

Abstract
Gemfibrozil is long known for its ability to reduce the level of triglycerides in the blood circulation and to decrease the risk of hyperlipidemia. However, a number of recent studies reveal that apart from its lipid-lowering effects, gemfibrozil can also regulate many other signaling pathways responsible for inflammation, switching of T-helper cells, cell-to-cell contact, migration, and oxidative stress. In this review, we have made an honest attempt to analyze various biological activities of gemfibrozil and associated mechanisms that may help to consider this drug for different human disorders as primary or adjunct therapy.
Keywords: Gemfibrozil, Inflammation, Signal transduction, Human disorders
Introduction
Gemfibrozil, commonly known as ‘Lopid’ in the pharmacy, is a FDA-approved fibrate drug, which is structurally an amphipathic carboxylic acid molecule (Fig. 1). It was designed first at the Parke Davis Research Laboratories, Detroit in the year of 1968 in order to lower serum lipid. After three years of intense research, in 1971, gemfibrozil was proposed as a new drug with its lipid lowering ability and had been sent for clinical trial [1]. In the year of 1976, gemfibrozil was successfully introduced in the market as a hypolipidemic drug with its profound ability to reduce plasma triglyceride level [2].
Figure 1. Structure of gemfibrozil.
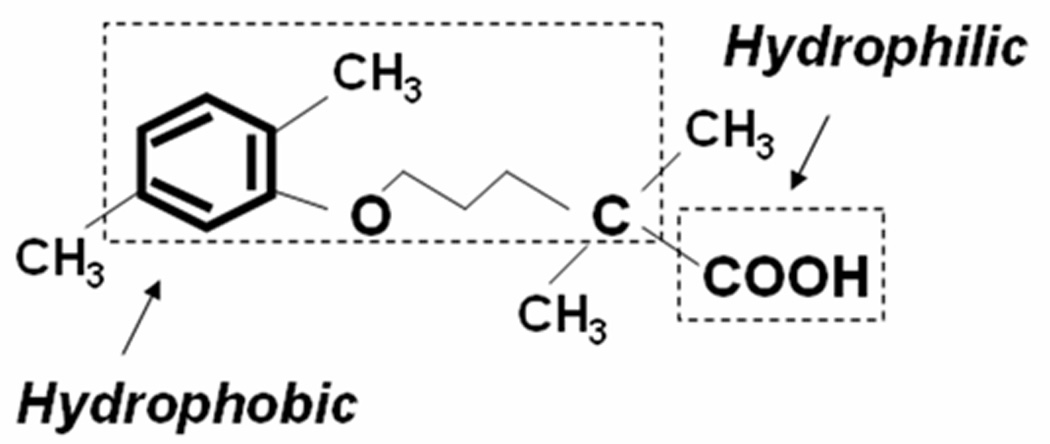
Gemfibrozil, an amphipathic carboxylic acid, is chemically known as, 2-dimethyl-5-(2, 5-dimethylphenoxy) pentanoic acid. This molecule is amphipathic because it carries a long hydrophobic backbone of an alkyl-benzene and a negatively charged carboxylic acid group in the other end.
Gemfibrozil has some general advantages over other lipid lowering drugs. First of all, it can be administered orally which is less painful and secondly, it generates relatively less amount of side effects compared to other lipid-lowering agents. Therefore, since its introduction to the lipid lowering field, many researches had been being conducted on lipid reducing ability of gemfibrozil. However, much later in 1992, Johan Auwerx first proposed its mode of action in his Belgian endocrine society lecture [3]. He proposed a class of nuclear hormone receptor known as peroxisome proliferators-activated receptor–α (PPAR-α) might be the target protein of fibrate drugs including gemfibrozil. After that, the significance of gemfibrozil in lipid lowering research has been revitalized with a clear picture of its signal transduction mechanism. In 2002, Xu et al reported the crystal structure of PPAR-α with a detailed description of its ligand binding pocket [4]. PPAR-α carries a large ligand binding pocket (1300Å) in its carboxy-terminal domain, which allows PPAR-α to establish stereospecific interactions with many endogenous as well as exogenous molecules. Endogenous ligands of PPAR-α are mainly fatty acid metabolites [5,6] whereas exogenous ligands are carboxylic acid molecules with long hydrophobic side chain.
Among its exogenous synthetic ligands, fibrates are most commonly used molecules [7]. Despite its stereospecific interaction, gemfibrozil is considered among low affinity ligands of PPAR-α. The affinity of gemfibrozil with PPAR-α is much lower (0.23 mM) than other fibrate drugs including clofibrate (0.7 mM) and benzafibrate (1 mM) [8]. The low affinity can be explained in terms of the conformational flexibility of 5 different amino acids in the ligand binding pocket. The flexibility had been observed in Phe282 (F282), Leu330 (L330), Phe363 (F363), Met364 (M364), Tyr473 (Y473) and Arg288 (R288) residues [9]. The moderate binding affinity of gemfibrozil with PPAR-α allows this drug to perform many other biological activities independent of PPAR-α. Although, the most common application of gemfibrozil is to reduce the plasma lipids, the critical impact of gemfibrozil on numerous diseases including atherosclerosis [10], diabetes [11], arthritis [12], cancer [13], and CNS disorders [14] can not be ignored. A number of basic, preclinical and clinical studies have proposed that gemfibrozil may be used as an immunomodulatory, anti-inflammatory and anti-migratory drug.
Hypolipidemic action of gemfibrozil
Hyperlipidemia refers to abnormal increase of lipid and lipoprotein in blood plasma that is one of the most common life-threatening metabolic disorders all over the world. Fibrates are widely accepted group of lipid-lowering drugs available in the market. Among different fibrate drugs, hypolipidemic function of gemfibrozil has been extensively documented.
The effect of gemfibrozil in cholesterol mobilization and storage
Cholesterol mobilization from late endosome to plasma membrane is controlled by the active participation of different endosomal proteins including Nieman Pick type C1 and C2 (NPC1 and NPC2) [15]. NPC-1 functions as a membrane bound permease [16], which allows the exit of phospholipids and cholesterol from endosomal vesicles to extracellular acceptor whereas NPC-2 may function as a chaperone to facilitate cholesterol insertion into the endosomal membrane [17]. Both NPC-1 and NPC-2 contain PPAR-α responsive element (PPRE) in their promoter (18). Gemfibrozil increases the expression of NPC-1 and NPC-2 via activation of PPAR-α (Fig. 2) and stimulates endosomal mobilization of cholesterol towards the plasma membrane [18,19].
Figure 2. PPAR-α-dependent hypolipidemic action of gemfibrozil.
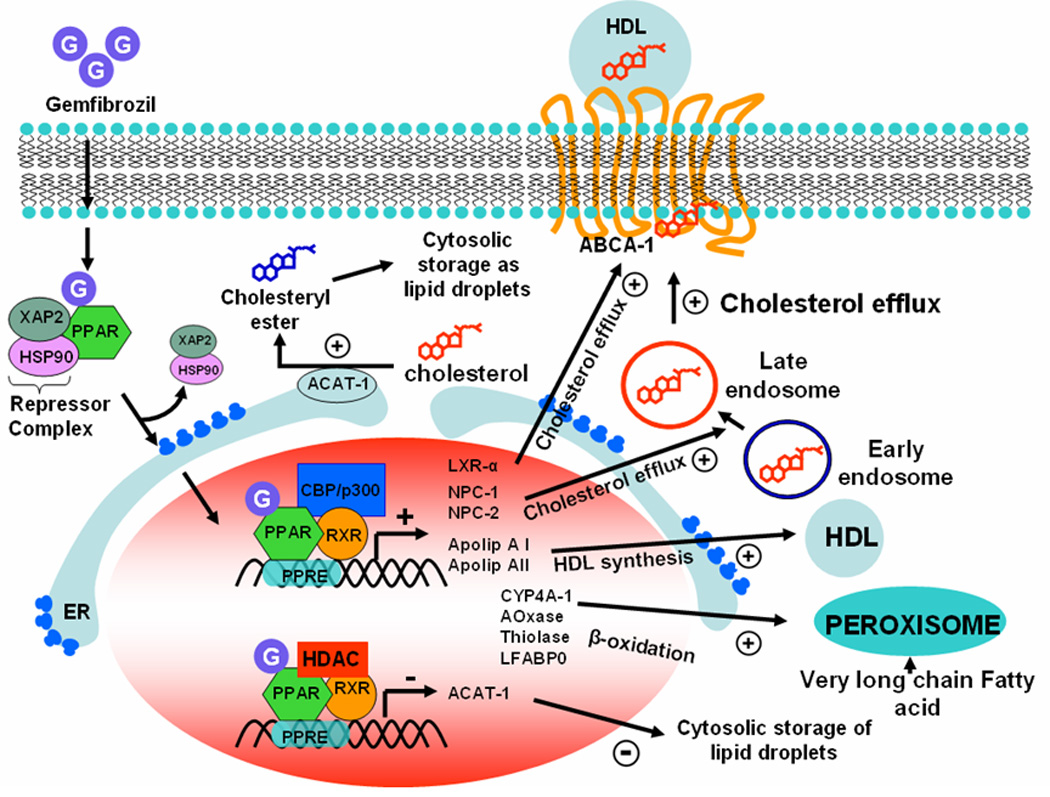
In cytosol PPAR-α forms an inactive complex with two different repressor proteins, HSP-90 and hepatitis virus B-X-associated protein-2 (XAP-2). In the presence of gemfibrozil, PPAR-α releases repressor complex and becomes active. Gemfibrozil-bound active PPAR-α then enters into nucleus and stimulates the expression of LXR-α, NPC-1 and 2 in order to suppress intracellular storage of cholesterol. On the other hand, it inhibits the expression of ACAT-1 to minimize the formation of cholesteryl ester, which is the intracellular storage form of cholesterol. The expression of LXR-α, NPC-1, NPC-2, and ACAT-1 is regulated by PPAR-α. Recruitment of CBP/p300 co-activator to the promoter stimulates the expression of genes associated with cholesterol efflux whereas recruitment of histone deacetylase (HDAC) co-repressor inhibts the expression of ACAT-1. Similarly, association of CBP/p300 with PPAR-α stimulates the expression of several HDL synthesis factors such as CYP4A-I, apolipoprotein AI & AII. Gemfibrozil also requires PPAR-α-mediated transcription of acyl-CoA oxidase, 2-enoyl-CoA hydratase and thiolase to stimulate peroxisomal β-oxidation.
