

Abstract
Diabetes mellitus (DM) is a multifactorial chronic metabolic disease characterized by hyperglycaemia. Several different mechanisms have been implicated in the development of the disease, including endoplasmic reticulum (ER) stress. ER stress is increasingly acknowledged as an important mechanism in the development of DM, not only for β-cell loss but also for insulin resistance. Accumulating evidence suggests that ER stress-induced apoptosis may be an important mode of β-cell loss and therefore important in the development of diabetes. Recent data also suggest a role of ER stress-induced apoptosis in liver and adipose tissue in relation to diabetes, but more extensive studies on human adipocyte and hepatocyte (patho)physiology and ER stress are needed to identify the exact interactions between environmental signals, ER stress and apoptosis in these organs.
Keywords: Diabetes, Endoplasmic reticulum stress, Apoptosis, Adipose tissue, Liver
Introduction
Diabetes mellitus (DM) is a multifactorial chronic metabolic disease characterized by hyperglycaemia. Several different mechanisms have been implicated in the development of the disease. Although the precise molecular events underlying the different forms of diabetes still remain unclear, it is generally accepted that the underlying defects include decreased secretion of insulin, its impaired signalling or both. Type 1 diabetes (T1DM) is known to result from an excessive loss of pancreatic β-cells, leading to insulin deficiency. Among other important causes, autoimmune and inflammatory processes have been reported to disrupt β-cells, cause insulin deficiency and hyperglycaemia and subsequently T1DM. Type 2 diabetes (T2DM), the most common form of diabetes, is characterized by impaired insulin action (insulin resistance) paralleled by impaired insulin secretion and a progressive decline in β-cell function. Insulin resistance, often associated with obesity and physical inactivity, is a major factor in the progression of T2DM. Obesity is a well-known risk factor for the development of T2DM. Importantly, obesity is not only associated with lipid accumulation in adipose tissue, but also in non-adipose tissues, such as liver and muscle. Lipid accumulation in non-adipose tissue, also known as ectopic lipid accumulation, has also been associated with the development of insulin resistance. Therefore, muscle, adipose tissue and liver are, beside pancreas, crucial tissues contributing to the development of insulin resistance and thus to the development of T2DM.
A relatively new player in the DM field is endoplasmic reticulum (ER) stress. ER stress and/or ER stress-induced apoptosis are increasingly acknowledged as important mechanisms in the development of DM, not only for β-cell loss but also for insulin resistance. Since the last decade, it has been generally accepted that ER stress plays an important role in β-cell function and loss [1]. This is for instance illustrated in Akita mice [2, 3], and the Wolcott-Rallison syndrome [4, 5]. Akita mice spontaneously develop diabetes with significant early loss of pancreatic β-cell mass resulting from a missense mutation (Cys96Tyr) in the insulin 2 gene that disrupts a disulfide bond between A and B chains of insulin [6]. The Wolcott-Rallison syndrome is a rare autosomal-recessive disorder characterized by the association of permanent neonatal or early-infancy insulin-dependent diabetes, and growth retardation, and other variable multisystemic clinical manifestations. The gene responsible for this syndrome is EIF2AK3 (PERK), the pancreatic eukaryotic initiation factor 2 (eIF2) kinase [4, 5]. More recently, it was acknowledged that high fat- and obesity-induced insulin resistance is also associated with ER stress in adipose tissue and liver [7, 8]. Remarkably, until now no studies have demonstrated a role for ER stress in skeletal muscle in relation to (the development of) obesity or diabetes [7, 9]. Besides, the role of ER stress in the development of diabetes that will be discussed in this paper, there is also evidence that diabetes can induce or aggravate ER stress and thereby affect the complications of diabetes, such as renal disease, retinopathy and vascular abnormalities [10–12].
In this review an overview of ER stress, the unfolded protein response (UPR), and ER stress induced apoptosis is given (see also refs [13–20]) with a further focus on the possible role of ER stress-induced apoptosis in the liver and adipose tissue in the onset of diabetes.
Endoplasmic reticulum stress-unfolded protein response
The endoplasmic reticulum (ER) is an important organelle required for cell survival and normal cellular function. In the ER, nascent proteins are folded with the assistance of ER chaperones (i.e. molecular chaperones and folding enzymes). Subsequently, correctly folded proteins are transported to the Golgi apparatus. Unfolded and misfolded proteins, on the other hand, are retained in the ER, retro-translocated to the cytoplasm by the machinery of ER associated degradation (ERAD), and degraded by the proteasome. As a major intracellular calcium storage compartment, the ER also plays a critical role towards maintenance of cellular calcium homeostasis. In addition, the ER also has a role in lipid biosynthesis, e.g. lipid membrane synthesis and controlling the synthesis of cholesterol and other membrane lipid components.
ER stress is caused by perturbations of any of the three homeostatic functions of the ER, i.e. functioning as a site for protein folding, for synthesis of unsaturated fatty acids (FA), sterols, and phospholipids and for intracellular Ca2+ storage. ER stressors include: (1) disturbances in cellular redox regulation caused by hypoxia, oxidants, or reducing agents, which interfere with disulfide bonding of proteins in the lumen of the ER, (2) glucose deprivation, probably by interfering with N-linked protein glycosylation in the ER, (3) disruption of Ca2+ metabolism causing impaired functions of Ca2+ dependent chaperones such as Grp78, Grp94 and calreticulin, (4) viral infections, which overload the ER with virus encoded proteins, (5) high fat diet, and (6) protein mutations that hamper adequate folding [17, 18]. The consequence of ER stress is an overwhelmed or compromised ability of the ER to properly fold proteins.
Accumulation of unfolded and/or misfolded proteins in the ER lumen is a hallmark of perturbation of any of the three functions of the ER and results in activation of the unfolded protein response (UPR). The UPR is a complex and coordinated adaptive signalling mechanism to re-establish homeostasis of ER functions (Fig. 1). ER stress sensors [IRE1 (inositol requiring 1), ATF6 (activated transcription factor 6) and PERK (ER-resident PKR-like eIF2α kinase)] detect the accumulation of unfolded and/or misfolded protein at the onset of ER stress and initiate the UPR. To re-establish homeostasis and normal ER function, the UPR initiates a global decrease in protein synthesis while increasing the production of ER chaperone proteins and ER-associated degradation (ERAD).
