

Abstract
The primary goal of reverse genetics, the identification of null mutations in targeted genes, is achieved through screening large populations of randomly mutagenized plants. T-DNA and transposon-based mutagenesis has been widely employed but is limited to species in which transformation and tissue culture are efficient. In other species, TILLING (for Targeting Induced Local Lesions IN Genomes), based on chemical mutagenesis, has provided an efficient method for the identification of single base pair mutations, only 5% of which will be null mutations. Furthermore, the efficiency of inducing point mutations, like insertion-based mutations, is dependent on target size. Here, we describe an alternative reverse genetic strategy based on physically induced genomic deletions that, independent of target size, exclusively recovers knockout mutants. Deletion TILLING (De-TILLING) employs fast neutron mutagenesis and a sensitive polymerase chain reaction-based detection. A population of 156,000 Medicago truncatula plants has been structured as 13 towers each representing 12,000 M2 plants. The De-TILLING strategy allows a single tower to be screened using just four polymerase chain reaction reactions. Dual screening and three-dimensional pooling allows efficient location of mutants from within the towers. With this method, we have demonstrated the detection of mutants from this population at a rate of 29% using five targets per gene. This De-TILLING reverse genetic strategy is independent of tissue culture and efficient plant transformation and therefore applicable to any plant species. De-TILLING mutants offer advantages for crop improvement as they possess relatively few background mutations and no exogenous DNA.
Due to advances in sequencing technology, the generation of genomic sequence data is no longer a limiting factor in the genetic dissection of plant development and physiology. Identification of new genes and verification of gene structure have also been facilitated by high-throughput characterization of RNA transcripts. Attempts to complement the massive availability of sequence data with automated computational annotation has been of only limited value, identifying only a proportion of functional gene products and producing high levels of inaccurate annotation (Yamada et al., 2003; Haas et al., 2005). Descriptive genomic approaches, such as gene expression analysis, massively parallel signature sequencing, and serial analysis of gene expression, supply basic information about gene expression but fall short of allowing us to assign gene function. Therefore, the development of these technologies has fuelled demand for reverse genetic platforms. The generation and analysis of mutants remains central to contemporary genetics.
The ability to infer gene function through homology and expression analysis leads biologists to directly test hypotheses by disrupting the activity of genes known only by their sequence. Forward genetics, starting from phenotypic screens, has historically underpinned plant genetics and remains a central and unbiased approach to genetic questions. However, even with the availability of dense genetic maps anchored to genomic sequence data, cloning genes on the basis of phenotype is still not a trivial task. Systematic reverse genetic platforms, allowing researchers to obtain plants mutated at any identified locus, have streamlined functional genomics in well-resourced model species. These platforms are generally based on insertional mutagenesis using T-DNA (Azpiroz-Leehan and Feldmann, 1997; Krysan et al., 1999; Sussman et al., 2000; Sessions et al., 2002; Alonso et al., 2003) or transposon tagging (Martienssen, 1998; Parinov et al., 1999; Speulman et al., 1999; Parinov and Sundaresan, 2000; Tadege et al., 2008), which, through the introduction of known sequences, greatly facilitates gene cloning and allows the high-throughput characterization of insertion sites. However, these approaches are not feasible in the majority of species where, although genomic sequence information may be extensive, transformation and tissue culture-based methods are not practical. In these species, TILLING (for Targeting Induced Local Lesions IN Genomes) is the most widely applied strategy (McCallum et al., 2000a, 2000b; Colbert et al., 2001). TILLING is based on the alkylating agent ethyl methanesulfonate (EMS), which introduces point mutations across the genome. These can be supported to very high densities without causing lethality. For this reason, EMS mutagenesis has been widely employed in both forward and reverse genetic screens where relatively small populations can yield multiple mutant alleles of a gene. As a consequence, TILLING mutants possess high numbers of background mutations. TILLING is also relatively labor intensive with PCR amplification and heteroduplex analysis generally carried out at low levels of pooling, requiring many hundreds of samples to be screened. In addition, in common with insertional mutagenesis, the recovery of mutations in genes of <1 kb is highly inefficient, a significant limitation in the light of research highlighting the importance of microRNAs and other small transcriptional products.
