

Abstract
Hypertrophic cardiomyopathy (HCM) is a disease of mutant sarcomeric proteins (except for phenocopy). Cardiac hypertrophy is the clinical diagnostic hallmark of HCM and a major determinant of morbidity and mortality in various cardiovascular diseases. However, there is remarkable variability in expression of hypertrophy, even among HCM patients with identical causal mutations. We hypothesized modifier genes are partly responsible for the variation in hypertrophic expressivity. To map the modifier loci, we typed 811 short-tandem repeat markers (~5 cMdense) in 100 members of an HCM family including 36 with the InsG791 mutation in MYBPC3. We performed oligogenic simultaneous segregation and linkage analyses using Markov Chain Monte Carlo methods and detected linkage on 3q26.2 (180 cM), 10p13 (41 cM), 17q24 (108 cM) with log of the posterior placement probability ratio (LOP) of 3.51, 4.86 and 4.17, respectively, and suggestive linkage (LOP of 2.40) on 16q12.2 (73 cM). The effect sizes varied according to the modifier locus, age and sex. It ranged from ~8 g shift in left ventricular mass for 10p13 locus heterozygosity for the common allele to ~90 g shift for 3q26.2 locus homozygosity for the uncommon allele. Refining the 10p13 locus restricted the candidate modifier genes to ITGA8, C10orf97 (CARP) and PTER. ITGA8 and CARP are biologically plausible candidates as they are implicated in cardiac fibrosis and apoptosis, respectively. Since cardiac hypertrophy is a major determinant of total and cardiovascular mortality and morbidity, regardless of the etiology, identification of the specific modifier genes could have significant prognostic and therapeutic implications for various cardiovascular diseases.
INTRODUCTION
Familial hypertrophic cardiomyopathy (HCM) is a monogenic disease with an autosomal dominant Mendelian pattern of inheritance. HCM is diagnosed clinically by detection of left ventricular hypertrophy in the absence of a known cause (1). Cardiac hypertrophy is the major cause of diastolic heart failure and sudden cardiac death (SCD) not only in patients with HCM but also in all cardiovascular disorders (2,3). A notable phenotypic feature of HCM is the presence of considerable variability in expression of cardiac hypertrophy, even among affected members of a given family. Factors that determine phenotypic variability of cardiac hypertrophy in HCM are largely unknown.
The molecular genetic basis of HCM has been partially elucidated and mutations in over a dozen genes encoding sarcomeric proteins (except for phenocopy) have been identified (reviewed in 4). Causal mutations, while essential for expression of the phenotype, do not fully explain the variability in hypertrophic expressivity. This is best illustrated in familial HCM, wherein the affected individuals who share a common causal mutation exhibit a variable degree of cardiac hypertrophy. We hypothesized genes other than the causal genes, i.e. the modifier genes contribute to phenotypic expression of cardiac hypertrophy in HCM.
To map the modifier genes for cardiac hypertrophy, we performed oligogenic simultaneous segregation-linkage analyses of quantitative traits utilizing Markov Chain Monte Carlo (MCMC) methods, as implemented in the computer program Loki (5). The methods are Bayesian in nature and employ MCMC techniques to estimate posterior probabilities of linkage over a large space of models. The analysis simultaneously estimates the number of genes contributing to a quantitative trait, the effects of those genes and the effects of any covariate factors. This is in contrast to most other methods that do not explicitly model the effects of known genes while looking for additional genes that affect a trait. Oligogenic segregation and linkage analysis has been successfully used to localize genes contributing to a variety of diseases and traits (6-11). We genotyped 100 members of a large family with HCM caused by the Ins791G mutation in myosin binding protein C (MyBP-C) and performed oligogenic simultaneous linkage and segregation analysis utilizing MCMC methods to map the genetic loci that modify expression of cardiac hypertrophy.
RESULTS
Variability in expression of cardiac hypertrophy
Family members with the InsG791 mutation exhibited considerable variability in phenotypic expression of cardiac hypertrophy (12). The characteristics of the family members who participated in the present study are shown in Table 1. The mean (±SD) left ventricular mass (LVM) was greater in males than in females [207.5 ± 89.6 (median 196.9) versus 169.0 ± 53.0 (median 170.5), P = 0.01]. In linear regression analysis allowing for familial correlation, LVM was associated with sex (F = 5.6, adjusted R2 = 4%, P = 0.02), age at the time of measurement (F = 24.0, adjusted R2 = 19%, P < 0.0001), height (F = 6.2, adjusted R2 = 9%, P = 0.02), weight (F = 38.1, adjusted R2 = 42%, P < 0.0001) and blood pressure (for systolic blood pressure: F = 24.7, adjusted R2 = 26%, P < 0.0001 and for diastolic blood pressure: F = 17.6, adjusted R2 = 20%, P < 0.0001). When we performed stepwise multiple regression analysis with all the above variables included, only body weight (P < 0.0001) and age (P = 0.09) showed association with LVM. However, weight was not available on all subjects, and without weight, both sex (P = 0.0016) and age (P < 0.0001) were independently associated with LVM.
Table 1.