Fig. 1.
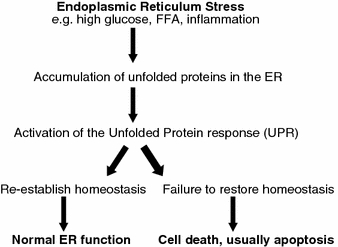
The relation between ER stress and ER stress induced apoptosis in the development of diabetes
The mammalian UPR with its signalling components is complex, diverse and flexible as has been described in great detail in recent reviews [16, 20]. In short, UPR signals through three pathways, that each utilizes one of the three ER stress sensors IRE1, ATF6 and PERK (Fig. 2). IRE1 is a transmembrane kinase/endoribonuclease (RNAse). Activation of IRE1 initiates the nonconventional splicing of Xbp-1 mRNA. Spliced Xbp-1 mRNA encodes a transcription activator that drives transcription of genes such as ER chaperones, whose products directly participate in ER protein folding. PERK is a transmembrane kinase that phosphorylates the eukaryotic translation initiation factor 2 subunit (eIF2). This leads to a reduced protein synthesis, which counteracts ER protein overload. ATF6 is an ER-resident transmembrane protein. Upon activation, the cytoplasmic domain of ATF6 is released from its membrane anchor by regulated proteolysis. The cleaved N-terminal fragment migrates to the nucleus, acts as an active transcription factor, and increases the expression of the genes encoding proteins that function to augment the ER protein folding capacity. The exact mechanism of UPR activation is unknown. One of the most described models is the competition model, in which the ER chaperone protein glucose regulated protein (Grp)78/BiP, is an UPR regulator and plays an essential role in the activation of IRE1, PERK and ATF6. In the inactive state, i.e. in resting cells, all three ER stress sensors (IRE-1, PERK and ATF6) are maintained in an inactive state through their association with the ER chaperone BiP (Fig. 2a). When the ER homeostasis is perturbed, i.e. upon ER stress, BiP is sequestered by unfolded and/or misfolded proteins that accumulate in the ER lumen (Fig. 2b, c). Dissociation of BiP from de ER stress sensors triggers the activation of IRE1, PERK and ATF-6 (Fig. 2d). Other models of UPR activation are the ligand-binding model in which unfolded and/or misfolded proteins directly interact with the ER stress-sensing domains of the ER stress sensors, and the probing model, in which newly synthesized stress-sensing proteins probe the efficiency of the ER-resident protein-folding machinery by presenting themselves as substrates to the folding machinery [20].
Fig. 2.
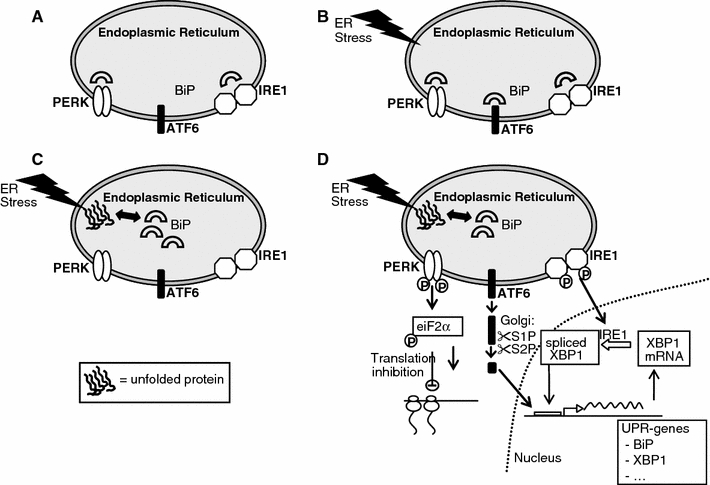
The unfolded proteins response and its signaling components. A simplified scheme of the initiation of the unfolded protein response (UPR). In the inactive state, i.e. in resting cells, all three ER stress sensors (IRE-1, PERK and ATF6) are maintained in an inactive state through their association with the ER chaperone BiP (a). Upon ER stress, BiP is recruited by the unfolded and/or misfolded proteins (b). This results in BiP dissociation from its conformational binding state to the transmembrane receptor proteins PERK, IRE1 and ATF6 (c). Dissociation results in activation of IRE1, PERK and ATF6 (d). The activated cytosolic domain of PERK phosphorylates the eukaryotic translation initiation factor 2 subunit (eIF2), inhibiting translation. The activated cytosolic domain of IRE1 initiates the nonconventional splicing of Xbp-1 mRNA, thereby cleaves a 252 bp intron from XBP1. Spliced Xbp-1 mRNA encodes a transcription activator that drives transcription of genes such as ER chaperones, whose products directly participate in ER protein folding. Activated ATF6 translocates to the Golgi, cleaved by proteases to form an active 50 kDa fragment. ATF6 p50 and XBP1 bind ER stress-response element (ERSE) promoters in the nucleus to produce up regulation of the proteins that function to augment the ER protein folding capacity
Endoplasmic reticulum stress—apoptosis
Under conditions of severe and prolonged ER stress, the UPR is unable to restore normal cellular function. Subsequently, cell death, usually occurring by apoptosis, is triggered (Fig. 1). Cell death results in loss of cell/tissue function and may be the primary reason for the manifestation of disease in several ER stress-related disorders. Indeed, cell death induced by ER stress has been implicated in a wide variety of diseases including ischemic injury (stroke, myocardial infarction), heart failure, several neurodegenerative diseases, diabetes and bipolar disorder [17, 18]. The mechanisms of apoptosis are highly complex, involving an energy-dependent cascade of molecular events. There are two main apoptotic pathways: the extrinsic or death receptor pathway and the intrinsic or mitochondrial pathway [21]. Current evidence suggests that these two pathways are linked and that molecules in one pathway can influence the other [22]. The extrinsic signalling pathways act via transmembrane receptor-mediated interactions. These involve death receptors that are members of the tumor necrosis factor (TNF) receptor gene superfamily [23]. The intrinsic signalling pathways involve a diverse array of non-receptor-mediated stimuli that produce intracellular signals that act directly on targets within the cell and are mitochondrial-initiated events. These non-receptor stimuli include radiation, toxins, hypoxia, hyperthermia, viral infections, and free radicals but also the absence of certain growth factors, hormones and cytokines [21].
