

Abstract
The N-terminally extended “synaptic” acetylcholinesterase variant N-AChE-S operates to promote apoptosis; however, the protein partners involved in this function remain unknown. Here, we report that when microinjected to fertilized mouse oocytes, N-AChE-S caused embryonic death as early as the zygotic stage. To identify the putative protein partners involved, we first tried yeast two hybrid screening, but this approach failed, probably because of the N-AChE-S-induced lethality. In contrast, sequence analysis and a corresponding peptide array revealed possible partners, which were validated by co-immunoprecipitation. These include the kinases GSK3, Aurora and GAK, the membrane integrin receptors, and the death receptor FAS. Each of these could potentially modulate N-AChE-S-induced apoptosis with possible therapeutic value for the treatment of Alzheimer’s disease.
Keywords: Acetylcholinesterase; Apoptosis, N-AChE-S; Neurodegeneration; Protein–protein interactions
Introduction
Molecular evidence points to two principal apoptotic pathways leading to neuron death in Alzheimer’s disease (AD). The amyloid cascade hypothesis attributes the neurodegenerative process observed in AD brains to a complex series of events triggered by abnormal processing of APP, the amyloid precursor protein of Aβ. This causes deposition of aggregates and neurotoxicity of the Aβ derivative of APP, disrupting calcium homeostasis. Production of free radicals and inflammation ensue leads to neuronal damage (Parihar and Hemnani 2003). However, neuron loss in AD is selective and occurs particularly in the cholinergic basal nuclei, hippocampus and neocortex (Coyle et al. 1983), and the amyloid hypothesis does not provide an explanation for the cholinergic loss in AD. Combination of the amyloidogenic and cholinergic pathways may thus provide an in-depth view of the processes leading to the initiation and progression of AD.
The cholinergic hypothesis states that the cognitive impairments in AD stem from the loss of cholinergic function (Terry and Buccafusco 2003).This hypothesis is supported by postmortem anatomical findings in AD patients, verifying substantial loss of cholinergic neurons. Dysfunction of the cholinergic system is indeed, by itself, sufficient to produce memory deficits in animal models, which are analogous symptoms to those observed in Alzheimer’s dementia (Gordon et al. 1995; Herholz et al. 2000, 2004; Ikarashi et al. 2004; Rees et al. 2003; Rees and Brimijoin 2003). Accordingly, AChE inhibitors serve to ameliorate symptoms by prolonging ACh availability (Mesulam 2004). Some argue for attenuation of the disease process under treatment with AChE inhibitors (Nordberg et al. 2006; Recanatini and Valenti 2004), others develop alternative AD therapeutics, including inhibitors of the Tau kinase, glycogen synthase kinase 3 (GSK3) (Bhat et al. 2004; Huang and Klein 2006), or other key proteins of the apoptotic pathway, but it is still unclear if these different approaches reflect a single targeted cascade and if so, what triggers this cascade.
Intriguingly, the known principal AD-associated mutations all converge in impaired Ca2+ homeostasis which is also predicted to activate the AChE promoter. Specifically, this involves changes in calcium signaling through the CCAAT motif located −1,270 to −1, 248 from the start codon. This motif binds to the CCAAT binding factor (CBF/NF-Y) (Wan et al. 2006; Zhu et al. 2007c). Also, NFATc3 and NFATc4 which are activated by calcineurin during apoptotic signaling are responsible for AChE expression (Zhu et al. 2007a, b, c). Reciprocally, under thapsigargin-induced calcium imbalance and apoptosis, N-AChE-S is upregulated, whereas its silencing by siRNA prevented thapsigargin-induced cell death. Moreover, over-expression of the N-terminally extended N-AChE-S variant induces apoptosis both in primary cortical cells and in different cell lines of various tissue and species origins, attributing to this protein a conserved and ubiquitous role as a general trigger of apoptosis (Toiber et al. 2008). N-AChE-S causes cell death and morphological impairments in primary brain cultures by activating an apoptotic pathway that includes activation of GSK3, Bax and caspases. Moreover, N-AChE-S overexpression was upregulated in AD brains and showed positive correlation with hyperphosphorylated Tau (Toiber et al. 2008). However, the mechanism through which N-AChE-S initiates the apoptotic pathway remains uncertain.
