

Abstract
The binding of cyclophilin A (CypA) to the human immunodeficiency virus type 1 (HIV-1) capsid protein (CA protein) is required soon after virus entry into natural target cells. In Jurkat T lymphocytes, disrupting CypA-CA interaction either by cyclosporine (Cs) treatment or by alteration (e.g., P90A) of the CA inhibits HIV-1 infection. In HeLa cells, however, treatment with Cs or Cs analogues minimally inhibits the early phase of HIV-1 infection but selects for a Cs-dependent virus with a change (A92E) in CA. To understand these phenomena, we examined the effects of the P90A and A92E changes in the HIV-1 CA protein on the stability of capsid complexes assembled in vitro and on capsid disassembly in the cytosol of virus-exposed target cells. The A92E change impaired CA-CA interactions in vitro and decreased the amount of particulate capsids in the cytosol of HeLa target cells. Reducing the binding of CypA to the A92E mutant capsid, either by Cs treatment or by an additional P90A change in the CA protein, increased the amount of particulate capsids and viral infectivity in HeLa cells. In contrast, reduction of the binding of CypA to HIV-1 capsids in Jurkat T lymphocytes resulted in a decrease in the amount of particulate capsids and infectivity. Thus, depending on the capsid and the target cell, CypA-CA binding either stabilized or destabilized the capsid, indicating that CypA modulates HIV-1 capsid disassembly. In both cell types examined, decreased stability of the capsid was associated with a decrease in the efficiency of HIV-1 infection.
The human immunodeficiency virus type 1 (HIV-1) RNA genome is encapsulated in a conical capsid core, which is delivered into the target cell cytosol as a result of the virus entry process. The HIV-1 capsid is composed of approximately 1,500 capsid (CA) proteins (11). The CA proteins oligomerize into hexamers by virtue of interactions among the N-terminal domains, and additional interactions involving the C-terminal and N-terminal domains promote interhexamer interactions that allow the capsid structure to be assembled (24, 35, 44). After virus entry, the conical capsid core undergoes morphological changes in the target cell cytosol (40), and most of the capsid proteins are disassociated from the viral genome, a process termed uncoating (20). Although not well understood, proper uncoating appears to be essential for efficient HIV-1 infection. For example, changes in the HIV-1 CA protein have been suggested to affect capsid stability, thus impairing an early step in the virus life cycle between entry and the reverse transcription of the viral genome (21). As-yet-unidentified host cellular factors have been proposed to activate HIV-1 uncoating and induce reverse transcription in vitro (4, 21). Other host factors present in particular mammalian species interrupt retrovirus infection at the uncoating step. For example, the capsid-interacting factor TRIM5α has been shown to restrict retroviral infection by promoting rapid, premature disassembly of the capsid (51). Some primates express a TRIM5-cyclophilin A (CypA) fusion protein called TRIMCyp that can bind the capsids of certain retroviruses, accelerate disassembly, and inhibit infection (15, 16, 41, 47).
The host cytosolic protein CypA binds the proline-rich “CypA-binding loop” in the N-terminal domain of the HIV-1 CA protein (22, 23, 26, 37, 52). The CypA-binding loop is exposed on the surface of the assembled HIV-1 capsid and thus is accessible to CypA in the cytosol of the newly infected cell. Residues glycine 89 and proline 90 in the CypA-binding loop are critical for CypA binding, and changes that disrupt CypA-CA interactions (e.g., G89V or P90A) result in decreased HIV-1 infection in certain cell types (8, 49, 53, 60). Cyclosporine (Cs) is an immunosuppressive drug that binds to the CypA active site and competitively inhibits the CypA-CA interaction. Cs and its analogues decrease the efficiency of HIV-1 replication in some human cells, including natural target cells (9, 45, 56). The effects of Cs treatment of target cells on HIV-1 infection can be recapitulated by homozygous deletion of the PPIA gene encoding CypA (10, 49, 53).
The exact role of CypA during HIV-1 infection remains elusive. CypA appears to be involved in an early postentry step of the HIV-1 life cycle (9, 45, 53). CypA interaction with the HIV-1 capsid in the target cell, rather than in the producer cell, is important for successful HIV-1 infection (9, 28, 49). Disruption of CypA-CA interactions by either Cs treatment or CA alteration results in a reduction in the accumulation of HIV-1 reverse transcripts (9). CypA is a prolyl isomerase and has been shown to catalyze the cis/trans isomerization of the glycine 89-proline 90 peptide bond on the HIV-1 CA protein (6, 7). Alteration of this peptide bond can cause conformational changes within and distal to the CypA-binding loop (6, 7, 26, 41); it has been hypothesized that such CypA-mediated changes might facilitate the disassembly of the HIV-1 capsid (6-9, 29, 38, 61). Indeed, it has been shown that the naturally dimeric TRIMCyp protein or artificially multimerized forms of CypA bind the HIV-1 capsid more efficiently than monomeric CypA (30, 59); some of these oligomeric CypA fusion proteins accelerate HIV-1 capsid disassembly and restrict viral infection (30, 59). CypA binding to the HIV-1 capsid can also increase the sensitivity of HIV-1 to Old World monkey TRIM5α proteins, which can promote premature capsid disassembly in virus-exposed cells (5, 31, 51). These observations hint at a role for CypA in HIV-1 uncoating.
Inhibiting CypA with Cs or its analogues can exert a range of consequences on HIV-1 replication, depending on the capsid and the host cell. Although Cs analogues have been reported to suppress replication of HIV-1 in HeLa cells (1, 45, 46), these agents exert minimal effects on the early phase of HIV-1 infection in HeLa cells (28, 42, 49, 50, 61). Nonetheless, passage of HIV-1 in a HeLa-CD4+ cell line in the presence of a nonimmunosuppressive analogue of Cs resulted in the generation of two virus variants, each with a distinct change, A92E or G94D, in the CypA-binding loop of CA (1, 8). In HeLa-CD4+ cells, these two mutants replicate efficiently in the presence of Cs or when additional changes that disrupt CypA-CA interactions are introduced into the CA protein (1, 8). Neither change alters the affinity of CA for CypA. Surprisingly, the efficient replication of the A92E and G94D mutants is dependent on the disruption of CypA-CA interactions in HeLa and H9 cells but not in Jurkat, TE617, or HOS cells (28, 38, 49, 60). The basis of this cell-type dependency is not understood. Some studies have suggested that the level of CypA expression in the target cell may affect the efficiency of infection of the A92E mutant virus (60, 61). Other studies have led to the proposal that the low level of replication of the A92E mutant in HeLa cells is due to the action of a CypA-dependent dominant restriction factor (38, 50).
In the present study, we investigated the effects of changes in the CypA-binding loop on HIV-1 capsid stability in vitro. We found that the assembly of the A92E CA protein was less efficient than that of the wild-type HIV-1 CA protein and that this defect was influenced by the presence of a proline residue at position 90 in the CypA-binding loop. In infected HeLa cells, the relative instability of the A92E capsid was dependent on CypA binding to the capsid. In Jurkat cells, CypA binding stabilized the wild-type and, to a lesser extent, the A92E capsids. In both cell types, a lower level of particulate, cytosolic capsids was associated with decreased infectivity. Our results suggest that CypA modulates HIV-1 capsid disassembly and that changes in capsid stability can influence HIV-1 sensitivity to the inhibition of CypA binding.
MATERIALS AND METHODS
Production of recombinant HIV-1 expressing GFP.
The pCMVΔenvpA packaging plasmids encoding the wild-type and mutant variants of the HIV-1 Gag/Pol polyprotein were constructed by PCR mutagenesis, as previously described (42). Vesicular stomatitis virus protein G (VSV-G)-pseudotyped HIV-1 expressing green fluorescent protein (GFP) was made by cotransfecting 293T cells with the HIV-1 reporter vector pHIvec2.GFP, wild-type or mutant pCMVΔP1ΔenvpA, a pHCMV-G plasmid expressing VSV-G, and a Rev-expressing plasmid, as described previously (42). Virus-like particles (VLPs) lacking the GFP-expressing vector were generated by cotransfecting 293T cells with all of the above plasmids except pHIvec2.GFP. VLPs without the VSV-G envelope glycoprotein were made by cotransfecting 293T cells with the pCMVΔP1ΔenvpA plasmid and a Rev-expressing plasmid. All virus- and VLP-containing supernatants were cleared of cell debris by low-speed centrifugation and were quantitated by measurement of the reverse transcriptase (RT) activity using an RT assay (Roche Applied Science, Indianapolis, IN). Virus-containing supernatants were stored in aliquots at −80°C.