Characteristics of the participant family members
| Phenotype | MYBPC3 InsG791 mutation | ||
|---|---|---|---|
| All | Yes | No | |
| N | 100 | 36 | 64 |
| Male/female | 51/49 | 21/15 | 30/34 |
| Age at diagnosis (years) | 34.6 ± 17.0 | 32.4 ± 14.7 | 35.8 ± 18.2 |
| Height (meter) | 1.65 ± 0.10 | 1.66 ± 0.10 | 1.64 ± 0.11 |
| Weight (Kg) | 68.97 ± 15.82 | 69.9 ± 13.5 | 68.2 ± 17.7 |
| Body mass index (Kg/m2) | 25.3 ± 5.1 | 25.5 ± 4.7 | 25.2 ± 5.5 |
| Heart rate (beat per minutes) | 71.6 ± 12.4 | 69.7 ± 11.9 | 73.2 ± 12.9 |
| Systolic blood pressure (mmHg) | 110.8 ± 20.7 | 110.1 ± 19.3 | 111.3 ± 22.4 |
| Diastolic blood pressure (mmHg) | 70.7 ± 13.1 | 72.6 ± 9.8 | 68.7 ± 15.7 |
| Septal thickness (mm) | 11.6 ± 4.3 | 13.8 ± 5.2 | 9.7 ± 2.0* |
| Posterior wall thickness (mm) | 9.3 ± 1.5 | 9.5 ± 1.6 | 9.2 ± 1.4 |
| LV end diastolic diameter (mm) | 43.9 ± 5.5 | 43.5 ± 4.9 | 44.2 ± 5.8 |
| LV fractional shortening (%) | 35.7 +7.2 | 37.8 +7.2 | 34.9 +7.2 |
| LVM (g) | 189.4 ± 76.8 | 225.1 ± 98.8 | 169.3 ± 52.0* |
All values are mean ± SD unless specified. LV, left ventricular, LVM, left ventricular mass.
P < 0.005.
Oligogenic simultaneous segregation and linkage analysis of quantitative traits
Strong linkage signals were detected on chromosomes 10p13 (peak between D10S1653 and D10S674) and 17q24 (peak closest to D17S949). The maximum L scores for each chromosome are recorded in Table 2 for genome-wide analyses for the runs with only MYBPC3 genotype as a covariate as well as for the runs with MYBPC3 genotype, sex and age as covariates. The 10p13 and 17q24 loci show very strong signals, as evidenced by very high L scores in both analyses. In a previous simulation study based on the present data set, the L score cut-off for an empirical chromosome-wide P-value of 95% was 7.3 (13). In both analysis runs, the L scores were over the above significance threshold on 10p13 and 17q24 loci. The 16q12.2 locus had a maximum L score of 16.62 when MYBPC3 genotype was the only covariate. The L score dropped to 2.03, when sex and age were added to MYBPC3 mutation as covariates, suggesting an interaction between the 16q12.2 locus and age/sex. The L score for chromosome 3q26.2 locus, however, increased from 4.79 to 16.04 when age and sex were added as covariates.
Table 2.
Maximum chromosome-wide scores from 500,000 iteration runs on all chromosomes
| Chromosome | MYPBC as covariate | MYBPC, sex and age as covariates | ||
|---|---|---|---|---|
| Max L score | Location, cM | Max L score | Location, cM | |
| 1 | 1.39 | 107.50 | 1.74 | 174.50 |
| 2 | 2.11 | 199.50 | 2.59 | 254.50 |
| 3 | 4.54 | 183.50 | 18.96 | 184.50 |
| 4 | 1.72 | 58.50 | 1.77 | 61.50 |
| 5 | 2.07 | 214.50 | 2.39 | 213.50 |
| 6 | 3.57 | 0.50 | 4.21 | 0.50 |
| 7 | 1.52 | 16.50 | 1.75 | 113.50 |
| 8 | 1.93 | 86.50 | 0.91 | 86.50 |
| 9 | 2.56 | 18.50 | 0.83 | 135.50 |
| 10 | 34.08 | 40.50 | 17.76 | 40.50 |
| 11 | 0.80 | 130.50 | 1.35 | 129.50 |
| 12 | 3.51 | 177.50 | 5.98 | 86.50 |
| 13 | 0.83 | 90.50 | 1.89 | 42.50 |
| 14 | 1.35 | 25.50 | 2.05 | 13.50 |
| 15 | 0.92 | 97.50 | 2.07 | 99.50 |
| 16 | 16.62 | 73.50 | 2.03 | 74.50 |
| 17 | 58.91 | 113.50 | 17.64 | 118.50 |
| 18 | 2.00 | 91.50 | 1.33 | 9.50 |
| 19 | 1.32 | 87.50 | 0.60 | 82.50 |
| 20 | 2.93 | 0.50 | 3.21 | 14.50 |
| 21 | 1.07 | 59.50 | 0.57 | 0.50 |
| 22 | 3.16 | 58.50 | 1.70 | 60.50 |
To better assess the linkage signals on the chromosomes with L score >7.3, we calculated the LOP, which is a more stable indicator of linkage (14). We used runs with 10 different pseudo chromosomes of 2 million iterations each, resulting in 20 million total iterations. Every fifth iteration was used in the analyses. The results are summarized in Table 3 and shown in Figure 1. Highly significant LOPs were detected on 10p13 (LOP = 4.86), and 17q24 (LOP = 4.17). The LOP on chromosome 3 (3.51) is also significant and even the highest LOP on chromosome 16 (2.40) was greater than the 95% chromosome-wide significance level of 2.23 found in our previous simulation study (13).
Table 3.
Maximum chromosome-wide LOP and L score from 20 million iteration runs on chromosomes with linkage signals
| Chromosome | MYBPC3 as covariate | MYBPC3, sex and age as covariates | ||||||
|---|---|---|---|---|---|---|---|---|
| LOP | Location, cM | L score | Location, cM | LOP | Location, cM | L score | Location, cM | |
| 3 | 3.41 | 177.5 | 4.79 | 183.50a | 3.51 | 179.5 | 16.04 | 183.50 |
| 10 | 4.95 | 40.5 | 47.76 | 40.50 | 4.86 | 40.5 | 25.86 | 40.50 |
| 16 | 3.73 | 73.5 | 17.84 | 73.50 | 2.40 | 72.5 | 2.55 | 73.50 |
| 17 | 4.52 | 113.5 | 60.49 | 113.5 | 4.17 | 107.5 | 16.21 | 118.50 |
L scores are less robust at the ends of the chromosomes because of lower information content (aedge effect L score of 4.91 at 0.50 cM).
Figure 1.
