

Abstract
Marek's disease virus (MDV) is a lymphotropic alphaherpesvirus that induces fatal rapid-onset T-cell lymphomas in chickens, its natural host. The MDV-encoded nuclear oncoprotein Meq is essential for lymphomagenesis and acts as a regulator of transcription. Meq has structural features, including a basic domain adjacent to a leucine zipper motif (B-ZIP), that suggest it is related to the Jun/Fos family of transcription factors. Via the leucine zipper, Meq can form homodimers or heterodimerize with c-Jun. Meq/Meq homodimers are associated with transrepression, and Meq/Jun heterodimers can transactivate target genes carrying an AP-1-like binding site. In order to determine the role of the leucine zipper and of Meq dimerization in T lymphomagenesis, specific point mutations were engineered into the highly oncogenic RB-1B strain of MDV to produce virus completely lacking a functional Meq leucine zipper (RB-1B MeqBZIP/BZIP) or virus encoding Meq that cannot homodimerize but can still bind to c-Jun and an AP-1-like site on DNA (RB-1B MeqHom/Hom). Both of these mutant viruses were capable of replication in cultured chicken embryo fibroblasts. However both mutations resulted in a complete loss of oncogenicity, since no lymphomas were produced up to 90 days postinfection in experimentally infected chicks. We conclude that the leucine zipper is necessary for the oncogenic activity of Meq and/or the efficient establishment of long-term MDV latency in T cells. Moreover, it appears that the ability to form homodimers is an absolute requirement and the ability to bind c-Jun alone is insufficient for the T-cell lymphomagenesis associated with virulent MDV.
Marek's disease (MD) is a common lymphoproliferative and neurotropic disease of poultry caused by the highly contagious alphaherpesvirus called Marek's disease virus (MDV). Because of its contagious nature, rapid disease onset, and persistence in both the host and environment, MDV is arguably one of the most economically significant pathogens of poultry. More than 5 billion doses of MDV vaccine are used annually in an attempt to control the disease (16). The pathogenesis of MD is very complex. Infection is via the respiratory route and is quickly followed by a cytolytic infection of mainly B cells in lymphoid organs. Subsequently, activated T cells (largely of the CD4+ phenotype) that are recruited to the site of cytolytic infection become latently infected and transformed. This leads to neoplastic T-cell lesions in visceral organs, and infiltrating lymphocytes can cause edema in peripheral nerves and produce paralysis (3, 26). Virus also replicates in the feather follicle epithelium, the site of a productive infection that allows shedding and horizontal spread. Although MDV is an alphaherpesvirus, biologically it more closely resembles the lymphotropic oncogenic gammaherpesviruses, such as Epstein-Barr virus, Kaposi's sarcoma-associated herpesvirus, and herpesvirus saimiri (7).
A gene within the Marek's EcoRI Q genomic fragment that forms part of both the terminal and internal repeats is designated Meq. The protein it encodes—Meq—has a nuclear localization, is one of the few viral genes expressed in lytic and latent infections, and is highly expressed in both MD tumor tissues and T-lymphoblastoid cell lines derived from cultured lymphoma explants. This 339-amino-acid protein has features suggesting that it functions as a DNA-binding transcription factor. It has a basic region/leucine zipper domain (B-ZIP) that closely resembles those found in c-Fos and c-Jun, and through the B-ZIP, it can homodimerize and also form functional heterodimers with c-Jun (Fig. 1A). Meq/Jun heterodimers bind with high affinity to DNA sequences (resembling tetradecanoyl phorbol acetate response elements and cyclic AMP-dependent response elements) called Meq-responsive elements (MERE I) and can activate linked reporter genes in in vitro assays through them. In contrast, Meq/Meq homodimers bind a second, unique class of binding site called MERE II (ACACA) and appear to act as repressors of transcription in in vitro assays (13, 23). Although the critical target genes in the chicken genome that are regulated through MEREs have yet to be identified, microarray expression profiling of Meq-expressing DF-1 chicken embryo fibroblasts (CEF) has revealed Meq-mediated activation of genes, such as those encoding JTAP-1, JAC, and HB-EGF, that are also downstream targets of the avian retroviral oncogene v-Jun (10). Meq heterodimers have been shown to be essential for lymphomagenesis; this was determined using a recombinant MDV containing a mutant Meq protein, with a substituted GCN4 leucine zipper, that cannot heterodimerize with cJun but remains able to homodimerize (25). Meq/Jun dimers can also transactivate the Meq promoter via an AP-1-like MERE I site, suggesting positive autoregulation might occur. Conversely, it has been proposed that Meq/Meq homodimers may inhibit MDV lytic replication and hence initiate and/or sustain latency (12, 20). This is because Meq homodimers bind MERE II sites located at the putative origin of lytic replication of the MDV genome (MDV Orilyt) and have been shown in transient reporter assays to repress the transcription of the flanking enhancer/bidirectional promoter for the genes encoding the early phosphoproteins pp38 and pp14 (11). Consistent with Meq acting as both a transactivator and a transrepressor of transcription, in addition to including a proline-rich C-terminal activation domain (23), the N terminus of Meq has a region that can interact with the CtBP corepressor complex via a consensus (20PLDLS24) CtBP-binding site (5, 8). The interaction of Meq and CtBP is essential for Meq function in MDV oncogenicity and suggests that Meq might recruit a corepressor complex that is capable of epigenetic silencing of a critical subset of target genes or modifying the function of CtBP in some way (5, 6, 9).
FIG. 1.

Dimerization mutations in the Meq gene. (A) Schematic representation of Meq as either a homodimer or a heterodimer with c-Jun and the DNA-binding specificity it has for either MERE I or MERE II DNA-binding sites. (B) Schematic of the B-ZIP dimer rule for the g-e pair and the hydrophobic “a” and “d” core. The structure of the alpha-helix of the B-ZIP protein is shown as an end view, revealing the hydrophobic core created by the “a” and “d” residues. (C) The amino acid of the MeqWT leucine zipper and the corresponding mutations that were introduced to produce the MeqBZIP and MeqHom constructs.