Cell culture and infectivity assays.
HeLa, HOS, and HEK 293 cells (American Type Culture Collection [ATCC]) were grown at 37°C in Dulbecco modified Eagle medium (Invitrogen). Jurkat T lymphocytes (ATCC) were grown at 37°C in RPMI 1640 (Invitrogen). All media were supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. For infection, HeLa, HOS, HEK 293, and Jurkat cells were seeded at a density of 104 cells/well in 96-well plates. HIV-1-GFP viruses were normalized by RT activity and added to cells in a total volume of 0.1 ml. Target cells were treated with either dimethyl sulfoxide (DMSO) or Cs (Sigma) 30 min prior to virus inoculation. In most experiments, a single concentration (5 μM) of Cs was used; in some experiments, a range of Cs concentrations (0 to 20 μM) was used. Cells were incubated with viruses at 37°C for ∼4 h before removal of the supernatants and the addition of 0.2 ml of fresh medium. Cells were harvested 48 h after infection and fixed in phosphate-buffered saline containing 4% formaldehyde. The percentage of infected, GFP-positive cells was determined by fluorescence-activated cell sorting (FACS) using FACScan (Becton Dickinson).
Viral protein analysis and Western blotting.
293T cells were transfected with a plasmid expressing either the wild-type or mutant HIV-1 Gag/Pol protein and a plasmid encoding a FLAG-tagged human CypA (CypA-FLAG) protein. In the control experiment, no plasmids were transfected. Cells were harvested and lysed 48 h after transfection. The expression levels of the cell-associated Gag proteins and the CypA-FLAG protein were analyzed by Western blotting with a monoclonal anti-HIV-1 p24 antibody and a horseradish peroxidase (HRP)-conjugated anti-FLAG antibody (Abcam, Cambridge, MA), respectively. Virions released into the transfected cell supernatants were filtered (0.45-μm pore size) and pelleted through a 20% sucrose cushion. Pelleted viral particles were normalized based on the RT activities and lysed. Lysates were analyzed by Western blotting with a monoclonal anti-HIV-1 p24 antibody and an HRP-conjugated anti-FLAG antibody (Abcam).
For detection of endogenous CypA expression, HeLa and Jurkat cells were lysed in hypotonic lysis buffer (10 mM Tris-HCl [pH 8.0]; 10 mM KCl; 1 mM EDTA) by homogenization. Cell lysates were normalized for total protein concentration and analyzed by Western blotting, with a mouse anti-CypA antibody (Abcam) and a control HRP-conjugated anti-GAPDH antibody (Abcam). Protein band densities were determined by using ImageJ software.
Electron microscopy of VLPs.
Ten ml of transfected cell supernatants containing wild-type and mutant VLPs were centrifuged at 4°C for 90 min at 27,000 rpm in a Beckman SW41 rotor. Pelleted viral particles were fixed with glutaraldehyde and analyzed by thin-section electron microscopy at the Harvard Medical School Electron Microscope Facility.
Quantitative real-time PCR of viral reverse transcripts.
Virus stocks derived from transfection of 293T cells were treated with 20 U of DNase I (NEB)/ml for 60 min at room temperature. Approximately 106 HeLa cells, treated with 5 μM Cs or DMSO, were infected with 20 ng of RT of the HIV-1-GFP virus or heat-inactivated (20 min at 80°C) virus as a control. Genomic DNA was isolated at 16 h after infection. The late HIV-1 reverse transcripts were quantified by real-time PCR analysis by using the primers MH531 and MH532 and the probe LRT-P as described previously (13).
Purification and assembly of HIV-1 CA proteins.
The pET-based plasmids for the expression of wild-type and mutant His-tagged HIV-1 CA proteins were generated by PCR mutagenesis, using a previously described (25) HIV-1 CA-expressing plasmid as a template. The expression of the His-tagged CA proteins in freshly transformed BL21(DE3) cells (Novagen) was induced by treatment with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside; Sigma). Cells were harvested 4 h after induction, and the His-tagged CA proteins were purified by Ni-NTA affinity chromatography (Qiagen) according to the manufacturer's protocol. CA proteins eluted from the columns were dialyzed into 20 mM Tris buffer (pH 8.0), concentrated to ∼400 μM, and stored at −80°C until use.
The assembly reactions were initiated by mixing 125 μl of 5 M NaCl with 125 μl of 88 μM purified CA proteins at room temperature. After the addition of NaCl, the increase in optical density at 340 nm was monitored every 10 s for 1 h. The optical densities at early times (0 to 120 s) were fitted to a straight line by least-squares approximation to calculate the initial rates of assembly for each CA protein. At least three independent assembly experiments were performed for each CA protein.
Fate-of-capsid assay.
The assay to examine the fate of HIV-1 capsids in the cytosol of newly infected HeLa cells was performed essentially as described previously (51). VLPs in ∼10 ml of cell supernatant were normalized by RT activities and incubated with 6 × 106 HeLa cells, with either DMSO or 5 μM Cs in the medium. The cells and VLPs were initially incubated for 30 min at 4°C and then shifted to 37°C. At 4 h after infection, the cells were washed with fresh medium and cultured at 37°C for another 12 h. The cells were then washed in ice-cold phosphate-buffered saline and lysed in 2.5 ml of hypotonic lysis buffer (10 mM Tris-HCl [pH 8.0], 10 mM KCl, 1 mM EDTA) by homogenization. After the cell debris was removed by centrifugation for 3 min at 2,000 × g, 2 ml of the clarified lysate was layered onto a 7-ml 50%-sucrose cushion and centrifuged at 125,000 × g for 2 h at 4°C. Prior to centrifugation, 100 μl of the cleared lysate was saved to analyze the total input level of HIV-1 capsid. After centrifugation, 100 μl of the topmost part of the supernatant was collected. The pellet was resuspended in 100 μl of 1× sodium dodecyl sulfate sample buffer. The input, supernatant, and pellet samples were subjected to Western blotting analysis of capsid proteins with a monoclonal anti-HIV-1 p24 antibody.
For the fate-of-capsid assay in Jurkat target cells, 30 ml of VLP-containing supernatant was concentrated by centrifugation at 4°C for 90 min at 27,000 rpm in a Beckman SW28 rotor. The pellet was resuspended in 1 ml of medium and normalized to 1,000 ng of RT/ml using an RT assay (Roche). Approximately 5 × 107 Jurkat cells were incubated with 0.5 ml of each concentrated VLP preparation, in the presence of either DMSO or 5 μM Cs. The medium also contained 10 μg of Polybrene (American Bioanalyticals)/ml. After 4 h of incubation at 37°C, the cells were washed and cultured at 37°C in 10 ml of fresh medium for another 12 h. The subsequent steps were identical to those described above for the fate-of-capsid assay in HeLa cells.
RESULTS
Effects of changes in the CypA-binding loop of CA on HIV-1 infectivity.
To assess the effect of changes in the CypA-binding loop of the HIV-1 CA on viral infectivity, we altered this loop in the Gag proteins of a recombinant, single-round HIV-1-GFP vector (Fig. 1A). The P90A change reduces CypA binding (22). The A92E change arose by selection of HIV-1 in HeLa cells treated with a Cs analogue and renders HIV-1 Cs dependent in HeLa cells (1, 8). In the P90A/A92E mutant, referred to here as the AE mutant, both of these changes in the CypA-binding loop of CA are combined. To examine the effect of two acidic residues at these positions in the HIV-1 CypA-binding loop, the P90E/A92E mutant, here called the EE mutant, was created.
FIG. 1.
