

Abstract
Nonhomogeneous Markov models of nucleotide substitution have received scant attention. Here we explore the possibility of using nonhomogeneous models to identify host shift nodes along phylogenetic trees of pathogens evolving in different hosts. It has been noticed that influenza viruses show marked differences in nucleotide composition in human and avian hosts. We take advantage of this fact to identify the host shift event that led to the 1918 ‘Spanish’ influenza. This disease killed over 50 million people worldwide, ranking it as the deadliest pandemic in recorded history. Our model suggests that the eight RNA segments which eventually became the 1918 viral genome were introduced into a mammalian host around 1882–1913. The viruses later diverged into the classical swine and human H1N1 influenza lineages around 1913–1915. The last common ancestor of human strains dates from February 1917 to April 1918. Because pigs are more readily infected with avian influenza viruses than humans, it would seem that they were the original recipient of the virus. This would suggest that the virus was introduced into humans sometime between 1913 and 1918.
Keywords: Influenza, Spanish flu, Swine flu, H1N1, Non-homogeneous model, CG content, Molecular dating
Introduction
Markov models of nucleotide substitution have now become widely used in phylogenetic analysis (Yang 2006; Felsenstein 2003). Markov models are defined by a substitution matrix that describes the pattern of changes that occur in a sequence as it evolves along a phylogenetic tree. If the pattern of nucleotide substitution is independent of time (i.e., it is the same along the whole tree), the process is said to be time homogeneous. In a homogeneous process, as time approaches infinity, the distribution of nucleotide frequencies in a sequence approaches a stationary or equilibrium distribution (usually denoted π). Most Markov evolutionary models assume that forward and backward evolution along a tree branch are indistinguishable at equilibrium. This reversibility property is simply a restriction that facilitates the mathematical treatment of the models (Yang 1994). One of the important properties of a reversible process at equilibrium is the so called ‘pulley effect’ (Felsenstein 1981) that prevents identification of the root of a stationary tree because the direction of evolution in such trees is not defined. Most models currently used in phylogenetic analysis assume homogeneity, stationarity, and reversibility.
The nucleotide frequencies of sequences belonging to distantly related species are generally quite different, a clear indicator that the homogeneity and stationarity assumptions are being violated (Yang and Roberts 1995). For trees including distantly related organisms, different models might be needed to describe the patterns of nucleotide substitution in different parts of the tree, and sometimes, even one model per branch might be needed to achieve a realistic representation of the evolutionary process (Yang and Roberts 1995). Such nonhomogeneous trees involve a large number of parameters that cannot be reliably estimated by maximum likelihood (ML) or that might become mathematically intractable. For this reason, despite their importance, relatively little work has been done on the use of nonhomogeneous models in phylogenetics (see for example Barry and Hartigan 1987; Boussau et al. 2008; Gu and Li 1998; Blanquart and Lartillot 2008; Yang and Roberts 1995; Galtier et al. 1999; Galtier and Gouy 1998; Lockhart et al. 1994). An interesting possibility that might lead to easily tractable nonhomogeneous models concerns the analysis of patterns of nucleotide substitution for viruses that have experienced well established host transfer events. If the intracellular environment of the new host is substantially different, this could lead to a shift in the substitution pattern of the virus in the new host (Fig. 1). The nucleotide frequencies of the viral genome would then drift toward new equilibrium values. Trees accommodating viral sequences isolated from different hosts could then be analyzed by assuming just one set of evolutionary parameters for each host clade. If one of the hosts serves as a natural reservoir, viral evolution within this host would be stationary. The process would be nonstationary in the new hosts. Branches linking different host clades would contain host shift nodes, and the positions of these nodes could be determined by maximum likelihood.
Fig. 1.

The hypothetical evolution of a virus after a cross species jump (host shift). Evolution along the new host branches is non-stationary. The inset figure shows a computer simulation of the frequency of an arbitrary nucleotide i along evolutionary time (d) after a host shift. The equilibrium frequency in the reservoir host is π i* and in the new host is πi
If the G + C content of human, avian, and swine influenza virus sequences are plotted against the isolation year, a conspicuous pattern of G + C composition decay is seen in the mammalian viruses (Fig. 2), indicating that different substitution patterns characterize the evolution of the viral segments in mammalian and avian hosts (Rabadan et al. 2006). The evolution of influenza viruses is therefore better represented by a nonhomogeneous Markov model where different substitution patterns would describe the evolution process in various parts of the virus phylogenetic tree. This raises the intriguing possibility that this change in substitution pattern might allow us to identify and study the point along the phylogenetic tree where host shifts have occurred.
Fig. 2.

Genome G + C content versus isolation year for influenza viruses. Black dots A/H1N1 waterfowl. Red dots A/H1N1 human. The empty dots are human viruses that reappeared after 1977, the isolation time for these viruses has been corrected for the period of evolutionary stasis (see text). Blue dots A/H1N1 classical swine. Gray dots A/H5N1 human. These are avian-like sequences that have not spread within the human population, and thus retain the avian nucleotide content. Green dots Influenza B. These viruses mainly infect humans, and they may have evolved from an avian reservoir at an unknown remote date (Gammelin et al. 1990). (Color figure online)
Influenza A is a negative-strand RNA virus with a segmented genome that causes annual epidemics of disease in humans and domestic animals. The natural reservoir of the influenza A virus is waterfowl, in which the virus replicates and spreads causing little or no disease (Webster et al. 1992). The eight negative-strand RNA segments that comprise the virus genome encode 11 proteins. Two of these, the hemagglutinin (HA) and neuraminidase (NA), are surface glycoproteins that interact with the host’s immune system. Influenza viruses are classified according to the antigenic properties of the HA and NA proteins. A total of 16 HA and 9 NA serotypes have been identified in wild waterfowl, whereas only three HA (H1, H2, and H3) and only two NA (N1 and N2) subtypes are known to have been involved in epidemic disease in humans.
Avian viruses usually do not infect humans as these viruses are not adapted to the human host. Periodically, however, human viruses might acquire gene segments from an avian source, perhaps through an intermediary host, resulting in global pandemics in immunologically naive human populations. Two of the three 20th century flu pandemics were caused by this process. The 1957–1958 (H2N2, Asian flu) and 1968–1969 (H3N2, Hong Kong flu) pandemics that caused substantial mortality in the human population, were the result of reassortant viruses that had acquired novel segments coding for HA or HA and NA, and a polymerase gene (PB1) from an avian-like source (reviewed in Hay et al. 2001). Whether the 1918–1919 pandemic (H1N1, ‘Spanish’ flu) was caused by a reassortant virus like the 1957 and 1968 viruses, or was the result of transfer of a whole virus from an avian reservoir has been hotly debated (Gorman et al. 1990; Gibbs and Gibbs 2006; Gammelin et al. 1990; Taubenberger et al. 2006; Reid et al. 2004; Taubenberger et al. 2005; Gorman et al. 1991; Antonovics et al. 2006). During each of these pandemics the preceding virus subtype became extinct and was replaced by the new reassortant. In 1977, the H1N1 virus subtype which had become extinct in 1957 reappeared in the human population, infecting mainly young people (<25 years) who had not been exposed to the H1N1 subtype circulating previously. Since then, both H1N1 and H3N2 viruses have co-circulated with influenza B in humans. A stable lineage of H1N1 influenza in North American pigs (classical swine) was noticed after the 1918 pandemic. It is though that this classical swine lineage originated from the human ‘Spanish’ virus (Taubenberger 2006).