The core domain of N-AChE-S is positioned extracellularly, whereas its extended N terminus protrudes into the cytoplasm (Toiber et al. 2008). Therefore, we hypothesized that N-AChE-S lethality stems from protein partners interacting with the extended N terminus. To challenge this hypothesis, we used two hybrid screening and peptide array screens, with co-immunoprecipitation for validation.
Materials and methods
Circular dichroism measurements
N-AChE peptide was dissolved in double-distilled water to a final concentration of 1 × 10−4 M. Direct CD spectra were recorded at room temperature using a CD J-810 spectropolarimeter (Jasco, Easton, MD) with a 100-QS 1-mm path-length quartz cuvette (Hellma, Müllheim, Germany). Recordings were at 0.5-nm intervals in the spectral range 185–260 nm.
Two-hybrid screen
The two-hybrid screen was carried out by Hybrigenics (Paris, France). A plasmid containing 66 amino acids of the unique N-AChE N-terminal was sent and cloned into their proprietary two-hybrid vectors.
Peptide array
The synthetic N-terminal peptide of mouse N-AChE was diluted in PBS to the final concentration of 500 μg/ml, and added to 96 wells in NUNC-IMMUNO PLATE (F96 MAXSORP Cat 442404) to a final volume of 100 μl, for overnight incubation. Supernatant (sup) was discarded and the plate washed three times in PBT [5 min, room temperature (RT)]. Protein extracts (100 μg) were added in a final volume of 100 μl for 2 h at RT. Following washes as above, blocking buffer (5% skim milk in PBT) was added (1 h at RT). Sup was discarded and primary antibody was added, diluted 1:100 in the blocking buffer (2 h, RT). Following an additional round of wash, secondary antibody diluted 1:10,000 in blocking buffer was added (2 h, RT). Detection followed repeated wash, with 100 μl 3,3′,5,5′-tetramethylbenzidine (TMB, light protected). When binding occurred, the TMB turned blue, reaction was stopped by addition of 50 μl 1 M H2SO4. Absorbance at 450 nm served to evaluate the binding efficiency.
Results
Lethality of N-AChE-S in microinjected mouse embryos
Transgenic animals are commonly used as models to study the effect of enforced over-expression of genes in the live organism. To substantiate the lethality of N-AChE-S, we microinjected fertilized mouse oocytes; this generated relatively small litters (2–3 mice per female), with none of the newborn carrying the transgene [compared to a minimum 8% of transgenic animals when microinjected with control, non-lethal genes (Steward et al. 1982)]. In search of the developmental stage(s) where embryonic death took place, we analyzed the microinjected embryos before implantation. Most of the N-AChE-S injected embryos did not develop further than the two-cell stage and only one of them developed to a blastocyst; however, it presented massive morphological impairments (Fig. 1 and data not shown). For comparison, water-injected embryos all survived to reach morula and blastocyst stages. Together, this indicated that N-AChE-S over-expression is already lethal at the zygotic stage. Because the shorter AChE-S is not lethal and yields viable transgenic mice (Beeri et al. 1995; Toiber et al. 2008), we suspected that the extended N terminus was the cause.
Fig. 1.
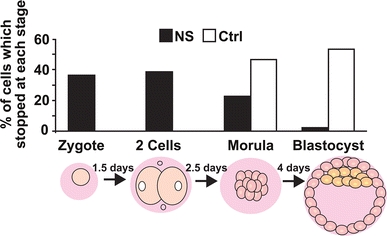
N-AChE-S induced cell death in early embryonic stage. Mouse embryos were microinjected with an N-AChE-S expression vector or DDW for control. Results are presented as percent of cells in each cellular stage. N-AChE-S N = 43, and Ctrl N = 30
Sequence considerations of the N-AChE terminus
The human exon hE1e encodes the N-AChE extended N terminus, 66 amino acids. Using SwissProt, this protein sequence was screened for homology against all known proteins, as well as for structure predictions and protein modification sites. No significant homology was found for this new protein sequence in the database. Intriguingly, the hE1e translation product includes 15 positively charged residues (11 arginines, 1 lysines, 3 histidines), making it an extremely positively charged peptide, with a pI of 11.76. Such high pI values are often found for histones and other nucleic acid binding proteins (http://www.expasy.org/tools/tagident.html).