Meq has been reported to possess transforming activity in cell culture, including the capacity to morphologically transform fibroblasts, induce anchorage-independent growth of rodent fibroblasts, and suppress apoptosis (13, 19). The precise molecular mechanisms underlying these phenotypes are not known, but it has been suggested that Meq/Jun-mediated activation of a v-Jun-like cascade of gene expression is central to the transformation process (10). In addition, anecdotal reports suggest that Meq may bind the tumor suppressor proteins p53 and pRb. Although there are no published data on either of these proposed interactions, the latter is consistent with the presence of an LXCXE pRb-binding motif in Meq (117LACHE121). Meq is clearly a good candidate for the role of a major transforming oncogene in MDV-mediated T-cell neoplasia. Recent studies, using recombinant forms of MDV carrying either deletions or mutations of Meq made in a bacterial artificial chromosome (BAC) system or cosmid system, have indeed confirmed that Meq is essential for the induction of T-cell lymphomas in chicks and that its interaction with CtBP is an absolute requirement in this lymphomagenesis (5, 14). Given that Meq can regulate viral and cellular genes, it seems likely that it is necessary for both the establishment of viral latency in T cells and the transformation of these cells and their progeny into malignant lymphomas.
The leucine zipper in Meq is essential for both homo- and heterodimerization and efficient DNA binding, and it has also been shown that an MDV containing a Meq that cannot heterodimerize, but can homodimerize, is unable to induce T-cell lymphomas (25). However, this does not exclude the possibility that Meq homodimerization is important for lymphomagenesis. Therefore, we asked whether Meq homodimers are also necessary for the induction of T-cell lymphomas in chicks. To investigate this, two mutants were constructed, in one of which alanine residues were substituted for three of the four leucines that constitute the hydrophobic face of the zipper motif (MeqBZIP). The “zipper” function in this mutant is completely disrupted and is therefore no longer capable of facilitating homodimerization or interacting with c-Jun. A second, more refined mutant (MeqHom) was also made; it fails to form homodimers but still binds c-Jun and DNA. Schematics showing these mutations can be seen in Fig. 1. When, after validation in vitro, the mutant genes were recombined into both copies of the Meq locus in the pRB-1B BAC and the resulting viruses were used to infect chicks, both recombinants appeared completely attenuated, since no tumors were detected and the infected birds remained perfectly healthy even after 90 days. These results showed that the formation of Meq dimers is essential for lymphomagenesis and are therefore consistent with the v-Jun model for transformation described above. However, more surprising was the demonstration of the absolute requirement for Meq/Meq homodimers in the induction of tumors. These results suggested that repression of a subset of viral and/or cellular target genes by Meq/Meq homodimers is also critical and that the interaction with c-Jun, although it may be necessary, is by itself insufficient for T-cell transformation.
MATERIALS AND METHODS
Vector construction.
The plasmid pGEMt-Meq2.2 contains cDNA comprising the Meq open reading frame from pRB-1B and 1.2 kb of flanking DNA. The plasmid was constructed by PCR amplification from pRB-1B using the primers Meq2.2_For and Meq2.2_Rev (Table 1). The pGEMt-MeqHom2.2 plasmid was constructed by PCR amplification from pRB-1B using the primers MeqAvrII_For with MeqHom_rev and MeqHom_for with MeqXbaI_rev. The products were digested with AvrII and BglII or BglII and KpnI, respectively, and ligated into pGEMt-Meq2.2 digested with AvrII and KpnI. The pGEMt-MeqBZIP2.2 vector was constructed as described above for pGEMt-MeqHom2.2 using the primers MeqBZIP_rev and MeqBZIP_for instead of MeqHom_rev and MeqHom_for. pcDNA3.1 derivatives were generated by PCR amplification from the appropriate constructs with the primers MeqAvrII_For and MeqXbaI_Rev. Myc-tagged derivatives were constructed by using MeqAvrII-Myc_For, and hemagglutinin (HA) derivatives were constructed by using MeqAvrII-HA_For. pGEX constructs were generated by PCR amplification of the region containing the first 147 amino acids of Meq protein from the appropriate constructs with MeqGSTEcoRI_For and MeqGSTBamHI_Rev. pST76K_SR derivatives were generated by subcloning them from pGEMt-Meq2.2 (or the appropriate mutant) using SphI and SacI. All constructs were validated by DNA sequence analysis.
TABLE 1.
Primers used for construction of vectors
| Primer | Sequence |
|---|---|
| Meq2.2_For | ATTGAGCTCCTGTCGTCTGCATTGTTC |
| Meq2.2_Rev | TAAGCATGCATATACCCCCCCTCCCC |
| MeqAvrII_For | CACTGATTCCTAGGCAGGCGTCTCT |
| MeqXbaI_rev | GCTCTAGAGCTCAGGGTCTCCCGTCACCTGGAAAC |
| MeqHom_for | TCGAGATCTGGAGACTGAGAAGACGTCCCTGGAGGTACAGTTG |
| MeqHom_rev | CTTAGATCTCGAATTTCCTTCTGTAGGTGTTCCTTCTCCCTTTCCAGCTCTTCTGTTTCTTCATG |
| MeqBZIP_for | CACGCGCGCAAGGAAATTCGAGATGCAAGGACTGAGTGC |
| MeqBZIP_rev | AAGGTACCCTTGCGCGCGTGTTCATTGGCCCTCTGCGCCTCTTCACA |
| MeqAvrII-Myc_For | ATACCTAGGGCCACCATGGAACAAAAACTCATCTCAGAAGAGGATCTGATGTCTCAGGAGCCAGAGCCG |
| MeqAvrII-HA_For | ATACCTAGGGCCACCATGGCATACCCATACGATGTTCCAGATTACGCTATGTCTCAGGAGCCAGAGCCG |
| MeqGSTEcoRI_Rev | GTGTAAAGGATCCTCTCAGGAGCCAGAGCC |
| MeqGSTBamHI_Rev | ACAGAATTCAGGTTGGGAACCGGAGCAATG |
Immunoprecipitations and Western blotting.
Immunoprecipitations and Western blot assays were performed essentially as described previously (8). The antibodies used were rabbit anti-Meq (raised against GST-MeqWT1-147 [5]at the Institute for Animal Health, Compton), rabbit-anti-HA (Covance), mouse-anti-Myc (Santa Cruz), mouse-anti-CtBP1 (BD Bioscience), mouse-anti-cJun (BD Bioscience), and rabbit-anti-Mouse (Dako).
Construction of mutant viruses.