The 1918–1919 ‘Spanish’ flu has been the most devastating epidemic disease in recorded human history. It killed an estimated 50 million people worldwide (Johnson and Mueller 2002), many more than the number of deaths caused by the First World War. Given the constant threat of new zoonotic pandemics, much research has tried to understand the origin of the 1918 pandemic. The strongest evidence for an avian origin for the Spanish flu came from analysis of the genome sequence of the 1918 virus, obtained from lung tissue from a victim buried in the Alaskan permafrost (Taubenberger 2006; Reid et al. 2004; Taubenberger et al. 2005). Analysis of the consensus amino acid sequence of polymerase genes from avian viruses showed very little differences when compared to those from the 1918 virus (Taubenberger et al. 2005), while subsequent lineages of classical swine and human viruses had accumulated a substantial number of amino acid substitutions. This intuitively suggested that the introduction of the H1N1 virus into humans occurred in a relatively ‘short’ period (up to several years; Taubenberger et al. 2006) before the pandemic. A similar lack of adaptive evolution was also observed in other proteins of the 1918 virus (Reid et al. 2004) providing evidence for a single host shift event. Interestingly, on the nucleotide level, the 1918 virus was closer to other mammalian virus sequences than known avian virus consensus sequences, suggesting an early divergence between the current avian and 1918 virus lineages. This observation led Taubenberger et al. (2005) to suggest that the donor of the 1918 virus was in evolutionary isolation from other known avian flu viruses. A number of authors have questioned this interpretation (Gibbs and Gibbs 2006; Antonovics et al. 2006). One issue is the reliance of Taubenberger et al. (2005) on simplistic evolutionary models, and their focus on changes at the protein level, making the analysis susceptible to statistical noise and possible systematic biases. A rigorous phylogenetic study including the genome sequence of the 1918 virus, where the host shift event is clearly identified along the phylogenetic tree, and where modern molecular dating techniques are applied, has not yet been carried out.
As suggested by Fig. 2, influenza is well suited for study as a nonhomogeneous evolutionary process. Here we explore the possibility of using such a nonhomogeneous model to study the evolution of H1N1 viruses in birds, pigs, and humans. We address the question of the origin of the 1918 virus and time of the putative host shift event that led to the introduction of this virus from an avian into a mammalian host. These results suggest that the segments that formed the 1918 virus were transmitted to a mammalian host some time within the interval 1882–1913, followed by subsequent divergence between the human and classical swine lineages around 1913–1915. The virus was likely introduced into the human population between 1913 and 1918. This suggests a minimum of 5 years evolution in mammals prior to 1918, and that the classical swine lineage did not originate from the pandemic virus of 1918.
Methods
Data and Tree Estimation
We analyzed 40 full genome sequences of H1N1 influenza viruses isolated from avian (15), human (15), and swine (10) hosts. The eight RNA segment sequences from each genome were concatenated into a super gene and aligned (Muscle v3.6; Edgar 2004). The alignment, 13,140 sites, was edited manually. The tree topology was estimated by ML (HKY85 + dΓ5, PhyML v2.4.4; Guindon and Gascuel 2003), and the reliability of the tree topology was tested by bootstrapping 1,000 times. The virus strains analyzed and the consensus tree are shown in Fig. 3. Currently, all full genome sequences of H1N1 waterfowl viruses available in GenBank have been isolated from North American birds. We repeated some of the analyses with waterfowl viruses from other parts of the world. The estimated evolutionary parameters (such as the equilibrium nucleotide frequencies) appear independent of the geographical origin. Thus, the results should not be affected if the virus from which the 1918 pandemic originated was of Eurasian, rather than American, origin.
Fig. 3.

Consensus tree for 1,000 bootstrap replicates. Support values for the mammalian virus clades are shown. The avian viruses are mostly from waterfowl except for a pigeon isolate. Estimating the tree under a Bayesian framework (MrBayes v3.1; Huelsenbeck et al. 2001) leads to essentially the same results. The tree is shown rooted for illustrative purposes only. The black dot indicates the position of the most recent common ancestor of the human clade (MRCAH)
Nonhomogeneous Models of Influenza Evolution
We used the Hasegawa et al. (1985) Markov model of nucleotide substitution (HKY85) to describe the local nucleotide substitution pattern along the branches of the avian and mammalian influenza virus tree. The evolutionary parameters (π = {πi} and transition/transversion rate parameter κ) and the branch lengths (d i) for a given tree topology were estimated by ML (Yang 2006). The HKY85 model offers a good compromise between accuracy, computational speed, and relatively low variance when compared to more general models of nucleotide substitution (Yang 1994).
Using different sets of π values to describe the evolution along different branches of the tree implies time heterogeneity in the substitution pattern. In this work, we considered three models of evolution in the human–swine–avian tree (Fig. 4). The first model (M1) assumed homogeneity and stationarity, with one set of equilibrium nucleotide frequencies describing the substitution process in all branches of the tree. The second model (M2) assumed that equilibrium nucleotide frequencies are different in mammalian and avian hosts. The third model (M3), assumed different sets of equilibrium nucleotide frequencies for avian, human, and swine hosts, with the initial avian to mammal host shift occurring either to swine (M3s) or to humans (M3h). In models M2 and M3, evolution along the avian clade is stationary. Models M1, M2, and M3 are nested, so their log-likelihoods can be compared with the likelihood ratio test (LRT) to select the best model. The three models described above assumed a single avian to mammal host shift event. A variation of the M2 model was also tested that assumes that influenza was transmitted independently from birds to humans and from birds to swine following the divergence of these two lineages (M2.2j, Fig. 4). This model is not nested with any of the other models so the LRT cannot be used to assess its adequacy; the Akaike Information Criterion (AIC) can be used instead (Akaike 1974). All the models were tested on the data above using a nonhomogeneous implementation of the HKY85 model (PAML v3.15; Yang 1997; Yang and Roberts 1995) that considers rate variation among sites as a discrete gamma distribution (Yang 1996). A single gamma shape parameter (α) was assumed for the whole tree. Consideration of rate variation is fundamental since nucleotide frequencies decay at different rates at different sites, and averaging over them would lead to underestimation of the branch linking the mammalian clade with the host shift event.