Signalling through the ER stress sensors can trigger pro-apoptotic signals during prolonged ER stress. However, the ER stress sensors do not directly cause cell death but rather initiate the activation of downstream molecules such as CHOP or JNK, which further push the cell down the path of death. This results in caspase activation, the execution phase of ER stress-induced apoptosis, and finally in the ordered and sequential dismantling of the cell. Caspases are cysteine proteases that exist within the cell as inactive pro-forms or zymogens and are cleaved to form active enzymes following the induction of apoptosis. ER stress activates both intrinsic and extrinsic apoptotic pathways [13, 14]. Currently, three main pathways of ER stress-induced apoptosis are identified (Fig. 3): (1) the proapoptotic pathway of CHOP/GADD153 transcription factor which is mainly induced via PERK/eIF2, (2) IRE1-mediated activation of apoptosis signal-regulating kinase 1 (ASK1)/c-Jun NH2-terminal kinase (JNK), and (3) activation of the ER localized cysteine protease, caspase 12 [15, 18, 19].
Fig. 3.
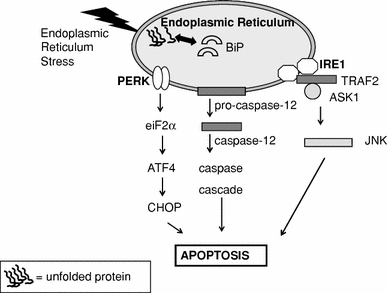
ER stress induced apoptosis. Three main pathways of ER stress-induced apoptosis are identified: (1) the proapoptotic pathway of CHOP/GADD153 transcription factor which is mainly induced via PERK/eIF2. CHOP down-regulates the anti-apoptotic factor B cell lymphoma-2 (Bcl-2), but also upregulates Ero-1, a thiol oxidase that promotes protein folding in the ER but also generates reactive oxygen species (ROS), and thereby promotes apoptosis, (2) IRE1-mediated activation of apoptosis signal-regulating kinase 1 (ASK1)/c-Jun NH2-terminal kinase (JNK). IRE1 interacts with TRAF2 (TNF receptor-associated factor-2) and ASK1. This leads to activation of ASK1 and JNK, followed by apoptosis, and (3) activation of the ER localized cysteine protease, caspase 12. Caspase 12 is activated by m-Calpain in the cytoplasm. Activation of m-Calpain is a consequence Ca2+ efflux out of the ER upon ER stress. These three pathways all end in caspase cascade activation, the execution phase of ER stress-induced apoptosis
ER stress, UPR and apoptosis in different organs and the development of diabetes
ER stress, UPR and apoptosis in the pancreas
β-cell loss plays a crucial role in the development of insulin deficiency and in the onset and/or progression of diabetes. Regulation of the β-cell mass involves a balance of β-cell replication and cell death. Accumulating evidence suggests that apoptosis may be the main mode of β-cell death in both types of diabetes. Recent studies point to a role of the ER in the sensing and transduction of apoptotic signals in β-cells as recently described in detail in excellent reviews [19, 24]. We now addressed the most relevant data of the last year, with a focus on ER stress and apoptosis in the pancreas, adipose tissue and the liver.
Several studies show evidence for a role of ER stress in β cell failure. Mutations in the primary sensors of the UPR or mutations that affect chaperone functions of the UPR, e.g. EIF2AK3, IRE1, P58IPK (DNAJC3) and EIF2α, impair β cell health and function [4, 25–28]. Moreover, mutations in proinsulin causing disruption of disulfide bond pairing result in misfolding and accumulation of proinsulin in the ER lumen of β cells. This accumulation can cause β cell failure [6, 29, 30]. In vivo data show that also pathological conditions like high glucose, free fatty acids, cytokines, and nitric oxide induce UPR gene expression and compromise β cell function [25, 31–33]. Moreover, in islets of T2DM patients, ER stress has been demonstrated by increased staining for ER chaperones and CHOP along with increased ER size [34–37].
However, the exact molecular mechanisms for the ER stress-induced apoptosis in β cells are not entirely clear. The most recent data support that the PERK-ATF4-CHOP stress signalling pathway is important in β-cell apoptosis (Fig. 3). This pathway plays a role in β-cell injury induced by oxidative stress and saturated fatty acids [38–42]. This is confirmed by the finding that CHOP deletion reduces oxidative stress, improves β cell function, and promotes cell survival in multiple mouse models of diabetes [39]. However, the PERK-ATF4-CHOP pathway is not the only pathway inducing apoptosis in β-cells. In contrast to apoptosis by high lipids, the PERK-ATF4-CHOP ER stress–signalling pathway is not necessary for cytokine-induced β-cell death [42]. Other data show that also the IRE1-JNK pathway is associated with the apoptosis in β cells [41] (Fig. 3). This pathway is also involved in ER stress-induced apoptosis caused by chronic high glucose, fatty acids, and Il-1β induced depletion of Ca2+ [41, 43–45].
ER stress, UPR and apoptosis in adipose tissue
The prevalence of obesity is increasing rapidly worldwide, especially in developing countries. An important consequence of obesity is an increased risk of developing impaired glucose tolerance and T2DM. Indeed, along with the increase in obesity, a parallel increase in the prevalence of T2DM, impaired glucose tolerance has occurred [46, 47]. The metabolic complications of obesity, usually referred to as the metabolic syndrome, consist of insulin resistance (often culminating in β-cell failure, impaired glucose tolerance and T2DM), dyslipidemia, hypertension, and premature heart disease. Our understanding of the role of adipose tissue in metabolic syndrome has continued to evolve with the identification of adipose tissue as a potent endocrine organ. Adipose tissue secretes large amounts of adipocyte-generated factors, such as adipokines, cytokines and complement components. Cells that are specialized for a high secretory capacity, such as mature B lymphocytes, liver cells and pancreatic β-cells, are known to expand and adopt their ER capabilities to meet an increased demand of protein synthesis [48]. It is, therefore, likely that ER stress plays a role in adipose tissue dysfunction and most probably also in cell death.