Mutagenesis of the BAC clone pRB-1B5 was carried out essentially as described previously (5). Mutant clones were generated by using a two-step markerless replacement technique (22). The single-copy mutant (pRB-1B-Meqmut/wt) was selected by restriction digestion analysis of PCR products. For the construction of BAC clones with mutations in both copies of Meq (pRB-1B-Meqmut/mut), the process was repeated with the same pST76K_SR construct. A full revertant (pRB-1B-mut/mutR) was constructed by twice repeating the process on the double mutant using the wild-type Meq shuttle vector. BAC integrity was confirmed by pulsed-field agarose gel electrophoresis analysis, and the mutations in each of the clones were confirmed by restriction digestion of PCR fragments and further DNA sequence analysis. Virus stocks were made as previously described (5).
GST fusion proteins.
Glutathione S-transferase (GST) protein was produced essentially as described previously (8).
EMSAs.
DNA-binding assays were performed in a 20-μl volume of electrophoretic mobility shift assay (EMSA) buffer (20 mM HEPES, pH 7.9, 20 mM NaCl, 2 mM MgCl2, 10% glycerol) supplemented with 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and 1 μg poly(dI-dC). One hundred nanograms of each GST protein was used, and each EMSA reaction mixture contained 35 fmol of the appropriate double-stranded oligonucleotide, derived from the MDV Orilyt (5′-TGC TCA TTT GCA TAC ACA TCA CGT GAT AGT-3′), the AP-1-like site in the Meq promoter (5′-ACG ATA GTC ATG CAT GAC GTG G-3′), or a canonical AP-1 site (5′-CGC TTG ATG AGT CAG CCG GAA-3′) (Promega, Madison, WI). Oligonucleotides were annealed by heating them to 95°C and cooling them for 2 h, followed by polyacrylamide gel electrophoresis (PAGE) purification. Each oligonucleotide also contained a 5′ GG overhang that was 32P labeled by “filling in” with dCTP using the Klenow polymerase. Protein-DNA complexes were formed at room temperature for 20 min and then loaded onto a 5% polyacrylamide gel cast in 0.3× Tris-borate-EDTA and run at 10 V/cm at 4°C for 2 h.
Animal experiments.
All experiments were carried out in accordance with the United Kingdom Home Office guidelines using the specific-pathogen-free inbred line P (B19/19) (4) and the outbred line Rhode Island Red (RIR) obtained from the Poultry Production Unit of the Institute for Animal Health. For pathogenicity studies, 14-day-old birds were infected with 1,000 PFU of the cell-associated virus stocks by the intra-abdominal route. The MDV-infected birds and the noninfected control birds were maintained in separate HEPA-filtered rooms. Blood (150 μl in 3% sodium citrate) and feather samples were collected at different times after infection to determine the in vivo replication rates of the viruses.
Determination of the incidence of MD.
All the infected birds were examined for gross lesions at postmortem, and representative tissue samples were collected in 10% buffered formalin for microscopic examination. The tissues were processed and embedded in paraffin wax, and 4- to 5-μm tissue sections were stained with hematoxylin and eosin and examined for histopathological changes.
qPCR analysis of virus replication in vivo and in vitro.
DNA was extracted from either infected CEF, peripheral blood leukocytes (PBL), or feather tips at the indicated time points using a DNEasy kit (Qiagen) according to the manufacturer's protocols. Quantitative PCR (qPCR) was carried out as described previously for PBL and feathers (1) and for infected CEF (2) with a Meq-specific probe.
RESULTS
Site-specific mutations that disrupt Meq B-ZIP function.
Site-specific mutations that disrupt B-ZIP function were introduced into Meq. These mutations were designed to favor the formation of heterodimers and prevent the formation of homodimers (MeqHom) or to completely disrupt dimerization (MeqBZIP).
The controlling elements in the dimerization specificities of two interacting leucine zipper proteins are the g-e interhelical interactions (Fig. 1B) (27). These interactions effectively determine whether Meq forms homo- or heterodimers. The mutations introduced into MeqHom (Fig. 1C) were primarily mutations in the g and e positions of the leucine zipper designed to create interhelical acidic-acidic interactions unfavorable to homodimerization, similar to those seen in the c-Fos leucine zipper (18). As in c-Fos, the interhelical interactions between MeqHom and c-Jun remain favorable (Fig. 2), so it retains its capacity to interact with c-Jun. Secondary mutations in the “a” positions of the first two heptad repeats and the fourth repeat were also created. The basic amino acids lysine and arginine were introduced into these positions to destabilize a homodimer and favor heterodimerization. The leucine zipper is stabilized by the internal hydrophobic core formed from the a-d interhelical interactions (Fig. 1B). Therefore, by replacing three of the four B-ZIP leucines in MeqHom with alanine, the hydrophobic core was disrupted and MeqBZIP was created (Fig. 1C). MeqBZIP was expected to lack the ability to dimerize with itself or any other leucine zipper proteins.
FIG. 2.

Prediction of the dimerization potential of MeqHom. The dimerization potential of the homodimer mutation was determined though the interacting g-e pairs. The lines with arrowheads indicate favorable interactions between charged amino acids, while the lines without arrowheads indicate unfavorable pairs. The interhelical interactions between MeqHom and c-Jun remain favorable; in contrast, an interaction with another copy of itself is not possible.
Validation of the mutations in protein-protein interaction assays.
For biochemical evidence that the site-specific mutations introduced into the Meq gene produced proteins that behaved as predicted, immunoprecipitation experiments were performed. Initially, in order to determine whether the mutants were capable of forming homodimers, Myc or HA epitope-tagged Meq expression constructs were used. Plasmid DNAs encoding tagged variants of each mutant were cotransfected into DF-1 cells, and the lysates were subjected to immunoprecipitation using an anti-HA antibody and a Western blot probed with anti-Myc (Fig. 3A). These experiments showed that while wild-type Meq (MeqWT) and MeqCtBP can form homodimers, both the MeqHom and MeqBZIP mutations interfered with the interaction and no homodimers were seen (Fig. 3A). It was noted that the MeqHom mutant had reduced mobility on sodium dodecyl sulfate (SDS)-PAGE analysis. The reason for this is unknown; however, restriction analysis of the expression constructs and complete DNA sequencing of the MeqHom plasmid revealed no insertions or mutations. The apparent size shift was present in multiple expression systems—35S-labeled in vitro translation (data not shown), transfected cell lysates probed by Western blotting (Fig. 3A), and GST expressed protein (Fig. 4C) —and must have resulted from the changes in amino acid sequence engineered into Meq. Reduced mobility of proteins during SDS-PAGE analysis has previously been reported as being caused by changes in charged residues (15). The size shift of MeqHom is probably due to the changed composition of charged amino acids in the protein.