According to recent reports, gemfibrozil has been shown to stimulate cholesterol efflux via upregulation of ATP-binding cassette transporter or ABCA-1 transporter protein [20]. ABCA-1 is a transmembrane protein which transfers intracellular cholesterol molecule to extracellular HDL particle and thereby lowers the intracellular burden of cholesterol molecules [21]. The expression of ABCA-1 is transcriptionally regulated by PPAR-α responsive transcription factor liver X receptor-α (LXR-α) [22]. Activation of PPAR-α by gemfibrozil has been reported to stimulate the expression of LXR-α and thus the expression of ABCA-1 indirectly [23]. Moreover, gemfibrozil is also reported to inhibit the intracellular storage of excess esterified cholesterol molecules. Excess cholesterol is esterified on the membrane of endoplasmic reticulum (ER) and is stored in cytosol as lipid droplets [24]. ACAT-1 is the membrane bound transferase enzyme of ER that actively participates with cholesterol esterification [25]. According to Chinetti et al [26], gemfibrozil-mediated PPAR-α activation inhibits the esterification of cholesterol via suppression of ACAT-1.
Effect of gemfibrozil on fatty acid β-oxidation
Activation of PPAR-α also stimulates the β-oxidation of very long chain fatty acids (VLCFA) in peroxisome. VLCFAs are major components of LDL and VLDL. Therefore, the increased catabolism of VLCFA in peroxisome is directly correlated with the decreased intracellular production of VLDL and LDL [27]. Gemfibrozil stimulates peroxisomal β-oxidation (Fig. 2) by up-regulating the expression of all three important peroxisomal β-oxidation enzymes (acyl-CoA oxidase, 2-trans-enoyl-CoA hydratase and thiolase) via PPAR-α-dependent pathways [28,29]. Gemfibrozil also stimulates the expression of other auxiliary proteins necessary for β-oxidation such as catalase, carnitine acyltransferase (CAT) and peroxisomal membrane protein-70 (PMP-70). Catalase removes the burden of H2O2 generated from acyl-CoA oxidase reaction [30], whereas both CAT and PMP-70 are membrane bound transporter molecules necessary for the transport of acetyl-CoA and very-long-chain acyl-CoA, respectively across the peroxisomal membrane [31,32]. It has been shown that gemfibrozil-mediated β-oxidation is inhibited by MAP kinase pathway [33] while stimulated by PI3-kinase and PKC pathways [34].
The role of gemfibrozil on HDL synthesis
Gemfibrozil also stimulates the production of HDL by upregulating the expression of apolipoprtein A-I and A-II [35,36]. Both of these molecules are two major constituents of high density lipoprotein or HDL particle. Apolipoprtein A-1 is the cofactor of the enzyme lecithin cholesterolacyltranferase (LCAT), which plays a key role on the efflux of cholesterol from extrahepatic tissue to liver for excretion. The decreased level of Apo-AI is associated with increased risk of CHD. Apo-AII is also an important lipoprotein component of HDL (Fig. 2). However, the exact biological role of Apo-AII is unclear, but polymorphism of Apo-AII gene is associated with visceral adiposity among African-American women [37].
The impact of gemfibrozil among hyperlipidemic patients
According to veteran affairs of high density lipoprotein and cholesterol intervention trial (VA HIT) study, gemfibrozil has been shown to reduce death from coronary heart disease by 24%. VA-HIT was 5 year follow-up study carried out among 2531 patients with mean age 64 ±7 yrs. After one year, gemfibrozil was able to reduce plasma triglyceride level by 31%, cholesterol level by 4% and increased HDL level by 6% [38]. In another 5 year follow-up study conducted by Helsinki heart study group, gemfibrozil was reported to reduce plasma triglyceride level by 35% and cholesterol level by 11% with mean increase of HDL by 11% [39]. After five years, the incidence of the coronary heart disease was significantly reduced by 34% and interestingly, the decline of the cardiac events became evident after two years of gemfibrozil treatment. These reports suggest that gemfibrozil significantly reduces the risk of coronary diseases by lowering the elevated level of circulating triglycerides and cholesterol as well as by stimulating the synthesis of HDL.
Anti-inflammatory activity of gemfibrozil
Gemfibrozil can suppress the inflammation and pain associated with chronic inflammatory diseases.
Suppression of proinflammatory molecules by gemfibrozil
Although, gemfibrozil is a FDA-approved lipid lowering drug, in 2002, we have reported anti-inflammatory property of gemfibrozil. In that article [45], we have demonstrated that gemfibrozil significantly inhibits the expression of iNOS mRNA and the production of nitric oxide in human astrocytes. Similar to gemfibrozil, other fibrate drugs such as clofibrate [40] and fenofibrate [41,42,43] also suppress the expression of proinflammatory molecules in human and mouse glial cells. However, synthetic PPAR-α ligand WY-14643 is more potent than gemfibrozil and other fibrate drugs in inhibiting proinflammatory molecules [42,43].
Attenuation of proinflammatory transcription factor activation by gemfibrozil
Because human iNOS promoter harbors consensus sequences for the binding of many transcription factors, including IFN-γ regulatory factor1 (IRF1) binding to interferon stimulated responsive element (ISRE), signal transducer and activator of transcription (STAT) binding to γ activation site (GAS), nuclear factor-κB (NF-κB), activator protein1 (AP1), and CCAAT/enhancer binding proteinβ (C/EBPβ), we investigated the effect of gemfibrozil on the activation of these transcription factors. The combination of IL-1β and IFN-γ induces the activation of NF-κB, AP1, C/EBPβ, and GAS, but not ISRE, suggesting that IRF1 may not be involved in cytokine-induced expression of iNOS in human astrocytes. Interestingly, gemfibrozil strongly inhibited the activation of NF-κB, AP1 and C/EBPβ, but not GAS, in cytokine-stimulated human astroglia suggesting that gemfibrozil attenuates the induction of iNOS probably by inhibiting the activation of NF-κB, AP1 and C/EBPβ.
Subsequently, other studies [41,42,44,45] have also demonstrated anti-inflammatory activity of gemfibrozil in astrocytes and microglia. Interestingly, all of these studies attempted to propose the anti-inflammatory action of gemfibrozil via PPAR-α–dependent pathways. In contrast; we have found that dominant-negative mutant of human PPAR-α (ΔhPPARα) is unable to abrogate gemfibrozil-mediated inhibition of iNOS in human astroglia [40] suggesting that gemfibrozil inhibits iNOS independent of PPAR-α. Recently by isolating brain cells from wild type and PPAR-α (−/−) mice, we have demonstrated that gemfibrozil does not require PPAR-α for its anti-inflammatory effect [43].
Activation of phosphatidylinositol-3 (PI-3) kinase by gemfibrozil
While investigating the underlying mechanisms further, we discovered that gemfibrozil induces the activation of phosphatidylinositol-3 kinase (PI3 Kinase) [43], a member of growth-supportive survival kinases and that PI-3 kinase is responsible for its anti-inflammatory activity. While a dominant-negative mutant of p85α and a kinase-dead mutant of PI-3 kinase abrogate the inhibitory effect of gemfibrozil on proinflammatory molecules, a constitutively-active PI-3 kinase stimulates gemfibrozil-mediated suppression of proinflammatory molecules suggesting that the activation of PI-3 kinase plays a crucial role in anti-inflammatory activity of gemfibrozil. Furthermore, gemfibrozil-induced PI3-kinase pathway also up-regulates the synthesis of IκBα, an anti-inflammatory molecule capable of arresting NF-κB complex in the cytosol (Fig. 3). These observations suggest that gemfibrozil exerts anti-inflammatory effect via activation of the PI-3 kinase pathway.
Figure 3. PPAR-α-independent action of gemfibrozil.