Although apoptosis of (pre)adipocytes has not been extensively studied, there is growing evidence that, under specific circumstances, decreases in adipose tissue mass in humans could result from a loss of fat cells through programmed cell death. The general idea is that in a normal healthy situation adipocyte number is relatively stable when the energy intake is less than the energy output. In this case, the adipose tissue mass only decreases as a result hypotrophy via mobilization of triglycerides [49]. On the other hand, conditions of pathological fat wasting can involve loss of adipocytes through apoptotic mechanisms. For example, apoptotic events were observed in fat tissue of patients with tumor cachexia and in the fat remodelling processes associated with highly active antiretroviral therapy, e.g. ritonavir, in HIV infected patients with lipodystrophy [50–52]. Ritonavir not only induces apoptosis and inhibits adipocyte differentiation, but also affects inflammatory mediators, ER stress and oxidative stress, as shown by gene profiling [53, 54].
Recent data suggest that ER stress may, via several mechanisms, also be involved in apoptosis of (pre)adipocytes in relation to the development of obesity/diabetes. In obese individuals, adipose tissue is poorly oxygenated [55, 56], which may lead to local hypoxia in adipose tissue. ER stress may form a link between hypoxia and apoptosis. Disturbances in cellular redox regulation caused by hypoxia interfere with disulphide bonding in the lumen of the ER, leading to unfolded and misfolded proteins. In 3T3-L1 adipocytes, hypoxia is associated with ER stress, as shown by increased levels of GRP78 and CHOP [57]. Yin et al. [58] described that hypoxia induces cell death, promotes free FA release and inhibits glucose uptake in adipocytes by inhibition of insulin signalling pathway. These metabolic effects of hypoxia may also add to the generation of ER stress, e.g. in addition to hypoxia itself, palmitate, a saturated fatty acid (FA), also activated UPR and induced apoptosis in preadipocytes. CHOP was one of the proteins that were influenced [59]. Moreover, very recently, three papers for the first time show ER stress in human adipose tissue [60–62]. Although none of these papers show direct evidence for a relation between obesity and ER stress-induced apoptosis, the results of Sharma et al. [61], are very suggestive for this. They used ATF4, GADD43 and ATF3 as markers of apoptosis pathways, and show a relation with obesity. Thus, although the data strongly suggest a role for ER stress in apoptosis of adipose tissue, experiments are needed to fully explore this pathway.
For all studies performed with adipose tissue biopsies, it should be emphasized that the precise identity of cells within adipose tissue that show ER stress, and possibly related apoptosis, is not clear. Adipocytes generally account for only 50% of the total number of cells in adipose tissue. Other cells within adipose tissue, e.g. preadipocytes, macrophages and vascular cells, can also secrete an extensive range of protein signals and factors linked to inflammatory response and may therefore also be sensitive for ER stress. This is of special interest since adipose tissue is more and more recognized as a tissue containing a molecular network that connects obesity, adipokine secretion, chronic inflammation and insulin resistance. Inflammation of adipose tissue is often observed in obesity and diabetes and is associated with the infiltration of macrophages into adipose tissue, which may be triggered by adipocyte death, adipokine secretion e.g. TNF-alpha and IL-6, and elaboration of chemokines by adipocyte e.g. monocyte chemo-attractant protein (MCP)-1 [63–65]. The mechanism via which adipocyte death stimulates macrophage infiltration has been proposed to occur via an alternative death pathway that share features of both necrosis and apoptosis [66]. This possibility is supported by the finding that macrophages are located around dead adipocytes in the adipose tissue [67]. Apoptosis of macrophages in adipose tissue may also be linked to diabetes. It has been suggested that macrophage cell death in adipose tissue is an important effect of pioglitazone treatment and this may play an essential role in the management of diabetes mellitus and metabolic syndrome [68]. Hypoxia and hypoxia related ER stress may also play a role in apoptosis of macrophages in adipose tissue. Hypoxia does not only stimulate the inflammatory response of macrophages [69, 70], but also induced apoptosis and cell cycle arrest at G0/G1 phase, via AKT and JNK [71]. To our knowledge no studies have been published on adipose tissue histology showing ER stress related apoptosis in a specific cell type.
ER stress, UPR and apoptosis in the liver
ER stress has been recognized in various models of liver injury and human liver diseases (as reviewed in [72]). The liver plays essential roles in metabolism, biosynthesis, excretion, secretion and detoxification. Comparable to adipose tissue, the liver contains a range of different cell types. The three main liver cell types are hepatocytes, resident macrophages (i.e. Kupffer cells), and endothelial cells. Apoptosis in the liver occurs in many forms of liver injury, e.g. chronic viral liver disease, nonalcoholic and alcoholic steatohepatitis [73–76].
Nonalcoholic fatty liver disease (NAFLD) results from metabolic hepatic dysregulation in metabolic syndrome and T2DM. NAFLD refers to a wide spectrum of liver disease ranging from simple fatty liver (steatosis), to nonalcoholic steatohepatitis (NASH), to cirrhosis (irreversible, advanced scarring of the liver). Several studies have shown that NAFLD predicts future development of T2DM (reviewed in [77]). The pathogenesis of NAFLD is thought to be a multiple-hit process involving insulin resistance, oxidative stress, apoptosis, and adipokines. In NASH, inflammation of the liver is associated with the accumulation of fat in the liver and additionally to different degrees of scarring, which may lead to severe liver scarring and cirrhosis. The general idea is that as consequence of both hepatic and peripheral insulin resistance, the hepatocellular accumulation of triglycerides, initially leads to an altered metabolism of glucose and free fatty acids in the liver. Increased expression of death receptors in response to this altered hepatic metabolism enhances the hepatocytes’ susceptibility for pro-apoptotic stimuli, thus eliciting excessive hepatocyte apoptosis and inflammation. Interestingly, hepatocyte apoptosis is significantly increased in patients with NASH and correlates with disease severity [75, 78].