FIG. 3.
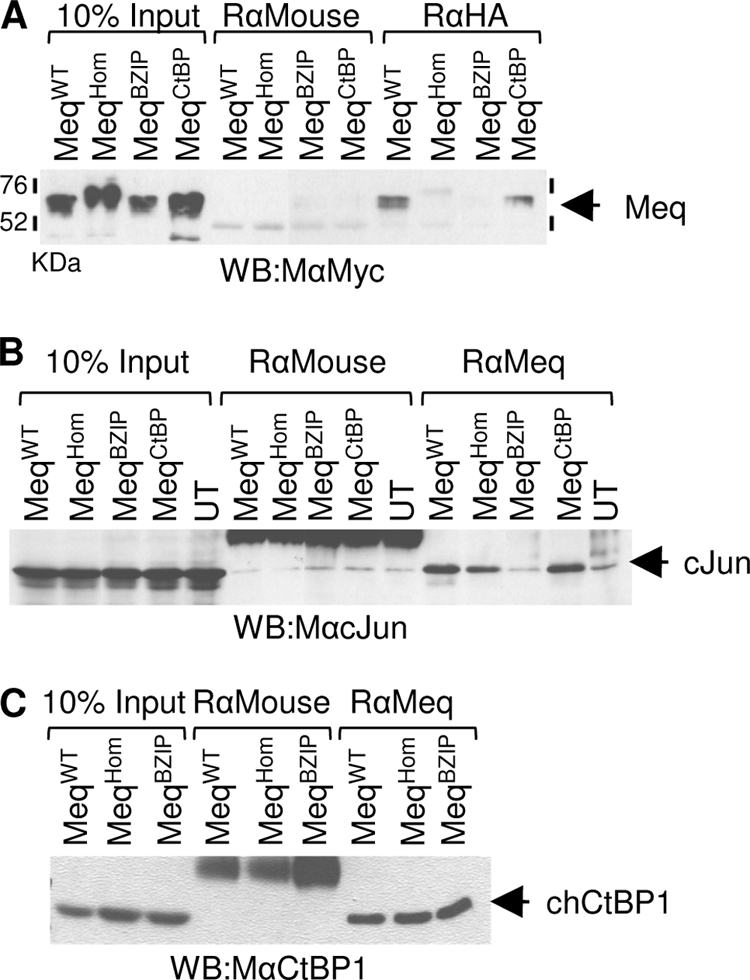
B-ZIP mutations alter the ability of Meq to form homo- and heterodimers. Analysis was performed after the transfection of DF-1 cells with the expression vectors indicated. (A) Meq proteins containing the putative dimerization mutations N-terminally tagged with either HA or Myc epitopes were subjected to immunoprecipitation with a rabbit anti-HA or a control antibody. After SDS-PAGE and Western blotting (WB), they were probed with an anti-Myc antibody. (B) Untagged Meq mutants were immunoprecipitated with rabbit anti-Meq or a control antibody and detected, after Western blotting, with anti-c-Jun antibody. UT indicates untransfected cells expressing no Meq. Only MeqBZIP failed to bind c-Jun. Because a large excess of control rabbit anti-mouse antibody was used, it is seen as a cross-reactive band in the five central lanes. (C) Similar analysis showed that B-ZIP dimerization mutations have no effect on the interaction between chicken CtBP (chCtBP1) and Meq. After transfection of MeqWT, MeqHom, or MeqBZIP, all proteins coimmunoprecipitated equally efficiently with endogenous CtBP.
FIG. 4.

B-ZIP mutations alter the ability of Meq to bind DNA. (A) EMSAs using GST or GST-Meq1-147 containing the appropriate mutations were used to shift a [32P]dCTP end-labeled oligonucleotide (Oligo) containing a MERE II binding site corresponding to a region found in the MDV Orilyt. MeqWT bound the probe, but both MeqHom and MeqBZIP failed to bind and created a band shift. (B) GST or GST-Meq1-147 containing the indicated mutations mixed with GST-c-Jun was used to shift a [32P]dCTP end-labeled oligonucleotide containing either a canonical AP-1 site or the AP-1-like site in the Meq promoter (see Materials and Methods). Both MeqWT and MeqHom formed complexes with and shifted the AP-1 probe DNA, but MeqBZIP produced no corresponding band shift. (C) Purified GST fusion proteins were resolved by SDS-PAGE and then visualized using Coomassie blue.
Further immunoprecipitations were performed using anti-Meq antibodies, followed by Western blots probed with anti-c-Jun, to assay for the formation of heterodimers. Lysates were made from DF-1 cells, which express c-Jun constitutively, 48 h after they had been transfected with the various Meq expression vectors. Immunoprecipitation with the Meq-specific polyclonal antibody showed that Meq can interact with the endogenous c-Jun and that this capacity was retained by MeqHom. In contrast, the dimerization mutant MeqBZIP was unable to bind c-Jun above background levels in these assays. Also included in the experiment was the previously described MeqCtBP mutant, which retained the capacity to form dimers with c-Jun (Fig. 3B).
Since we previously described how Meq binds to CtBP through the conserved PLDLS motif (5) and this interaction is essential for MDV oncogenicity, we established that the B-ZIP mutations induced no secondary effects preventing CtBP binding (Fig. 3C) that might complicate the interpretation of the in vivo analyses described below.
Validation of mutations by DNA EMSAs.
In order to determine whether the dimerization mutations had the predicted effect on the ability of Meq to recruit a binding partner and bind specific DNA sequences, EMSAs were carried out. To investigate the DNA-binding characteristics of Meq homodimers, an oligonucleotide probe corresponding to the MDV Orilyt region that includes a MERE II site was used. The EMSAs showed that while GST-MeqWT1-147 bound to the probe, in contrast, GST, GST-MeqHom1-147, and GST-MeqBZIP1-147 were all unable to bind (Fig. 4A).