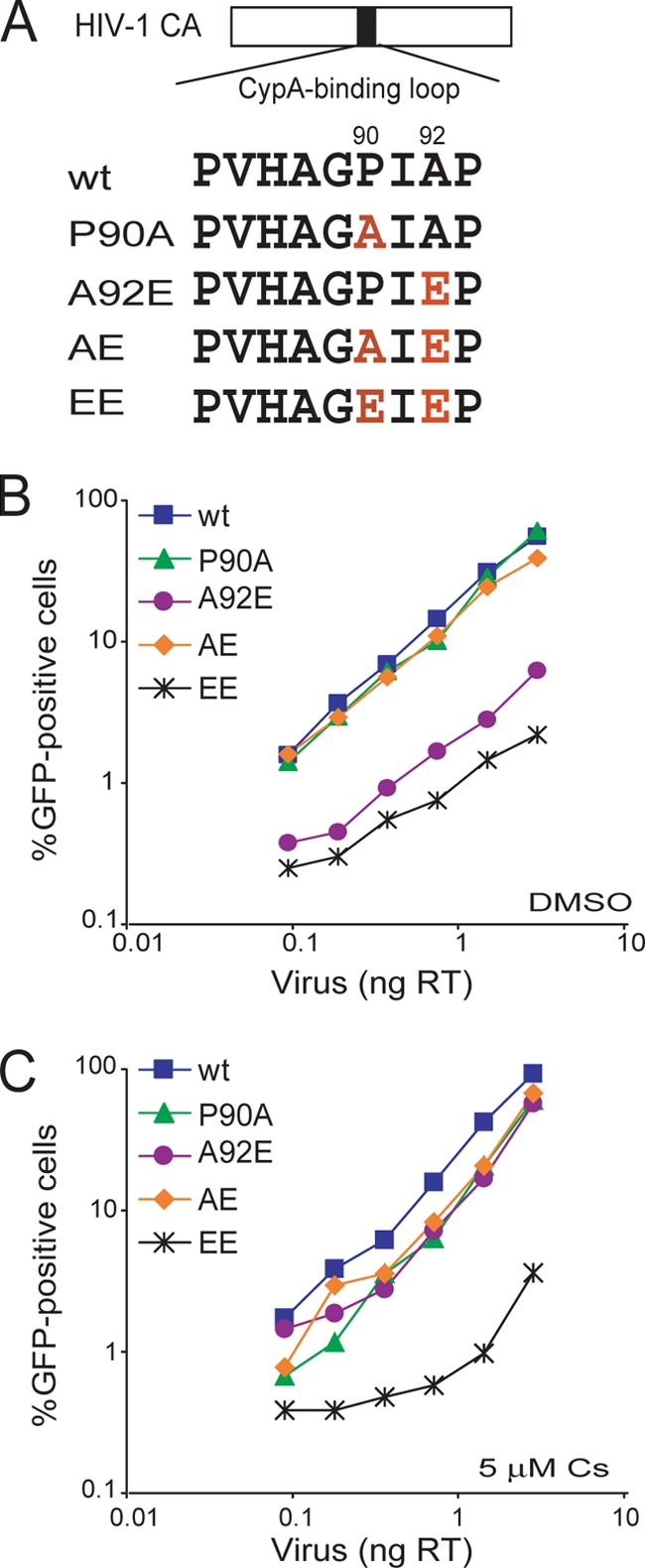
Infectivity of HIV-1 CA mutants in HeLa cells. (A) The amino acid sequences of the CypA-binding loop of the wild-type and mutant HIV-1 CA proteins is shown. (B and C) HeLa cells were infected in the presence of DMSO (B) or 5 μM Cs (C) with the indicated amounts of wild-type or mutant HIV-1-GFP. After 48 h, the percentage of GFP-positive cells was determined by FACS. The results of a single experiment typical of those obtained in three independent experiments are shown. wt, wild type.
HeLa cells were challenged with various amounts of wild-type and mutant HIV-1-GFP in the presence or absence of Cs. Consistent with previous reports (28, 50, 61), the A92E mutant exhibited an ∼10-fold lower infectivity than the wild-type virus in HeLa cells (Fig. 1B). Cs treatment or the additional P90A alteration increased the infectivity of viruses with the A92E change to near-wild-type levels (Fig. 1B and C). Interestingly, the EE mutant exhibited substantially lower infectivity than the wild-type virus with or without Cs treatment (Fig. 1B and C). Thus, in HeLa cells, the replication defect resulting from the A92E change is dependent on CypA-CA binding, as previously observed (1, 28, 49, 50, 61). In contrast, the infectivity of the EE mutant is defective regardless of Cs treatment.
Expression and processing of the mutant Gag proteins.
To investigate the mechanism of the decreased infectivity associated with the A92E and EE mutants, we examined whether the expression and processing of the HIV-1 Gag polyprotein were affected by these changes. 293T cells were transfected with plasmids encoding the wild-type or mutant HIV-1 Gag/Pol proteins. At 48 h after transfection, both cell-associated and virion-associated Gag proteins were analyzed by Western blotting with a monoclonal anti-HIV-1 p24 antibody. The expression level, virion incorporation, and proteolytic cleavage of the mutant Gag proteins were similar to those of the wild-type protein (Fig. 2). The results suggest that the changes introduced into CA do not significantly interfere with Gag expression, processing, or virion formation.
FIG. 2.

Effect of HIV-1 CA changes on Gag processing. 293T cells were transfected with plasmids encoding the wild-type (wt) or mutant HIV-1 Gag/Pol polyproteins. Cells were harvested 48 h later, and total cellular proteins were subjected to Western blotting analysis with a monoclonal anti-HIV-1 p24 antibody (left panel). The positions of the molecular-weight-marker proteins are indicated. The virion-containing cell supernatants were centrifuged. Solubilized virion pellets were analyzed by Western blotting with a monoclonal anti-HIV-1 p24 antibody (right panel).
Some changes in the HIV-1 CA protein have been shown to alter the characteristic conical shape of the mature core; aberrant core morphology was often associated with diminished infectivity (27, 55). To examine whether the changes introduced into the HIV-1 CA interfere with core formation, we analyzed the morphology of wild-type and mutant VLPs by thin-section electron microscopy. Mature, conical cores were observed in the wild-type and all four mutant VLPs (Fig. 3A). The frequency of classical conical cores in the sectioned VLPs was similar among wild-type and mutant virions (Fig. 3B). Apparently, the formation and morphology of the mature virion-associated core are not significantly affected by the introduced changes in the CA protein.
FIG. 3.

Thin-section electron microscopy of HIV-1 virions. Virions were purified from the supernatant of 293T cells transfected with plasmids encoding the wild-type or mutant HIV-1 Gag/Pol proteins and fixed with glutaraldehyde for thin-section electron microscopy analysis. (A) Examples of virions with evident conical cores are shown. (B) The indicated number (n) of virion particles was examined for each of the HIV-1 variants. The percentage of virions in which classical conical cores were evident was calculated for each variant. wt, wild type.
Binding of CypA to Gag and incorporation into HIV-1 virions.
The incorporation of CypA into HIV-1 virions is dependent upon its interaction with the CypA-binding loop of the Gag polyprotein (22, 23). We tested whether the introduced CA changes affected CypA-CA interactions by examining the incorporation of CypA into virions. 293T cells were cotransfected with plasmids encoding the wild-type or mutant HIV-1 Gag/Pol proteins and a plasmid encoding a FLAG-tagged CypA protein. Virions were concentrated from the supernatant of the transfected 293T cells and were analyzed by Western blotting. Cell lysates prepared in parallel showed similar levels of CypA expression (Fig. 4). The amounts of CypA associated with the P90A, AE, and EE mutant virions were lower than that associated with the wild-type virions, a finding consistent with the importance of proline 90 on CA for CypA binding (9, 23). In contrast, the amounts of CypA associated with the A92E and wild-type virions were similar (Fig. 4). No correlation between CypA incorporation and viral infectivity in HeLa cells was observed.
FIG. 4.

CypA incorporation into HIV-1 virions. 293T cells were cotransfected with plasmids encoding the wild-type (wt) or mutant HIV-1 Gag/Pol proteins and with a plasmid encoding a FLAG-tagged CypA (CypA-FLAG) protein. In the mock experiment, no viral or CypA-FLAG DNA was transfected. After 48 h, the transfected cells were lysed, and equal amounts of the total cellular proteins were subjected to Western blot analysis; the CypA-FLAG protein was detected with an HRP-conjugated anti-FLAG antibody (upper panel). Virions were concentrated from the supernatant of the transfected 293T cells by pelleting through a 20% sucrose cushion. Solubilized virion pellets were analyzed by Western blotting; the blots were probed with an HRP-conjugated anti-FLAG antibody (middle panel) and a monoclonal anti-HIV-1 p24 antibody (lower panel).