Fig. 4.

Non-homogeneous models of influenza evolution. All model trees are unrooted. The real root is assumed to lie somewhere along the avian branches, however, its position is irrelevant since stationary evolution of the virus in the avian host is being assumed. Model M1 is homogeneous and the host shift event (HSE) cannot be determined. In models M2 and M3 the HSE is assigned avian equilibrium frequencies. Different shadings indicate that different rate matrices (equilibrium nucleotide frequencies) are used to describe evolution along the corresponding branches. With current data it is not possible to distinguish whether the HSE was avian to human, or avian to swine, so model M3 is in reality two models according to whether the branch linking the human–swine split (HSS) and the HSE is assigned human (M3h) or swine (M3s) equilibrium frequencies. Model M2.2J assumes two independent host shifts bird to mammal (see text)
Molecular Dating
The tree fitted under the best nonhomogeneous model has branch lengths in substitutions per site. We time calibrated the tree using a fully relaxed clock model under a penalized likelihood scheme (r8s v1.71; Sanderson 2003; Langley and Fitch 1974). Nonhomogeneous model fitting and time calibration was repeated for each of the 1,000 bootstrapped trees and their corresponding alignments. Isolation dates for most of the sequences analyzed are available to within 1 year. To correct for this level of uncertainty, the ages of the viruses in the bootstrap analysis were drawn from a random uniform distribution for the corresponding interval, i.e., if a virus is reported as isolated in 1957, its bootstrap distribution of age was sampled from the uniform distribution with boundaries [1957.0–1958.0). Hence the uncertainties in tree topology, branch lengths, and virus isolation times were carried through the analyses. The earliest human isolate is dated November 1918. The bootstrap confidence intervals for the evolutionary parameters and the node ages were calculated as described elsewhere (Venables and Ripley 2002, p 136). Data manipulation and basic statistics were carried out with the R environment for statistical computing (www.r-project.org). As an additional analysis, the third codon sites from the alignment (4,256 sites) were extracted, tree topology estimated, best nonhomogeneous model fitted, and the tree time calibrated. The results were essentially identical to the whole alignment case, albeit with wider confidence intervals.
Results
ML Estimation of Branch Lengths and Evolutionary Parameters Under Models M1, M2, and M3
We used the consensus tree topology estimated above to fit by ML the three M models (M1, M2, and M3) and assess the suitability of the different hypotheses concerning the homogeneity of the evolution of influenza viruses. Assuming nonhomogeneous evolution of the virus gene segments significantly improves the model fit when compared to a fully homogeneous model (LRT, M1 vs. M2,  , P ≪ 0.001, Table 1). Allowing for different substitution patterns in humans and swine does not significantly improve the model fit (LRT, M2 vs. M3h ≈ M3s,
, P ≪ 0.001, Table 1). Allowing for different substitution patterns in humans and swine does not significantly improve the model fit (LRT, M2 vs. M3h ≈ M3s,  , P ≤ 0.31, Table 1). This indicates that the shift in substitution patterns is a property of the evolution of the virus in mammalian hosts. The branch lengths for models M1 and M2 are highly correlated, but the homogeneous model slightly overestimates long branches (d
M2 = 0.96d
M1, r > 0.999). Model M2.2j, which assumes two independent bird to mammal host shifts, has a lower likelihood than M2 (Table 1). These two models are not nested, so the LRT cannot be used. The Akaike information criterion supports M2 as the best model overall (AIC, Table 1). Our results, while not definitive, support a single jump from birds to mammals, a conclusion consistent with the more frequently observed inter-mammalian host shifts than shifts between avian and mammal species.
, P ≤ 0.31, Table 1). This indicates that the shift in substitution patterns is a property of the evolution of the virus in mammalian hosts. The branch lengths for models M1 and M2 are highly correlated, but the homogeneous model slightly overestimates long branches (d
M2 = 0.96d
M1, r > 0.999). Model M2.2j, which assumes two independent bird to mammal host shifts, has a lower likelihood than M2 (Table 1). These two models are not nested, so the LRT cannot be used. The Akaike information criterion supports M2 as the best model overall (AIC, Table 1). Our results, while not definitive, support a single jump from birds to mammals, a conclusion consistent with the more frequently observed inter-mammalian host shifts than shifts between avian and mammal species.
Table 1.
Likelihoods and model comparison
| Model | lnℓ | np | P-value | AIC |
|---|---|---|---|---|
| M1 | −83,751 | 82 | – | 167,668 |
| M 2 | −83,670 | 86 | 0.001 | 167,514 |
| M3 | −83,668 | 89 | 0.31 | 167,516 |
| M2.2j | −83,672 | 87 | – | 167,520 |
np Number of estimated parameters
Models M3h and M3s have essentially the same likelihood. The bold values highlight the statistically best model
Table 2 shows the ML estimates of the evolutionary parameters for model M2 and their 95% confidence intervals (CI) from the bootstrap analysis. It is clear that the relative rates of G → A and C → U transition substitutions are accelerated in mammalian  when compared to avian
when compared to avian  viruses. This shift in G → A and C → U transition rates is responsible for the G + C composition decay observed in mammalian viruses (Fig. 2). Reasons for this shift in substitution rates are not clear. A few hypotheses of how this substitution pattern might have come about in human compared to avian hosts have been discussed (Greenbaum et al. 2008; Rabadan et al. 2006). It seems experimental work is needed to address this issue. The ML method is, however, blind to the causes of the substitution shift and simply identifies the most likely location of the host shift. Here we are content with using this substitution pattern shift to time the ancestor of human and swine H1N1 viruses rather than with the causes of the substitution pattern itself.
viruses. This shift in G → A and C → U transition rates is responsible for the G + C composition decay observed in mammalian viruses (Fig. 2). Reasons for this shift in substitution rates are not clear. A few hypotheses of how this substitution pattern might have come about in human compared to avian hosts have been discussed (Greenbaum et al. 2008; Rabadan et al. 2006). It seems experimental work is needed to address this issue. The ML method is, however, blind to the causes of the substitution shift and simply identifies the most likely location of the host shift. Here we are content with using this substitution pattern shift to time the ancestor of human and swine H1N1 viruses rather than with the causes of the substitution pattern itself.
Table 2.