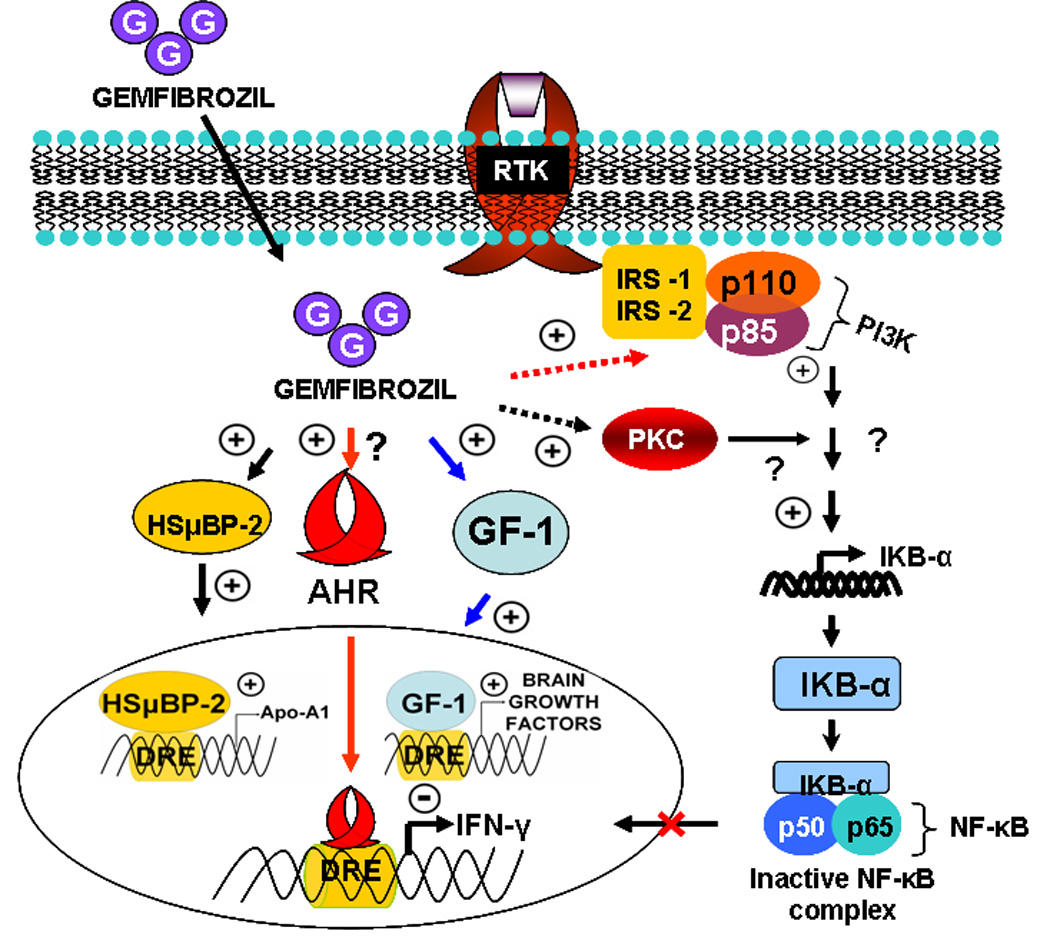
Anti-inflammatory, anti-oxidative and immuno-modulatory activities of gemfibrozil do not depend on PPAR-α. In one mechanism, gemfibrozil binds and stimulates the recruitment of human S μ binding protein-2 (HSµBP-2), glial factor-1 (GF-1), and possibly aryl hydrocarbon receptor (Ahr) at the drug responsive element (DRE) of numerous anti-inflammatory as well as neurotrophic genes. In another pathway, gemfibrozil is able to stimulate phosphatidylinositol-3 (PI-3) kinase via an unknown PPAR-α-independent mechanism. Once PI-3 kinase is activated, it stimulates the expression of inhibitory kappa B-alpha (IκBα) molecule, which in turn binds with functionally active NF-κB molecule and arrests the nuclear migration of NF-κB complex.
The diverse application of gemfibrozil as an anti-inflammatory drug
Long term treatment of gemfibrozil has been shown to reduce inflammatory events associated with coronary heart disease [46], rheumatoid arthritis [47], diabetes [48], murine experimental autoimmune encephalomyelitis (EAE) [14], and many other chronic inflammatory conditions.
Gemfibrozil ameliorates EAE
In MS and in its animal model EAE, different soluble neurotoxic factors released from myelin-laden microglia, macrophages and hypertrophic scarring astrocytes contribute significantly to the disease progression. Upon activation, both microglia and astrocytes release many inflammatory molecules including inflammatory cytokines TNF-α, IL-1β, IL-6, IL-12; chemokines including CCL2, 4, 6, 8 and CXCL10; or even, small diffusible chemical compounds including NO and ROS. These molecules participate in wide range of inflammatory reactions contributing to the pathology of MS and EAE (49,50). Because gemfibrozil inhibits the expression of these neurotoxic and inflammatory molecules from both microglia and astrocytes; we and others examined the effect of this lipid lowering drug on the disease process of EAE and reported suppression of EAE at multiple steps [14,40,41,44]. Gemfibrozil switches the immune response from Th1 to Th2, attenuates infiltration of mononuclear cells into the CNS and prevents demyelination in mice with relapsing-remitting EAE [14].
Because gemfibrozil is an amphipathic molecule, we also examined the entry of gemfibrozil into the brain after oral feeding. After 7 days of feeding of chow containing 0.2% gemfibrozil, the level of gemfibrozil in the brain reaches to 17.2 ± 5.09 µg/gm tissue [14]. On the other hand, we detected no gemfibrozil in either plasma or brain of mice receiving normal chow containing only the vehicle. These results suggest that gemfibrozil is capable of entering into the brain.
Among glia-derived inflammatory molecules, nitric oxide (NO) and ROS stimulate both microglia and astrocytes by upregulating the expression of their activation marker protein CD11b [51] and GFAP [52], respectively. The enhanced expression of CD11b is associated with severe activation of microglia around MS plaques. On the other hand, elevated expression of GFAP is also associated with hypertrophic, “scarring” astrocytes near chronic MS lesion. Our studies reported that NO plays crucial role in the activation of both astrocytes and microglia. Scavenging NO or suppression of iNOS enzymic activity significantly inhibited the expression of CD11b and GFAP in microglia and astrocytes respectively. In the previous section, we described that gemfibrozil is capable of inhibiting the gene expression of inducible nitric oxide synthase (iNOS) in activated glia. Because iNOS is the major nitric oxide producing enzyme in activated glia, gemfibrozil also could be involved in the suppression of NO-mediated gliosis in different neuroinflammatory diseases including MS and EAE. Activated glial cells are also associated with the expression of other inflammatory cytokines such as TNF-α and IL-1β. Gemfibrozil and other fibrate drugs inhibit the mRNA and protein expression of these genes [53]. Apart from NF-κB, gemfibrozil also inhibits the activation of AP-1 and C/EBP-β in glia to inhibit the expression of microglia-derived IL-1β, TNF-α, NO [43] and different other neurotoxic molecules, which play a role in EAE.
Gemfibrozil in arthritis
Rheumatoid arthritis (RA) is associated with massive synovial infiltration and proliferation of inflammatory cells followed by the destruction of cartilage and bone. Activated macrophages and inflammatory Th1 cells are known to release TNF-α, IL-6 and IL-1 which play important role in the pathogenesis of RA. The activation of NF-κB and COX-2 enzymic activity is often correlated with the disease severity of RA. Fibrate groups of drugs inhibit the expression of COX-2 and prostaglandin synthesis by interfering NF-κB activation [54]. Furthermore, gemfibrozil, known to switch Th1 to Th2 (14), may also lower the burden of Th1 cytokines in RA. Elevated concentration of uric acids in serum and its deposition in the articular joint often generate pain and inflammatory response among arthritic patients. This phenomenon is called hyperuricaemia and the associated arthritis is named as gout. Gemfibrozil treatment significantly improves the arthritic pain among gout patients. Different clinical studies confirmed that gemfibrozil was able to lower the serum level of uric acid more significantly than any other arthritic drugs [55] assuring its application as a potential drug in hyperuricaemia and gout.
Gemfibrozil reduces inflammation in atherosclerotic patients
The treatment of gemfibrozil over longer period of time significantly reduces the size and swelling of atherosclerotic lesions in aortic blood vessel [56]. Elevated expression of C-reactive protein (CRP), TNF-α and IL-6 has been found to be associated with the formation of chronic inflammatory lesions in the arterial wall of heart. Several studies have shown that CRP and TNF-α exert a proinflammatory effect by increasing the expression of monocytic chemo-attractant protein-1 (MCP-1), intercellular adhesion molecule 1 (ICAM-1), and plasminogen activator inhibitor 1 (PAI-1) in endothelial cells of blood vessels. Gemfibrozil is reported to reduce the production of CRP, TNF-α and IL-6 in peripheral blood mononuclear cells (PBMC) of atherosclerotic patients. Gemfibrozil is able to reduce the plasma CRP concentration among hyperlipidemic patients, significantly by 30% after six months of daily oral administration [57]. Stales et al have observed that fibrate drugs are able to inhibit the production of cyclooxygenase-2 (COX-2) and prostaglandins in smooth muscle cells around atherogenic lesions [57]. COX-2 is an inducible and inflammatory enzyme which converts arachidonic acid to prostaglandins. Prostaglandins binds with a diverse group of membrane bound and G-protein-coupled receptors to induce pain and inflammatory response in the target tissue. COX-2 is absent in normal tissue but can be induced by any inflammatory response near the sites of inflammation. Taken together, gemfibrozil reduces the risk of atherosclerosis by inhibiting the synthesis of adhesion molecules and inflammatory factors near lesion area.
Immuno-modulatory effect of gemfibrozil
Multiple sclerosis (MS) is a Th1 dominant chronic inflammatory disease of central nervous system (CNS) where an auto-reactive and inflammatory population of Th1 cells participates in demyelination of CNS white matter. There is another subtype of helper T cells in MS patients, known as Th2 that is less in number and anti-inflammatory in nature. The stimulation of Th2 cell-driven response and suppression of Th1 cell-mediated response at the same time has been confirmed to ameliorate the symptoms of MS.