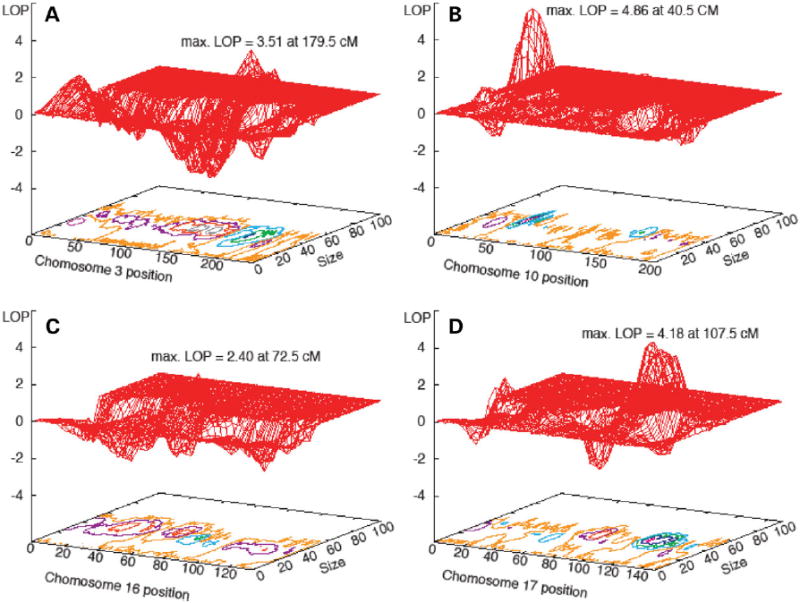
The LOP values at the mapped loci. The X-axis shows the chromosomal position, the Y axis the effect size and the Z axis the LOP. All computations include InsG791 mutation, age and sex as the covariates. Contour plots (shown in detail in Fig. 2) are shown below the LOP surface. Maximum LOP values on each chromosome are indicated. All four analyses were run for 20 million iterations: 2 million iterations with 10 different pseudo chromosomes for each chromosome (every fifth iteration retained). (A) Chromosome 3; (B) Chromosome 10; (C) Chromosome 16 and (D) Chromosome 17.
The details of the localizations on each chromosome are shown in LOP contour plots (Fig. 2). We only present plots for the runs with sex and age as covariates since the regions localized were the same in both sets of analyses. The 10p13 locus was identified in an early stage of the analysis, which led us to type additional markers in this region. This is illustrated in Figure 2, showing denser marker spacing in the linked region. The initial mapped region was ~15 cM (from D10S547 to D10S548), which was reduced to a ~5 cM region between D10S223 and D10S674. Likewise, the primary region of interest was reduced to a ~2 cM region between D10S1653 and D10S674, which comprises ~800 kb of genomic DNA. While the LOP peaks on both 10p13 and 17q24 are >4, the region on 17q24 with LOP > 3 is wider because of the sparser density of typed markers.
Figure 2.
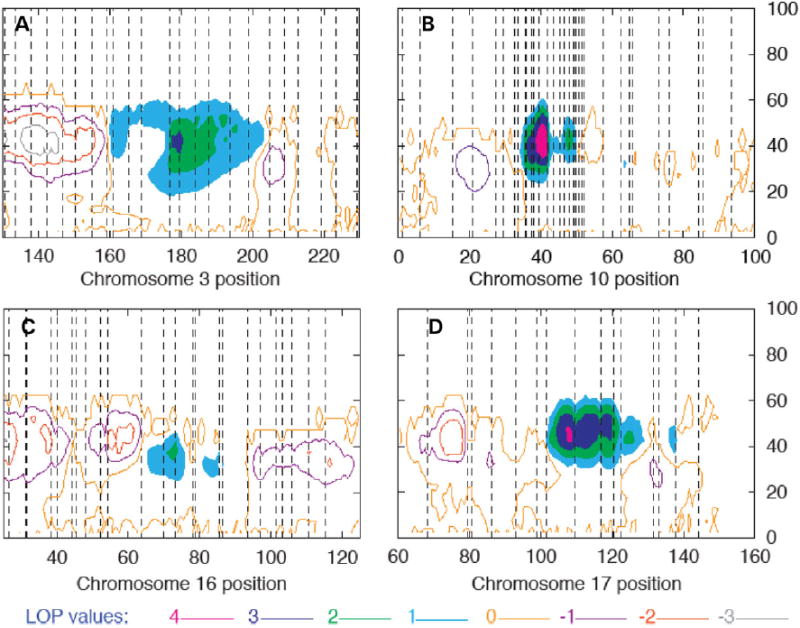
Details of LOP contour plots from Figure 2. One unit LOP contours are shown for a 100 cM region about the linkage peak. The highest contour level (seen on chromosomes 10 and 17) is >4. Regions between LOP contours >1 are shaded. The X-axis indicates the position along the chromosome in Haldane cM. The Y axis indicates the mean effect size of the QTL localized. The vertical lines indicate marker positions on each chromosome as follows: (A) Chromosome 3: D3S1278, D3S1558, D3S1267, D3S3606, D3S1292, D3S3637, D3S1309, D3S1569, D3S1593, D3S1555, D3S1279, D3S3668, D3S1614, D3S3725, D3S1565, D3S3715, D3S3609, D3S1262, D3S3686, D3S1580, D3S1601, D3S2748, D3S1265; (B) Chromosome 10: D10S249, D10S1745, D10S591, D10S189, D10S1649, D10S547, D10S2325, D10S570, D10S1725, D10S223, D10S1664, D10S191, D10S1653, D10S674, D10S504, D10S548, D10S203, D10S211, D10S550, D10S1749, D10S586, D10S572, D10S197, D10S213, D10S208, D10S1780, D10S578, D10S196, D10S1790, D10S1652, D10S581, D10S537; (C): Chromosome 16: D16S404, D16S3075, D16S3102, D16S500, D16S3103, D16S3041, D16S3046, D16S403, D16S3068, D16S3100, D16S3136, D16S3034, D16S415, D16S3057, D16S3140, D16S514, D16S503, D16S3066, D16S515, D16S3049, D16S516, D16S3040, D16S505, D16S3091 and (D): Chromosome 17: D17S927, D17S1868, D17S1795, D17S787, D17S957, D17S944, D17S1816, D17S949, D17S1862, D17S1807, D17S785, D17S1847, D17S836, D17S784, D17S928.