ML estimates of evolutionary parameters for the HKY85 M2 model
| Host | Par | Value (95% CI) |
|---|---|---|
| All |
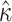
|
12.5 (11.8, 13.8) |
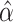
|
0.226 (0.216, 0.237) | |
| Avian |
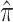 U
U
|
0.235 (0.228, 0.242) |
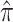 C
C
|
0.207 (0.200, 0.213) | |
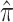 A
A
|
0.329 (0.322, 0.337) | |
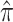 G
G
|
0.229 (0.222, 0.236) | |
| Mammalian |
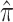 U
U
|
0.253 (0.239, 0.267) |
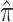 C
C
|
0.178 (0.167, 0.188) | |
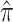 A
A
|
0.399 (0.385, 0.415) | |
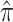 G
G
|
0.170 (0.159, 0.179) |
Note: the substitution rate from nucleotide i to j, q ij, can be calculated from this table as q ij = cκπj for transitions and q ij = cπj for transversions, where c is a proportionality constant (for details see chap 1 in Yang 2006)
Stability of the Host Shift Node
An important property of nonhomogeneous, nonstationary models is their theoretical ability to identify the position where changes in the substitution pattern have occurred. The drift in base frequencies towards different equilibrium values along the tree branches should give, in theory, enough information to the maximum likelihood method to be able to identify the position of those nodes. In our case, it should allow the identification of the location where the host shift occurred. Figure 5 shows the likelihood surface for the branch projecting from the host shift towards the mammalian clade (d
ma) versus the branch projecting from the host shift towards the waterfowl clade (d
wf). The likelihood surface appears highly correlated along the d
ma + d
wf line, as are the estimated branch lengths from the bootstrap analysis (Fig. 5). The bootstrapping exercise is essentially equivalent to sampling trees from the likelihood surface (a parametric bootstrap gives essentially the same results). For comparison, Fig. 5 also shows the likelihood surface for the two branches projecting forward from the human–swine split (d
hu and d
sw). The estimation of these branches is far more accurate, and their estimates are uncorrelated (Fig. 5). The correlation in the likelihood surface seen in Fig. 5 translates into wide confidence intervals for the lengths of the branches projecting from the host shift (e.g., 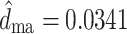 , 95% CI: 0.0, 0.0626). It is interesting to note that the sum of these branches, can be estimated much more reliably (
, 95% CI: 0.0, 0.0626). It is interesting to note that the sum of these branches, can be estimated much more reliably (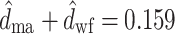 , 95% CI: 0.147, 0.175). The correlation observed between d
ma and d
wf is directly related to the pulley effect that precludes the identification of the root in a reversible, stationary tree (Felsenstein 1981).
, 95% CI: 0.147, 0.175). The correlation observed between d
ma and d
wf is directly related to the pulley effect that precludes the identification of the root in a reversible, stationary tree (Felsenstein 1981).
Fig. 5.

Stability of the maximum likelihood estimates of branch lengths for model M2. The plot shows the log-likelihood profiles (top) and bootstrap sample estimates (bottom) for selected pairwise branch comparisons. The inset tree, is the tree optimized under the HKY85 M2 model, showing the waterfowl (Wf), human (Hu), and swine (Sw) clades, the host shift event (HSE) and the human–swine split (HSS). The two branches protruding from host shift event are d wf and d ma, and the two branches protruding forward from the human–swine split are d sw and d hu
Tree Calibration and the Origin of the 1918 Pandemic Virus
The HKY85 M2 tree optimized by ML has branch lengths in substitutions per site, as substitution rate and real time are confounded factors that cannot be estimated independently without additional information (Yang 1994). To estimate the date of the host shift event we calibrated the tree using Langley and Fitch’s (LF) molecular clock model (Langley and Fitch 1974) and timed the nodes along the human–swine portion of the HKY85 M2 optimized tree. We used an implementation that uses a negative binomial correction to account for rate heterogeneity among sites and that considers local variations in the clock rate (r8s; Sanderson 2002, 2003). Substitution rates for each branch (a fully relaxed clock) and the ages of internal nodes were then estimated by penalized likelihood under the corrected LF model. This procedure was repeated for each one of the 1,000 bootstrap trees, as to assess the variability of substitution rates and age estimates under variable branch lengths and tree topologies.
Before fitting the LF model to date the host shift event, two oddities concerning the data analyzed need to be addressed (Fig. 6). First, human viruses isolated between 1933 and 57 have been passaged an undefined number of times in the laboratory before sequence determination, thus accumulating a substantial amount of nucleotide substitutions (Bush et al. 2000). Including these lab-adapted virus sequences in the estimation of the tree topology above is, however, not expected to lead to any errors since only the corresponding tips in the tree are expected to be elongated. These sequences provide valuable information for estimation of the evolutionary parameters and help reduce the variance of estimated internal branch lengths. However, including these sequences in the tree calibration would certainly lead to overestimation of the substitution rate, so the eight human viruses isolated between 1933 and 57 were not considered for the LF analysis. The 1918 Brevig Mission virus sequence was obtained directly from tissue of an Inuit woman buried in the Alaskan permafrost (Taubenberger 2006), and has no passage history, so it was included. The other oddity in the data is that the H1N1 viruses that reappeared in the human population in 1977 were very similar to the extinct strains circulating around 1950 (Nakajima et al. 1978). The reasons for this evolutionary stasis are not clear (Kilbourne 2006), prompting the speculation that these were the product of a lab accident, perhaps involving the release of a frozen strain (Palese 2004). We estimated the phylogenetic age of the modern H1N1 viruses by maximizing the likelihood of the LF model assuming variable intervals of evolutionary stasis. A time gap of 24.6 years is the most likely, indicating that the 1977 strain originated around 1953 (95% CI: 1948–1956) in agreement with previous studies (Nakajima et al. 1978; Raymond et al. 1986). The average branch substitution rate per site per year in human and classical swine viruses is 2.44 × 10−3 year−1 (95% CI: 2.29 × 10−3, 2.58 × 10−3).
Fig. 6.

Branch length versus year of isolation for human and swine H1N1 viruses. The total branch length from each tip to the human–swine split is plotted against the isolation year. Red dots human, blue dots classical swine. The empty dots show the corrected ages for the human viruses that reappeared in 1977. The regression slope is the approximated substitution rate. Some of the human viruses isolated between 1933 and 1957 deviate from the regression line due to extensive lab passing. The effect is negligible for the early swine viruses (1931–1957). (Color figure online)
The human and swine lineages are estimated to have diverged between March 1913 and October 1915 (Table 3). The divergence time of this node seems reliable as the likelihood surface is well developed (Fig. 5). The most recent common ancestor of human viruses (MRCAH, Fig. 3) dates back to between February 1917 and April 1918 (Table 3). The host shift is estimated to have happened around 1882–1912. This assumes that the virus evolved at the average mammalian rate just after the host shift. However, accelerations of up to 50% in rate have been observed in swine viruses from recent avian origin (Ludwig et al. 1995). Assuming such increased substitution rate throughout the genome, would place the host shift around 1893–1913. Because the estimates of the length of the two branches projecting from the host shift are correlated (Fig. 5), a large CI for the host shift date cannot be avoided (Table 3).