The extended N-terminal domain translated from hE1e is in frame with the signal peptide (MRPPQCLLHTPSLASPLLLLLLWLLGGGVGA, position 1-31) of the regular AChE protein. This signal peptide is normally cleaved off during protein maturation (Fournier et al. 1992). The addition of the extended N terminus masks the signal peptide, potentially preventing its cleavage. This would result in a protein larger by 96 (66 + 30) amino acid residues, compared to the regular AChE. The very hydrophobic signal peptide would then become an alternative trans-membrane region, similar to the neurexin and cyclooxygenase proteins (Dean et al. 2003). The resultant N-AChE protein would then possess a 66 residues long protrusion at the other side of the plasma membrane from the enzyme’s core domain, similar to neuroligins (Ichtchenko et al. 1996).
Sequence analysis revealed that the N-extended terminus of AChE carries multiple consensus sites for predicted protein–protein interactions and/or modifications that potentially participate in signal transduction. These include an SH3 interaction domain as well as binding sites for GSK3, cyclin, 14-3-3, etc. (Fig. 2). These interaction motifs served for planning experiments to find new possible partners.
Fig. 2.
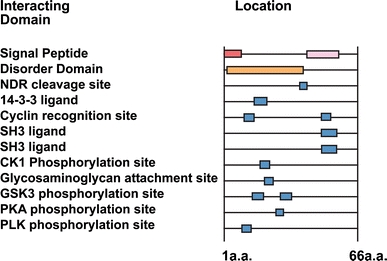
Predicted protein motifs of the N extended AChE. The N-extended AChE tail forecasts interaction with cell signaling proteins involved in different cellular pathways (eukaryotic linear motif) http://elm.eu.org. Peptide sequence: MSCPDRTLVTKVRSHPSGNQHRPTRGGSRSFHCRRGVRPRPAALRVLPRCPAFSDAA
The N-extended protein is naturally unfolded
The N-extended terminus was predicted to be a naturally unfolded peptide, as was demonstrated by circular dichroism (CD). The secondary structure of a synthetic peptide carrying this sequence was determined by CD spectroscopy in the “far-uv” spectral region (190–250 nm). At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. CD signals reflect an average of the entire molecular population and may report the existence of alpha-helix, beta-sheet and random coil structures, each with a characteristic shape and magnitude of the CD spectrum. The tested peptide emerged as a naturally unfolded protein (Fig. 3), which implies “promiscuous” protein–protein interaction features because of its flexibility.
Fig. 3.
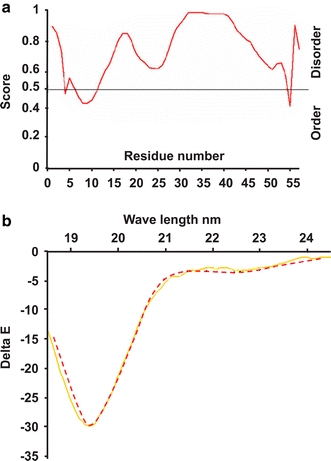
Circular dichroism evidence for natural unfolding of the murine extended N terminus. a Prediction of the disorder domain of the N-AChE terminus. b Shown is the profile of the 46 amino acids of the N-extended terminus of mouse AChE. Note the characteristic shape of an unfolded protein. Y axis: delta E (Delta e = e L − e R), showing the difference in extinction coefficients for left- and right-handed circularly polarized light. X axis: wavelength (yellows, predicted unfolded structure, red N-peptide)
N-AChE protein–protein interactions
To discover the protein partners interacting with the N terminus, we first attempted a yeast two hybrid screening; however, yeast cells expressing N-AChE terminus were unable to survive. Next, we used an optimized two-hybrid screening which again yielded very few possible protein partners with weak binding properties, possibly due to the lethality of this protein (Table 1). That none of those found partners showed tight interactions, called for alternative search technologies. The peptide array we employed next differs substantially from two-hybrid screening, which uses a chimeric protein, in a nuclear environment. Nuclear interactions with the chimeric domains (in both, the bait and the prey) may yield false positives, whereas, cytoplasmic interactions may be overlooked. In the case of toxic peptides, very few protein interactions would be observed in this in vivo system. The peptide array overcomes these limitations, because the synthetic environment is indifferent to toxic effects. False positive interactions were excluded by the use of negative controls, leading to some plausible partners for the N-AChE terminus.
Table 1.