Effect size of the mapped loci
In addition to providing estimates of gene position, oligogenic segregation and linkage analysis affords the opportunity to estimate the effects of other model parameters. As expected, age, sex and the InsG791 mutation in MYBPC3 were significant determinants of cardiac mass (Table 4). Carrying one copy of the causal mutation was estimated to increase LVM by ~50 g, while age was associated with an increase of >1 g per year, and sex was found to have a ~20 to ~40 g shift in LVM in different models. As shown in Table 4, the differences in the effect estimates vary according to the combination of the covariates, suggestive of complex interactions between the parameters. In particular, the effect of MYBPC3 mutation on LVM is significantly decreased when modifier loci are included in the analysis (by nearly 20 g when age and sex are included in the analysis). The findings suggest an interaction between the causal mutation and the modifier genes.
Table 4.
Estimated effects on LVM of different variables according to the parameters included in the model
| Variables | Without modifier loci | With modifier loci | ||||||
|---|---|---|---|---|---|---|---|---|
| MYBPC3 | MYBPC3, age | MYBPC3, sex | MYBPC3, sex/age | MYBPC3 | MYBPC3, age | MYBPC3, sex | MYBPC3, sex/age | |
| MYBPC3 het. effect | 80.73 (14.84) | 71.21 (14.53) | 73.40 (14.70) | 62.38 (14.14) | 55.45 (13.14) | 50.37 (12.09) | 51.56 (12.51) | 43.54 (12.03) |
| Age | – | 1.56 (0.42) | – | 1.70 (0.40) | – | 1.11 (0.28) | – | 1.33 (0.31) |
| Sex | – | – | −35.72 (14.53) | −41.86 (13.49) | – | – | −21.54 (10.68) | −27.58 (10.15) |
The numbers in parenthesis are SD.
The estimates of genotype effect of each locus on LVM are plotted in Figure 3 and shown in Table 5. The findings suggest that a single copy of the less common allele on chromosomes 10 and 17 modifies HCM in a deleterious direction. For the chromosome 10 locus, the estimates showed a common allele frequency of ~0.80 and a heterozygote effect of increasing LVM by ~8–10 g, regardless of inclusion of age and sex in the analysis. Likewise, the frequency of the common modifier allele on 17q24 locus was estimated at ~0.80 and a heterozygote effect at an ~8 g increase in LVM, when age and sex were not included in the model. However, when age and sex were included, the estimate of the heterozygote effect increased to ~24 g, suggesting a joint locus and age/sex effect. The effect of homozygosity for the minor allele is based on only a small number of homozygous individuals (as evidenced by the large SDs) for 10p13 and 17q24 loci and hence, should be interpreted with caution.
Figure 3.
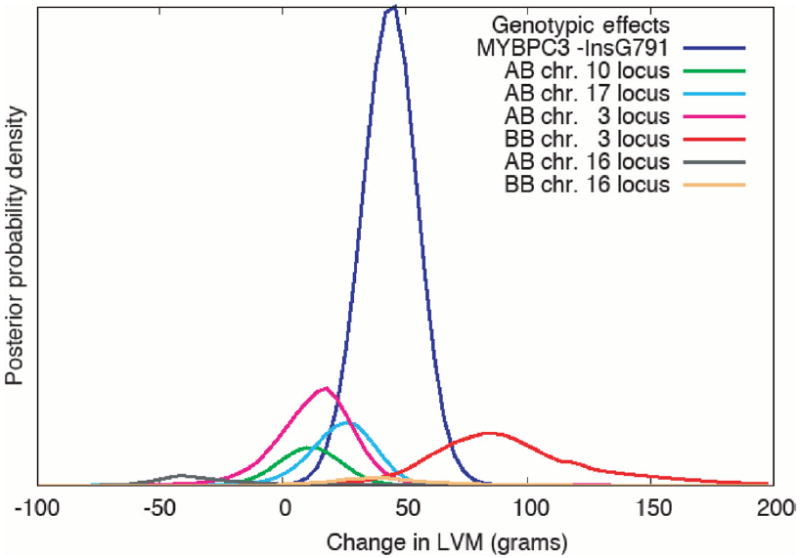
Posterior probability distributions for the estimated effects of MYPBC3 mutation and modifier loci genotypes on the LVM. ‘A’ and ‘B’ represent the two alleles at each locus. The effects at each locus are estimated relative to the AA genotype (the common homozygote), which is fixed at 0. The effects of the BB genotypes for the loci on chromosomes 10 and 17 or the MYBPC3 are not plotted since those genotypes are too rare for our sample to provide a meaningful estimate. The area under each curve approximates the posterior probability of a QTL at each of the loci. The area under the MYBPC3 curve is 1 since it is always in the model. The areas under the chromosome 3 locus curves are larger than those under the chromosomes 10 and 17 curves because the potential region for the chromosome 3 locus is much larger (see Fig. 2). The chromosome 10 and 17 loci are better characterized, as reflected in the LOP values and generally smaller standard deviations in allele frequency and effect estimates.
Table 5.