Table 3.
Estimated dates for the host shift, human–swine split, and MRCAH
| Node | Date (95% CI) |
|---|---|
| Host shift | 1901.1 (1882.8, 1912.2) |
| Host shifta | 1905.8 (1893.2, 1913.0) |
| Human–swine split | 1914.6 (1913.2, 1915.8) |
| MRCAH | 1917.8 (1917.2, 1918.3) |
aAssuming an accelerated substitution rate, 1.5 times faster the average rate
Reliability of the LF Local Clock Model Calibration
To test the reliability of the LF local clock calibration, we set the isolation date of the 1918 sequence as an unknown parameter and re-estimated it. We repeated this procedure for every sequence (except for the early, lab-adapted human isolates, 1933–57). We recovered the isolation date to within −1.30–1.52 years for all sequences (mean error = 0.013 years, SD = 0.64 years). The pandemic virus, dating from November 1918, was dated as June 1918, a 5 month error. Because the tip ages are highly correlated with the ages of the corresponding subtending nodes, and the variances of the estimated tip ages are larger than the variance of the corresponding node ages, it seems that the LF relaxed clock gives a robust calibration of the tree. We also re-analyzed the third codon sites from the whole alignment. Using only these sites we were able to retrieve the tree topology, the evolutionary parameters under model M2, and all the node ages.
A limitation of the LF model is that it assumes the substitution process is Poissonian (or negative binomial when rate variation is considered). This is true under simple nucleotide substitution models such as Jukes and Cantor; however, for more complicated models like HKY85 the process is not Poissonian (Yang 2006), although the deviations do not seem important. Also, the use of the ML branch lengths as proxy for the observed number of substitutions in the LF calibration, instead of re-estimating the branch lengths under a clock model and a full substitution matrix implies a loss of information from the data. We used an implementation of the TipDate model (PAML; Yang 1997; Rambaut 2000) to re-estimate the ages of all internal nodes under the HKY85 model, which should address the concerns about the LF model above. The current TipDate implementation assumes stationarity, however, this does not seem to generate any noticeable discrepancies as the estimated ages for the internal nodes are nearly identical for both methods (r > 0.999).
There is a subtle but important point to the penalized likelihood and bootstrap approach used here. Although the bootstrap correctly accounts for uncertainties in branch length estimates, it does not take into account variations in the relaxed clock rates and divergence times, even if the branch lengths were perfectly known (Thorne and Kishino 2005). The result is that the uncertainties in divergence times are underestimated. Applying a Bayesian MCMC approach with an independent log-normal relaxed clock (Drummond and Rambaut 2007; Drummond et al. 2006), we find a divergence time for human and swine viruses between 1911.7–1916.1 and 1916.3–1918.1 for the MRCAH. This approach assumes homogeneity and stationarity so it cannot be used to date the host shift. Furthermore, the independence assumption is likely to overestimate the uncertainty in date estimates as it overlooks the different substitution rates in the human and swine lineages (Ludwig et al. 1995).
Discussion
Rabadan et al. (2006) noticed the differences in nucleotide composition between avian and human influenza viruses. Here we show that these differences extend to classical swine viruses and that they can be modeled as a nonhomogeneous process along the waterfowl–mammalian phylogenetic tree. Analysis of the posterior site rates from the discrete gamma distribution (Yang and Kumar 1996), show that the mostly synonymous third codon sites evolve over 5 times faster than first and second sites. Most of the G + C decay signal comes from these third sites. Moreover, when the whole analysis was repeated using third sites alone, essentially all results were reproduced. This would suggest that the G + C decay is the consequence of a selectively neutral process (although see Greenbaum et al. 2008). Rabadan et al. (2006) used the increase in U frequency observed in two human strains (1918 and 1933) to calculate the earliest date for the introduction of the polymerase genes into a mammalian virus, estimating this at roughly 1910. This point estimate falls within our estimated CI for the host shift; however, we disagree with the conclusion of those authors that this is the earliest possible date for the host shift, as they neither considered the variance of their estimate, nor the effect of rate variation among sites.
Our analysis was performed on the concatenated set of gene segments. Is this approach justified? The estimated topology for the concatenated set of eight RNA segments for the mammalian part of the tree is fully resolved (Fig. 1). However, this is not the case when the topology is estimated for each gene segment separately. Analysis of the individual segments show similar results, with four of the segments (PA, HA, NP, NA) supporting this topology. The segments encoding the PB2 and PB1 proteins place the 1918 sequence at the bottom of the swine virus lineage with high, but inconclusive, bootstrap support (52 and 77%). The segment encoding the M and NS genes, the smallest of the eight segments, do not hold enough phylogenetic information to resolve the position of the 1918 sequence relative to the human–swine split node. The uncertainty in the position of the 1918 sequence for these segments is most likely an artifact of the long branches linking this sequence with the rest of the tree. The 1918 sequence itself is confidently placed at the bottom of the human branch when the full concatenated set is considered (100% bootstrap support). If we take the gene trees literally, the only possibility is that there were two different strains circulating in 1918 that reassorted to form the 1918 pandemic virus. This reassortant would have been replaced later by a non-reassortant some time before the earliest post-1918 human isolates of the 1930s. While this is an intriguing possibility, in the absence of more convincing statistical support we agree with Worobey’s (2008) view that the 1918 sequence is much more reasonably placed on the human lineage. There does, however, seem to be reassortment occurring on the avian part of the tree, but the topology and timing of this part of the tree is not used in the analysis, and such reassortment does not affect the estimation of evolutionary parameters. Analysis of the individual genes gives similar values for the evolutionary parameters for all eight gene segments, as well as the concatenated gene set, especially in nucleotide frequencies, indicating that our values are robust to errors in tree topology in the avian part of the tree (Fig. 1)
There still exists, however, the possibility that the segments that formed the 1918 virus were the product of sequential reassortment events involving avian-like viruses in a mammalian host before the split of the human and swine lineages. For example, a mammalian virus might have reassorted with an avian virus to produce a hybrid reassortant (such as in the 1957 and 1968 pandemics; Kawaoka et al. 1989), this hybrid might in turn have reassorted again one or more times losing the original segments and resulting in an avian-like virus with different segments introduced at different times and showing different levels of nucleotide composition decay. We performed a similar analysis on each of the eight H1N1 RNA segments, and obtained individual host transfer dates for each segment varying from 1840 to 1912. In particular, the HA and NP segments seem to have been introduced earlier (pre-1890) than the polymerase genes (post-1900). We intentionally avoid given specific ages to the individual segments, as the branches projecting from the host shift node are highly correlated (Fig. 7), making the estimation of the individual host transfer dates highly uncertain. Concatenating the segments reduces the variance of date estimates, at the expense of assuming a single host shift event. The pulley effect that precludes the identification of the root in a stationary tree is a pervasive effect that is still present, and hampers the identification of the substitution pattern shift node along a nonhomogeneous tree. With the current data and analysis it is not possible to distinguish between a single host shift event or a successive series of host transfer/reassortment events. Disentangling the ages of the individual gene segments that formed the 1918 virus is difficult and will require further analysis.