Evidence is mounting for an important role for ER stress-induced apoptosis in NAFLD. In relation to the onset of diabetes, most in vivo and in vitro studies on the relation between ER stress-induced apoptosis and fatty liver focus on saturated FA. Saturated FA induce ER stress and apoptosis at physiologic concentrations and with a relatively rapid time course in H4IIE liver cells [79, 80], as illustrated by the induction of ER stress response genes and apoptosis which occurred after 4 h and between 6 and 16 h, respectively [79]. Ozcan et al. [7] showed that chronic excessive nutrient intake activated the UPR both in liver and in adipose tissue. A very recent study used transgenic mice carrying the XBP-1-delta-DBD-venus expression gene, which acts as an ER stress-activated indicator (ERAI). In these transgenic mice, the gene encoding venus, a variant of green fluorescent protein, is fused as a reporter downstream of a partial sequence of human XBP-1 including the ER stress-specific intron. The XBP-1/venus fusion protein is produced in cells under ER stress conditions, and cells under ER stress can be detected by monitoring the generation of fluorescence. They showed in the liver of the ERAI transgenic mice that ERAI fluorescence was observed as early as 4 weeks after treatment with a high fat, high sucrose (HF/HS) diet, whereas it was not detected in the fat and muscle, even after 12 weeks of HF/HS diet treatment [9]. It is important to realize that not all FA activate the UPR. Only livers and hepatocytes from rats on a high saturated fat diet, but not high polyunsaturated fat diet, were characterized by the presence of spliced XBP-1 mRNA and increased GRP78 and CHOP protein [81]. This not only suggests that the UPR may sense and respond to the fatty acid environment but also that the ratio of saturated to unsaturated FA may be an important determinant of hepatic ER homeostasis. Although not directly shown in hepatocytes, several mechanisms have been proposed for fatty acid induced ER stress. One possible mechanism is the rapid incorporation of palmitate into lipid components of the rough ER followed by disruption of ER structure and function [82]. Another mechanism of palmitate-induced ER stress is the generation of reactive oxygen species (ROS). ROS by itself can induce ER stress. Prolonged or severe ER stress, which may occur in the presence of excess palmitate, can lead to further ROS accumulation, potentially amplifying the apoptotic/cell death response [83]. Alternatively, as described in β cells, palmitate can lead to an early and sustained depletion of ER Ca2+ stores, which may trigger ER stress via impaired protein folding [41].
ER stress—UPR—insulin resistance
ER stress and the UPR are not only associated with apoptosis in of β-cells, hepatocytes and adipocytes but also with metabolic derangements, especially with insulin resistance. In adipose tissue and liver, the relation of ER stress with insulin resistance is actually more evident than its relation with apoptosis. The general idea is that ER stress interferes with the signalling of the insulin receptor via JNK (Fig. 4). Therefore JNK can not only be a link between ER stress and apoptosis (Fig. 3) but also between ER stress and insulin resistance. A major site of regulation of insulin signalling, both positive and negative, is phosphorylation of the important insulin receptor docking protein insulin receptor protein-1 (IRS-1), whereby phosphorylation of the tyrosine (Tyr) residues in IRS-1 induces phosphorylation of the serine (Ser) residues in IRS-1 and hampers insulin signal transduction (reviewed in [84]). Although the exact mechanisms that lead to Ser phosphorylation of IRS-1 are not yet known, it is apparent that several intracellular serine kinases, e.g. IκB kinase (IKK) and JNK, mTOR and PKC-θ are involved. A wide variety of factors, including nutrients such as FA and amino acids, have been found to induce insulin resistance at least in part through inhibitory IRS-1 Ser phosphorylation. Insulin resistant states (e.g obesity, T2DM) are associated with activation of JNK and/or IKK leading to Ser phosphorylation of IRS1 and hence induction of insulin resistance [85–88]. Activation of JNK in obesity may be a particular consequence of ER stress since IRE-1 has, apart from endoribonuclease activity, also kinase activity that activates JNK (Fig. 4). The liver and adipose tissue of genetic and high-fat diet-induced mouse models of obesity demonstrated increased levels of several ER stress markers as well as induction of insulin resistance via increased Ser phosphorylation/decreased Tyr phosporylation IRS-1. It is of interest that JNK and IKK are also potential links between ER stress and inflammation [89]. Other evidence for a link between ER stress and insulin resistance comes from studies using chaperones, such as 4-phenyl butyric acid (PBA), trimethylamine N-oxide dihydrate (TMAO), and dimethyl sulfoxide or oxygen regulated protein 150kD (ORP150). These chaperones protect cells from ER stress, e.g via stabilization of protein conformation, improvement of ER folding capacity, and therefore enhance the adaptive capacity of the ER. Introduction of chaperones increased insulin sensitivity in the liver of obese diabetic mice [8, 90]. Moreover, an in vitro model of hepatocytes (HepG2) shows that triglycerides induce the expression of endogenous ER stress markers, including GRP 78, IRE-1alpha, XBP-1, p-eIF2alpha, CHOP, and p-JNK. ER stress, in turn, leads to the suppression of insulin receptor signalling through increase in serine phosphorylation and decrease of tyrosine phosphorylation of insulin receptor substrate-1 (IRS-1), and therefore insulin resistance [91]. More evidence for a link between insulin resistance and ER stress is shown in a study using a mouse model that is hypersensitive to insulin (i.e. liver-specific-PTP1B deficient mice). The livers of these mice are both insulin sensitive and protected against a high fat diet-induced ER stress response [92].
Fig. 4.