Estimated allele frequencies and effects on LVM of modifier loci with different covariates
| Chromosome | Covariates | Frequency, A | Effect, AB | Effect, BB |
|---|---|---|---|---|
| 10p13 | MYBPC3 | 0.786 (0.072) | 7.78 (17.02) | 180.30 (38.70) |
| 10p13 | MYBPC3, sex, age | 0.790 (0.071) | 10.56 (14.00) | 173.31 (28.99) |
| 17q24 | MYBPC3 | 0.789 (0.067) | 8.08 (15.17) | 177.49 (23.53) |
| 17q24 | MYBPC3, sex, age | 0.810 (0.066) | 24.19 (13.50) | 174.62 (24.72) |
| 3q26 | MYBPC3 | 0.735 (0.132) | 13.10 (20.64) | 111.803 (35.38) |
| 3q26 | MYBPC3, sex, age | 0.657 (0.149) | 13.79 (17.20) | 92.59 (33.19) |
| 16q12 | MYBPC3 | 0.689 (0.120) | −36.23 (19.52) | 53.61 (16.78) |
| 16q12 | MYBPC3, sex, age | 0.550 (0.184) | −28.10 (26.15) | 41.33 (20.58) |
The numbers in parenthesis are SD.
The range of models for the 3q locus is wider, while the modeling on 16q suggests a complex locus (Table 5 and Fig. 3). At the 3q26.2 locus, the estimated frequency of the common allele was ~0.75 when age and sex were not included in the model, and ~0.65 when they were included. The heterozygote increased LVM by ~13 g with or without age and sex. The frequency of homozygote individuals for the minor allele is small for a conclusive estimate, but the estimates for this locus include the possibilities of a recessive or multiplicative model. Furthermore, we examined a restricted set of models that localized modifier loci on chromosomes 3, 10 and 17 simultaneously and included the causal mutation, age and sex. In these models, the 3q26.2 locus was modeled as being recessive with the minor allele homozygote increasing LVM by ~50 g. The modeling of the 16q12.2 locus suggests a complex effect. The common allele frequency was estimated at ~0.70 without age and sex, and ~0.55 with age and sex as covariates. The model at this locus was an over-dominant. The heterozygote is protective (lower LVM), whereas the minor allele homozygote is deleterious (higher LVM). This pattern of over-dominance was the same whether or not sex and age were included. However, the effect sizes and allele frequencies differed suggesting a joint effect with age/sex. We note the evidence for linkage, although beyond the 95% chromosome-wide significance level, was relatively modest on 16q12.2 locus. Thus, we cannot exclude the possibility of random segregation. On the other hand, the strength of the linkage signals at each locus is in accord with the expectations if the modeling is correct, i.e. modifiers that act in a dominant fashion produce the strongest signals (10p13 and 17q24), a modifier locus that acts somewhat recessive locus (3q26.2) produces a moderate signal and the complex modifier effect (16q12.2) produces the smallest signal.
DISCUSSION
We have performed a large-scale genome scan to map the modifier loci for human HCM and identified four loci that affect expression of cardiac hypertrophy caused by the InsG791 mutation in MYBPC3. The evidence for linkage is very strong for the 10p13 and 17q24, moderate on 3q26.2 and modest on 16q12.2. The effect of each modifier locus is large enough to be considered clinically significant, as an increase in LVM, regardless of the etiology, is associated with a significant increase in cardiovascular morbidity and mortality (15-18). However, as it would be expected the effect is less than that of the causal mutation. The data indicate that several genes, in addition to MYBPC3, contribute to variation in expression of cardiac hypertrophy in HCM. The findings substantiate the presence of modifier genes for HCM, an archetypical single-gene disorder, and document that phenotypic expression of single-gene disorders is determined not only by the causal mutation but also by modifier genes.
Mapping and identification of the modifier genes is a more complex problem than mapping the primary causative genes, hence, requiring complex statistical methods. Accordingly, it is necessary to detect and analyze not only the effects of the causal gene/mutation but also the effects of other known covariates, such as age and sex. To our knowledge, the present study is the first genome-wide linkage analysis for modifier loci of a single-gene disorder with a Mendelian pattern of inheritance. We computed the L scores to perform the initial genome scan and substantiate the linkage by computing the LOP, which is considered more robust (14). The LOP remained >4 for chromosomes 10, and 17 loci, even after inclusion of sex, age and MYBPC3 mutation as covariates. In contrast, evidence of linkage to chromosome 16 locus remained marginal, which may reflect marginal power to map this locus in this family or a false positive result. We have previously shown that a LOP of >4 had an empirical P-value of <0.001; i.e. there were no values of LOP > 4 in the empirical null distribution (14). Thus, LOP of >4 is considered to be strong evidence of linkage. Because LOP uses unlinked ‘pseudo chromosomes’ to control for differing levels of information content across the true chromosome, it can provide a more stable indicator of linkage than the L score. Collectively, the data provide strong evidence of modifying effects for chromosome 10 and 17 loci and moderate evidence for chromosome 3.
To facilitate identification of the modifier genes, we have refined the 10p13 locus by typing of additional markers to genomic region. The refined region with the highest LOP (>4.0) comprises only three genes, namely, ITGA8, C10orf97 (aka CARP) and PTER. ITGA8 is a biologically plausible candidate to affect expression of cardiac hypertrophy. ITGA8 encodes the subunit α8 of the integrin family, which is a ubiquitously expressed group of cell surface receptors that link the extracellular matrix to the intracellular cytoskeleton (19). The subunit associates exclusively with the β1 subunit to form an RGD-binding integrin, which interacts with fibronectin, vitronectin, osteopontin and others (19). ITGA8 is also expressed in myofibroblasts and smooth muscle cells of myocardium (20). Furthermore, expression of ITGA8 is upregulated in response to pro-hypertrophic and pro-fibrotic agent angiotensin II (19). Moreover, increased expression of ITGA8 is also associated with increased cardiac interstitial fibrosis (21), which is a pathological feature of human HCM (22). Thus, ITGA8 is the prime candidate to modify expression of cardiac hypertrophy in HCM. CARP is also a biologically plausible candidate to influence cardiac hypertrophic response. The gene encodes a CARD-containing protein involved in recruitment of caspases and apoptosis (23). It is most abundantly expressed in the heart (23). However, its role in the heart remains to be determined. It is notable that suppression of expression of CARP results in proliferation in several mammalian cell lines and its over-expression leads to apoptosis and inhibition of proliferation in tumor cell lines (23). Thus, CARP is also an attractive candidate to affect expression of cardiac phenotype in HCM. The third region in the high priority refined 10p13 locus (LOP > 4.0) is PTER, which encodes phosphotriesterase-related protein. PTER is a member of zinc metalloenzymes that catalyze the hydrolysis of a range of phosphotriester compounds. It is diffusely expressed including in the heart. However, its biological function in the heart remains unexplored.