Two other oligonucleotides, one corresponding to a canonical AP-1 site and the other to part of the Meq promoter, were used to investigate whether MeqHom retained its ability to bind AP-1 and AP-1-like sites as a heterodimer with c-Jun. EMSAs were carried out using GST, GST-MeqWT1-147, or the dimerization mutants GST-MeqHom1-147 and GST-MeqBZIP1-147. Each binding reaction also contained the binding partner GST-c-Jun (Fig. 4B). The Meq-cJun complexes bound to the probe, and it was clearly demonstrated that MeqHom retained its ability to bind DNA in a complex with c-Jun. In contrast, MeqBZIP failed to bind DNA, and the probe was not shifted (Fig. 4B). The difference in mobility between the band shifts of MeqWT-cJun and MeqHom-cJun corresponds to the difference previously shown by SDS-PAGE (Fig. 3A). The GST fusion proteins that were used in the assays are shown in Fig. 4C, and again, the difference in mobility is apparent.
Construction and validation of mutant RB-1B BAC clones.
Having established that the dimerization mutants of Meq behaved in vitro as predicted, we examined whether the formation of dimers was important in MDV-induced oncogenicity. Starting with the infectious BAC clone (pRB-1B5) of the highly oncogenic RB-1B strain of MDV (21), reverse genetics was used to construct viruses carrying the mutations in Meq. We used the markerless replacement technique (22) with the pRB-1B5 clone to construct four MDV mutants. In the first virus, the homodimerization-null mutant (MeqHom) gene was inserted into both copies of the Meq locus; this BAC construct was called pRB-1B-MeqHom/Hom. The procedure was repeated to make the full revertant pRB-1B-MeqHom/HomR, in which the mutation at each Meq locus was repaired to wild-type sequence. The MeqBZIP dimerization-null mutant gene was also introduced into both copies of Meq, making pRB-1B-MeqBZIP/BZIP. Subsequently a full revertant of this was also made, pRB-1B-MeqBZIP/BZIPR. A schematic showing the sequence of events in the construction of all the BAC clones is shown in Fig. 5A. The gross integrity of each of the BAC DNAs was established by digestion with the restriction enzyme BamHI or EcoRI and pulsed-field agarose gel electrophoresis (Fig. 5B). To confirm that the mutations did not affect the replication of MDV in vitro, CEF cultures were transfected with the BAC DNA to allow reconstitution of the virus. MDV-specific plaques were detected in cells transfected with each of the BAC DNA clones, and Meq was detected in each by immunofluorescence microscopy (data not shown). Comparison of the in vitro replication of the pRB1B-derived mutants and revertants to that of the parental BAC virus by TaqMan real-time qPCR also showed no significant differences between the viruses (Fig. 5C).
FIG. 5.

Insertion of the mutations into pRB-1B5. (A) Schematic showing the construction of the BAC clones used in the animal experiments: the wild type (pRB-1B5), a double-homodimerization mutant (pRB-1BHom/Hom), and its full revertant (pRB-1BHom/HomR). Also shown are the double-dimerization-null mutant clone (pRB-1BBZIP/BZIP) and its full revertant (pRB-1BBZIP/BZIPR). The intermediate constructs that were not used in the animal experiments are also illustrated (pRB-1BWT/Hom, pRB-1BHom(R)/Hom, pRB-1BWT/BZIP, and pRB-1BBZIP(R)/BZIP). (B) To determine whether the markerless recombination had made any significant second-site mutations or rearrangements, BAC DNA was digested with BamHI or EcoRI restriction enzyme and analyzed by ethidium bromide-stained 1% agarose pulsed-field gel electrophoresis. All of the recombinant MDV BACs were indistinguishable from the wild type. (C) To compare the in vitro replication of the mutant and revertant viruses with that of the parental pRB1B BAC, the genome copy number per 10,000 cells was determined at the indicated times by TaqMan real-time qPCR after inoculation of 100 PFU of each virus into primary CEF. This showed there were no significant differences between the parental and recombinant viruses. The standard error of the mean for each group is shown.
Infection of chicks reveals an essential role for the B-ZIP in T-cell lymphomagenesis.
We next determined the oncogenic potential of the dimerization-defective Meq mutant pRB-1B5 BAC-derived viruses. Initially, experimental infection of 14-day-old inbred congenic line P (B19/19) chicks (n = 12) with pRB-1B5 BAC-derived virus produced tumors in 100% of the birds, and they were killed within 70 days of infection. In contrast, both the dimerization- and homodimerization-null mutations in the Meq gene of pRB-1B5 resulted in a complete loss of oncogenicity, as could be seen when similar numbers of birds were infected with pRB-1B-MeqHom/Hom or pRB-1B-MeqBZIP/BZIP virus and no tumors were produced (Fig. 6A and Table 2). The loss of oncogenicity of the pRB-1B-MeqHom/Hom and pRB-1B-MeqBZIP/BZIP viruses was not restricted to the genetically susceptible inbred line P strain of chickens, because similar results were obtained in more genetically diverse RIR birds (n = 9) (Fig. 6B and Table 2). However, the RIR chicks showed more modest differences in survival because the more resistant chicken genotype resulted in delayed mortality for all the viruses. Examination of the birds postmortem to determine the MD incidence showed that in both chicken lines the mutant viruses were completely attenuated in comparison to wild-type and revertant viruses. No gross or microscopic lesions were detected during a systematic postmortem examination; the birds infected with pRB-1BHom/Hom or pRB-1BBZIP/BZIP were indistinguishable from uninfected controls. In contrast, although revertant viruses were generally less virulent than the wild-type RB-1B BAC, it is quite clear that the reintroduction of MeqWT in the revertants restored the oncogenic phenotype. A full breakdown of MD incidence is shown in Table 2. The results clearly indicated that the leucine zipper is crucial for the development of T-cell lymphomas. Furthermore, the complete attenuation of pRB-1BHom/Hom indicated that the formation of heterodimers between Meq and c-Jun alone is insufficient for MDV-induced oncogenicity. Rescue of oncogenicity in the revertant pRB-1B-MeqHom/HomR and pRB-1B-MeqBZIP/BZIPR viruses ruled out any major contribution from second-site mutations to the attenuation of the B-ZIP mutants; this is discussed in more detail below.
FIG. 6.

Analysis of the survival of chickens infected with the BAC-derived MDVs carrying B-ZIP mutations in the Meq gene. Groups of 14-day-old line P (n = 12) or RIR (n = 9) birds were infected with 1,000 PFU of the RB-1B strain of MDV, BAC-derived pRB-1B5, or the mutant viruses pRB-1BHom/Hom and pRB-1BBZIP/BZIP and their revertant viruses. All birds were infected by intra-abdominal injection with the viruses. Shown are line P (A) and RIR (B) birds that did not suffer fatal MD expressed as a percentage of infected individuals.