Switching of T-helper cells
Switching of auto-immune Th1 to Th2 cells has been found to be beneficial in improving symptoms of MS relapses. The immunomodulation is associated with the suppression of cell-surface marker and cytokine profile of auto-reactive Th1 populations while stimulating the expression of Th2 cell specific surface proteins and releasing factors. Different immuno-modulatory drugs are available in the market for ameliorating MS symptoms such as avonex, betaseron, copaxone, novantrone, and rebif. Recently, gemfibrozil has been reported to suppress the invasion of inflammatory Th1 cells into the spinal cord and stimulate the production of IL-4, IL-10 in the CNS of EAE mice (14,58). These events underline the immunomodulating ability of gemfibrozil. In the splenocytes of EAE mice, gemfibrozil inhibits the secretion of Th1-specific cytokine IFN-γ and promotes the secretion of Th2-specific cytokine IL-4. We have also found that gemfibrozil inhibits the expression and DNA-binding activity of T-bet, a key regulator of interferon-γ (IFN-γ) expression and stimulates the expression and DNA-binding activity of GATA3, a key regulator of IL-4 [14]. Moreover, we have also demonstrated that the differential effect of gemfibrozil on the expression of T-bet and GATA3 is due to its inhibitory effect on NO production. While excess NO favors the expression of T-bet, scavenging of NO stimulates the expression of GATA-3. Therefore, by suppressing the production of NO, gemfibrozil decreases the number of T-bet-positive T cells and increases the number of GATA3-positive T cells in the spleen of EAE mice.
The inflammatory processes also play a crucial role in atherogenesis, which is reflected by the presence of large amounts of inflammatory cells, mainly monocytes/macrophages and T-lymphocytes, within atherosclerotic plaques. The major subtype of T cells in the atherogenic plaques is also Th1 in nature. Fibrate drugs have been found to suppress the Th1 cell phenotypes near atherosclerotic lesion area and stimulate the production of Th2-specific cytokines such as IL-4, IL-5 and IL-10. In all above cases, the immunomodulatory effect of gemfibrozil does not require the activation of PPAR-α. Treatment with a potent and specific PPAR-α ligand GW7647 does not augment IL-4 production in splenocytes [59]. Moreover gemfibrozil induces IL-4 expression in splenocytes from PPAR-α knockout mouse (14), suggesting that the immuno-modulatory effect of gemfibrozil is not dependent on PPAR-α.
Modulating contact activity of T cells
MS and its animal model EAE are Th1 dominant disease where neuroantigen-primed inflammatory Th1 cells infiltrate into the CNS and stimulate various inflammatory pathways. Previously, we have shown that MBP-primed Th1 cells stimulate the expression of different inflammatory molecules such as NO, TNF-α and IL-1β in microglia. While investigating underlying mechanisms, we have observed that cell to cell contact between inflammatory Th1 cells with antigen presenting cells plays a crucial role in amplifying the inflammatory response in MS and EAE [60]. The increased expression of α4β1 integrin complex or very-late antigen-4 (VLA-4) is observed on the surface of inflammatory autoimmune Th1 cells. The elevated expression of VLA-4 is associated with the trans-endothelial migration of T cells as well as the production of different inflammatory and neurotoxic molecules from microglia [60]. The interaction between VLA-4 of T cells and VCAM-1 of activated microglia stimulates the microglial production of nitric oxide (NO), tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) which contribute significantly to the disease progression. Gemfibrozil was also shown to modify the expression profile of surface molecules in autoimmune T cells, which modulates T cell-to-glia contact activity [61]. Gemfibrozil upregulated the surface expression of αvβ3 integrin complex in autoimmune T cell population which stimulated the production of neurotrophic factors in both microglia and astrocytes [61]. The contact between αvβ3 integrin complex of T cells and PDGFR-beta on the surface of activated glia induced the transcription of neurotrophic factors in glial cells via activation of cAMP responsive element binding protein (CREB) [61].
Anti-oxidative action of gemfibrozil
In the last two decades, reactive oxygen species (ROS) have emerged as important signaling molecules in the regulation of various cellular processes. They can be generated by the electron transport chain in mitochondria and activation of polymorphonuclear leukocytes (PMN) during inflammatory conditions. Excessive generation of ROS may result in attack and damage to most intracellular and extracellular components in living organism.
Effect of Gemfibrozil on ROS controlling cellular proteins
The production of superoxide molecule is mainly regulated by the enzymatic activity of NADPH oxidase. NADPH oxidase, a membrane-bound enzyme, generates superoxide radical by transferring electron from intracellular NADPH to molecular oxygen. It has five “phox” subunits including gp91phox, p22phox, p40phox, p47phox, and p67phox. Interestingly, gemfibrozil suppresses the expression of the genes encoding the NADPH oxidase subunits p47phox and gp91phox [62]. These two subunits of NADPH oxidase form a complex with gp67phox and small G-protein Rac-1 and the resulting complex is responsible for the generation of superoxide [63,64]. Gemfibrozil is known to inhibit the ROS-mediated inflammation by inhibiting the expression of Rac-1 [62]. Gemfibrozil induces the expression of heat shock proteins (HSPs) [64], which are required for the correct folding of nascent polypeptides and for the repairing of misfolded proteins after damage from chemicals that induce ROS. Recently, gemfibrozil treatment has also been shown to stimulate the activity of paraxonase, a ubiquitously expressed membrane bound anti-oxidant protein, among patients with type-2 diabetic [65,66]. In summary, gemfibrozil reduces the burden of superoxide, lipid peroxidation products by down-regulating the expression of p47phox and gp91 phox gene and Rac-1. On the other hand; it also fortifies the cellular defense by stimulating the activity of anti-oxidant proteins such as paraxonase.
Free radical scavenging ability of gemfibrozil
Gemfibrozil is associated with free radical scavenging ability as well as metal ion chelation. Metal ion-mediated superoxide production from glucose is common among diabetic patients [67]. Glucose can produce superoxide and hydrogen peroxide in the presence of copper ion (Cu2+) [68]. Para-hydroxy metabolite of gemfibrozil has been shown to reduce the burden of ROS by scavenging copper-ion in plasma of diabetic patients [65].
Gemfibrozil inhibits LDL-peroxidation
Intracellular burden of ROS stimulates the peroxidation of low density lipoproteins to generate lipid aldehydes such as hexanal, MDA and 4- hydroxy-nonenal [69]. These oxidized molecules are atherogenic which cause arterial cell death, accumulation of inflammatory cells in the arterial wall and induction of cytokine release .ROS driven lipid peroxidation is also common during the relapse of MS[70]. Patients with acute exacerbation of multiple sclerosis have significantly higher concentrations of pentane compared to patients with remission [71]. Pentane is one of the major end products of lipid peroxidation. ROS generated from lipid peroxidation actively participate in the axonal degeneration of demyelinated nerve fibers in progressive multiple sclerosis and in age-related neurodegenerative disorders including Alzheimer’s disease (AD). Gemfibrozil has been observed to attenuate plaque area and superoxide production in the aortic wall of atherosclerotic patients. Orally administered fibrate drugs are reported to decrease lipid peroxidation products such as malonyldialdehyde and 4-hydroxydialkenal in plasma of Wister rats. According to a double-blind, placebo-controlled intervention trial study, gemfibrozil reduces LDL peroxidation among hyperlipidemic patients by 33% after 8 week of treatment [72]. In addition, gemfibrozil prevents HDL-lipid peroxidation by preserving HDL-paraoxonase enzyme activity [66,73]. Paraoxonase is an enzyme associated with high-density lipoprotein (HDL) that protects the oxidation of low-density lipoprotein (LDL) and hence attenuates the risk of coronary artery disease. There are three isotypes of paraoxonase encoding genes named as pon-1, pon-2 and pon-3. Gemfibrozil is reported to upregulate the expression of pon-2 gene to stimulate the production of paraoxonase enzyme on HDL particles [73].
Anti-migratory effect of gemfibrozil
Effect of gemfibrozil on the expression of cell-surface adhesion molecules
Infiltration of peripheral mononuclear cells through the blood brain barrier (BBB) is observed in MS. Trans-endothelial migration of peripheral immune cells is regulated by the expression of adhesion molecules such as ICAM-1, VCAM-1 and selectins on the surface of endothelial cells as well as on infiltrating mononuclear cells. The expression of these adhesion molecules also regulates the infiltration of inflammatory cells into the lesion area of different chronic inflammatory diseases including MS, arthritis and atherosclerosis. Gemfibrozil is reported to inhibit the migration of monocytes in the arterial wall of atherosclerotic patients. Gemfibrozil inhibits the transendothelial migration of lymphocytes and macrophages into the CNS of EAE mice (14). Different PPAR-α activators reduce cytokine-induced expression of VACM-1 and ICAM-1 in human carotid artery endothelial cells in culture [74]. In another clinical study, fibrates decrease the expression of VCAM-1 and ICAM-1 levels among hypertriglyceridemic patients in the fasting state [75].
Effect of gemfibrozil on the expression of soluble migratory factors
Monocyte chemotactic protein-1 (MCP-1), a member of the small inducible gene (SIG) family, stimulates the recruitment of monocytes to sites of injury and infection. Pasceri et al [76] have reported that fibrates down-regulate CRP-induced expression of MCP-1 in human umbilical vein endothelial cells. The down-regulation of MCP-1 is also observed in patients of type IIb dyslipidemia [77]. Migration and proliferation of and smooth muscle cells (SMC) play an important role in atherosclerosis. Activation of small G proteins, such as Ras and Rho, is known to promote SMC migration and proliferation. Ras promotes cell cycle progression via activation of the MAP kinase pathway [78], whereas Rho/Rho kinase induces cell proliferation via destabilization of the inhibitor of cyclin-dependent kinase, p27kip1. Because gemfibrozil inhibits the activation of Ras and Rho, it also suppresses SMC migration and proliferation. Activated SMC is also an important source of inflammatory cytokines and express wide range of adhesion molecules near the lesion of vascular injury. Adhesion molecules expressed on SMC contributes to the retention of inflammatory cells within the vessel wall, and further promotes the inflammatory response within atherosclerotic lesions. Fibrates inhibit the expression of adhesion molecules and cytokines by inhibiting NF-κB and AP-1 signaling pathway [79]. The enhanced expression of IL-8 has been reported to induce inflammation, monocyte recruitment, angiogenesis, and VSMC migration and/or proliferation. LDL-particles stimulate human aortic smooth muscle cells (hAoSMC) to up-regulate IL-8 via activation of p-38 MAPK and AP-1 [80]. Activation of PPAR-α by fibrate drugs inhibits the upregulation of LDL-induced IL-8 expression via inhibition of AP-1 in human aortic smooth muscle cells [81]. Therefore, in summary, gemfibrozil may function as an anti-migratory agent either by down-regulating the expression of different soluble chemokines including MCP-1 and IL-8 or by inhibiting the expression of different cell surface proteins including ICAM-1, VCAM-1 and selectin.