Fig. 7.

Bootstrap distribution of the branches projecting from the host shift node (d ma and d wf) for the HA gene. Both branch parameters are highly correlated, making the estimation of the age of the HA gene in mammals unreliable
Even before the genome sequence of the 1918 virus became available, several authors had already suggested that the ancestor of the 1918 virus was of avian origin (Gorman et al. 1990, 1991; Gammelin et al. 1990). Gammelin et al. (1990) cautiously suggested an origin for the mammalian virus around 1837. Because they used the divergence between mammalian and avian viruses as the reference point in the NP phylogenetic tree to propose their date, this should be regarded as the earliest possible date. Gorman et al. (1991) also used a phylogenetic tree based on the NP segment. They noticed that the NP proteins from early human and classical swine viruses (~1930s) were very similar to those from avian viruses, and argued (similarly to Reid et al. 2004; Taubenberger et al. 2005) that the host shift event must have been coincident roughly with the divergence of these lineages, an event that they calculated as occurring around 1912–1913 (close to our estimate of the date of the human–swine split) or 1918 (after considering the possibility of an accelerated substitution rate between 1918 and the 1930s). The accelerated substitution rate was suggested to explain how the host shift event could have occurred in 1918, allowing the simultaneous epidemics of swine and humans to be caused by a single event. With the availability of the 1918 sequence, the phylogenetic tree becomes much more resolved and this possibility is eliminated. Both of these studies implicitly assumed that the host shift happened at internal bifurcating nodes in the tree. Here we show that this is not necessarily so, as the host shift is more likely to have occurred before the divergence of the human and swine lineages.
Previous work (Taubenberger et al. 2005; Gorman et al. 1991; Gammelin et al. 1990) has highlighted the difficulty in piecing together evolutionary scenarios based solely on phylogenetic trees. Ideally we would want an internal clock that starts to tick when the host shift event occurs. Previous researchers have used the amino acid substitutions that distinguish mammalian and avian influenza (Taubenberger et al. 2005; Gorman et al. 1991). There are numerous reasons to suspect the validity of such calculations, as amino acid substitutions are relatively few in number and subject to idiosyncratic timing caused both by substitutions that might influence the probabilities of host shifts and by the evolutionary pressure to accept these substitutions in the new host. In contrast, we have analyzed the changes that occur in nucleotide frequency, representing host-specific substitution rates rather than adaptive changes. For instance, when only the mostly synonymous, third codon sites from the concatenated alignment were used, we were still able to retrieve the tree topology, the evolutionary parameters, and all the node timings, including the host shift.
Because most nucleotide changes seem to be selectively neutral, and since they occur at numerous locations along the entire sequence, we were not only able to make a reasonable estimate of host shift event, but we were also able to use sophisticated nonstationary evolutionary models and perform the type of rigorous statistical analysis that has been lacking in previous work. Our results are hence more likely to be robust to the different effects that occur with different locations under different degrees of selective pressure at the amino acid level in varying size populations. The nonhomogeneous method we propose here should have wider applications beyond influenza.
It has been suggested that the H1N1 classical swine lineage of influenza originated from a human source during the 1918–1919 outbreak (Taubenberger 2006). Our results, however, strongly indicate that this lineage split from the human one about 4 years before the pandemic. There are at least three possible hypotheses concerning the origin of the human and classical swine lineages of influenza: (a) an avian virus infected an unknown mammal, where it evolved for several years before infecting humans. It then infected swine around 1918 (Taubenberger et al. 2006); (b) an avian virus infected a human population where it evolved for several years before diverging into the classical swine and human lineages around 1914. Sometime after this date, the virus was introduced into the swine population; (c) an avian virus was transmitted to a swine population (Ludwig et al. 1995) where it evolved for several years, and sometime after 1913, but before early 1918, it crossed into humans leading to the 1918 pandemic. The problem with the first hypothesis is that the molecular data strongly supports a human–swine split between 1913 and 1916, inconsistent with the idea that classical swine originated from the 1918 human epidemic. The problem with the second hypothesis is that avian viruses are less well adapted to the human than the swine host. Avian hemagglutinin (including avian H1) bind preferentially to SAα-2,3Gal type avian receptors (Rogers and Paulson 1983), whereas human-adapted viruses (H1N1, H3N2, H2N2) bind preferentially SAα-2,6Gal type receptors expressed in the upper respiratory tract in humans. Thus, avian viruses (such as H5N1) that have infected humans directly, have not spread in the human population (Subbarao and Katz 2000). On the other hand, pigs express both SAα-2,6Gal and SAα-2,3Gal receptors and can readily be infected with avian and mammalian influenza viruses. This characteristic of the swine host led to the proposal of swine as mixing vessels for the reassortment of avian and mammalian influenza viruses (e.g. Scholtissek et al. 1985). Avian H1N1 viruses that became established in pigs in Europe (Brown et al. 1997) have subsequently caused occasional infections in humans (Gregory et al. 2003). More significantly, the emerging 2009 H1N1 pandemic is due to a reassortant virus which acquired its eight genes from different swine virus lineages, some of which originated from avian and human hosts (Dawood et al. 2009). There is still the problem of explaining the nearly simultaneous epidemics in swine and humans in 1918, given that the classical swine and human lineages had diverged years earlier. One possible explanation is that the swine epidemic was not noted until a similar epidemic appeared in humans in 1918. Alternatively, it is possible that the outbreaks of disease observed in swine during 1918 (Taubenberger 2006) were not due to a virus of the classical swine lineage but were caused by the human pandemic virus. This scenario is supported by the observation of human H1N1 viruses occasionally infecting swine (e.g., Neumeier et al. 1994), and by the recent infection of pigs in Canada by the 2009 H1N1 virus from a human source.