The weakening of the linkage signal on 16q12.2 when age and sex were included as covariates suggests an interaction with age and/or sex. To examine a potential interaction, we conducted two additional analyses, one with age and MYBPC3 as covariates, and one with sex and MYBPC3 as covariates. In both analyses, the L scores were reduced as compared to that with MYBPC3 as a covariate, although the analysis with age had a greater reduction (L score of 2.51 at 72.5 cM) than the analysis with sex (L score of 7.71 at 74.5 cM). Thus, it would appear that interactions, if any, are greater with age than with sex. We note, however, that the linkage signal on 16q12.2 locus may be spurious from a random marker segregation pattern. Alternatively, in the case of true linkage, the lower score may reflect the power to map in this particular family. As a result, inclusion of the estimates of two additional parameters could reduce the power to detect the chromosome 16 locus. Furthermore, the QTL modeling at the chromosome 16 locus was the most complex of the four loci, with an over-dominant model. It would not be a surprise if the power to detect linkage in this sample was the lowest for the gene with the most complex model. Further study of this region in a larger group of HCM families is warranted.
The parameter estimates for the modifier loci could provide insight into the mechanisms by which the mapped loci affect expression of cardiac hypertrophy. However, these estimates should be used with caution. While the estimates of gene location with these methods have been found to be robust, bias in genotype effects and allele frequency can arise from ascertainment (24). Thus, the effect estimates should be considered as guidelines rather than the exact effect of the gene on the trait. The estimated effects can differ according to the variables included in the model, as illustrated in Table 4. This is to be expected since these estimates are adjusted for the other effects included in the model. In addition, more complex interactions between these factors than our analysis model allowed could also contribute to differences.
Two-dimensional echocardiography is the conventional modality for the diagnosis of HCM and the area-length method is an established and most commonly used technique for the measurement of LVM in patients with HCM (25-30). The echocardiographic measurement of the LVM assumes certain cardiac geometry, which may not be fully applicable to a subset of patients with HCM who have asymmetric septal hypertrophy. We have recognized this potential limitation and determined the reproducibility of measurement of the LVM by the area-length method. Accordingly, the 95% confidence interval of intraobserver variability of LVM was found to be within −12 to 14% (31). We also note that the phenotypic data were collected in a single time point. The prevalence of clinical events, such as cardiac arrhythmias, syncope and death was relatively low at the time of clinical evaluation, as described previously (12). Longitudinal follow up data would be necessary to assess the effects of the modifier loci on additional clinical phenotype. Likewise, whether different modifier genes are involved in different stages of development and progression of HCM remains to be determined. Furthermore, the analysis was performed in members of a HCM family caused by the InsG791 mutation in MYBPC3. Thus, generalization of the findings to other HCM families caused by mutations in other causal genes remains to be established.
While the statistical methods employed in the oligogenic segregation and linkage analysis are sophisticated, they are not without limits and cautions. First, while these methods provide very strong evidence that several loci modify the expression of HCM, the estimates of the exact number of loci should be viewed with some caution. Second, it reflects the number of loci for which there is evidence, and for which there is power to find in this sample. Smaller effect loci will be more difficult to detect and will require a larger sample. Finally, as with any model, the analysis model may not fully reflect the biology. For example, the analysis model assumes diallelic modifier loci, but if the system is multiallelic with alleles that have different effects, the results could be affected. A multiallelic locus could explain the over-dominant model found on chromosome 16. Another limitation is the availability of weight data only in a subset. Inclusion of weight in the analysis reduces the power to detect any modifier genes because of missing values. Likewise, as in any other modeling, adding more covariates to the analysis means additional estimated parameters. While inclusion of covariates can potentially lead to better modeling of the trait, the estimation of the additional parameters also has the potential to reduce the power. Nonetheless, the presence or absence of covariates had a negligible effect on estimates of gene location. For example, the regions with LOP>3 were virtually identical whether or not age and sex were included as covariates. Thus, we suggest evidence for linkage of cardiac hypertrophy to mapped loci is robust.
In conclusion, we have mapped, utilizing simultaneous oligogenic segregation and linkage analysis, four modifier loci, 3q26.2, 10p13, 16q12.2 and 17q24 that influence expression of cardiac hypertrophy in members of a large family with HCM caused by the InsG791 mutation in MYBPC3. The refined 10p13 locus comprises two biologically plausible genes ITGA8 and C10Orf97 (CARP) at the high priority region. The genetic findings beckon experimentations to validate the effects of the mapped genes on expression of cardiac hypertrophy and to delineate the molecular mechanisms. The findings indicate that phenotypic expression of HCM, an archetypical single-gene disorder with Mendelian inheritance, is determined not only by the causal mutation but also by the modifier genes. Since cardiac hypertrophy is a common phenotype of various cardiovascular diseases and a major determinant of diastolic heart failure and SCD (2,3), the findings could have considerable clinical and biological implications. Thus, experiments are needed to identify the actual modifier genes and elucidate the molecular mechanisms by which they influence cardiac hypertrophic response. The results could lead to identification of new diagnostic and prognostic markers and therapeutic targets for cardiac hypertrophy.
MATERIAL AND METHODS
Demographics, clinical characteristics and causal gene/mutation
Demographic and clinical characteristics of 153 family, mapping of the causal gene to chromosome 11q11 (maximum LOD score of 7.21) and identification of the causal mutation as an insertion mutation G791 in exon 25 of the MyBP-C, encoded by MYBPC3, have been published (12). The family is comprised of 244 members. One hundred family members (51 males and 49 females), including 36 with the InsG791 mutation in MYBPC3 were genotyped to map modifier genes.