Two-hybrid screening
| (A) Gene or clone ID | # of clones | Category | Function | Processes | Component | Reference |
|---|---|---|---|---|---|---|
| HCN1 | 3 | B | Sodium channel, voltage gated | Apical protein, ion transport | Cell membrane, axons, dendrites | GeneID: 348980 |
| ASH1L | 1 | D | DNA binding, RNA polyII transcription factor activity, metal binding | Chromatin modification | Nucleus, Golgi | GeneID: 55870 |
| EIF4A2 | 1 | D | Translation Initiation factor, ATP binding | Translation | Cytosolic | GeneID: 1974 |
| GAK | 1 | D | Kinase activity, ATP binding, transferase activity | cell cycle, protein phosphorylation | Cytoplasm, Golgi, Cell junction | GeneID: 2580 |
| PPP3CA | 2 | D | Phosphatase activity, calcium binding, metal binding | Cell cycle, synaptic transmission, stress response | Nucleus | GeneID: 5530 |
| COPS5 | 15 | E | Peptidase activity, Transcription co-activator, metal binding | Cell cycle, translation | Cytoplasm, Signalosome | GeneID: 10987 |
| RNF123 | 5 | E | Ligase activity, metal binding | Ubiquitin catabolic process | Cytoplasm | GeneID: 63891 |
| SNAPAP | 7 | E | soluble N-ethylmaleimide-sensitive factor attached protein receptor (SNARE) binding | Exocytosis, Synaptic transmission | Synaptic vesicles, synaptosome | GeneID: 23557 |
| SRI | 2 | E | Calcium channel regulator activity, calcium binding | Signal transduction, Heart development, transport | Cytoplasm | GeneID: 6717 |
| ST13 | 7 | E | Protein binding, bridging | Protein folding | Cytoplasm | GeneID: 6767 |
| UBQLN1 | 5 | E | Kinase binding | Protein modification process | Cytoplasm, Nucleus | GeneID: 29979 |
| B. Gene or clone ID | # of clones | Category | Gene or clone ID | # of clones | Category | |
|---|---|---|---|---|---|---|
| GID: 13445029 | 1 | C | GID: 21955084 | 1 | D | |
| GID: 22024561 | 1 | D | GID: 2337879 | 1 | D | |
| GID: 15668106 | 1 | D | GID: 22038612 | 1 | D | |
| GID: 21955084 | 1 | D | GID: 19774385 | 2 | D | |
| GID: 21427685 | 1 | D | GID: 46395361 | 3 | D | |
| GID: 21622701 | 1 | D | GID: 2815548 | 4 | D | |
| GID: 7768673 | 7 | D | GID: 22024561 | 1 | D | |
| GID: 6670859 | 1 | D | GID: 13445029 | 1 | C | |
| GID: 23462906 | 1 | D | GID: 13445029 | 1 | C | |
| GID: 21955084 | 1 | D | GID: 22024561 | 1 | D | |
| GID: 9857986 | 1 | D | GID: 15668106 | 1 | D | |
| GID: 21954909 | 1 | D | GID: 21955084 | 1 | D | |
| GID: 21427685 | 1 | D |
(A) Known transcripts, (B) unknown transcripts, for further details, see Entrez Genome NCBI database http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene. The category column notes the reliability of the results, as follows: A very high, B high, C good, D moderate confidence in the interaction identified through one unique prey fragment (singleton), E these interactions involve at least one domain of interaction that might be a potential false positive
ELISA-based reactions primarily tested N-AChE interactions with those target proteins predicted by the sequence analysis. A synthetic peptide comprising the mouse N-AChE terminal 46 amino acid residues was tested for its possible interactions with three candidate proteins: integrin, involved in signal transduction from the cell membrane, for example in AD (ffrench-Constant and Colognato 2004; Ho et al. 2005); Aurora kinase members, involved in cell cycle regulation (Kollareddy et al. 2008; Taylor and Peters 2008), and finally Apaf, a proapoptotic protein, part of the apoptosome complex (Fig 4) (Bao and Shi 2007). Integrin, Aurora kinase and APAF showed interaction, whereas three different proteins did not interact (not shown). Integrin which showed a promising interaction and other proteins known to be involved in the apoptotic signaling cascade, i.e., GSK3, GAK (cyclin G associated kinase), JAB1/CSN5, integrin and Fas (TNF receptor super-family, member 6) were further subjected to co-immunoprecipitation. Positive co-precipitation (Fig. 5) served as a validating test, supporting the notion of co-interaction with these tested partner proteins.
Fig. 4.