It is apparent that avian H1N1 viruses have become established in swine, while no instances of avian H1N1 viruses becoming established directly in humans have been observed. Considering this, we suggest an avian virus infected a swine host around 1883–1913, where it evolved for some time before acquiring the capacity to infect and spread in humans. This virus then entered the human population sometime after 1913 but before early-1918, when it initiated the pandemic. It is unlikely that the H1N1 virus was widespread in the human population before 1918. Seroarchaeological studies suggest that an H3 subtype was circulating worldwide at the time (Dowdle 1999). What happened to the virus during 1913–1918 is not clear; analysis of archaeoviral samples predating 1918 might shed some light on this issue. We might never get a definite answer to what happened during the years preceding 1918, but the possibility of potentially hazardous viruses smoldering in an isolated host population (whether human or swine), stresses the importance of extensive worldwide surveillance of influenza.
While the current article was in review, Smith et al. also concluded that the common ancestor of the classical swine and human H1N1 lineages was likely a few years before the pandemic of 1918 (Smith et al. 2009a), inconsistent with the Classical Swine lineage originating from the human 1918 outbreak and consistent with the identification of swine as a possible intermediate host.
While this manuscript was in preparation, the emergent pandemic H1N1 2009 virus was identified (Dawood et al. 2009). This is the first example, with the possible exception of 1918, that a virus of swine origin has become established in the human population to cause a pandemic. Certain parallels are apparent between the 1918 and 2009 pandemics, especially the possible role of swine as an intermediate host. The role of swine as a mixing vessel of different lineages, an important feature of the 2009 Swine-origin virus (Smith et al. 2009b), is less clear with the ‘Spanish flu’ pandemic; while we find limited evidence that the 1918 human pandemic was the result of a human/swine reassortment, Scholtissek (2008) and Smith et al. (2009a) both argue that this might have occurred for some of the segments. The possibility that the 2009 pandemic virus might increase in pathogenicity emphasizes the importance of understanding how the 1918 virus emerged and the basis of its extreme pathogenicity.
Acknowledgments
Thanks to John McCauley, Ziheng Yang and Rod Daniels for helpful comments. Thanks to Seena Shah for programming advice. This work was supported by the Medical Research Council, UK, and the European Union FP6 FLUPOL project number 044263.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Akaike H. A new look at the statistical model identification. IEEE Trans Autom Control. 1974;19:716–723. doi: 10.1109/TAC.1974.1100705. [DOI] [Google Scholar]
- Antonovics J, Hood ME, Baker CH. Molecular virology: was the 1918 flu avian in origin? Nature. 2006;440:E9. doi: 10.1038/nature04824. [DOI] [PubMed] [Google Scholar]
- Barry D, Hartigan JA. Statistical analysis of hominoid molecular evolution. Stat Sci. 1987;2:191–210. doi: 10.1214/ss/1177013353. [DOI] [Google Scholar]
- Blanquart S, Lartillot N. A site- and time-heterogeneous model of amino acid replacement. Mol Biol Evol. 2008;25:842–858. doi: 10.1093/molbev/msn018. [DOI] [PubMed] [Google Scholar]
- Boussau B, Blanquart S, Necsulea A, Lartillot N, Gouy M. Parallel adaptations to high temperatures in the Archaean eon. Nature. 2008;456:942–945. doi: 10.1038/nature07393. [DOI] [PubMed] [Google Scholar]
- Brown IH, Ludwig S, Olsen CW, Hannoun C, Scholtissek C, Hinshaw VS, Harris PA, McCauley JW, Strong I, Alexander DJ. Antigenic and genetic analyses of H1N1 influenza A viruses from European pigs. J Gen Virol. 1997;78:553–562. doi: 10.1099/0022-1317-78-3-553. [DOI] [PubMed] [Google Scholar]
- Bush RM, Smith CB, Cox NJ, Fitch WM. Effects of passage history and sampling bias on phylogenetic reconstruction of human influenza A evolution. Proc Natl Acad Sci USA. 2000;97:6974–6980. doi: 10.1073/pnas.97.13.6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med. 2009;360:2605–2615. doi: 10.1056/NEJMoa0903810. [DOI] [PubMed] [Google Scholar]
- Dowdle WR. Influenza A virus recycling revisited. Bull World Health Organ. 1999;77:820–828. [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Ho SY, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PloS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies. Sunderland, USA: Sinauer Associates; 2003. [Google Scholar]
- Galtier N, Gouy M. Inferring pattern and process: maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Mol Biol Evol. 1998;15:871–879. doi: 10.1093/oxfordjournals.molbev.a025991. [DOI] [PubMed] [Google Scholar]
- Galtier N, Tourasse N, Gouy M. A nonhyperthermophilic common ancestor to extant life forms. Science. 1999;283:220–221. doi: 10.1126/science.283.5399.220. [DOI] [PubMed] [Google Scholar]
- Gammelin M, Altmuller A, Reinhardt U, Mandler J, Harley VR, Hudson PJ, Fitch WM, Scholtissek C. Phylogenetic analysis of nucleoproteins suggests that human influenza A viruses emerged from a 19th-century avian ancestor. Mol Biol Evol. 1990;7:194–200. doi: 10.1093/oxfordjournals.molbev.a040594. [DOI] [PubMed] [Google Scholar]
- Gibbs MJ, Gibbs AJ. Molecular virology: was the 1918 pandemic caused by a bird flu? Nature. 2006;440:E8. doi: 10.1038/nature04823. [DOI] [PubMed] [Google Scholar]
- Gorman OT, Donis RO, Kawaoka Y, Webster RG. Evolution of influenza A virus PB2 genes: implications for evolution of the ribonucleoprotein complex and origin of human influenza A virus. J Virol. 1990;64:4893–4902. doi: 10.1128/jvi.64.10.4893-4902.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman OT, Bean WJ, Kawaoka Y, Donatelli I, Guo YJ, Webster RG. Evolution of influenza A virus nucleoprotein genes: implications for the origins of H1N1 human and classical swine viruses. J Virol. 1991;65:3704–3714. doi: 10.1128/jvi.65.7.3704-3714.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum BD, Levine AJ, Bhanot G, Rabadan R. Patterns of evolution and host gene mimicry in influenza and other RNA viruses. PLoS Pathog. 2008;4:e1000079. doi: 10.1371/journal.ppat.1000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory V, Bennett M, Thomas Y, Kaiser L, Wunderli W, Matter H, Hay A, Lin YP. Human infection by a swine influenza A (H1N1) virus in Switzerland. Arch Virol. 2003;148:793–802. doi: 10.1007/s00705-002-0953-9. [DOI] [PubMed] [Google Scholar]