Left ventricular mass
We calculated the LVM by the area-length method from two-dimensional echocardiographic images according to the recommendations of the American Society of Echocardiography (32) and as described (25).
Genotyping
We genotyped the family members for 811 short-tandem repeat (STR) DNA markers, positioned at ~5 cM intervals across the human genome (Linkage Mapping Set v2.5, HD5, from Applied Biosystems, Inc.). STR markers were genotyped by PCR using fluorescently-labeled oligonucleotide primers and capillary electrophoresis on an ABI 3130xl system. Initial allele calls were made using GeneMapper software v. 3.7. The alleles were then checked for pedigree-wide Mendelian consistency with both the PedCheck (33) and Loki software packages (5). Inconsistencies were resolved via graphical analysis of the raw genotypes and examination of the pedigree data. If a marker genotype for a particular genotype could not be resolved, it was deleted. If a marker or individual was found to have a large number of inconsistencies and missed calls, that marker or individual was removed from further analysis.
MCMC segregation and linkage analysis
We applied the MCMC segregation and linkage analysis methods implemented in the Loki software package to estimate the number, effects and location of genetic loci, in addition to MYBPC3, contributing to variation in LVM. These methods allow for covariate effects, and the trait model is given by , where μ is the baseline trait value, X is the incidence matrix for covariate effects, β is the vector of covariate effects, Qi is the incidence matrix for the effects of QTLi, αi is the vector of effects for QTLi, e is the normally distributed residual effect and k is the number of QTL currently estimated (k ≥ 0). The MCMC process samples μ, β, αi and k as well as parameters such as unobserved marker and trait genotypes. The parameter values are sampled proportional to their posterior probability, and thus are consistent with the data observed. We retain every fifth iteration in our analyses for reasons of computational efficiency. After the number of sampling iterations is sufficiently large, the sampled values provide an estimate of the posterior probability distribution over the space of possible parameter configurations.
To evaluate evidence for linkage, first we computed ‘L scores’ estimated from 500 000 iteration analysis runs over 1 cM-wide bins along the chromosomes. An L score is simply the posterior probability of linkage divided by the prior probability. In the absence of any data, a Bayesian analysis should have posterior probability equal to the prior probability. Thus, an L score of 1 indicates that the data contains no information for or against linkage. An L score <1 indicates evidence against linkage, whereas an L score >1 indicates evidence for linkage. In regions in which we found L scores >7.3 (an empirical threshold for 95% chromosome-wide significance) (13), we computed the LOP (Log of placement posterior probability ratio) as described (14). The LOP has a number of properties that make it preferable to the L score, but it is computationally more intensive to estimate. Thus, we used the L score as a preliminary test to identify chromosomes on which to estimate LOP. The LOP compares evidence for linkage on the real chromosome with information on simulated pseudo chromosomes, which are generated to match the information of the markers on the real chromosomes, but are unlinked. LOP is scored over both QTL position and square root of the phenotypic variance attributed to the QTL, i.e. the mean effect size. LOP gives an indication of both the position on the chromosome and importance of a locus. The LOP values were computed for 20 million iterations using 10 different pseudo chromosomes, which is expected to provide stable estimates.
Acknowledgments
This work was supported in part by grants from the National Heart, Lung and Blood Institute (R01-HL68884), Clinician-Scientist Award in Translation Research from Burroughs Wellcome Fund (#1005907) and Greater Houston Community Foundation (TexGen).
Footnotes
Conflict of Interest statement. There is no direct or indirect conflict of interest for any of the authors for the data presented in this manuscript in whole or in part.
References
- 1.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 2.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 3.Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000;342:1778–1785. doi: 10.1056/NEJM200006153422403. [DOI] [PubMed] [Google Scholar]
- 4.Marian AJ. Clinical and molecular genetic aspects of hypertrophic cardiomyopathy. Current Cardiol Rev. 2005;1:53–63. [Google Scholar]
- 5.Heath SC. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am J Hum Genet. 1997;61:748–760. doi: 10.1086/515506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daw EW, Kumm J, Snow GL, Thompson EA, Wijsman EM. Monte Carlo Markov chain methods for genome screening. Genet Epidemiol. 1999;17(Suppl 1):S133–S138. doi: 10.1002/gepi.1370170723. [DOI] [PubMed] [Google Scholar]
- 7.Daw EW, Liu X, Wu CC. Age-of-onset of hypertension vs. a single measurement of systolic blood pressure in a combined linkage and segregation analysis. BMC Genet. 2003;4(Suppl 1):S80. doi: 10.1186/1471-2156-4-S1-S80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wijsman EM, Rothstein JH, Thompson EA. Multipoint linkage analysis with many multiallelic or dense diallelic markers: Markov chain-Monte Carlo provides practical approaches for genome scans on general pedigrees. Am J Hum Genet. 2006;79:846–858. doi: 10.1086/508472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Igo RP, Jr, Chapman NH, Berninger VW, Matsushita M, Brkanac Z, Rothstein JH, Holzman T, Nielsen K, Raskind WH, Wijsman EM. Genomewide scan for real-word reading subphenotypes of dyslexia: novel chromosome 13 locus and genetic complexity. Am J Med Genet B Neuropsychiatr Genet. 2006;141:15–27. doi: 10.1002/ajmg.b.30245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gagnon F, Jarvik GP, Badzioch MD, Motulsky AG, Brunzell JD, Wijsman EM. Genome scan for quantitative trait loci influencing HDL levels: evidence for multilocus inheritance in familial combined hyperlipidemia. Hum Genet. 2005;117:494–505. doi: 10.1007/s00439-005-1338-4. [DOI] [PubMed] [Google Scholar]