Mode of Action
PPAR-α-dependent signaling pathways of gemfibrozil
Binding and activation of PPAR-α is believed as a major signaling event of gemfibrozil (Fig. 2). The hypolipidemic activities of gemfibrozil are mainly controlled by the activation of PPAR-α.
The possible receptor-ligand interaction between gemfibrozil and PPAR-α
Although PPAR-α-dependent signaling pathways of fibrate drugs are well studied, the interaction between PPAR-α and fibrate compounds has not been established so far. In stead, the interaction between PPAR-α and other fibrate-like compounds such as AZ242, GW409544 has been depicted by X-ray crystallography and NMR studies. According to X-ray crystallography studies, PPAR-α is a 466 amino acid long protein with five functionally different domains including N-terminal variable domain (A/B), DNA binding domain (DBD), hinge region, C-terminal ligand binding domain (LBD), and activation domain (AF2). C-terminal ligand binding domain (LBD) is made of five helices named as H-3, -5, -7, -11 and -12 with a large T-shaped ligand binding pocket in the center [9,82]. In the center of the five helices, carboxylic acid group of fibrate-like compounds form a strong hydrogen bond interaction with tyrosine 464 and an electrostatic interaction with histidine 440. On the other hand, π electron cloud of their cyclic resonating head structure constructs strong Vander Wall interactions with two other phenylalanine residues F273 and F351 [4–6]. The extreme carboxy-terminal helix or helix 12 of AF-2 participates in the heterodimerization with RXR. The binding of a PPAR agonist holds the AF-2 helix in the active conformation and enhances the formation of the functional PPAR:RXR heterodimer complex. The active conformation of AF-2 helix stabilizes the interaction of its Tyr477 residue with helices 7 and 10 of RXR. AF-2 helix also provides the allosteric binding sites for other co-activator molecules such as steroid receptor co-activator (SRC), PPAR-binding protein (PBP), PPAR-interacting protein (PRIP), and PRIP-interacting protein with methyltransferase domain (PIMT) [4–6].
PPAR-α mediated signaling event
PPAR-α is present in the cytoplasm as an inactive complex with heat-shock protein 90 (HSP-90) and hepatitis virus B-X-associated protein-2 (XAP-2). The biological consequence of this interaction remained unknown until a recent study describes HSP90 and XAP-2 as repressors of PPAR-α [82]. HSP90 interacts with ligand binding domain of PPAR-α and may inhibit the PPAR-α -ligand interaction. Fibrate drugs replace HSP90 repressor complex and helps to rescue the transcriptional activity of PPAR-α. However, it is not clear whether the binding of fibrate with PPAR occurs in cytosol or nucleus. In the nucleus, PPAR-α forms a heterodimeric complex with retinoic acid-X-receptor-α (RXR-α). Within the nucleus, PPAR-α:RXR heterodimer is bound to other repressor molecules such as “nuclear receptor co-repressor” (NCoR), “silencing mediator for retinoid and thyroid hormone receptor” (SMRT) and histone deacetylase (HDAC). In the presence of ligand, NCoR, SMRT and HDAC are released from the complex followed by the recruitment of histone acetyltransferase (CBP/p300), SRC, PBP, PRIP, and PMT (Fig. 2). Subsequently, the entire active complex binds to PPRE present in the promoter of peroxisomal fatty acid β-oxidizing enzymes. PPRE is composed of the hexameric direct repeat sequence –AGGRCA- separated by a single nucleotide. The recruitment of co-activator molecules triggers the chromatin remodeling of target gene, which stimulates the subsequent gene expression.
Although lipid-metabolizing activity of various fibrate drugs including gemfibrozil depends on PPAR-α, other PPARs may also contribute to lipid metabolism as well. For example, fenofibrate, bezafibrate, gemfibrozil, and LY518674 increase the expression of ATP-binding cassette transporter A1 (ABCA1) and stimulate the biogenesis of HDL dependent on PPAR-α in association with the liver X receptor α (LXR-α) upregulation [83]. While fenofibrate and LY518674 show exclusive dependency on PPAR-α for these activities, bezafibrate and gemfibrozil exhibit dependency on PPAR-β/δ and PPAR-γ as well [83].
PPAR-α-independent signaling pathway of gemfibrozil
Recently, gemfibrozil has been reported to control biological pathways independent of PPAR-α. Both anti-inflammatory and antioxidative mechanism of gemfibrozil is believed to occur via PPAR-α-independent pathways (Fig. 3).
Anti-inflammatory functions of gemfibrozil requires PPAR-α-independent signaling pathways
In 2002, we have demonstrated that gemfibrozil does not require the involvement of PPAR-α to suppress the expression of iNOS and the production of NO in human astroglia [40]. In our recent study [43], we have found that gemfibrozil inhibits the expression of proinflammatory molecules in primary microglia isolated from both wild type and PPAR-α knockout mice suggesting a PPAR-α-independent anti-inflammatory role of gemfibrozil. Because lipid-metabolizing activity of gemfibrozil may depend partly on PPAR-β/δ and PPAR-γ [83], gemfibrozil may exhibit anti-inflammatory activity via PPAR-β/δ and/or PPAR-γ. However, this possibility has not been tested. Furthermore, a series of studies have reported that activation of PPAR-α often augments inflammatory response in different cell types. Tordjman et al have demonstrated that there is decreased incidence of atherosclerosis and vascular inflammation in PPAR-α null mouse suggesting the chronic inflammatory role of PPAR-α [84]. Activation of PPAR-α has been also shown to augment the expression of cyclo-oxygenase 2 (COX-2) in colorectal cells [85] among colorectal cancer patients.
The possible role of drug-responsive transcription factors in PPAR-independent action of gemfibrozil
Recent studies indicated that gemfibrozil stimulated the DNA binding ability of transcription factor human S μ binding protein-2 (HSμBP-2) and glial factor-1 (GF-1) without the involvement of PPAR-α [86] (Fig. 3). HSμBP-2 is a transcription factor with a potential ATPase and helicase activity. It contains a transactivation domain which might be involved with gemfibrozil-induced DNA binding. Glial factor-1 (GF1), an incomplete version of HSμBP2 lacking the first 494 and the last 128 amino acids, is also expressed in a wide range of human cells with a potential transactivation domain. Recently, another transcription factor has been reported which could be involved with fibrate-mediated gene expression. Aryl hydrocarbon receptor (Ahr) is that transcription factor which has been shown to be activated in the presence of different chemicals with aryl hydrocarbon residues. Activation of Ahr is possible without the involvement of PPAR-α. All different fibrate drugs might be involved with the activation of Ahr independent of PPAR-α activation [87]. Moreover, activation of HSμBP-2, GF-1 and Ahr leads to the stimulation of anti-inflammatory pathways (Fig. 3). After ligand binding, these transcription factors translocate into the nucleus and bind with specific drug responsive element (DRE) located in the promoter of anti-inflammatory genes (Fig. 3).
The risk of gemfibrozil for human application
Sometimes, daily use of gemfibrozil may produce side effects such as gastro intestinal disturbances, nausea, depression, dizziness, and allergy. However, the amount and intensity of side effects is much less compared to other lipid lowering drugs including statins [88,89]. Gemfibrozil is usually prescribed after non-drug treatment options such as dietary restriction or extensive physical exercise have not been fully successful at lowering lipids [90]. This drug should not be prescribed for patients with hepatic or renal dysfunction [91] and should be used cautiously among pregnant women [92].
Furthermore, gemfibrozil may increase the risk of malignancies [93], gall bladder disease and incidence of noncoronary mortality in human [94]. However, gemfibrozil appears to have a lower propensity for causing these untoward effects compared to other lipid-lowering drugs. Moreover, fibrate drugs like gemfibrozil, clofibrate, and fenofibrate induce the proliferation of peroxisomes in rats and mice [94–96]. Continuous administration of fibrate drugs to the rats and mice for 40–50 weeks leads to the formation of hepatic tumor. However, induction of hepatic tumor promotion by fibrate drugs has not been demonstrated in human, other primates, and guinea pig, species that have lost their ability to synthesize ascorbate due to the inherent loss of the gulonolactone oxidase gene. Because ascorbate synthesis is accompanied by H2O2 production, and consequently its induction can be potentially harmful, Braun et al [96] have recently reported that the evolutionary loss of the gulonolactone oxidase gene may contribute to the missing carcinogenic effect of peroxisome proliferators in humans. In addition to that, recent studies have also revealed that humans have considerably lower levels of PPAR-α in liver than rodents [97], and this difference may, in part, explain the species differences in the carcinogenic response to fibrates. Nevertheless, the debate continues between the benefit and risk involved with the use of gemfibrozil as a therapeutic agent in human disorders.