TABLE 2.
Incidence of MD and distribution of lymphoid neoplastic lesions in different organs in two genetic lines of birds experimentally infected with different MDVs
| Birds | Virus | Incidence (no./total) in: |
MD incidence [no./total (%)] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Liver | Heart | Spleen | Kidney | Proventriculus | Gonads | Bursa | Nerves | Muscle | |||
| P line | pRB1B | 7/11 | 3/11 | 0/11 | 8/11 | 3/11 | 4/11 | 0/11 | 3/11 | 0/11 | 11/11 (100) |
| pRB1BHom/Hom | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 (0) | |
| pRB1BHom/HomR | 9/12 | 3/12 | 3/12 | 9/12 | 2/12 | 6/12 | 1/12 | 4/12 | 2/12 | 11/12 (91.7) | |
| pRB1BBZIP/BZIP | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 | 0/12 (0) | |
| pRB1BBZIP/BZIPR | 7/12 | 2/12 | 0/12 | 7/12 | 1/12 | 2/12 | 0/12 | 3/12 | 2/12 | 9/12 (75) | |
| RIR | pRB1B | 6/9 | 2/9 | 1/9 | 3/9 | 0/9 | 2/9 | 1/9 | 2/9 | 0/9 | 8/9 (88.9) |
| pRB1BHom/Hom | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 (0) | |
| pRB1BHom/HomR | 4/9 | 0/9 | 0/9 | 2/9 | 0/9 | 0/9 | 0/9 | 1/9 | 0/9 | 4/9 (44.4) | |
| pRB1BBZIP/BZIP | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 | 0/9 (0) | |
| pRB1BBZIP/BZIPR | 6/9 | 2/9 | 0/9 | 5/9 | 1/9 | 1/9 | 0/9 | 0/9 | 0/9 | 5/9 (55.6) | |
The replication of wild-type and mutant viruses in vivo was assessed using a Meq-specific real-time qPCR that measured the MDV genome copy number in PBL or feather tips of experimentally infected chickens (1). In line P birds, MDV genome copy numbers in the PBL from all of the control groups showed a steady increase during the course of infection, with the pRB-1B5, pRB-1B-MeqHom/HomR, and pRB-1B-MeqBZIP/BZIPR groups all registering 105 to 106 copies per million cells 35 days after infection. Birds from the groups infected with pRB-1B-MeqHom/Hom and pRB-1B-MeqBZIP/BZIP carried a much lower virus load of 102 to 103 copies per million cells, indicating that these mutations influence the replication of MDV in vivo (Fig. 7A). The pattern of replication of the viruses in PBL of the more genetically diverse RIR birds was very similar (Fig. 7B). However, it is interesting that although both mutants showed reduced replication in peripheral blood, the pRB-1B-MeqBZIP/BZIP mutant had the greater defect, since its genome copy number tended to decrease between 5 and 20 days. This indicates that Meq dimerization is necessary for replication in PBL and that there is a feature of MDV replication or persistence in vivo that depends on Meq homodimers.
FIG. 7.

In vivo replication of MDVs carrying B-ZIP mutations in Meq. Shown is the replication of MDV in the PBL of line P (A) and RIR (B) chickens infected with the pRB-1B BAC viruses as described in the legend to Fig. 5. A qPCR method (1) was used to determine the copy number of MDV genomes in DNA extracted from PBL samples taken at the times indicated throughout the experiment. Samples were taken from the same six birds from each group throughout the experiment. The mean genome copy number for the six birds is shown at each time point, with the bars representing the 95% confidence interval. (C) Analysis similar to that shown in panel A using DNA extracted from the feather tips of infected line P birds. The standard error of the mean for each group is shown.
Viral-genome levels were also analyzed in the feather tip DNA from the line P chicks, and this produced results that suggested fewer B-ZIP mutants than wild-type viruses replicated in the feather follicles. This is consistent with the reduced viral load in PBL and reflects the reduced number of virions reaching the feather follicles. Although there was an almost 2-log-unit reduction in the starting genome copy number, the similar rates of increase in all of the BAC-derived MDVs at this site between 5 and 20 days postinfection indicate that replication in the epithelium is probably not impaired by the mutations in Meq (Fig. 7C).
DISCUSSION
Meq is one of the few MDV proteins expressed during both lytic and latent infections. It has a nuclear distribution and can function as a homodimer or a heterodimer with cellular B-ZIP proteins to regulate transcription (17). The ability to form homodimers alone is insufficient for MDV-mediated oncogenesis, suggesting an important role for heterodimers (25). Here, we asked whether the ability to form homodimers is equally important in the biology of MDV and the pathology associated with MD of infected chickens. Two types of mutations were engineered into the Meq leucine zipper; they were predicted to ablate only the homodimerization of Meq or to prevent dimer formation with any leucine zipper-containing protein. In vitro biochemical analysis of protein-protein and protein-DNA interactions confirmed that the Meq molecules carrying these designed mutations behaved as predicted. The mutant Meq genes were therefore introduced into both genomic loci encoding Meq (in the terminal and internal repeats) in the highly virulent oncogenic pRB-1B5. Revertant viruses were made in order to exclude phenotypic effects due to second-site mutations. These viruses were all used to explore the role of Meq dimerization in the course and outcome of MDV infection of recently hatched chickens. Chicks were infected at 2 weeks of age in order to make early monitoring of virus in feather follicles (which develop after about 12 days) possible. In our experience, infections at 1 day and at 2 weeks produce the same course of disease progression and outcome (our unpublished data).
We and others have shown previously that although Meq is expressed during lytic replication in CEF, it is not necessary for virus production (5, 14). Consistent with this, there were no apparent differences in the abilities of the B-ZIP mutant viruses to replicate in CEF, as shown by the appearance of MDV plaques and growth curves similar to those produced by pRB-1B5 or the revertant viruses. When MDV infects chickens, there is an initial burst of lytic replication, predominantly in B cells, lasting up to 5 to 7 days (19). Measurement of the viral load in PBL suggested that the Meq leucine zipper mutations do not significantly affect lytic replication. Moreover, later in the course of infection, when the virus has been distributed to feather follicles, there is further virus replication in terminally differentiated epithelium prior to shedding of the virus into the environment. Consistent with the behavior of the mutant viruses in fibroblasts in vitro and B cells immediately following infection in vivo, the rate of replication of RB-1BHom/Hom and RB-1BBZIP/BZIP in feather tips indicated that these mutations in Meq do not significantly compromise virus lytic replication at this site, either. Any roles of Meq and its dimerization partners in CEF, B cells, and FFE remain to be determined.