Effect of CA changes on HIV-1 reverse transcription.
We wanted to determine whether the CA changes affect viral cDNA synthesis in target cells. HeLa cells were infected with equal amounts (normalized by virion-associated RT) of wild-type and mutant HIV-1-GFP, in the presence or absence of Cs. Total DNA was purified from the cells 16 h after infection, and the level of late reverse transcripts was measured by quantitative real-time PCR. Without Cs treatment, the A92E and EE mutants exhibited significantly lower amounts of late RT products compared to wild-type HIV-1-GFP (Fig. 5A). There was no significant difference among the levels of late reverse transcripts in the cells infected with the wild-type virus or the P90A and AE mutants. In the presence of Cs, the level of reverse transcripts for the A92E mutant, but not the EE mutant, increased significantly. Thus, the level of late RT products correlated with viral infectivity. These data suggest that the A92E and EE changes result in HIV-1 replication defects prior to the completion of reverse transcription.
FIG. 5.

Levels of HIV-1 reverse transcripts in infected HeLa cells. (A) VSV-G-pseudotyped HIV-1-GFP viruses were normalized by RT activity and incubated with HeLa cells in the presence of DMSO or 5 μM Cs. DNA was isolated from the target cells 16 h later and the levels of viral late reverse transcription products were analyzed by real-time PCR. (B) HeLa cells were infected under the conditions described in panel A and harvested 48 h after infection. The percentage of GFP-positive cells was determined by FACS. The results of a typical experiment of two independent experiments are shown.
The in vitro assembly kinetics of mutant HIV-1 CA proteins.
Purified HIV-1 CA proteins assemble in vitro into helical tubes and cones, which resemble authentic mature capsids (35). The assembly is mediated by CA-CA interactions, and therefore the efficiency of assembly indicates the strength of the intersubunit forces that stabilize the capsid (19, 33, 35). To examine the effects of the CA changes on self-association, we compared the in vitro assembly kinetics of wild-type and mutant HIV-1 CA proteins, using an approach modified from Lanman et al. (33). His-tagged HIV-1 CA proteins carrying different changes in the CypA-binding loop were expressed in bacteria and purified to near homogeneity (Fig. 6A). Each CA protein at a concentration of 44 μM was incubated in a high-salt (2.5 M NaCl) buffer and allowed to assemble into capsid-like structures. The resulting increase in optical density at 340 nm was monitored every 10 s (Fig. 6B). The initial rate of assembly for each CA protein was estimated by fitting the optical densities measured at the early time points (0 to 120 s) to a straight line, using a least-squares approximation (Fig. 6C). The P90A mutant CA protein assembled almost as efficiently as the wild-type protein, although early P90A CA assembly was slightly delayed compared to that of wild-type CA. The AE mutant CA protein initially assembled at a lower rate than that of the wild-type CA protein but reached a final optical density similar to that of the wild-type protein. The A92E and EE mutant CA proteins assembled less efficiently than the wild-type CA protein. Of note, the final amount of the mutant HIV-1 CA proteins assembled in vitro correlated with the infectivity of viruses with these CA changes in untreated HeLa cells. This observation suggested that alteration of the intrinsic stability of the mutant capsids could influence HIV-1 infectivity and prompted an examination of the uncoating of these viral capsids in infected cells.
FIG. 6.

Assembly kinetics of purified HIV-1 CA proteins. (A) His-tagged wild-type or mutant HIV-1 p24 CA proteins were expressed in bacteria and purified by Ni-NTA column chromatography. Portions (20 μg) of each purified CA protein were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Coomassie blue staining. (B) Each CA protein, at a concentration of 44 μM, was assembled in 2.5 M NaCl. The optical density at 340 nm was monitored every 10 s. (C) The initial rate of assembly for each CA protein was estimated by fitting the increase in optical density at early times (0 to 120 s) to a straight line, using a least-squares approximation. Error bars represent the standard deviations of three independent estimations. wt, wild type.
Effects of CA changes on HIV-1 uncoating in HeLa cells.
To gain insight into the infectivity defects caused by the CA changes, we studied the uncoating of the wild-type and mutant viruses by using a previously developed fate-of-capsid assay (51). This assay allows us to measure the amounts of particulate and soluble forms of the HIV-1 capsid present in the cytosol of infected cells. The particulate, cytosolic capsids measured in this assay have been shown to be associated with viral RNA (43) and exhibit densities that closely match those of cores prepared by detergent treatment of virions (21, 32, 43, 51, 57). Moreover, the appearance of particulate capsids in the cytosol depends upon successful virus entry and inversely correlates with restriction potency in target cells expressing TRIM5 or TRIMCyp variants (16, 17, 30, 43, 51). Previous studies (16, 51) indicate that VSV-G-mediated virus entry continues for at least 16 h after the exposure of the target cell to the virus, and therefore the steady-state levels of cytosolic capsids measured in this assay reflect both entry and decay of the capsids. Because virus entry is not influenced by the particular capsid variant, comparison of the steady-state levels of cytosolic, particulate capsids allows an assessment of the conversion of particulate capsids to soluble capsids, as well as the decay of these two forms of CA proteins.
Initially, we compared the disassembly of the A92E mutant and wild-type virus capsids, and the effect of Cs on this process, in infected HeLa cells. Equal amounts of VSV-G-pseudotyped wild-type and A92E mutant HIV-1, normalized by virion-associated RT activity, were incubated with HeLa cells in the absence and presence of 5 μM Cs. As a control, wild-type HIV-1 lacking the VSV-G envelope glycoprotein was incubated with HeLa cells under the same conditions. Four hours later, the cells were washed and incubated at 37°C in fresh medium for another 12 h. After lysis of the target cells, cytoplasmic lysates (“Input”) were separated over a 50% sucrose cushion into supernatant (“Sup”) and particulate (“Pellet”) fractions. The fractions were analyzed by Western blotting with a monoclonal anti-HIV-1 p24 antibody. No cytosolic p24 CA protein was observed in the HeLa cells incubated with the control virus lacking the VSV-G envelope glycoprotein (Fig. 7A). The amounts of HIV-1 p24 CA protein in the input and supernatant fractions were similar for the wild-type and A92E viruses pseudotyped with the VSV-G glycoprotein, suggesting that similar numbers of these viruses entered the target cells (Fig. 7A). In the absence of Cs treatment, the amount of pelletable cytosolic capsid for the A92E mutant was lower than that detected for the wild-type virus. Remarkably, with Cs treatment, similar amounts of pelletable capsid were detected for the wild-type and A92E viruses (Fig. 7A). Thus, the A92E change resulted in a decrease in the amount of particulate HIV-1 capsid in the cytosol of HeLa target cells, and this decrease can be rescued by Cs treatment.
FIG. 7.

Fate of the capsid in HeLa cells infected with HIV-1 CA variants. (A) VSV-G-pseudotyped (Env+) wild-type (wt) or A92E mutant HIV-1 virions were incubated with HeLa cells in the presence or absence of 5 μM Cs. As a control, virions lacking envelope glycoproteins (Env−) were also incubated with HeLa cells under the same conditions. At 4 h after infection, the cells were washed and incubated at 37°C in fresh medium for another 12 h. After lysis of the target cells, cytoplasmic lysates were analyzed directly (“Input”) or were separated over a 50% sucrose cushion into supernatant (“Sup”) and particulate (“Pellet”) fractions. The fractions were analyzed by Western blotting, with a monoclonal anti-HIV-1 p24 antibody. The experiment was repeated twice with similar results. (B) HIV-1 CA mutants were used in the same experiment as that described in panel A, in the absence of Cs. (C) HIV-1 CA mutants were used in the same experiment as that described in panel A, in the presence of 5 μM Cs. The results of two independent experiments (Exp 1 and 2) are shown in B and C. wt, wild type.
Next, we examined all of the CA mutants using the fate-of-capsid assay. In the absence of Cs, we observed decreased amounts of particulate cytosolic capsid for the A92E and EE mutants, relative to the amount seen for the wild-type virus (Fig. 7B). In contrast, the amount of pelletable capsid protein detected for the P90A and AE mutants was closer to that of the wild-type virus. In the presence of Cs, the levels of particulate capsid observed for the wild-type virus and the A92E, P90A and AE mutant viruses were similar; however, the particulate EE mutant capsids remained at undetectable levels (Fig. 7C). Thus, there is a correlation between infectivity and the recovery of particulate HIV-1 capsids from the cytosol of HeLa cells. More importantly, the amount of particulate capsid containing the A92E change in the cytosol of HeLa cells was increased by reducing CypA-CA interactions, suggesting that CypA promotes disassembly of the A92E capsid in these cells.