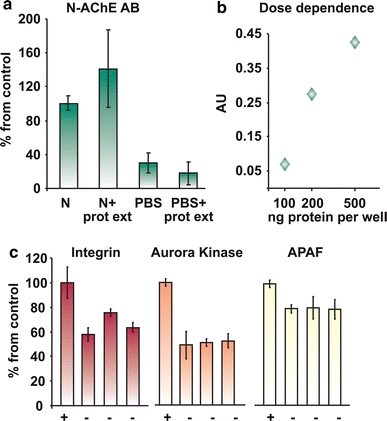
Peptide array for protein interactions: a Positive control. Note the first two bars detection compared to the negative controls. b Dose dependence. Different concentrations of the synthetic peptide were added to the plate, and detected by the N-specific antibody, showing the sensitivity of the assay. c Positive interaction
Fig. 5.
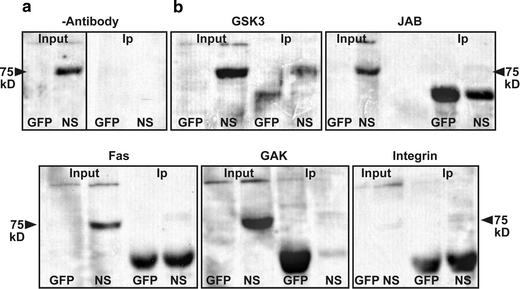
Co-immunoprecipitation 293 cells were transfected with GFP or N-AChE-S and the protein extract used for Co-Ip a Negative control was without antibody. In each experiment four samples were used GFP input, N-AChE-S (NS) Input and GFP co-IP, NS Co-IP. b Westerns were probed with anti-AChE which yields a specific band at 75 kD. The IP was considered specific if the band appeared in the NS IP and not GFP IP. The lower stronger band is nonspecific. Antibodies that yielded positive interactions: GSK3, JAB1(CSN5), Fas, GAK and integrin
N-AChE-S protein multimers with integrin receptors were also observed. Integrin receptors are known to interact with many other cellular receptors and be involved in cell signaling, including the observed GSK3 activation and dephosphorylation of GSK3 by activation of Protein Phosphatase 2A (Ivaska et al. 2002). In addition, N-AChE-S showed a specific interaction with GSK3, fortifying the importance of this enzyme in the N-AChE-S apoptotic pathway (Toiber et al. 2008).
Discussion
Unusual AChE forms were reported in AD (Navaratnam et al. 1991) and in dementia (Shen 1997). In addition, some of the commercially available anti-AChE antibodies yield double bands around 66–70 kDa [see, for example (Belbeoc’h et al. 2003; Brenner et al. 2003; Meshorer et al. 2004; Morel et al. 2001)]. Sucrose gradient analyses also reveal yet unexplained diversity of the various AChE isoforms (Massoulie 2002). These observations could potentially reflect the new, longer member(s) of the AChE family; and the potent labeling of brain AChE-like protein with anti-N antibodies (Meshorer et al. 2004) suggests abundance of such variants.
The novel human N-AChE protein variant shows similarity to the murine protein, supporting the notion that it is physiologically relevant. In addition to the N-terminal extension, the new protein spans the very hydrophobic sequence of the former signal peptide, which becomes a trans-membrane domain in N-terminally extended AChE variants. Parallel examples include the asialoglycoprotein receptor (Spiess and Lodish 1986), the bovine adrenal dopamine β-monooxygenase (Taljanidisz et al. 1989), α-neurexin (an extended form of β-neurexin) (Ushkaryov et al. 1994), and cyclooxygenase (Chandrasekharan et al. 2002). For the extended N-AChE variants, positioning of the “conservative” signal peptide in the membrane would enable direct docking of AChE to the cell membrane, independently of the structural PRiMA, or ColQ proteins necessary to adhere AChE-S tetramers to the synapse (Massoulie 2002) (Krejci et al. 1997).
Together with the N-terminal extension, the AChE protein is enlarged by 20%. However, its catalytic activity is unaffected, suggesting that the extended N terminus forms a structurally independent domain, distinct from the globular AChE structure (Sussman et al. 1991) and separated from it by the conservative signal peptide. This further implies that the role of the N terminus is not related to AChE’s catalytic characteristics.