In contrast, the most striking phenotype associated with Meq is confined to the stage of infection when MDV is thought to become latent in T cells and to induce their transformation into T-cell lymphomas (5, 14). Meq is essential for lymphomagenesis and may be essential for the establishment of latency in the T-cell compartment. Currently, there are no markers to discriminate between latently infected and transformed cells in MDV-infected birds. However, the results presented here demonstrate that a functional leucine zipper in Meq is an absolute requirement for lymphomagenesis and perhaps for the concomitant MDV latency in transformed T cells.
Experiments were performed in two genetically distinct strains of chicken. Eleven or 12 P-line birds were used for each type of virus, and 9 RIR birds were used for each type of virus. The data comparing a wild-type MDV BAC with both types of B-ZIP mutant were unequivocal (Table 2): wild-type MDV BACs produced lymphomas in 11/11 (100%) P-line birds and 8/9 (88.9%) RIR birds. In contrast, neither of the B-ZIP mutants produced any lymphomas (0%) in either strain of bird. As a control for the effects of second-site mutations, revertants of the two mutant viruses were constructed. The nononcogenic B-ZIP mutants were reconstituted to wild type by sequentially repairing both alleles of the Meq gene. When these revertant viruses were tested in the P-line birds, 11/12 (91.7%) and 9/12 (75%) produced tumors (Table 2). In the out-bred RIR chickens (in which the pathogenicity of wild-type MDV is generally less severe), four of nine (44%) and five of nine (55%) infections produced tumors.
We therefore conclude that restoration of MeqWT in the revertants restores the ability to induce lymphomas in the vast majority of infected birds, but the revertants are generally less virulent, indicating that during their production, “second-site” mutations that reduced the viruses' fitness to produce disease in chickens were probably introduced into the viral genome. Nevertheless, reversion restored oncogenicity in 75 to 91.7% of infections in P-line birds and about 50% of infections of RIR birds. Thus, for example, in the P-line birds, no more than 8% of MeqHom and about 25% of MeqBZIP infections might have failed to produce tumors, perhaps due to unplanned random mutations elsewhere in the genome. Since the revertant genomes went through two more rounds of recombination in Escherichia coli than the original mutants, even these values are likely to be a significant overestimate of the actual effect of second-site mutations on lymphomagenesis.
The mutations that prevent the formation of homodimers but still allow interaction with c-Jun tell us at least two things. First, homodimerization, and presumably binding to MERE II sites, is essential for T-cell transformation and/or latency. Second, while Meq heterodimers formed with c-Jun (or perhaps other, similar cellular B-ZIP proteins) are crucial for lymphomagenesis (25), they alone are unable to initiate and/or sustain the transformed phenotype in MDV-infected T cells if Meq homodimers are absent. Since Meq/Meq dimers bind MERE II sites and repress transcription (10), the requirement for homodimers is consistent with two hypotheses that are not mutually exclusive. If Meq dimers are unavailable, repression of Orilyt (and the lytic genes pp38 and pp14) may not occur, and this could lead to entry by default into the lytic cycle in T cells. As a consequence, the establishment of a latent infection in T cells by pRB-1BHom/Hom or pRB-1BBZIP/BZIP might not be possible, and transformation of the cells could not take place. Alternatively (but conceivably in addition), MERE II sites in the promoter regions of important chicken genes could be the target of Meq/Meq dimers, and the repression of some of these genes is necessary for either the establishment of virus latency and/or transformation of T cells into autonomously proliferating T lymphomas. All these putative activities of Meq homodimers could of course complement the actions of Meq/c-Jun heterodimers, which appear to be essential for lymphomagenesis (25).
In conclusion, it is worth considering how the MeqHom/Hom phenotype relates to the MeqCtBP/CtBP phenotype. CtBP is a highly conserved corepressor and forms complexes on chromatin that include histone deacetylase and methyl transferase enzymes that can initiate the repression of transcription and epigenetic silencing of target genes (6, 24). Point mutations in the canonical PLDLS CtBP-binding site in Meq prevent it from binding to CtBP and produce an MDV phenotype in chickens that is almost indistinguishable from that of MeqHom/Hom described here (Fig. 6 and 7) (5). If the MeqHom mutation exerts its effect because of a failure to repress gene expression through MERE II sites, then it is possible that this involves CtBP, which raises the question of whether the profound defect associated with both MeqHom and MeqCtBP could result from a failure to target and repress the same set of viral and/or cellular genes. Hopefully, a combined investigation of the MERE II sites in the MDV genome and microarray transcription profiling of cellular gene expression in infected chicken T cells will provide some insights.
Acknowledgments
We thank Charles Vinson (Center for Cancer Research, NIH, Bethesda, MD) for his advice on the construction of the B-ZIP mutants.
This work was supported by the Biotechnology and Biological Sciences Research Council, United Kingdom.
Footnotes
Published ahead of print on 19 August 2009.