Effects of CA changes on HIV-1 infectivity in Jurkat T lymphocytes.
Some changes in the CypA-binding loop of the HIV-1 CA protein result in target cell-dependent infectivity phenotypes (2, 28, 39, 49, 60). Consistent with this, differences in the relative infectivities and Cs sensitivities of our panel of HIV-1 CA mutants were observed in HeLa, Jurkat, HOS, and HEK 293 target cells (see Fig. S1 in the supplemental material). The infectivity phenotypes of the HIV-1 CA mutants were more similar in the Jurkat, HOS, and HEK 293 cells, whereas the phenotypes in HeLa cells, with respect to both the behavior of the A92E mutant and CypA dependence, were distinct.
We further explored the effect of the CA changes on infectivity in Jurkat T lymphocytes, which resemble primary T lymphocytes with respect to the Cs sensitivity of HIV-1 infection (45, 56, 60). Jurkat cells were challenged with increasing amounts of wild-type and mutant HIV-1-GFP, and GFP-positive cells were counted 48 h later. The P90A, AE, and EE mutants all exhibited decreased infectivity relative to that of wild-type HIV-1-GFP (Fig. 8A, left panel). Interestingly, the A92E mutant exhibited an intermediate level of infectivity in Jurkat cells; the infectivity of the A92E mutant was higher than that of the P90A mutant, a pattern opposite to that observed in HeLa cells. Cs treatment decreased the infectivity of the wild-type and A92E viruses; thus, in the presence of Cs, none of the viruses infected the Jurkat cells efficiently (Fig. 8A, right panel). The results indicate that disruption of the CypA-CA interaction is detrimental to HIV-1 infection in Jurkat lymphocytes.
FIG. 8.

Infectivity and fate of the capsid of HIV-1 CA variants in Jurkat T lymphocytes. (A) Jurkat cells were infected in the presence of DMSO or 2.5 μM Cs with the indicated amounts of wild-type or mutant HIV-1-GFP. The results shown are typical of those obtained in three independent experiments. (B) The fate-of-capsid assay was optimized for the study of HIV-1 infection of Jurkat cells. VSV-G-pseudotyped (Env+) wild-type or P90A HIV-1 virions were concentrated, normalized by total RT activity and then incubated with Jurkat cells. As a control, virions lacking envelope glycoproteins (Env−) were studied in parallel. Four hours after infection, the cells were washed and incubated at 37°C in fresh medium for another 12 h. After lysis of the target cells, cytoplasmic lysates were analyzed directly (“Input”) or were separated over a 50% sucrose cushion into supernatant (“Sup”) and particulate (“Pellet”) fractions. The fractions were analyzed by Western blotting, with a monoclonal anti-HIV-1 p24 antibody. (C) An experiment similar to that described in panel B was carried out, except that 5 μM Cs was added to the Jurkat cell medium as indicated. (D) HIV-1 CA variants were analyzed in an experiment similar to that described in panel B in the presence of DMSO (upper panel) or 5 μM Cs (lower panel). Note that the lower panel was exposed longer than the upper panel. The results of a single experiment are shown; the experiment was repeated twice, with comparable results. wt, wild type.
Effects of CA changes on HIV-1 uncoating in Jurkat lymphocytes.
Compared to HeLa cells (Fig. 1B), Jurkat cells were less efficiently transduced by the same amount of wild-type HIV-1-GFP (Fig. 8A). To evaluate viral uncoating in Jurkat cells, we optimized the fate-of-capsid assay by increasing the concentration of recombinant viruses and the number of cells used. Thirty ml of VSV-G-pseudotyped wild-type or P90A viruses were concentrated, and each virus suspension was adjusted to contain the equivalent of 500 ng of RT. The concentrated viruses were then incubated with 5 × 107 Jurkat cells in a 500-μl volume. As a control, wild-type viruses lacking the VSV-G envelope glycoprotein were also processed under the same conditions. Sixteen hours after infection, the cells were lysed and analyzed by a fate-of-capsid assay similar to that described above for HeLa cells. High levels of the HIV-1 CA protein were detected in the input lysates derived from cells exposed to viruses lacking the VSV-G glycoprotein (Fig. 8B); this observation suggests that, under these conditions, many HIV-1 particles are endocytosed into the Jurkat cells. Endosomes are of insufficient density to sediment through a 50% sucrose cushion (3, 14, 18) and are thus eliminated from the pellet. Indeed, although a significant amount of the p24 capsid protein was detected in the pellets derived from cells incubated with VSV-G-pseudotyped wild-type viruses, only a very low level of p24 capsid protein was detected in the pellets derived from cells incubated with the virions lacking the VSV-G glycoprotein (Fig. 8B and C). The amount of pelletable capsid detected for the P90A mutant was much less than that of the wild-type virus (Fig. 8B). Cs treatment reduced the amount of particulate capsid in Jurkat cells exposed to wild-type HIV-1 virions (Fig. 8C). These results suggest that, in contrast to what we observed in HeLa cells, reducing the CA-CypA interaction in Jurkat cells decreases HIV-1 infectivity and the amount of particulate cytosolic capsid.
All of the CA mutants were then studied in the fate-of-capsid assay in Jurkat T lymphocytes. In the absence of Cs, a substantial decrease was observed in the amount of particulate cytosolic capsid protein for the P90A, AE, and EE mutants compared to that seen for the wild-type virus (Fig. 8D). The particulate capsid level for the A92E mutant was between that of the wild-type virus and those of the other mutant viruses. In the presence of Cs, similar low levels of particulate capsids were observed for wild-type, P90A, A92E, and AE viruses; a lower amount of pelletable capsid was observed for the EE mutant (Fig. 8D). Although the effects of reducing CypA-CA interactions in Jurkat cells were quite different from those observed in HeLa cells, a strong correlation between the recovery of the cytosolic capsid particles and HIV-1 infectivity was evident in both target cell types. Disruption of the CypA-CA interaction resulted in decreases in the amount of particulate HIV-1 capsids in the cytosol of Jurkat cells; thus, in Jurkat cells, CypA binding stabilizes the HIV-1 capsid and improves infectivity.
Effect of Cs titration on HIV-1 infectivity in Jurkat and HeLa cells.
We wished to examine the effects of incremental reductions of CypA-CA binding on the infectivity of wild-type and A92E HIV-1 in Jurkat and HeLa target cells. To accomplish this objective, Jurkat and HeLa cells were incubated with equivalent amounts of wild-type and A92E HIV-1-GFP in the presence of increasing concentrations of Cs. The percentage of infected, GFP-positive cells was determined by FACS analysis 48 h after infection. In HeLa cells, the A92E virus exhibited lower basal levels of infectivity than wild-type HIV-1-GFP (Fig. 9A). With the addition of increasing concentrations of Cs, both A92E and wild-type HIV-1-GFP infectivity increased and plateaued. In Jurkat cells, the infectivity of wild-type HIV-1-GFP was greatest in the absence of Cs treatment; the infectivity of the A92E virus was lower than that of the wild-type virus in the untreated target cells (Fig. 9B). Treatment with Cs decreased the infectivity of both viruses in the Jurkat cells. These results indicate that saturating concentrations of Cs were used in our experiments. Moreover, in both cell types, the Cs dose-response curves were monophasic.
FIG. 9.