The potential partners for the extended AChE N terminus, as deduced from the sequence analysis, tested in the peptide array or by co-immunoprecipitation experiments included Cyclin G associated kinase (GAK) which contributes to cell signaling and internalization of clathrin-coated vesicles, and which is probably involved in the mechanism by which N-AChE-S is internalized. Also included was JAB1/CSN5, which is involved in the regulation of p53, and could have a role in preventing N-AChE-S-induced cell death (Lee et al. 2006). GSK was previously shown by us (Toiber et al. 2008) to be involved with N-AChE apoptosis.
Parallel heterodimers formation with other cell membrane receptors such as p75, TRK or FAS family members, can likewise, activate additional signaling mechanisms. FAS, which also interacts with N-AChE-S is a known death receptor, which interacts with caspases and with death ligands, inducing cell death from a variety of stimuli. The interaction of N-AChE-S with Fas could induce mutual activation, leading to known apoptotic cascades. Together, this data suggests possible protein partners initiating the N-AChE-S-induced apoptotic cascade and/or protection from it. Blockade of the apoptogenic partners to prevent the neuronal associated N-AChE-S cell death in AD should be considered.
Acknowledgments
This research was supported by the European Union’s Network of Excellence (LSH-2004-1.1.5-3) and STREP (LSHG-CT-2006-037277), The German Ministry of Science and German-Israel Project, The German Israel Project DIP-G 3.2, the Israel Science Foundation (Grant 399/07) and The Hebrew University’s Eric Roland Center for Neurodegenerative Diseases, the Interdisciplinary Center for Neuronal Computation (ICNC) and The ROSETREES Foundation, UK.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
D. Toiber and D.S. Greenberg contributed equally to this work.
Contributor Information
David S. Greenberg, Email: gdavid@cc.huji.ac.il
Hermona Soreq, Phone: +972-2-6585109, FAX: +972-2-6520258, Email: soreq@cc.huji.ac.il.
References
- Bao Q, Shi Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. 2007;14(1):56–65. doi: 10.1038/sj.cdd.4402028. [DOI] [PubMed] [Google Scholar]
- Beeri R, Andres C, Lev-Lehman E, Timberg R, Huberman T, Shani M, Soreq H. Transgenic expression of human acetylcholinesterase induces progressive cognitive deterioration in mice. Curr Biol. 1995;5(9):1063–1071. doi: 10.1016/S0960-9822(95)00211-9. [DOI] [PubMed] [Google Scholar]
- Belbeoc’h S, Massoulie J, Bon S. The C-terminal T peptide of acetylcholinesterase enhances degradation of unassembled active subunits through the ERAD pathway. EMBO J. 2003;22(14):3536–3545. doi: 10.1093/emboj/cdg360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RV, Budd Haeberlein SL, Avila J. Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem. 2004;89(6):1313–1317. doi: 10.1111/j.1471-4159.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- Brenner T, Hamra-Amitay Y, Evron T, Boneva N, Seidman S, Soreq H. The role of readthrough acetylcholinesterase in the pathophysiology of myasthenia gravis. FASEB J. 2003;17(2):214–222. doi: 10.1096/fj.02-0609com. [DOI] [PubMed] [Google Scholar]
- Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci USA. 2002;99(21):13926–13931. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219(4589):1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P. Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci. 2003;6(7):708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ffrench-Constant C, Colognato H. Integrins: versatile integrators of extracellular signals. Trends Cell Biol. 2004;14(12):678–686. doi: 10.1016/j.tcb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Fournier D, Mutero A, Rungger D. Drosophila acetylcholinesterase. Expression of a functional precursor in Xenopus oocytes. Eur J Biochem. 1992;203(3):513–519. doi: 10.1111/j.1432-1033.1992.tb16577.x. [DOI] [PubMed] [Google Scholar]