The real culprits
The wide range of side effects can be explained by the reactive nature of gemfibrozil metabolites. Nine different gemfibrozil metabolites have been identified so far. Although, gemfibrozil itself is a non-reactive molecule, its metabolites have increased tendency to form adduct with cellular proteins and DNA. The major derivatives of gemfibrozil are acylglucuronides that are generated in its hydroxylated benzene ring by the glycosidic conjugation reaction with glucuronic acids. Moreover, glucuronide component of acylglucuronides further undergoes intramolecular rearrangement to give rise six other metabolites. All the metabolites are glucuronidase resistant and highly reactive. The process of intramolecular rearrangement allows those molecules to form an open chain conjugate with free aldehyde group. The open ring aldehyde group can form stable covalent bonds with -NH2, -SH and –OH functional groups of cellular proteins. Acylglucuronides are reported to form adduct with many metabolic enzymes including glutathione, selenium binding protein, protein disulfide isomerase, aldehyde dehydrogenase, triosephosphate isomerase, and kidney aminoacylase [98]. It appears that by forming covalent bonds with proteins, these metabolites are able to function as haptens, resulting in hypersensitivity reactions. It is also believed that the formation of adducts with other organ macromolecules may produce additional cellular dysfunctions, possibly including cancer [99].
Acknowledgements
This work has been supported by NS39940-09, NS39940-10, and NS48923-03 and National Multiple Sclerosis Society (RG3422A1/1).
References
- 1.Hodges RM. Gemfibrozil--a new lipid lowering agent. Proceedings of the Royal Society of Medicine. 1976;69 Suppl 2:1–2. [PMC free article] [PubMed] [Google Scholar]
- 2.Betteridge DJ, Higgins MJ, Galton DJ. Properties of sterol biosynthesis in human leukocytes, effects of gemfibrozil. Proceedings of the Royal Society of Medicine. 1976;69 Suppl 2:104–106. [PMC free article] [PubMed] [Google Scholar]
- 3.Auwerx J. Regulation of gene expression by fatty acids and fibric acid derivatives, an integrative role for peroxisome proliferator activated receptors. The Belgian Endocrine Society Lecture 1992. Hormone research. 1992;38(5–6):269–277. doi: 10.1159/000182557. [DOI] [PubMed] [Google Scholar]
- 4.Xu HE, Lambert MH, Montana VG, et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(24):13919–13924. doi: 10.1073/pnas.241410198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hostetler HA, Petrescu AD, Kier AB, Schroeder F. Peroxisome proliferator-activated receptor alpha interacts with high affinity and is conformationally responsive to endogenous ligands. The Journal of biological chemistry. 2005;280(19):18667–18682. doi: 10.1074/jbc.M412062200. [DOI] [PubMed] [Google Scholar]
- 6.Pawar A, Jump DB. Unsaturated fatty acid regulation of peroxisome proliferator-activated receptor alpha activity in rat primary hepatocytes. The Journal of biological chemistry. 2003;278(38):35931–35939. doi: 10.1074/jbc.M306238200. [DOI] [PubMed] [Google Scholar]
- 7.Gottlicher M, Demoz A, Svensson D, Tollet P, Berge RK, Gustafsson JA. Structural and metabolic requirements for activators of the peroxisome proliferator-activated receptor. Biochemical pharmacology. 1993;46(12):2177–2184. doi: 10.1016/0006-2952(93)90607-x. [DOI] [PubMed] [Google Scholar]
- 8.Scatena R, Bottoni P, Martorana GE, et al. Mitochondria; ciglitazone and liver, a neglected interaction in biochemical pharmacology. European journal of pharmacology. 2007;567(1–2):50–58. doi: 10.1016/j.ejphar.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 9.Zoete V, Grosdidier A, Michielin O. Peroxisome proliferator-activated receptor structures, ligand specificity; molecular switch and interactions with regulators. Biochimica et biophysica acta. 2007;1771(8):915–925. doi: 10.1016/j.bbalip.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Nash DT. Hyperlipoproteinemia; atherosclerosis and gemfibrozil. Angiology. 1982;33(9):594–602. doi: 10.1177/000331978203300905. [DOI] [PubMed] [Google Scholar]
- 11.DeSalcedo I, Gorringe AL, Silva JL, Santos JA. Gemfibrozil in a group of diabetics. Proceedings of the Royal Society of Medicine. 1976;69(Suppl 2):64–70. [PMC free article] [PubMed] [Google Scholar]
- 12.Miller-Blair D, White R, Greenspan A. Acute gout involving the acromioclavicular joint following treatment with gemfibrozil. The Journal of rheumatology. 1992;19(1):166–168. [PubMed] [Google Scholar]
- 13.Dellavalle RP, Nicholas MK, Schilling LM. Melanoma chemoprevention, a role for statins or fibrates? American journal of therapeutics. 2003;10(3):203–210. doi: 10.1097/00045391-200305000-00007. [DOI] [PubMed] [Google Scholar]
- 14.Dasgupta S, Roy A, Jana M, Hartley DM, Pahan K. Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-alpha. Molecular pharmacology. 2007;72(4):934–946. doi: 10.1124/mol.106.033787. [DOI] [PubMed] [Google Scholar]
- 15.Strauss JF, Liu P, Christenson LK, Watari H. Sterols and intracellular vesicular trafficking, lessons from the study of NPC1. Steroids. 2002;67(12):947–951. doi: 10.1016/s0039-128x(02)00042-9. [DOI] [PubMed] [Google Scholar]
- 16.Ioannou YA. Multidrug permeases and subcellular cholesterol transport. Nature Reviews Molecular Cell Biology. 2001;2(9):657–668. doi: 10.1038/35089558. [DOI] [PubMed] [Google Scholar]
- 17.Schmitz G, Grandl M. Lipid homeostasis in macrophages - implications for atherosclerosis. Rev Physiol Biochemical pharmacology. 2008;160:93–125. doi: 10.1007/112_2008_802. [DOI] [PubMed] [Google Scholar]
- 18.Chinetti-Gbaguidi G, Rigamonti E, Helin L, et al. Peroxisome proliferator-activated receptor alpha controls cellular cholesterol trafficking in macrophages. Journal of Lipid Research. 2005;46(12):2717–2725. doi: 10.1194/jlr.M500326-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPAR-alpha; PPAR-gamma; and LXRs in mice and men. Arteriosclerosis, thrombosis, and vascular biology. 2008;28(6):1050–1059. doi: 10.1161/ATVBAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- 20.Hossain MA, Tsujita M, Gonzalez FJ, Yokoyama S. Effects of fibrate drugs on expression of ABCA1 and HDL biogenesis in hepatocytes. Journal of cardiovascular pharmacology. 2008;51(3):258–266. doi: 10.1097/FJC.0b013e3181624b22. [DOI] [PubMed] [Google Scholar]
- 21.Oram JF, Lawn RM. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. Journal of lipid research. 2001;42(8):1173–1179. [PubMed] [Google Scholar]
- 22.Venkateswaran A, Laffitte BA, Joseph SB, et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(22):12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forcheron F, Cachefo A, Thevenon S, Pinteur C, Beylot M. Mechanisms of the triglyceride-and cholesterol-lowering effect of fenofibrate in hyperlipidemic type 2 diabetic patients. Diabetes. 2002;51(12):3486–3491. doi: 10.2337/diabetes.51.12.3486. [DOI] [PubMed] [Google Scholar]
- 24.van Meer G. Caveolin; cholesterol; and lipid droplets? The Journal of cell biology. 2001;152(5):F29–F34. doi: 10.1083/jcb.152.5.f29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee O, Chang CC, Lee W, Chang TY. Immunodepletion experiments suggest that acyl-coenzyme A,cholesterol acyltransferase-1 (ACAT-1) protein plays a major catalytic role in adult human liver; adrenal gland; macrophages; and kidney; but not in intestines. Journal of lipid research. 1998;39(8):1722–1727. [PubMed] [Google Scholar]
- 26.Chinetti G, Lestavel S, Fruchart JC, Clavey V, Staels B. Peroxisome proliferator-activated receptor alpha reduces cholesterol esterification in macrophages. Circulation research. 2003;92(2):212–217. doi: 10.1161/01.res.0000053386.46813.e9. [DOI] [PubMed] [Google Scholar]
- 27.Mandel H, Getsis M, Rosenblat M, Berant M, Aviram M. Reduced cellular cholesterol content in peroxisome-deficient fibroblasts is associated with impaired uptake of the patient's low density lipoprotein and with reduced cholesterol synthesis. Journal of lipid research. 1995;36(6):1385–1391. [PubMed] [Google Scholar]
- 28.Hashimoto F, Hamada S, Hayashi H. Effect of gemfibrozil on centrifugal behavior of rat peroxisomes and activities of peroxisomal enzymes involved in lipid metabolism. Biological & pharmaceutical bulletin. 1997;20(4):315–321. doi: 10.1248/bpb.20.315. [DOI] [PubMed] [Google Scholar]
- 29.Aoyama T, Peters JM, Iritani N, et al. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha) The Journal of biological chemistry. 1998;273(2):5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- 30.Jones DP. Intracellular catalase function, analysis of the catalatic activity by product formation in isolated liver cells. Archives of biochemistry and biophysics. 1982;214(2):806–814. doi: 10.1016/0003-9861(82)90087-x. [DOI] [PubMed] [Google Scholar]