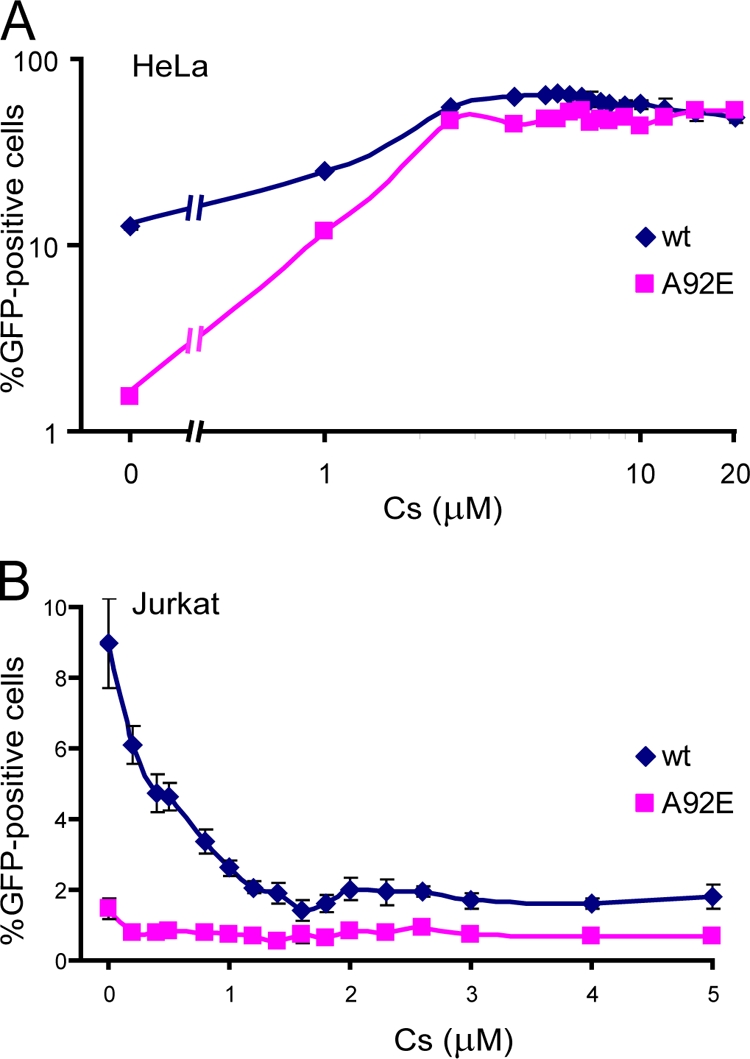
Effect of Cs titration on HIV-1 infectivity. (A) HeLa cells were infected with wild-type or A92E HIV-1-GFP in the presence of the indicated concentrations of Cs. After 48 h, the percentage of GFP-positive cells was determined by FACS. (B) Jurkat cells were used as target cells in the experiment described in panel A. Error bars represent the standard deviations obtained in at least three independent experiments. wt, wild type.
CypA expression levels in HeLa and Jurkat cells.
Since the level of CypA in different cell types has been implicated in modulating HIV-1 infection (2, 39, 60, 61), we examined the endogenous CypA level in both HeLa and Jurkat cells. Cell lysates were prepared from these two cell lines and were subjected to Western blotting analysis, with a mouse anti-CypA antibody and an HRP-conjugated anti-GAPDH antibody (Fig. 10A). Quantification of the unsaturated CypA protein bands, after normalization to the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) bands, indicated that the CypA protein level in HeLa cells is ∼3-fold higher than that in Jurkat cells (Fig. 10B). This difference in cellular CypA level may contribute to the difference in the response of HIV-1 to the disruption of CypA-CA interactions in these target cell types.
FIG. 10.

CypA expression level in HeLa and Jurkat cells. (A) Cell lysates from HeLa and Jurkat cells were normalized by total protein concentrations and subjected to Western blotting analysis. The blots were probed with an anti-CypA antibody and an HRP-conjugated anti-GAPDH antibody. The results from two independent experiments (Exp 1 and Exp. 2) are shown. (B) The ratio between CypA protein levels in HeLa cells and Jurkat cells, after normalizing to the corresponding GAPDH levels, was calculated based on quantification of protein band densities. The results from two independent experiments (Exp 1 and Exp. 2) are shown.
DISCUSSION
The importance of CypA-CA interactions to HIV-1 replication in biologically relevant target cells has been demonstrated (45, 56). Disruption of CypA-CA binding by changes in CA or by Cs analogues results in a block to HIV-1 replication after virus entry but prior to reverse transcription (9). Changes in the CA protein alter HIV-1 sensitivity to Cs, in some cases without detectably affecting CypA binding to the capsid (1, 8, 28, 58). These observations have given rise to the hypothesis that CypA plays a role in capsid uncoating (8, 9, 49). By studying the in vitro assembly of HIV-1 CA mutants and utilizing assays that measure the level of retroviral capsids in the cytosol of infected cells, we tested this hypothesis.
The rate and efficiency with which purified HIV-1 CA proteins assemble into capsid-like structures in vitro reflect the strength of the intermolecular associations that contribute to capsid stability (19, 33, 35). We found that some changes in the CypA-binding loop affected the efficiency of HIV-1 CA assembly in vitro. Of note, all three CA proteins with substitutions of acidic residues in the CypA-binding loop exhibited decreases in the efficiency of the in vitro assembly reactions. The CypA-binding loop is located on the peripheral face of each CA spoke in the capsid hexamer; therefore, in the assembled capsid, the CypA-binding loop is positioned near the trimeric pseudosymmetry axis at the interface of three adjacent hexamers (35, 44, 55). The introduction of acidic residues into the CypA-binding loop could contribute to electrostatic repulsion between the hexamers and thereby destabilize the assembled capsidlike structures. The observation that the EE mutant, with two acidic substitutions, assembles less efficiently than the A92E mutant, with one acidic substitution, is consistent with this model. Charged residues on the HIV-1 CA protein have previously been shown to influence capsid stability (19, 34).
Of interest, relative to the A92E mutant, the AE mutant CA protein exhibited an increased initial assembly rate in vitro and achieved greater (near-wild-type) levels of final products; thus, the P90A substitution partially compensated for the assembly defects associated with the A92E substitution. The glycine 89-proline 90 peptide bond in the free HIV-1 CA protein is thought to be in the cis conformation in 14% of the molecules and in the trans conformation in 86% of the molecules (6). This peptide bond in the P90A mutant would be expected to reside almost completely in the trans conformation, as in the case for most peptide bonds in native proteins. The resulting difference in conformation of the CypA-binding loop may alter the degree of electrostatic repulsion among the A92E CA hexamers and provide additional stability to the AE mutant.
Application of the fate-of-capsid assay to the study of the role of CypA in HIV-1 infection allowed measurement of the levels of particulate HIV-1 capsids in the cytosol of newly infected cells, which in some cases were treated with Cs. The levels of particulate capsids in both HeLa and Jurkat cytosols correlated with HIV-1 infectivity, implying that the modulation of HIV-1 capsid disassembly represents a major mechanism whereby CypA in the target cell influences HIV-1 infection.
The consequences of disruption of the CypA-CA interaction on HIV-1 uncoating and infectivity differed depending on the target cell. The Jurkat CD4+ T lymphocytes used in the present study resemble primary T lymphocytes with respect to the replication requirements of HIV-1 for CypA-CA binding (45, 56). The wild-type HIV-1 CA protein has presumably evolved to bind the amount of CypA that is optimal for stability and infectivity in these cell types. In the untreated Jurkat cells, the intrinsically less stable A92E mutant exhibited lower levels of particulate cytosolic capsids and lower infectivity than wild-type HIV-1. No particulate capsids were detected for the very unstable EE mutant, which replicated very inefficiently. These results are consistent with the suggestion that HIV-1 capsid mutants that exhibit decreased stability in the host cell will manifest defects in infectivity (12, 21, 22).
In Jurkat lymphocytes, the binding of CypA apparently leads to stabilization of the HIV-1 capsid. Disruption of the CypA-CA interaction in these cells resulted in accelerated capsid disassembly and decreased infectivity of the wild-type and A92E viruses. Premature uncoating has been associated with the restriction of retroviruses, including HIV-1, by TRIM5α and TRIMCyp (15-17, 30, 43, 51). Rapid uncoating may result in the loss of key components from the reverse transcription complex, or may prematurely expose viral elements to damaging host factors. The demonstration that, in relevant target cells, Cs inhibits HIV-1 infection by accelerating capsid disassembly underscores the potential to intervene at this stage of the retroviral life cycle.