- Gordon I, Grauer E, Genis I, Sehayek E, Michaelson DM. Memory deficits and cholinergic impairments in apolipoprotein E-deficient mice. Neurosci Lett. 1995;199(1):1–4. doi: 10.1016/0304-3940(95)12006-P. [DOI] [PubMed] [Google Scholar]
- Herholz K, Bauer B, Wienhard K, Kracht L, Mielke R, Lenz MO, Strotmann T, Heiss WD. In vivo measurements of regional acetylcholine esterase activity in degenerative dementia: comparison with blood flow and glucose metabolism. J Neural Transm. 2000;107(12):1457–1468. doi: 10.1007/s007020070009. [DOI] [PubMed] [Google Scholar]
- Herholz K, Weisenbach S, Zundorf G, Lenz O, Schroder H, Bauer B, Kalbe E, Heiss WD. In vivo study of acetylcholine esterase in basal forebrain, amygdala, and cortex in mild to moderate Alzheimer disease. Neuroimage. 2004;21(1):136–143. doi: 10.1016/j.neuroimage.2003.09.042. [DOI] [PubMed] [Google Scholar]
- Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr Drug Targets Inflamm Allergy. 2005;4(2):247–256. doi: 10.2174/1568010053586237. [DOI] [PubMed] [Google Scholar]
- Huang HC, Klein PS. Multiple roles for glycogen synthase kinase-3 as a drug target in Alzheimer’s disease. Curr Drug Targets. 2006;7(11):1389–1397. doi: 10.2174/1389450110607011389. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Nguyen T, Sudhof TC. Structures, alternative splicing, and neurexin binding of multiple neuroligins. J Biol Chem. 1996;271(5):2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- Ikarashi Y, Harigaya Y, Tomidokoro Y, Kanai M, Ikeda M, Matsubara E, Kawarabayashi T, Kuribara H, Younkin SG, Maruyama Y, Shoji M. Decreased level of brain acetylcholine and memory disturbance in APPsw mice. Neurobiol Aging. 2004;25(4):483–490. doi: 10.1016/S0197-4580(03)00122-2. [DOI] [PubMed] [Google Scholar]
- Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kahari VM, Heino J. Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol Cell Biol. 2002;22(5):1352–1359. doi: 10.1128/MCB.22.5.1352-1359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollareddy M, Dzubak P, Zheleva D, Hajduch M. Aurora kinases: structure, functions and their association with cancer. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2008;152(1):27–33. doi: 10.5507/bp.2008.004. [DOI] [PubMed] [Google Scholar]
- Krejci E, Thomine S, Boschetti N, Legay C, Sketelj J, Massoulie J. The mammalian gene of acetylcholinesterase-associated collagen. J Biol Chem. 1997;272(36):22840–22847. doi: 10.1074/jbc.272.36.22840. [DOI] [PubMed] [Google Scholar]
- Lee EW, Oh W, Song J. Jab1 as a mediator of nuclear export and cytoplasmic degradation of p53. Mol Cells. 2006;22(2):133–140. [PubMed] [Google Scholar]
- Massoulie J. The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals. 2002;11(3):130–143. doi: 10.1159/000065054. [DOI] [PubMed] [Google Scholar]
- Meshorer E, Toiber D, Zurel D, Sahly I, Dori A, Cagnano E, Schreiber L, Grisaru D, Tronche F, Soreq H. Combinatorial complexity of 5′ alternative acetylcholinesterase transcripts and protein products. J Biol Chem. 2004;279(28):29740–29751. doi: 10.1074/jbc.M402752200. [DOI] [PubMed] [Google Scholar]
- Mesulam M. The cholinergic lesion of Alzheimer’s disease: pivotal factor or side show? Learn Mem. 2004;11(1):43–49. doi: 10.1101/lm.69204. [DOI] [PubMed] [Google Scholar]
- Morel N, Leroy J, Ayon A, Massoulie J, Bon S. Acetylcholinesterase H and T dimers are associated through the same contact. Mutations at this interface interfere with the C-terminal T peptide, inducing degradation rather than secretion. J Biol Chem. 2001;276(40):37379–37389. doi: 10.1074/jbc.M103192200. [DOI] [PubMed] [Google Scholar]
- Navaratnam DS, Priddle JD, McDonald B, Esiri MM, Robinson JR, Smith AD. Anomalous molecular form of acetylcholinesterase in cerebrospinal fluid in histologically diagnosed Alzheimer’s disease. Lancet. 1991;337(8739):447–450. doi: 10.1016/0140-6736(91)93391-L. [DOI] [PubMed] [Google Scholar]
- Nordberg A, Eriksdotter-Jonhagen M, Garlind A, Grut M, Freund-Levi Y, Cornelius C, Ekstrom A, Fastbom J, Sedvall M, Viitanen M. Administration of symptom-relieving drugs in Alzheimer disease is beneficial. Lakartidningen. 2006;103(6):369–371. [PubMed] [Google Scholar]