REFERENCES
- 1.Baigent, S. J., L. J. Petherbridge, K. Howes, L. P. Smith, R. J. Currie, and V. K. Nair. 2005. Absolute quantitation of Marek's disease virus genome copy number in chicken feather and lymphocyte samples using real-time PCR. J. Virol. Methods 123:53-64. [DOI] [PubMed] [Google Scholar]
- 2.Baigent, S. J., L. J. Petherbridge, L. P. Smith, Y. Zhao, P. M. Chesters, and V. K. Nair. 2006. Herpesvirus of turkey reconstituted from bacterial artificial chromosome clones induces protection against Marek's disease. J. Gen. Virol. 87:769-776. [DOI] [PubMed] [Google Scholar]
- 3.Biggs, P. M. 1997. The Leeuwenhoek lecture, 1997. Marek's disease herpesvirus: oncogenesis and prevention. Phil. Trans. R. Soc. Lond. B 352:1951-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Briles, W. E., H. A. Stone, and R. K. Cole. 1977. Marek's disease: effects of B histocompatibility alloalleles in resistant and susceptible chicken lines. Science 195:193-195. [DOI] [PubMed] [Google Scholar]
- 5.Brown, A. C., S. J. Baigent, L. P. Smith, J. P. Chattoo, L. J. Petherbridge, P. Hawes, M. J. Allday, and V. Nair. 2006. Interaction of MEQ protein and C-terminal-binding protein is critical for induction of lymphomas by Marek's disease virus. Proc. Natl. Acad. Sci. USA 103:1687-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chinnadurai, G. 2007. Transcriptional regulation by C-terminal binding proteins. Int. J. Biochem. Cell Biol. 39:1593-1607. [DOI] [PubMed] [Google Scholar]
- 7.Epstein, M. A. 2001. Historical background. Phil. Trans. R. Soc. Lond. B 356:413-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hickabottom, M., G. A. Parker, P. Freemont, T. Crook, and M. J. Allday. 2002. Two nonconsensus sites in the Epstein-Barr virus oncoprotein EBNA3A cooperate to bind the co-repressor carboxyl-terminal-binding protein (CtBP). J. Biol. Chem. 277:47197-47204. [DOI] [PubMed] [Google Scholar]
- 9.Kuppuswamy, M., S. Vijayalingam, L. J. Zhao, Y. Zhou, T. Subramanian, J. Ryerse, and G. Chinnadurai. 2008. Role of the PLDLS-binding cleft region of CtBP1 in recruitment of core and auxiliary components of the corepressor complex. Mol. Cell. Biol. 28:269-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy, A. M., O. Gilad, L. Xia, Y. Izumiya, J. Choi, A. Tsalenko, Z. Yakhini, R. Witter, L. Lee, C. J. Cardona, and H. J. Kung. 2005. Marek's disease virus Meq transforms chicken cells via the v-Jun transcriptional cascade: a converging transforming pathway for avian oncoviruses. Proc. Natl. Acad. Sci. USA 102:14831-14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levy, A. M., Y. Izumiya, P. Brunovskis, L. Xia, M. S. Parcells, S. M. Reddy, L. Lee, H. W. Chen, and H. J. Kung. 2003. Characterization of the chromosomal binding sites and dimerization partners of the viral oncoprotein Meq in Marek's disease virus-transformed T cells. J. Virol. 77:12841-12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu, J. L., and H. J. Kung. 2000. Marek's disease herpesvirus transforming protein MEQ: a c-Jun analogue with an alternative life style. Virus Genes 21:51-64. [PubMed] [Google Scholar]
- 13.Liu, J. L., Y. Ye, L. F. Lee, and H. J. Kung. 1998. Transforming potential of the herpesvirus oncoprotein MEQ: morphological transformation, serum-independent growth, and inhibition of apoptosis. J. Virol. 72:388-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lupiani, B., L. F. Lee, X. Cui, I. Gimeno, A. Anderson, R. W. Morgan, R. F. Silva, R. L. Witter, H. J. Kung, and S. M. Reddy. 2004. Marek's disease virus-encoded Meq gene is involved in transformation of lymphocytes but is dispensable for replication. Proc. Natl. Acad. Sci. USA 101:11815-11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matagne, A., B. Joris, and J. M. Frere. 1991. Anomalous behaviour of a protein during SDS/PAGE corrected by chemical modification of carboxylic groups. Biochem. J. 280:553-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nair, V. 2004. Successful control of Marek's disease by vaccination. Dev. Biol. 119:147-154. [PubMed] [Google Scholar]
- 17.Nair, V., and H. J. Kung. 2004. Marek's disease virus oncogenicity: molecular mechanisms, p. 32-47. In F. Davison and V. Nair (ed.), Marek's disease: an evolving problem. Elsevier Academic Press, London, United Kingdom.
- 18.O'Shea, E. K., R. Rutkowski, W. F. Stafford III, and P. S. Kim. 1989. Preferential heterodimer formation by isolated leucine zippers from fos and jun. Science 245:646-648. [DOI] [PubMed] [Google Scholar]
- 19.Osterrieder, N., J. P. Kamil, D. Schumacher, B. K. Tischer, and S. Trapp. 2006. Marek's disease virus: from miasma to model. Nat. Rev. Microbiol. 4:283-294. [DOI] [PubMed] [Google Scholar]
- 20.Parcells, M. S., V. Arumugaswami, J. T. Prigge, K. Pandya, and R. L. Dienglewicz. 2003. Marek's disease virus reactivation from latency: changes in gene expression at the origin of replication. Poultry Sci. 82:893-898. [DOI] [PubMed] [Google Scholar]
- 21.Petherbridge, L., A. C. Brown, S. J. Baigent, K. Howes, M. A. Sacco, N. Osterrieder, and V. K. Nair. 2004. Oncogenicity of virulent Marek's disease virus cloned as bacterial artificial chromosomes. J. Virol. 78:13376-13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Posfai, G., V. Kolisnychenko, Z. Bereczki, and F. R. Blattner. 1999. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res. 27:4409-4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian, Z., P. Brunovskis, F. Rauscher III, L. Lee, and H. J. Kung. 1995. Transactivation activity of Meq, a Marek's disease herpesvirus bZIP protein persistently expressed in latently infected transformed T cells. J. Virol. 69:4037-4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi, Y., J. Sawada, G. Sui, B. el Affar, J. R. Whetstine, F. Lan, H. Ogawa, M. P. Luke, and Y. Nakatani. 2003. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422:735-738. [DOI] [PubMed] [Google Scholar]
- 25.Suchodolski, P. F., Y. Izumiya, B. Lupiani, D. K. Ajithdoss, O. Gilad, L. F. Lee, H. J. Kung, and S. M. Reddy. 2009. Homodimerization of Marek's disease virus-encoded Meq protein is not sufficient for transformation of lymphocytes in chickens. J. Virol. 83:859-869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venugopal, K. 2000. Marek's disease: an update on oncogenic mechanisms and control. Res. Vet. Sci. 69:17-23. [DOI] [PubMed] [Google Scholar]
- 27.Vinson, C., A. Acharya, and E. J. Taparowsky. 2006. Deciphering B-ZIP transcription factor interactions in vitro and in vivo. Biochim. Biophys. Acta 1759:4-12. [DOI] [PubMed] [Google Scholar]