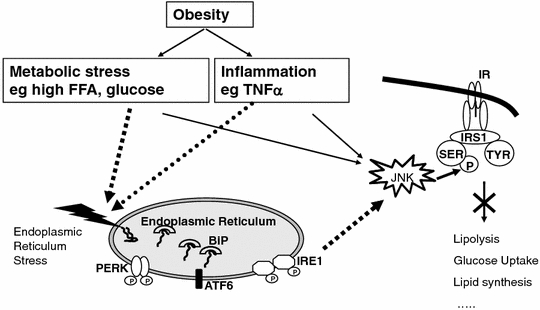
The role of ER stress in obesity related insulin resistance. JNK is a link between ER stress and insulin resistance. Inflammation and metabolic stress cause activation of the UPR. Activation of IRE1 results in JNK activation, leading to Ser phosphorylation of IRS1 and hence induction of insulin resistance
Conclusion
Taken together, these data indicate that ER stress plays a role in diabetes by affecting at least two major events: β-cell failure and generation of insulin resistance. Although most of the current understanding of the known mediators of the ER stress pathway comes from other experimental systems, it is clear that ER stress-induced apoptosis of β cells plays a role in the development of diabetes. Data obtained in liver and adipose tissue suggest that also ER stress-induced apoptosis in these tissues is important in the development of diabetes. In contrast to apoptosis of β cells, which will primarily affect insulin production, ER induced apoptosis in liver and adipose tissue will rather lead to increased insulin resistance. More extensive studies with human adipocytes and hepatocytes are needed to identify the exact interactions between environmental signals and ER stress.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- ASK1
Apoptosis signal-regulating kinase 1
- ATF3
Activated transcription factor 3
- ATF4
Activated transcription factor 4
- ATF6
Activated transcription factor 6
- Bcl-2
Factor B cell lymphoma-2
- BiP/GRP78
Glucose regulated protein 78/binding immunoglobulin protein
- CHOP/GADD153
C/EBP-homologous protein/growth arrest-and DNA damage-inducible gene GADD153
- DM
Diabetes mellitus
- EIF2AK3/PERK
ER-resident PKR-like eIF2α kinase/eukaryotic translation initiation factor 2-alpha kinase 3
- EIF2α
Eukaryotic translation initiation factor 2-alpha
- ERAD
ER associated degradation
- ERAI
ER stress activator indicator
- FA
Fatty acids
- GADD34
Growth arrest and DNA damage inducible protein (also known as PPP1R1A = protein phosphatase 1, regulatory (inhibitor) subunit 15A)
- GADD153/CHOP
C/EBP-homologous protein/growth arrest-and DNA damage-inducible gene GADD153
- GRP78/BiP
Glucose regulated protein 78/binding immunoglobulin protein
- GRP94
Glucose regulated protein 94
- HF/HS
High fat/high sucrose
- HIV
Human immunodeficiency virus
- IKK
IκB kinase
- IL-1β
Interleukin 1β
- IL-6
Interleukin 6
- IRE1
Inositol requiring 1
- IRS-1
Insulin receptor substrate 1
- JNK
c-Jun N-terminal kinase
- MCP-1
Monocyte chemo-attractant protein 1
- NAFLD
Nonalcoholic fatty liver disease
- NASH
Non-alcoholic steatohepatitis
- ORP150
Oxygen regulated protein (150 kD)
- PERK/EIF2AK3
ER-resident PKR-like eIF2α kinase/Eukaryotic translation initiation factor 2-alpha kinase 3
- PKC
Protein kinase C
- PTP1B
Protein tyrosine phosphatase 1B
- ROS
Reactive oxygen species
- Ser
Serine
- mTOR
Mammalian target of rapamycin
- T1DM
Type 1 diabetes mellitus
- T2DM
Type 2 diabetes mellitus
- TNFα
Tumor necrosis factor α
- TRAF2
TNF receptor-associated factor 2
- Tyr
Tyrosine
- UPR
Unfolded protein response
- XBP-1
X-box binding protein 1
References
- 1.Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51:S455–S461. doi: 10.2337/diabetes.51.2007.S455. [DOI] [PubMed] [Google Scholar]
- 2.Herbach N, Rathkolb B, Kemter E, Pichl L, Klaften M, de Angelis MH, Halban PA, Wolf E, Aigner B, Wanke R. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe β-cell loss in Munich Ins2C95S mutant mice. Diabetes. 2007;56:1268–1276. doi: 10.2337/db06-0658. [DOI] [PubMed] [Google Scholar]
- 3.Oyadomari S, Koizumi A, Takeda K, Gotoh T, Akira S, Araki E, Mori M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- 5.Senee V, Vattem KM, Delepine M, Rainbow LA, et al. Wolcott-Rallison syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes. 2004;53:1876–1883. doi: 10.2337/diabetes.53.7.1876. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, Izumi T. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103:27–37. doi: 10.1172/JCI4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 8.Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, Matsuhisa M. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–851. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- 9.Yoshiuchi K, Kaneto H, Matsuoka T-A, Kohno K, Iwawaki T, Nakatani Y, Yamasaki Y, Hori M, Matsuhisa M. Direct monitoring of in vivo ER stress during the development of insulin resistance with ER stress-activated indicator transgenic mice. Biochem Biophys Res Commun. 2008;366:545–550. doi: 10.1016/j.bbrc.2007.11.182. [DOI] [PubMed] [Google Scholar]
- 10.Oshitari T, Hata N, Yamamoto S. Endoplasmic reticulum stress and diabetic retinopathy. Vasc Health Risk Manag. 2008;4:115–122. doi: 10.2147/vhrm.2008.04.01.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kitamura M. Endoplasmic reticulum stress in the kidney. Clin Exp Nephrol. 2008;12:317–325. doi: 10.1007/s10157-008-0060-7. [DOI] [PubMed] [Google Scholar]
- 12.Lindenmeyer MT, Rastaldi MP, Ikehata M, Neusser MA, Kretzler M, Cohen CD, Schlondorff D. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol. 2008;19:2225–2236. doi: 10.1681/ASN.2007121313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferri KF, Kroemer G. Organelle-specific initiation of cell death pathways. Nat Cell Biol. 2001;3:E255–E263. doi: 10.1038/ncb1101-e255. [DOI] [PubMed] [Google Scholar]
- 14.Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 15.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 18.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 19.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocr Rev. 2008;29:317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schröder M. Endoplasmic reticulum stress responses. Cell Mol Life Sci (CMLS) 2008;65:862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- 23.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/S0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 24.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29:42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 25.Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, Rossini AA, Urano F. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006;4:245–254. doi: 10.1016/j.cmet.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Inoue H, Tanizawa Y, Wasson J, Behn P, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome) Nat Genet. 1998;20:143–148. doi: 10.1038/2441. [DOI] [PubMed] [Google Scholar]