- Gu X, Li WH. Estimation of evolutionary distances under stationary and nonstationary models of nucleotide substitution. Proc Natl Acad Sci USA. 1998;95:5899–5905. doi: 10.1073/pnas.95.11.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hay AJ, Gregory V, Douglas AR, Lin YP. The evolution of human influenza viruses. Philos Trans R Soc Lond B Biol Sci. 2001;356:1861–1870. doi: 10.1098/rstb.2001.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP. Bayesian inference of phylogeny and its impact on evolutionary biology. Science. 2001;294:2310–2314. doi: 10.1126/science.1065889. [DOI] [PubMed] [Google Scholar]
- Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull Hist Med. 2002;76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- Kawaoka Y, Krauss S, Webster RG. Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J Virol. 1989;63:4603–4608. doi: 10.1128/jvi.63.11.4603-4608.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbourne ED. Influenza pandemics of the 20th century. Emerg Infect Dis. 2006;12:9–14. doi: 10.3201/eid1201.051254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley CH, Fitch WM. An examination of the constancy of the rate of molecular evolution. J Mol Evol. 1974;3:161–177. doi: 10.1007/BF01797451. [DOI] [PubMed] [Google Scholar]
- Lockhart PJ, Steel MA, Hendy MD, Penny D. Recovering evolutionary trees under a more realistic model of sequence evolution. Mol Biol Evol. 1994;11:605–612. doi: 10.1093/oxfordjournals.molbev.a040136. [DOI] [PubMed] [Google Scholar]
- Ludwig S, Stitz L, Planz O, Van H, Fitch WM, Scholtissek C. European swine virus as a possible source for the next influenza pandemic? Virology. 1995;212:555–561. doi: 10.1006/viro.1995.1513. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Desselberger U, Palese P. Recent human influenza A (H1N1) viruses are closely related genetically to strains isolated in 1950. Nature. 1978;274:334–339. doi: 10.1038/274334a0. [DOI] [PubMed] [Google Scholar]
- Neumeier E, Meier-Ewert H, Cox NJ. Genetic relatedness between influenza A (H1N1) viruses isolated from humans and pigs. J Gen Virol. 1994;75(Pt 8):2103–2107. doi: 10.1099/0022-1317-75-8-2103. [DOI] [PubMed] [Google Scholar]
- Palese P. Influenza: old and new threats. Nat Med. 2004;10:S82–S87. doi: 10.1038/nm1141. [DOI] [PubMed] [Google Scholar]
- Rabadan R, Levine AJ, Robins H. Comparison of avian and human influenza A viruses reveals a mutational bias on the viral genomes. J Virol. 2006;80:11887–11891. doi: 10.1128/JVI.01414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. Estimating the rate of molecular evolution: incorporating non-contemporaneous sequences into maximum likelihood phylogenies. Bioinformatics. 2000;16:395–399. doi: 10.1093/bioinformatics/16.4.395. [DOI] [PubMed] [Google Scholar]
- Raymond FL, Caton AJ, Cox NJ, Kendal AP, Brownlee GG. The antigenicity and evolution of influenza H1 haemagglutinin, from 1950–1957 and 1977–1983: two pathways from one gene. Virology. 1986;148:275–287. doi: 10.1016/0042-6822(86)90325-9. [DOI] [PubMed] [Google Scholar]
- Reid AH, Taubenberger JK, Fanning TG. Evidence of an absence: the genetic origins of the 1918 pandemic influenza virus. Nat Rev Microbiol. 2004;2:909–914. doi: 10.1038/nrmicro1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GN, Paulson JC. Receptor determinants of human and animal influenza virus isolates: differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology. 1983;127:361–373. doi: 10.1016/0042-6822(83)90150-2. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ. Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol Biol Evol. 2002;19:101–109. doi: 10.1093/oxfordjournals.molbev.a003974. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19:301–302. doi: 10.1093/bioinformatics/19.2.301. [DOI] [PubMed] [Google Scholar]
- Scholtissek C. History of research on avian influenza. In: Klenk H-D, Matrosovic MN, Stech J, editors. Avian influenza. Basel: Karger; 2008. pp. 101–117. [Google Scholar]
- Scholtissek C, Burger H, Kistner O, Shortridge KF. The nucleoprotein as a possible major factor in determining host specificity of influenza H3N2 viruses. Virology. 1985;147:287–294. doi: 10.1016/0042-6822(85)90131-X. [DOI] [PubMed] [Google Scholar]
- Smith GJD, Bahl J, Vijaykrishna D, Zhang J, Poon LLM, Chen H, Webster RG, Malik Peiris JS, Guan Y. Dating the emergence of pandemic influenza viruses. Proc Natl Acad Sci USA. 2009;106:11709–11712. doi: 10.1073/pnas.0904991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GJD, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Malik Peiris JS, Guan Y, Rambaut A. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009;459:1122–1125. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- Subbarao K, Katz J. Avian influenza viruses infecting humans. Cell Mol Life Sci. 2000;57:1770–1784. doi: 10.1007/PL00000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK. The origin and virulence of the 1918 “Spanish” influenza virus. Proc Am Philos Soc. 2006;150:86–112. [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG. Characterization of the 1918 influenza virus polymerase genes. Nature. 2005;437:889–893. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin Guozhong, Fanning TG. Molecular virology: was the 1918 pandemic caused by a bird flu? Was the 1918 flu avian in origin? (Reply) Nature. 2006;440:e9–e10. doi: 10.1038/nature04825. [DOI] [PubMed] [Google Scholar]
- Thorne JL, Kishino H. Estimation of divergence times from molecular sequence data. In: Rasmus N, editor. Statistical methods in molecular evolution. New York, USA: Springer; 2005. pp. 235–256. [Google Scholar]
- Venables WN, Ripley BD. Modern Applied Statistics with S. New York, USA: Springer; 2002. [Google Scholar]
- Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56:152–179. doi: 10.1128/mr.56.1.152-179.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey M. Phylogenetic evidence against evolutionary stasis and natural abiotic reservoirs of influenza A virus. J Virol. 2008;82:3769–3774. doi: 10.1128/JVI.02207-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Estimating the pattern of nucleotide substitution. J Mol Evol. 1994;39:105–111. doi: 10.1007/BF00178256. [DOI] [PubMed] [Google Scholar]
- Yang Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol Evol. 1996;11:367–372. doi: 10.1016/0169-5347(96)10041-0. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- Yang Z. Computational molecular evolution. Oxford: Oxford University Press; 2006. [Google Scholar]
- Yang Z, Kumar S. Approximate methods for estimating the pattern of nucleotide substitution and the variation of substitution rates among sites. Mol Biol Evol. 1996;13:650–659. doi: 10.1093/oxfordjournals.molbev.a025625. [DOI] [PubMed] [Google Scholar]
- Yang Z, Roberts D. On the use of nucleic acid sequences to infer early branchings in the tree of life. Mol Biol Evol. 1995;12:451–458. doi: 10.1093/oxfordjournals.molbev.a040220. [DOI] [PubMed] [Google Scholar]