- 11.Shmulewitz D, Heath SC, Blundell ML, Han Z, Sharma R, Salit J, Auerbach SB, Signorini S, Breslow JL, Stoffel M, Friedman JM. Linkage analysis of quantitative traits for obesity, diabetes, hypertension, and dyslipidemia on the island of Kosrae, Federated States of Micronesia. Proc Natl Acad Sci USA. 2006;103:3502–3509. doi: 10.1073/pnas.0510156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, Seidman JG, Seidman CE. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–1257. doi: 10.1056/NEJM199804303381802. [DOI] [PubMed] [Google Scholar]
- 13.Daw EW, Shete S. Identifying secondary loci in existing genome scan data. Genet Epidemiol. 2007;31:468–469. [Google Scholar]
- 14.Daw EW, Wijsman EM, Thompson EA. A score for Bayesian genome screening. Genet Epidemiol. 2003;24:181–190. doi: 10.1002/gepi.10230. [DOI] [PubMed] [Google Scholar]
- 15.Brogan WC, III, Hillis LD, Flores ED, Lange RA. The natural history of isolated left ventricular diastolic dysfunction. Am J Med. 1992;92:627–630. doi: 10.1016/0002-9343(92)90781-6. [DOI] [PubMed] [Google Scholar]
- 16.Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med. 1991;114:345–352. doi: 10.7326/0003-4819-114-5-345. [DOI] [PubMed] [Google Scholar]
- 17.Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998;32:1454–1459. doi: 10.1016/s0735-1097(98)00407-0. [DOI] [PubMed] [Google Scholar]
- 18.Krumholz HM, Larson M, Levy D. Prognosis of left ventricular geometric patterns in the Framingham Heart Study. J Am Coll Cardiol. 1995;25:879–884. doi: 10.1016/0735-1097(94)00473-4. [DOI] [PubMed] [Google Scholar]
- 19.Hynes RO. Integrins: Bidirectional, Allosteric Signaling Machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 20.Ekwa-Ekoka C, Diaz GA, Carlson C, Hasegawa T, Samudrala R, Lim Kc, Yabu JM, Levy B, Schnapp LM. Genomic organization and sequence variation of the human integrin subunit [alpha]8 gene (ITGA8) Matrix Biol. 2004;23:487–496. doi: 10.1016/j.matbio.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Bouzeghrane F, Mercure C, Reudelhuber TL, Thibault G. [alpha]8[beta]1 integrin is upregulated in myofibroblasts of fibrotic and scarring myocardium. J Mol Cell Cardiol. 2004;36:343–353. doi: 10.1016/j.yjmcc.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Shirani J, Pick R, Roberts WC, Maron BJ. Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000;35:36–44. doi: 10.1016/s0735-1097(99)00492-1. [DOI] [PubMed] [Google Scholar]
- 23.Liu B, Liu Y, Chen J, Wei Z, Yu H, Zhen Y, Lu L, Hui R. CARP is a novel caspase recruitment domain containing pro-apoptotic protein. Biochem Biophys Res Commun. 2002;293:1396–1404. doi: 10.1016/S0006-291X(02)00379-0. [DOI] [PubMed] [Google Scholar]
- 24.Ma J, Amos CI, Daw EW. Ascertainment correction for Markov Chain Monte Carlo segregation and linkage analysis of a quantitative trait. Genet Epidemiol. 2007 doi: 10.1002/gepi.20231. [DOI] [PubMed] [Google Scholar]
- 25.Lechin M, Quinones MA, Omran A, Hill R, Yu QT, Rakowski H, Wigle D, Liew CC, Sole M, Roberts R. Angiotensin-I converting enzyme genotypes and left ventricular hypertrophy in patients with hypertrophic cardiomyopathy. Circulation. 1995;92:1808–1812. doi: 10.1161/01.cir.92.7.1808. [DOI] [PubMed] [Google Scholar]
- 26.Marian AJ, Yu QT, Workman R, Greve G, Roberts R. Angiotensin-converting enzyme polymorphism in hypertrophic cardiomyopathy and sudden cardiac death. Lancet. 1993;342:1085–1086. doi: 10.1016/0140-6736(93)92064-z. [DOI] [PubMed] [Google Scholar]
- 27.Patel R, Lim DS, Reddy D, Nagueh SF, Lutucuta S, Sole MJ, Zoghbi WA, Quinones MA, Roberts R, Marian AJ. Variants of trophic factors and expression of cardiac hypertrophy in patients with hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2000;32:2369–2377. doi: 10.1006/jmcc.2000.1267. [DOI] [PubMed] [Google Scholar]
- 28.Abchee A, Marian AJ. Prognostic significance of beta-myosin heavy chain mutations is reflective of their hypertrophic expressivity in patients with hypertrophic cardiomyopathy. J Invest Med. 1997;45:191–196. [PubMed] [Google Scholar]
- 29.Nagueh SF, Bachinski L, Meyer D, Hill R, Zoghbi WA, Tam JW, Quinones MA, Roberts R, Marian AJ. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with familial hypertrophic cardiomyopathy and provides a novel means for an early diagnosis prior to an independent of hypertrophy. Circulation. 2001;104:128–130. doi: 10.1161/01.cir.104.2.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagueh SF, McFalls J, Meyer D, Hill R, Zoghbi WA, Tam JW, Quinones MA, Roberts R, Marian AJ. Tissue Doppler imaging predicts the development of hypertrophic cardiomyopathy in subjects with subclinical disease. Circulation. 2003;108:395–398. doi: 10.1161/01.CIR.0000084500.72232.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazur W, Nagueh SF, Lakkis NM, Middleton KJ, Killip D, Roberts R, Spencer WH., III Regression of left ventricular hypertrophy after nonsurgical septal reduction therapy for hypertrophic obstructive cardiomyopathy. Circulation. 2001;103:1492–1496. doi: 10.1161/01.cir.103.11.1492. [DOI] [PubMed] [Google Scholar]
- 32.Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr. 1989;2:358–367. doi: 10.1016/s0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]
- 33.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]