- 27.Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, MacAuley A, Goodman AG, LeBoeuf RC, Katze MG. Pancreatic {beta}-cell failure and diabetes in mice with a deletion Mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes. 2005;54:1074–1081. doi: 10.2337/diabetes.54.4.1074. [DOI] [PubMed] [Google Scholar]
- 28.Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JW, Tsukamoto K, Ribick M, Schuit FC, Kaufman RJ. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11:757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- 29.Støy J, Edghill EL, Flanagan SE, Ye H, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci. 2007;104:15040–15044. doi: 10.1073/pnas.0707291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colombo C, Porzio O, Liu M, Massa O, et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J Clin Invest. 2008;118:2148–2156. doi: 10.1172/JCI33777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology. 2004;145:5087–5096. doi: 10.1210/en.2004-0478. [DOI] [PubMed] [Google Scholar]
- 32.Cardozo AK, Ortis F, Storling J, Feng Y-M, Rasschaert J, Tonnesen M, Van Eylen F, Mandrup-Poulsen T, Herchuelz A, Eizirik DL. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005;54:452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- 33.Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada I, Akira S, Araki E, Mori M. Nitric oxide-induced apoptosis in pancreatic β cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci. 2001;98:10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- 35.Marchetti P, Bugliani M, Lupi R, Marselli L, Masini M, Boggi U, Filipponi F, Weir G, Eizirik D, Cnop M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia. 2007;50:2486–2494. doi: 10.1007/s00125-007-0816-8. [DOI] [PubMed] [Google Scholar]
- 36.Hartman MG, Lu D, Kim M-L, Kociba GJ, Shukri T, Buteau J, Wang X, Frankel WL, Guttridge D, Prentki M, Grey ST, Ron D, Hai T. Role for activating transcription factor 3 in stress-induced β-cell apoptosis. Mol Cell Biol. 2004;24:5721–5732. doi: 10.1128/MCB.24.13.5721-5732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang C-J, Lin C-Y, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56:2016–2027. doi: 10.2337/db07-0197. [DOI] [PubMed] [Google Scholar]
- 38.Ariyama Y, Tanaka Y, Shimizu H, Shimomura K, Okada S, Saito T, Yamada E, Oyadomari S, Mori M, Mori M. The role of CHOP messenger RNA expression in the link between oxidative stress and apoptosis. Metabolism. 2008;57:1625–1635. doi: 10.1016/j.metabol.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 39.Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. 2008;118:3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diakogiannaki E, Welters HJ, Morgan NG. Differential regulation of the endoplasmic reticulum stress response in pancreatic beta-cells exposed to long-chain saturated and monounsaturated fatty acids. J Endocrinol. 2008;197:553–563. doi: 10.1677/JOE-08-0041. [DOI] [PubMed] [Google Scholar]
- 41.Cunha DA, Hekerman P, Ladriere L, Bazarra-Castro A, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci. 2008;121:2308–2318. doi: 10.1242/jcs.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akerfeldt MC, Howes J, Chan JY, Stevens VA, Boubenna N, McGuire HM, King C, Biden TJ, Laybutt DR. Cytokine-induced β-cell death is independent of endoplasmic reticulum stress signaling. Diabetes. 2008;57:3034–3044. doi: 10.2337/db07-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez SC, Tanabe K, Cras-Meneur C, Abumrad NA, Bernal-Mizrachi E, Permutt MA. Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes. 2008;57:846–859. doi: 10.2337/db07-0595. [DOI] [PubMed] [Google Scholar]
- 44.Lipson KL, Ghosh R, Urano F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS ONE. 2008;3:e1648. doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Q, Zhang H, Zhao B, Fei H. IL-1beta caused pancreatic beta-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca(2+) release through the c-Jun N-terminal kinase pathway. Mol Cell Biochem. 2009;324:183–190. doi: 10.1007/s11010-008-9997-9. [DOI] [PubMed] [Google Scholar]
- 46.Fourth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice M, Graham I, Atar D, Borch-Johnsen K, et al. (2007) European guidelines on cardiovascular disease prevention in clinical practice: executive summary: fourth joint task force of the European society of cardiology and other societies on cardiovascular disease prevention in clinical practice (Constituted by representatives of nine societies and by invited experts). Eur Heart J 28:2375–2414. doi: 10.1093/eurheartj/ehm316 [DOI] [PubMed]
- 47.Carey VJ, Walters EE, Colditz GA, Solomon CG, Willet WC, Rosner BA, Speizer FE, Manson JE. Body fat distribution and risk of non-insulin-dependent diabetes mellitus in women: the nurses’ health study. Am J Epidemiol. 1997;145:614–619. doi: 10.1093/oxfordjournals.aje.a009158. [DOI] [PubMed] [Google Scholar]
- 48.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 49.Arner P. Control of lipolysis and its relevance to development of obesity in man. Diabetes Metab Rev. 1988;4:507–515. doi: 10.1002/dmr.5610040507. [DOI] [PubMed] [Google Scholar]
- 50.Villarroya F, Domingo P, Giralt M. Lipodystrophy associated with highly active anti-retroviral therapy for HIV infection: the adipocyte as a target of anti-retroviral-induced mitochondrial toxicity. Trends Pharmacol Sci. 2005;26:88–93. doi: 10.1016/j.tips.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 51.Domingo P, Matias-Guiu X, Pujol RM, Francia E, Lagarda E, Sambeat MA, Vazquez G. Subcutaneous adipocyte apoptosis in HIV-1 protease inhibitor-associated lipodystrophy. AIDS. 1999;13:2261–2267. doi: 10.1097/00002030-199911120-00008. [DOI] [PubMed] [Google Scholar]
- 52.Prins JB, Walker NI, Winterford CM, Cameron DP. Human adipocyte apoptosis occurs in malignancy. Biochem Biophys Res Commun. 1994;205:625–630. doi: 10.1006/bbrc.1994.2711. [DOI] [PubMed] [Google Scholar]
- 53.Kim RJ, Wilson CG, Wabitsch M, Lazar MA, Steppan CM. HIV protease inhibitor-specific alterations in human adipocyte differentiation and metabolism. Obesity. 2006;14:994–1002. doi: 10.1038/oby.2006.114. [DOI] [PubMed] [Google Scholar]