In contrast to the situation in Jurkat lymphocytes, Cs did not detectably inhibit HIV-1 infection in HeLa cells. Consistent with this observation are the wild-type levels of infectivity in HeLa cells seen for the P90A and AE mutant viruses, which bind CypA poorly. Passage of HIV-1 in HeLa cells treated with a Cs analogue yielded two capsid variants, A92E and G94D (1). These mutants are dependent on Cs for efficient replication in these cells. In both mutants, an acidic residue was substituted in the CypA-binding loop; however, CypA binds efficiently to both mutant CA proteins (8, 28). Based on our observations of the inefficient in vitro assembly of HIV-1 CA variants with acidic residues in the CypA-binding loop, the A92E capsid appears to be inherently less stable than the wild-type capsid. We speculate that the G94D capsid complexes also exhibit decreased intrinsic stability compared to those of the wild-type CA protein. In untreated HeLa cells, the A92E mutant capsids that entered the cytosol disassembled more rapidly than wild-type HIV-1 capsids. Presumably as a result of this accelerated uncoating, the infectivity of the A92E mutant virus was inefficient in these cells. Likewise, the very unstable EE mutant virus infected HeLa cells poorly and demonstrated no detectable particulate capsids in the cytosol. Remarkably, Cs treatment of HeLa cells rescued the appearance of particulate A92E mutant capsids in the HeLa cytosol and restored the infectivity of this mutant. Thus, for the intrinsically unstable A92E mutant, CypA binding promotes capsid disassembly.
Why does CypA-CA binding in Jurkat lymphocytes lead to stabilization of the HIV-1 capsid, whereas in HeLa cells, CypA-CA binding destabilizes the A92E capsid? Based on the phenotypes observed in heterokaryons, it has been proposed that a dominant, cell-specific restriction factor that is dependent on CypA-CA binding inhibits A92E virus infection in HeLa cells (38, 50). Taking into account our observations, such a factor might destabilize the HIV-1 capsid in HeLa cells. The relative instability of the A92E capsid may render it more susceptible to such a factor than the wild-type capsid. In one version of this model, the factor is CypA itself, expressed at high levels. The higher level of CypA expression in HeLa cells compared to that in Jurkat cells is consistent with previous suggestions (60, 61) that CypA concentration in the cytosol might explain the observed host cell dependence of the A92E and G94D phenotypes. In this case, to account for the data, CypA would need to modulate uncoating and infectivity in a biphasic manner. At low concentrations, CypA would primarily stabilize the HIV-1 capsid, whereas at high concentrations, the capsid would be destabilized. Although we did not observe biphasic Cs dose-response curves in the present study, these have been reported for Cs analogues (39). Moreover, some evidence hints that capsid destabilization might result from CypA binding at high stoichiometry. Multimerization of CypA, either in the context of TRIMCyp or artificial CypA oligomers, increases capsid-binding avidity and leads to accelerated HIV-1 capsid disassembly and inhibition of HIV-1 infection (16, 30, 59).
Another model consistent with the data is that a dominant-acting, capsid-stabilizing factor is expressed at higher levels in HeLa cells than in Jurkat cells. In this model, CypA binding to the capsid would antagonize this factor. Moreover, the capsid-stabilizing effect of the binding of the factor would have to exceed that of CypA itself. Future work will be required to determine the validity of these models.
Although initially associated with HIV-1, CypA is now known to interact with the capsid proteins of several retroviruses (16, 36, 48, 54). Thus, CypA may be utilized by retroviruses in addition to HIV-1 for the purposes of modulating the uncoating process. Given the critical role of uncoating in HIV-1 infection, further investigation could reveal possibilities for intervention.
Supplementary Material
Acknowledgments
We thank Yvette McLaughlin and Elizabeth Carpelan for manuscript preparation.
This study was supported by the National Institutes of Health (AI063987 and AI076094), Center for AIDS Research Award AI06354, the International AIDS Vaccine Initiative, and the late William F. McCarty-Cooper.
Footnotes
Published ahead of print on 5 August 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Aberham, C., S. Weber, and W. Phares. 1996. Spontaneous mutations in the human immunodeficiency virus type 1 gag gene that affect viral replication in the presence of cyclosporins. J. Virol. 70:3536-3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackerson, B., O. Rey, J. Canon, and P. Krogstad. 1998. Cells with high cyclophilin A content support replication of human immunodeficiency virus type 1 Gag mutants with decreased ability to incorporate cyclophilin A. J. Virol. 72:303-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson, K. B., H. A. Diep, and A. Zedeler. 2006. Murine leukemia virus transmembrane protein R-peptide is found in small virus core-like complexes in cells. J. Gen. Virol. 87:1583-1588. [DOI] [PubMed] [Google Scholar]
- 4.Auewarakul, P., P. Wacharapornin, S. Srichatrapimuk, S. Chutipongtanate, and P. Puthavathana. 2005. Uncoating of HIV-1 requires cellular activation. Virology 337:93-101. [DOI] [PubMed] [Google Scholar]
- 5.Berthoux, L., S. Sebastian, E. Sokolskaja, and J. Luban. 2005. Cyclophilin A is required for TRIM5α-mediated resistance to HIV-1 in Old World monkey cells. Proc. Natl. Acad. Sci. USA 102:14849-14853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosco, D. A., E. Z. Eisenmesser, S. Pochapsky, W. I. Sundquist, and D. Kern. 2002. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. USA 99:5247-5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosco, D. A., and D. Kern. 2004. Catalysis and binding of cyclophilin A with different HIV-1 capsid constructs. Biochemistry 43:6110-6119. [DOI] [PubMed] [Google Scholar]
- 8.Braaten, D., C. Aberham, E. K. Franke, L. Yin, W. Phares, and J. Luban. 1996. Cyclosporine A-resistant human immunodeficiency virus type 1 mutants demonstrate that Gag encodes the functional target of cyclophilin A. J. Virol. 70:5170-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braaten, D., E. K. Franke, and J. Luban. 1996. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 70:3551-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braaten, D., and J. Luban. 2001. Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J. 20:1300-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briggs, J. A., M. N. Simon, I. Gross, H. G. Krausslich, S. D. Fuller, V. M. Vogt, and M. C. Johnson. 2004. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11:672-675. [DOI] [PubMed] [Google Scholar]
- 12.Brun, S., M. Solignat, B. Gay, E. Bernard. L. Chaloin, D. Fenard, C. Devaux, N. Chazal, and L. Briant. 2008. VSV-G pseudotyping rescues HIV-1 CA mutations that impair core assembly or stability. Retrovirology 5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler, S. L., M. S. Hansen, and F. D. Bushman. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631-634. [DOI] [PubMed] [Google Scholar]
- 14.Debanne, M. T., and E. Regoeczi. 1981. Subcellular distribution of human asialotransferrin type 3 in the rat liver. J. Biol. Chem. 256:11266-11272. [PubMed] [Google Scholar]
- 15.Diaz-Griffero, F., N. Vandegraaff, Y. Li, K. McGee-Estrada, M. Stremlau, S. Welikala, Z. Si, A. Engelman, and J. Sodroski. 2006. Requirements for capsid-binding and an effector function in TRIMCyp-mediated restriction of HIV-1. Virology 351:404-419. [DOI] [PubMed] [Google Scholar]
- 16.Diaz-Griffero, F., A. Kar, M. Lee, M. Stremlau, E. Poeschla, and J. Sodroski. 2007. Comparative requirements for the restriction of retrovirus infection by TRIM5α and TRIMCyp. Virology 369:400-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diaz-Griffero, F., M. Perron, K. McGee-Estrada, R. Hanna, P. V. Maillard, D. Trono, and J. Sodroski. 2008. A human TRIM5α B30.2/SPRY domain mutant gains the ability to restrict and prematurely uncoat B-tropic murine leukemia virus. Virology 378:233-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dickson, R. B., L. Beguinot, J. A. Hanover, N. D. Richert, M. C. Willingham, and I. Pastan. 1983. Isolation and characterization of a highly enriched preparation of receptosomes (endosomes) from a human cell line. Proc. Natl. Acad. Sci. USA 80:5335-5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douglas, C. C., D. Thomas, J. Lanman, and P. E. Prevelige, Jr. 2004. Investigation of N-terminal domain charged residues on the assembly and stability of HIV-1 CA. Biochemistry 43:10435-10441. [DOI] [PubMed] [Google Scholar]