- Parihar MS, Hemnani T. Phenolic antioxidants attenuate hippocampal neuronal cell damage against kainic acid induced excitotoxicity. J Biosci. 2003;28(1):121–128. doi: 10.1007/BF02970142. [DOI] [PubMed] [Google Scholar]
- Recanatini M, Valenti P. Acetylcholinesterase inhibitors as a starting point towards improved Alzheimer’s disease therapeutics. Curr Pharm Des. 2004;10(25):3157–3166. doi: 10.2174/1381612043383313. [DOI] [PubMed] [Google Scholar]
- Rees TM, Brimijoin S. The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. Drugs Today (Barc) 2003;39(1):75–83. doi: 10.1358/dot.2003.39.1.740206. [DOI] [PubMed] [Google Scholar]
- Rees T, Hammond PI, Soreq H, Younkin S, Brimijoin S. Acetylcholinesterase promotes beta-amyloid plaques in cerebral cortex. Neurobiol Aging. 2003;24(6):777–787. doi: 10.1016/S0197-4580(02)00230-0. [DOI] [PubMed] [Google Scholar]
- Shen ZX. An CSF anamalous molecular form of acetylcholinesterase in demented and non-demented subjects. NeuroReport. 1997;8(15):3229–3232. doi: 10.1097/00001756-199710200-00009. [DOI] [PubMed] [Google Scholar]
- Spiess M, Lodish HF. An internal signal sequence: the asialoglycoprotein receptor membrane anchor. Cell. 1986;44(1):177–185. doi: 10.1016/0092-8674(86)90496-4. [DOI] [PubMed] [Google Scholar]
- Steward TA, Wagner EF, Mintz B. Human beta-globin gene sequences injected into mouse eggs, retained in adults, and transmitted to progeny. Science. 1982;217(4564):1046–1048. doi: 10.1126/science.6287575. [DOI] [PubMed] [Google Scholar]
- Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 1991;253(5022):872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- Taljanidisz J, Stewart L, Smith AJ, Klinman JP. Structure of bovine adrenal dopamine beta-monooxygenase, as deduced from cDNA and protein sequencing: evidence that the membrane-bound form of the enzyme is anchored by an uncleaved signal peptide. Biochemistry. 1989;28(26):10054–10061. doi: 10.1021/bi00452a026. [DOI] [PubMed] [Google Scholar]
- Taylor S, Peters JM. Polo and Aurora kinases: lessons derived from chemical biology. Curr Opin Cell Biol. 2008;20(1):77–84. doi: 10.1016/j.ceb.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Terry AV, Jr, Buccafusco JJ. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther. 2003;306(3):821–827. doi: 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- Toiber D, Berson A, Greenberg D, Melamed-Book N, Diamant S, Soreq H. N-acetylcholinesterase-induced apoptosis in Alzheimer’s disease. PLoS ONE. 2008;3(9):e3108. doi: 10.1371/journal.pone.0003108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushkaryov YA, Hata Y, Ichtchenko K, Moomaw C, Afendis S, Slaughter CA, Sudhof TC. Conserved domain structure of beta-neurexins. Unusual cleaved signal sequences in receptor-like neuronal cell-surface proteins. J Biol Chem. 1994;269(16):11987–11992. [PubMed] [Google Scholar]
- Wan DC, Zhang X, Siow NL, Xie HQ, Tsim KW. Chick acetylcholinesterase promoter regulation. J Mol Neurosci. 2006;30(1–2):33–34. doi: 10.1385/JMN:30:1:33. [DOI] [PubMed] [Google Scholar]
- Zhu H, Gao W, Jiang H, Jin QH, Shi YF, Tsim KW, Zhang XJ. Regulation of acetylcholinesterase expression by calcium signaling during calcium ionophore A23187- and thapsigargin-induced apoptosis. Int J Biochem Cell Biol. 2007;39(1):93–108. doi: 10.1016/j.biocel.2006.06.012. [DOI] [PubMed] [Google Scholar]
- Zhu H, Gao W, Jiang H, Wu J, Shi YF, Zhang XJ. Calcineurin mediates acetylcholinesterase expression during calcium ionophore A23187-induced HeLa cell apoptosis. Biochim Biophys Acta. 2007;1773(4):593–602. doi: 10.1016/j.bbamcr.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Zhu H, Gao W, Shi YF, Zhang XJ. The CCAAT-binding factor CBF/NF-Y regulates the human acetylcholinesterase promoter activity during calcium ionophore A23187-induced cell apoptosis. Biochim Biophys Acta. 2007;1770(10):1475–1482. doi: 10.1016/j.bbagen.2007.07.007. [DOI] [PubMed] [Google Scholar]