- 31.Brady PS, Ramsay RR, Brady LJ. Regulation of the long-chain carnitine acyltransferases. Faseb Journal. 1993;7(11):1039–1044. doi: 10.1096/fasebj.7.11.8370473. [DOI] [PubMed] [Google Scholar]
- 32.Kamijo K, Taketani S, Yokota S, Osumi T, Hashimoto T. The 70-kDa peroxisomal membrane protein is a member of the Mdr (P-glycoprotein)-related ATP-binding protein superfamily. The Journal of biological chemistry. 1990;265(8):4534–4540. [PubMed] [Google Scholar]
- 33.Latruffe N, Cherkaoui Malki M, Nicolas-Frances V, Clemencet MC, Jannin B, Berlot JP. Regulation of the peroxisomal beta-oxidation-dependent pathway by peroxisome proliferator-activated receptor alpha and kinases. Biochemical pharmacology. 2000;60(8):1027–1032. doi: 10.1016/s0006-2952(00)00416-0. [DOI] [PubMed] [Google Scholar]
- 34.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochimica et biophysica acta. 2007;1771(8):952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin FY, Kamanna VS, Chuang MY, Morgan K, Kashyap ML. Gemfibrozil stimulates apolipoprotein A-I synthesis and secretion by stabilization of mRNA transcripts in human hepatoblastoma cell line (Hep G2) Arteriosclerosis, thrombosis, and vascular biology. 1996;16(8):1052–1062. doi: 10.1161/01.atv.16.8.1052. [DOI] [PubMed] [Google Scholar]
- 36.Saku K, Gartside PS, Hynd BA, Kashyap ML. Mechanism of action of gemfibrozil on lipoprotein metabolism. The Journal of clinical investigation. 1985;75(5):1702–1712. doi: 10.1172/JCI111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao J, Zhang F, Wiltshire S, et al. The apolipoprotein AII rs5082 variant is associated with reduced risk of coronary artery disease in an Australian male population. Atherosclerosis. 2008;199(2):333–339. doi: 10.1016/j.atherosclerosis.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 38.Robins SJ, Collins D, Wittes JT, et al. Relation of gemfibrozil treatment and lipid levels with major coronary events, VA-HIT, a randomized controlled trial. Jama. 2001;285(12):1585–1591. doi: 10.1001/jama.285.12.1585. [DOI] [PubMed] [Google Scholar]
- 39.Frick MH, Elo O, Haapa K, et al. Helsinki Heart Study, primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment; changes in risk factors; and incidence of coronary heart disease. The New England journal of medicine. 1987;317(20):1237–1245. doi: 10.1056/NEJM198711123172001. [DOI] [PubMed] [Google Scholar]
- 40.Pahan K, Jana M, Liu X, Taylor BS, Wood C, Fischer SM. Gemfibrozil; a lipid-lowering drug; inhibits the induction of nitric-oxide synthase in human astrocytes. The Journal of biological chemistry. 2002;277(48):45984–45991. doi: 10.1074/jbc.M200250200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J, Racke MK, Drew PD. Peroxisome proliferator-activated receptor-alpha agonist fenofibrate regulates IL-12 family cytokine expression in the CNS, relevance to multiple sclerosis. Journal of neurochemistry. 2007;103(5):1801–1810. doi: 10.1111/j.1471-4159.2007.04875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu J, Chavis JA, Racke MK, Drew PD. Peroxisome proliferator-activated receptor-alpha and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. Journal of neuroimmunology. 2006;176(12):95–105. doi: 10.1016/j.jneuroim.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 43.Jana M, Jana A, Liu X, Ghosh S, Pahan K. Involvement of phosphatidylinositol 3-kinase-mediated up-regulation of I kappa B alpha in anti-inflammatory effect of gemfibrozil in microglia. Journal of Immunology. 2007;179(6):4142–4152. doi: 10.4049/jimmunol.179.6.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y, Gocke AR, Lovett-Racke A, Drew PD, Racke MK. PPAR Alpha Regulation of the Immune Response and Autoimmune Encephalomyelitis. PPAR research. 2008 doi: 10.1155/2008/546753. 2008,546753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drew PD, Xu J, Storer PD, Chavis JA, Racke MK. Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int. 2006;49(2):183–189. doi: 10.1016/j.neuint.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Bloomfield, Rubins H, Davenport J, Babikian V, et al. Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol, The Veterans Affairs HDL Intervention Trial (VA-HIT) Circulation. 2001;103(23):2828–2833. doi: 10.1161/01.cir.103.23.2828. [DOI] [PubMed] [Google Scholar]
- 47.Cunard R. The potential use of PPARalpha agonists as immunosuppressive agents. Current Opinion in Investigational Drugs. 2005;6(5):467–472. [PubMed] [Google Scholar]
- 48.Calkin AC, Cooper ME, Jandeleit-Dahm KA, Allen TJ. Gemfibrozil decreases atherosclerosis in experimental diabetes in association with a reduction in oxidative stress and inflammation. Diabetologia. 2006;49(4):766–774. doi: 10.1007/s00125-005-0102-6. [DOI] [PubMed] [Google Scholar]
- 49.Watkins BA, Dorn HH, Kelly WB, et al. Specific tropism of HIV-1 for microglial cells in primary human brain cultures. Science. 1990;249(4968):549–553. doi: 10.1126/science.2200125. [DOI] [PubMed] [Google Scholar]
- 50.Pencalet P, Serguera C, Corti O, Privat A, Mallet J, Gimenez y, Ribotta M. Integration of genetically modified adult astrocytes into the lesioned rat spinal cord. Journal of neuroscience research. 2006;83(1):61–67. doi: 10.1002/jnr.20697. [DOI] [PubMed] [Google Scholar]
- 51.Roy A, Fung YK, Liu X, Pahan K. Up-regulation of microglial CD11b expression by nitric oxide. The Journal of biological chemistry. 2006;281(21):14971–14980. doi: 10.1074/jbc.M600236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brahmachari S, Fung YK, Pahan K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. Journal of Neuroscience. 2006;26(18):4930–4939. doi: 10.1523/JNEUROSCI.5480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pahan K, Sheikh FG, Liu X, Hilger S, McKinney M, Petro TM. Induction of nitric-oxide synthase and activation of NF-kappaB by interleukin-12 p40 in microglial cells. The Journal of biological chemistry. 2001;276(11):7899–7905. doi: 10.1074/jbc.M008262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jackson TC, Mi Z, Bastacky SI, McHale T, Melhem MF, Sonalker PA, Tofovic SP, Jackson EK. PPAR alpha agonists improve renal preservation in kidneys subjected to chronic in vitro perfusion: interaction with mannitol. Transplant international. 2007;20(3):277–290. doi: 10.1111/j.1432-2277.2006.00431.x. [DOI] [PubMed] [Google Scholar]
- 55.Okamoto H, Kamatani N. Successful treatment with fenofibrate; a peroxisome proliferator activated receptor alpha ligand; for a patient with rheumatoid arthritis. Annals of the rheumatic diseases. 2004;63(8):1002–1003. doi: 10.1136/ard.2003.015008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deans KA, Sattar N. “Anti-inflammatory” drugs and their effects on type 2 diabetes. Diabetes technology & therapeutics. 2006;8(1):18–27. doi: 10.1089/dia.2006.8.18. [DOI] [PubMed] [Google Scholar]
- 57.Neve BP, Fruchart JC, Staels B. Role of the peroxisome proliferator-activated receptors (PPAR) in atherosclerosis. Biochemical pharmacology. 2000;60(8):1245–1250. doi: 10.1016/s0006-2952(00)00430-5. [DOI] [PubMed] [Google Scholar]
- 58.Dasgupta S, Zhou Y, Jana M, Banik NL, Pahan K. Sodium phenylacetate inhibits adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice at multiple steps. Journal of Immunology. 2003;170(7):3874–3882. doi: 10.4049/jimmunol.170.7.3874. [DOI] [PubMed] [Google Scholar]
- 59.Racke M, Gocke A, Muir M, Diab A, Drew P, Lovett-Racke A. Nuclear Receptors and Autoimmune Disease: The Potential of PPAR Agonists to Treat Multiple Sclerosis. The journal of nutrition. 2006;136:700–703. doi: 10.1093/jn/136.3.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dasgupta S, Jana M, Liu X, Pahan K. Myelin basic protein-primed T cells induce nitric oxide synthase in microglial cells. Implications for multiple sclerosis. The Journal of biological chemistry. 2002;277(42):39327–39333. doi: 10.1074/jbc.M111841200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roy A, Liu X, Pahan K. Myelin basic protein-primed T cells induce neurotrophins in glial cells via alphavbeta3 [corrected] integrin. The Journal of biological chemistry. 2007;282(44):32222–32232. doi: 10.1074/jbc.M702899200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heyworth PG, Bohl BP, Bokoch GM, Curnutte JT. Rac translocates independently of the neutrophil NADPH oxidase components p47phox and p67phox. Evidence for its interaction with flavocytochrome b558. The Journal of biological chemistry. 1994;269(49):30749–30752. [PubMed] [Google Scholar]
- 63.Kleinberg ME, Malech HL, Mital DA, Leto TL. p21rac does not participate in the early interaction between p47-phox and cytochrome b558 that leads to phagocyte NADPH oxidase activation in vitro. Biochemistry. 1994;33(9):2490–2495. doi: 10.1021/bi00175a018. [DOI] [PubMed] [Google Scholar]