- 54.Adler-Wailes DC, Guiney EL, Koo J, Yanovski JA. Effects of ritonavir on adipocyte gene expression: eidence for a stress-related response. Obesity. 2008;16:2379–2387. doi: 10.1038/oby.2008.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Virtanen KA, Lonnroth P, Parkkola R, Peltoniemi P, Asola M, Viljanen T, Tolvanen T, Knuuti J, Ronnemaa T, Huupponen R, Nuutila P. Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. J Clin Endocrinol Metab. 2002;87:3902–3910. doi: 10.1210/jc.87.8.3902. [DOI] [PubMed] [Google Scholar]
- 56.Fleischmann E, Kurz A, Niedermayr M, Schebesta K, Kimberger O, Sessler DI, Kabon B, Prager G. Tissue oxygenation in obese and non-obese patients during laparoscopy. Obes Surg. 2005;15:813–819. doi: 10.1381/0960892054222867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 58.Yin J, Gao G, He Q, Zhou D, Guo Z, Ye J. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab. 2009;296:E333–E342. doi: 10.1152/ajpendo.90760.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo W, Wong S, Xie W, Lei T, Luo Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3–L1 and rat primary preadipocytes. Am J Physiol Endocrinol Metab. 2007;293:E576–E586. doi: 10.1152/ajpendo.00523.2006. [DOI] [PubMed] [Google Scholar]
- 60.Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P, Merali S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57:2438–2444. doi: 10.2337/db08-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma NK, Das SK, Mondal AK, Hackney OG, Chu WS, Kern PA, Rasouli N, Spencer HJ, Yao-Borengasser A, Elbein SC. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab. 2008;93:4532–4541. doi: 10.1210/jc.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Samuel K. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Di Gregorio GB, Yao-Borengasser A, Rasouli N, Varma V, Lu T, Miles LM, Ranganathan G, Peterson CA, McGehee RE, Kern PA. Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes. 2005;54:2305–2313. doi: 10.2337/diabetes.54.8.2305. [DOI] [PubMed] [Google Scholar]
- 66.Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, Obin MS. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–2355. doi: 10.1194/jlr.M500294-JLR200. [DOI] [PubMed] [Google Scholar]
- 67.Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, II, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 68.Bodles AM, Varma V, Yao-Borengasser A, Phanavanh B, Peterson CA, McGehee RE, Jr, Rasouli N, Wabitsch M, Kern PA. Pioglitazone induces apoptosis of macrophages in human adipose tissue. J Lipid Res. 2006;47:2080–2088. doi: 10.1194/jlr.M600235-JLR200. [DOI] [PubMed] [Google Scholar]
- 69.Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999;66:889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- 70.Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. 2008;14:1225–1230. doi: 10.2174/138161208784246153. [DOI] [PubMed] [Google Scholar]
- 71.Fong C–C, Zhang Q, Shi Y-F, Wu RSS, Fong W–F, Yang M. Effect of hypoxia on RAW264.7 macrophages apoptosis and signaling. Toxicology. 2007;235:52–61. doi: 10.1016/j.tox.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 72.Ji C. Dissection of endoplasmic reticulum stress signaling in alcoholic and non-alcoholic liver injury. J Gastroenterol Hepatol. 2008;23:S16–S24. doi: 10.1111/j.1440-1746.2007.05276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ribeiro PS, Cortez-Pinto H, Sola S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME, Rodrigues CMP. Hepatocyte apoptosis, expression of death receptors, and activation of NF-κB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 74.Natori S, Rust C, Stadheim LM, Srinivasan A, Burgart LJ, Gores GJ. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol. 2001;34:248–253. doi: 10.1016/S0168-8278(00)00089-1. [DOI] [PubMed] [Google Scholar]
- 75.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/S0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 76.Papakyriakou P, Tzardi M, Valatas V, Kanavaros P, Karydi E, Notas G, Xidakis C, Kouroumalis E. Apoptosis and apoptosis related proteins in chronic viral liver disease. Apoptosis. 2002;7:133–141. doi: 10.1023/A:1014472430976. [DOI] [PubMed] [Google Scholar]
- 77.Kotronen A, Yki-Jarvinen H. Fatty Liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:27–38. doi: 10.1161/ATVBAHA.107.147538. [DOI] [PubMed] [Google Scholar]
- 78.Wieckowska A, Zein N, Yerian L, Lopez A, McCullough A, Feldstein A. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology. 2006;44:27–33. doi: 10.1002/hep.21223. [DOI] [PubMed] [Google Scholar]
- 79.Wei Y, Wang D, Pagliassotti MJ. Saturated fatty acid-mediated endoplasmic reticulum stress and apoptosis are augmented by trans-10, cis-12-conjugated linoleic acid in liver cells. Mol Cell Biochem. 2007;303:105–113. doi: 10.1007/s11010-007-9461-2. [DOI] [PubMed] [Google Scholar]
- 80.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 81.Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 82.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 83.Borradaile NM, Buhman KK, Listenberger LL, Magee CJ, Morimoto ETA, Ory DS, Schaffer JE. A critical role for eukaryotic elongation factor 1A–1 in lipotoxic cell death. Mol Biol Cell. 2006;17:770–778. doi: 10.1091/mbc.E05-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gual P, Le Marchand-Brustel Y, Tanti J-F. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 85.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li Z-W, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 86.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 87.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 88.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 89.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim D-S, Jeong S-K, Kim H-R, Kim D-S, Chae S-W, Chae H-J. Effects of triglyceride on ER stress and insulin resistance. Biochem Biophys Res Commun. 2007;363:140–145. doi: 10.1016/j.bbrc.2007.08.151. [DOI] [PubMed] [Google Scholar]
- 92.Delibegovic M, Zimmer D, Kauffman C, Rak K, Hong E–G, Cho Y-R, Kim JK, Kahn BB, Neel BG, Bence KK. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes. 2009;58:590–599. doi: 10.2337/db08-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]