simplex virus type 2 UL56 interacts with the ubiquitin ligase Nedd4 and increases its ubiquitination. J. Virol. 82:5220-5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.VerPlank, L., F. Bouamr, T. J. LaGrassa, B. Agresta, A. Kikonyogo, J. Leis, and C. A. Carter. 2001. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 98:7724-7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Schwedler, U. K., M. Stuchell, B. Muller, D. M. Ward, H. Y. Chung, E. Morita, H. E. Wang, T. Davis, G. P. He, D. M. Cimbora, A. Scott, H. G. Krausslich, J. Kaplan, S. G. Morham, and W. I. Sundquist. 2003. The protein network of HIV budding. Cell 114:701-713. [DOI] [PubMed] [Google Scholar]
- 35.Williams, R. L., and S. Urbe. 2007. The emerging shape of the ESCRT machinery. Nat. Rev. Mol. Cell. Biol. 8:355-368. [DOI] [PubMed] [Google Scholar]
- 36.Wollert, T., C. Wunder, J. Lippincott-Schwartz, and J. H. Hurley. 2009. Membrane scission by the ESCRT-III complex. Nature 458:172-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zamborlini, A., Y. Usami, S. R. Radoshitzky, E. Popova, G. Palu, and H. Gottlinger. 2006. Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc. Natl. Acad. Sci. USA 103:19140-19145. [DOI] [PMC free article] [PubMed] [Google Scholar]