- 64.Kervinen H, Huittinen T, Vaarala O, et al. Antibodies to human heat shock protein 60; hypertension and dyslipidemia. A study of joint effects on coronary risk. Atherosclerosis. 2003;169(2):339–344. doi: 10.1016/s0021-9150(03)00229-6. [DOI] [PubMed] [Google Scholar]
- 65.Aviram M, Rosenblat M, Bisgaier CL, Newton RS. Atorvastatin and gemfibrozil metabolites; but not the parent drugs; are potent antioxidants against lipoprotein oxidation. Atherosclerosis. 1998;138(2):271–280. doi: 10.1016/s0021-9150(98)00032-x. [DOI] [PubMed] [Google Scholar]
- 66.Balogh Z, Seres I, Harangi M, Kovacs P, Kakuk G, Paragh G. Gemfibrozil increases paraoxonase activity in type 2 diabetic patients. A new hypothesis of the beneficial action of fibrates? Diabetes & metabolism. 2001;27(5):604–610. [PubMed] [Google Scholar]
- 67.Mowri HO, Frei B, Keaney JF. Glucose enhancement of LDL oxidation is strictly metal ion dependent. Free radical biology & medicine. 2000;29(9):814–824. doi: 10.1016/s0891-5849(00)00379-8. [DOI] [PubMed] [Google Scholar]
- 68.Wolff SP, Jiang ZY, Hunt JV. Protein glycation and oxidative stress in diabetes mellitus and ageing. Free radical biology & medicine. 1991;10(5):339–352. doi: 10.1016/0891-5849(91)90040-a. [DOI] [PubMed] [Google Scholar]
- 69.Requena JR, Fu MX, Ahmed MU, et al. Quantification of malondialdehyde and 4-hydroxynonenal adducts to lysine residues in native and oxidized human low-density lipoprotein. The Biochemical journal. 1997;322(Pt 1):317–325. doi: 10.1042/bj3220317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hunter MI, Nlemadim BC, Davidson DL. Lipid peroxidation products and antioxidant proteins in plasma and cerebrospinal fluid from multiple sclerosis patients. Neurochemical research. 1985;10(12):1645–1652. doi: 10.1007/BF00988606. [DOI] [PubMed] [Google Scholar]
- 71.Toshniwal PK, Zarling EJ. Evidence for increased lipid peroxidation in multiple sclerosis. Neurochemical research. 1992;17(5):205–207. doi: 10.1007/BF00966801. [DOI] [PubMed] [Google Scholar]
- 72.Yoshida H, Ishikawa T, Ayaori M, et al. Beneficial effect of gemfibrozil on the chemical composition and oxidative susceptibility of low density lipoprotein, a randomized; double-blind; placebo-controlled study. Atherosclerosis. 1998;139(1):179–187. doi: 10.1016/s0021-9150(98)00062-8. [DOI] [PubMed] [Google Scholar]
- 73.Gouedard C, Koum-Besson N, Barouki R, Morel Y. Opposite regulation of the human paraoxonase-1 gene PON-1 by fenofibrate and statins. Molecular pharmacology. 2003;63(4):945–956. doi: 10.1124/mol.63.4.945. [DOI] [PubMed] [Google Scholar]
- 74.Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99(24):3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marchesi S, Lupattelli G, Lombardini R, et al. Effects of fenofibrate on endothelial function and cell adhesion molecules during post-prandial lipemia in hypertriglyceridemia. Journal of clinical pharmacy and therapeutics. 2003;28(5):419–424. doi: 10.1046/j.0269-4727.2003.00512.x. [DOI] [PubMed] [Google Scholar]
- 76.Pasceri V, Cheng JS, Willerson JT, Yeh ET. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation. 2001;103(21):2531–2534. doi: 10.1161/01.cir.103.21.2531. [DOI] [PubMed] [Google Scholar]
- 77.Han SH, Quon MJ, Koh KK. Beneficial vascular and metabolic effects of peroxisome proliferator-activated receptor-alpha activators. Hypertension. 2005;46(5):1086–1092. doi: 10.1161/01.HYP.0000187900.36455.4c. [DOI] [PubMed] [Google Scholar]
- 78.Pahan K. Lipid-lowering drugs. Cellular and Molecular Life Sciences. 2006;63:1165–1178. doi: 10.1007/s00018-005-5406-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ye HJ, Zhao SP. Anti-atherogenic properties of fibrates may be largely due to their anti-inflammatory effects. Medical hypotheses. 2006;66(3):495–500. doi: 10.1016/j.mehy.2005.09.036. [DOI] [PubMed] [Google Scholar]
- 80.Ryoo SW, Kim DU, Won M, et al. Native LDL induces interleukin-8 expression via H2O2; p38 Kinase; and activator protein-1 in human aortic smooth muscle cells. Cardiovascular research. 2004;62(1):185–193. doi: 10.1016/j.cardiores.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 81.Ryoo S, Won M, Kim DU, et al. PPARalpha activation abolishes LDL-stimulated IL-8 production via AP-1 deactivation in human aortic smooth muscle cells. Biochemical and biophysical research communications. 2004;318(2):329–334. doi: 10.1016/j.bbrc.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 82.Sumanasekera WK, Tien ES, Davis JW, Turpey R, Perdew GH, Vanden Heuvel JP. Heat shock protein-90 (Hsp90) acts as a repressor of peroxisome proliferator-activated receptor-alpha (PPARalpha) and PPARbeta activity. Biochemistry. 2003;42(36):10726–10735. doi: 10.1021/bi0347353. [DOI] [PubMed] [Google Scholar]
- 83.Hossain MA, Tsujita M, Gonzalez FJ, Yokoyama S. Effects of fibrate drugs on expression of ABCA1 and HDL biogenesis in hepatocytes. Journal of Cardiovascular Pharmacology. 2008;51(3):258–266. doi: 10.1097/FJC.0b013e3181624b22. [DOI] [PubMed] [Google Scholar]
- 84.Tordjman K, Bernal-Mizrachi C, Zemany L, et al. PPARalpha deficiency reduces insulin resistance and atherosclerosis in apoE-null mice. The Journal of clinical investigation. 2001;107(87):1025–1034. doi: 10.1172/JCI11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oshio H, Abe T, Onogawa T, et al. Peroxisome proliferator-activated receptor alpha activates cyclooxygenase-2 gene transcription through bile acid transport in human colorectal cancer cell lines. Journal of gastroenterology. 2008;43:538–549. doi: 10.1007/s00535-008-2188-3. [DOI] [PubMed] [Google Scholar]
- 86.Mohan WS, Chen ZQ, Zhang X, et al. Human S mu binding protein-2 binds to the drug response element and transactivates the human apoA-I promoter, role of gemfibrozil. Journal of lipid research. 1998;39(2):255–267. [PubMed] [Google Scholar]
- 87.Maher JM, Cheng X, Slitt AL, Dieter MZ, Klaassen CD. Induction of the multidrug resistance-associated protein family of transporters by chemical activators of receptor-mediated pathways in mouse liver. Drug metabolism and disposition. 2005;33(7):956–962. doi: 10.1124/dmd.105.003798. [DOI] [PubMed] [Google Scholar]
- 88.Shek A, Ferrill MJ. Statin-fibrate combination therapy. The Annals of pharmacotherapy. 2001;35(7–8):908–917. doi: 10.1345/aph.10315. [DOI] [PubMed] [Google Scholar]
- 89.Broeders N, Knoop C, Antoine M, Tielemans C, Abramowicz D. Fibrate-induced increase in blood urea and creatinine, is gemfibrozil the only innocuous agent? Nephrology Dialysis Transplantation. 2000;15(12):1993–1999. doi: 10.1093/ndt/15.12.1993. [DOI] [PubMed] [Google Scholar]
- 90.Dube M, Fenton M. Lipid abnormalities. Clinical Infectious Disease. 2003;36(Suppl 2):S79–S83. doi: 10.1086/367562. [DOI] [PubMed] [Google Scholar]
- 91.Knauf H, Kolle EU, Mutschler E. Gemfibrozil absorption and elimination in kidney and liver disease. Klinische Wochenschrift. 1990;68(13):692–698. doi: 10.1007/BF01667018. [DOI] [PubMed] [Google Scholar]
- 92.Saadi HF, Kurlander DJ, Erkins JM, Hoogwerf BJ. Severe hypertriglyceridemia and acute pancreatitis during pregnancy, treatment with gemfibrozil. Endocr Pract. 1999;5(1):33–36. doi: 10.4158/EP.5.1.33. [DOI] [PubMed] [Google Scholar]
- 93.Freeman SR, Drake AL, Heilig LF, et al. Statins; fibrates; and melanoma risk, a systematic review and meta-analysis. Journal of the National Cancer Institute. 2006;98(21):1538–1546. doi: 10.1093/jnci/djj412. [DOI] [PubMed] [Google Scholar]
- 94.Leiss O, von Bergmann K, Gnasso A, Augustin J. Effect of gemfibrozil on biliary lipid metabolism in normolipemic subjects. Metabolism. 1985;34(1):74–82. doi: 10.1016/0026-0495(85)90064-2. [DOI] [PubMed] [Google Scholar]
- 95.Reddy JK, Mannaerts GP. Peroxisomal lipid metabolism. Annual review of nutrition. 1994;14:343–370. doi: 10.1146/annurev.nu.14.070194.002015. [DOI] [PubMed] [Google Scholar]
- 96.Braun L, Mile V, Schaff Z, et al. Induction and peroxisomal appearance of gulonolactone oxidase upon clofibrate treatment in mouse liver. FEBS Letter. 1999;458(3):359–362. doi: 10.1016/s0014-5793(99)01184-9. [DOI] [PubMed] [Google Scholar]
- 97.Gonzalez FJ, Peters JM, Cattley RC. Mechanism of action of the nongenotoxic peroxisome proliferators, role of the peroxisome proliferator-activator receptor alpha. Journal of the National Cancer Institute. 1998;90(22):1702–1709. doi: 10.1093/jnci/90.22.1702. [DOI] [PubMed] [Google Scholar]
- 98.Asif AR, Armstrong VW, Voland A, Wieland E, Oellerich M, Shipkova M. Proteins identified as targets of the acyl glucuronide metabolite of mycophenolic acid in kidney tissue from mycophenolate mofetil treated rats. Biochimie. 2007;89(3):393–402. doi: 10.1016/j.biochi.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 99.Thomas BF, Burgess JP, Coleman DP, Scheffler NM, Jeffcoat AR, Dix KJ. Isolation and identification of novel metabolites of gemfibrozil in rat urine. Drug metabolism and disposition, the biological fate of chemicals. 1999;27(1):147–157. [PubMed] [Google Scholar]