- 20.Fassati, A., and S. P. Goff. 2001. Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 75:3626-3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forshey, B. M., U. von Schwedler, W. I. Sundquist, and C. Aiken. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667-5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franke, E. K., H. E. Yuan, and J. Luban. 1994. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372:359-362. [DOI] [PubMed] [Google Scholar]
- 23.Gamble, T. R., F. F. Vajdos, S. Yoo, D. K. Worthylake, M. Houseweart, W. I. Sundquist, and C. P. Hill. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285-1294. [DOI] [PubMed] [Google Scholar]
- 24.Ganser-Pornillos, B. K., A. Cheng, and M. Yeager. 2007. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131:70-79. [DOI] [PubMed] [Google Scholar]
- 25.Ganser, B. K., S. Li, V. Y. Klishko, J. T. Finch, and W. I. Sundquist. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80-83. [DOI] [PubMed] [Google Scholar]
- 26.Gatanaga, H., D. Das, Y. Suzuki, D. D. Yeh, K. A. Kussain, A. K. Ghosh, and H. Mitsuya. 2006. Altered HIV-1 Gag protein interactions with cyclophilin A (CypA) on the acquisition of H219Q and H219P substitutions in the CypA binding loop. J. Biol. Chem. 281:1241-1250. [DOI] [PubMed] [Google Scholar]
- 27.Gurer, C., A. Hoglund, S. Hoglund, and J. Luban. 2005. ATPγS disrupts human immunodeficiency virus type 1 virion core integrity. J. Virol. 79:5557-5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hatziioannou, T., D. Perez-Caballero, S. Cowan, and P. D. Bieniasz. 2005. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 79:176-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howard, B. R., F. F. Vajdos, S. Li, W. I. Sundquist, and C. P. Hill. 2003. Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Biol. 10:475-481. [DOI] [PubMed] [Google Scholar]
- 30.Javanbakht, H., F. Diaz-Griffero, W. Yuan, D. F. Yeung, X. Li, B. Song, and J. Sodroski. 2007. The ability of multimerized cyclophilin A to restrict retrovirus infection. Virology 367:19-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keckesova, Z., L. M. Ylinen, and G. J. Towers. 2006. Cyclophilin A renders human immunodeficiency virus type 1 sensitive to Old World monkey but not human TRIM5α antiviral activity. J. Virol. 80:4683-4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kotov, A., J. Zhou, P. Flicker, and C. Aiken. 1999. Association of Nef with the human immunodeficiency virus type 1 core. J. Virol. 73:8824-8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanman, J., J. Sexton, M. Sakalian, and P. E. Prevelige, Jr. 2002. Kinetic analysis of the role of intersubunit interactions in human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol. 76:6900-6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leschonsky, B., C. Ludwig, K. Bieler, and R. Wagner. 2007. Capsid stability and replication of human immunodeficiency virus type 1 are influenced critically by charge and size of Gag residue 183. J. Gen. Virol. 88:207-216. [DOI] [PubMed] [Google Scholar]
- 35.Li, S., C. P. Hill, W. I. Sundquist, and J. T. Finch. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407:409-413. [DOI] [PubMed] [Google Scholar]
- 36.Lin, T. Y., and M. Emerman. 2006. Cyclophilin A interacts with diverse lentiviral capsids. Retrovirology 3:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luban, J., K. L. Bossolt, E. K. Franke, G. V. Kalpana, and S. P. Goff. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067-1078. [DOI] [PubMed] [Google Scholar]
- 38.Luban, J. 2007. Cyclophilin A, TRIM5, and resistance to human immunodeficiency virus type 1 infection. J. Virol. 81:1054-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuoka, S., E. Dam, D. Lecossier, F. Clavel, and A. J. Hance. 2009. Modulation of HIV-1 infectivity and cyclophilin A-dependence by Gag sequence and target cell type. Retrovirology 6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakai, M., and T. Goto. 1996. Ultrastructure and morphogenesis of human immunodeficiency virus. J. Electron Microsc. 45:247-257. [DOI] [PubMed] [Google Scholar]
- 41.Nisole, S., C. Lynch, J. P. Stoye, and M. W. Yap. 2004. A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc. Natl. Acad. Sci. USA 101:13324-13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owens, C. M., B. Song, M. J. Perron, P. C. Yang, M. Stremlau, and J. Sodroski. 2004. Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J. Virol. 78:5423-5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perron, M. J., M. Stremlau, M. Lee, H. Javanbakht, B. Song, and J. Sodroski. 2007. The human TRIM5α restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J. Virol. 81:2138-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pornillos, O., B. K. Ganser-Pornillos, B. N. Kelly, Y. Hua, F. G. Whitby, C. D. Stout, W. I. Sundquist, C. P. Hill, and M. Yeager. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenwirth, B., A. Billich, R. Datema, P. Donatsch, F. Hammerschmid, R. Harrison, P. Hiestand, H. Jaksche, P. Mayer, P. Peichl, et al. 1994. Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM 811, a nonimmunosuppressive cyclosporine analog. Antimicrob. Agents Chemother. 38:1763-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saphire, A. C., M. D. Bobardt, and P. A. Gallay. 2002. Trans-complementation rescue of cyclophilin A-deficient viruses reveals that the requirement for cyclophilin A in human immunodeficiency virus type 1 replication is independent of its isomerase activity. J. Virol. 76:2255-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sayah, D. M., E. Sokolskaja, L. Berthoux, and J. Luban. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430:569-573. [DOI] [PubMed] [Google Scholar]
- 48.Schaller, T., L. M. Ylinen, B. L. Webb, S. Singh, and G. J. Towers. 2007. Fusion of cyclophilin A to Fv1 enables cyclosporine-sensitive restriction of human and feline immunodeficiency viruses. J. Virol. 81:10055-10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sokolskaja, E., D. M. Sayah, and J. Luban. 2004. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol. 78:12800-12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song, C., and C. Aiken. 2007. Analysis of human cell heterokaryons demonstrates that target cell restriction of cyclosporine-resistant human immunodeficiency virus type 1 mutants is genetically dominant. J. Virol. 81:11946-11956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stremlau, M., M. Perron, M. Lee, Y. Li, B. Song, H. Javanbakht, F. Diaz-Griffero, D. J. Anderson, W. I. Sundquist, and J. Sodroski. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. USA 103:5514-5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thali, M., A. Bukovsky, E. Kondo, B. Rosenwirth, C. T. Walsh, J. Sodroski, and H. G. Gottlinger. 1994. Functional association of cyclophilin A with HIV-1 virions. Nature 372:363-365. [DOI] [PubMed] [Google Scholar]
- 53.Towers, G. J., T. Hatziioannou, S. Cowan, S. P. Goff, J. Luban, and P. D. Bieniasz. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9:1138-1143. [DOI] [PubMed] [Google Scholar]
- 54.Virgen, C. A., Z. Kratovac, P. D. Bieniasz, and T. Hatziioannou. 2008. Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc. Natl. Acad. Sci. USA 105:3563-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Schwedler, U. K., K. M. Stray, J. E. Garrus, and W. I. Sundquist. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439-5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wainberg, M. A., A. Dascal, N. Blain, L. Fitz-Gibbon, F. Boulerice, K. Numazaki, and M. Tremblay. 1988. The effect of cyclosporine A on infection of susceptible cells by human immunodeficiency virus type1. Blood 72:1904-1910. [PubMed] [Google Scholar]
- 57.Welker, R., H. Hohenberg, U. Tessmer, C. Huckhagel, and H. G. Kräusslich. 2000. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J. Virol. 74:1168-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang, R., and C. Aiken. 2007. A mutation in alpha helix 3 of CA renders human immunodeficiency virus type 1 cyclosporine-resistant and dependent: rescue by a second-site substitution in a distal region of CA. J. Virol. 81:3749-3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yap, M. W., G. B. Mortuza, I. A. Taylor, and J. P. Stoye. 2007. The design of artificial retroviral restriction factors. Virology 365:302-314. [DOI] [PubMed] [Google Scholar]
- 60.Yin, L., D. Braaten, and J. Luban. 1998. Human immunodeficiency virus type 1 replication is modulated by host cyclophilin A expression levels. J. Virol. 72:6430-6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ylinen, L. M., T. Schaller, A. Price, A. J. Fletcher, M. Noursadeghi, L. C. James, and G. J. Towers. 2008. Cyclophilin A levels dictate infection efficiency of A92E and G94D HIV-1 capsid escape mutants. J. Virol. 83:2044-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.