Fast neutrons are a form of high-energy radiation that has been shown to induce a broad range of deletions and other chromosomal mutations in plants. Several sources of fast neutrons are potentially available for mutagenesis, including particle accelerator spallation sources and nuclear research reactors, the latter type being used in this study. Fast neutrons produced by nuclear fission reactors are accompanied by gamma radiation, but the contribution is adjustable. The emission rate achieved with nuclear reactors is in general much higher compared to spallation sources, reducing irradiation time from days to hours. Importantly, the neutron energy should be in the range of approximately 500 keV to 5 MeV to generate the short-range secondary particles within the cell nucleus that mediate strand breakage (J. Palfalvi, personal communication).
Bruggemann et al. (1996) isolated and characterized 20 independent null alleles of the HY4 locus in a forward screen of 300,000 M2 fast neutron-irradiated Arabidopsis (Arabidopsis thaliana) plants. Deletions ranging from 300 bp to >8 kb were identified by Southern analysis, although larger or smaller deletions and more complex rearrangements could not be distinguished using this method. An indication of the size range of deletions induced by fast neutron bombardment has also been made through a review of the mutants catalogued by the National Science Foundation Arabidopsis Information Resource (TAIR; www.arabidopsis.org). This revealed 114 fast neutron alleles. Of the 53 sufficiently characterized, 43 (81%) were deletions and 10 were other types of mutation, including combinations of insertions, deletions, substitutions, and rearrangements. The deletions ranged from a single base pair to a 60-kb deletion spanning 12 genes. Strikingly, fast neutron-induced deletions spanning megabases have not been identified in Arabidopsis or to our knowledge in Medicago truncatula or tomato (Solanum lycopersicum). In contrast, deletions of this size seem to be common in wheat (Triticum aestivum). This may be because hexaploid wheat can support the removal of essential genes through the presence of duplicate gene copies on homeologous chromosomes (Roberts et al., 1999). Fast neutron bombardment therefore provides a nontransgenic and facile method of mutagenesis creating DNA lesions of a size amenable to direct PCR detection potentially at high levels of pooling compared to conventional TILLING.
Despite a long history of use as a mutagen in forward genetics, fast neutron bombardment has not been exploited extensively in the development of reverse genetic platforms. One exception to this is the work of Li et al. (2001), who developed a fast neutron-based reverse genetics platform in Arabidopsis known as delete-a-gene (Li et al., 2001, 2002). This platform employed a PCR strategy for detecting deletions using shortened extension times to limit amplification of longer wild-type sequence, allowing short deletion-containing alleles to be preferentially amplified. Using extension time suppression alone, it was demonstrated that this strategy could be used to selectively amplify a known deletion in pools of 1,000 plants. However, this strategy is limited to detecting deletions that remove a large proportion of the amplified region. Extension time suppression depends upon a large difference between the wild type and mutant amplicons. This is an intrinsic weakness because a randomly selected pair of primers >5 kb apart are unlikely to surround a deletion that removes >80% of the fragment. If smaller deletions could be detected within such amplicons, the recovery of mutants would be more efficient. The relatively high pooling depths in delete-a-gene are only possible when detecting large deletions, while the probability of closely flanking such large deletions with random primers is low. Therefore, the goal of this study was to develop a method whereby small deletions could be detected in large amplicons at high pooling depths.
Here, we describe the development of a novel reverse genetic strategy in the model legume M. truncatula, which exploits a large population of plants harboring chromosomal deletions and a highly efficient screening strategy for the discovery of deletions within targeted regions. Deletion based TILLING (De-TILLING) combines fast neutron mutagenesis with PCR-based screening and a three-dimensional (3D) pooling strategy for the efficient recovery of knockout mutants. This method can provide a useful alternative strategy for species in which T-DNA or transposon tagging resources are limited and for providing a targeted approach for identifying mutants in smaller or otherwise untagged genes of all plant species.
RESULTS
Screening Strategy
In a deletion-based system, pooling of plants means that wild-type sequences are preferentially amplified over rare deletion-containing alleles, even at relatively low pooling depths. To increase efficiency of mutant detection in a fast neutron mutagenized population, it was desirable to screen large pools of mutants. This creates the challenge to identify specific deletion alleles in a large background of wild-type alleles and, therefore, the need to suppress amplification from wild-type sequences. This was achieved by Li et al. (2001) by extension time suppression, but this required targeting large wild-type regions, which challenged the processivity of the polymerase under these condition and large percentage deletions to enable amplification of the small deletion allele. We tested the efficacy of delete-a-gene across a range of dilutions and using a range of target sizes. For this analysis, we used a previously characterized M. truncatula mutant, dmi1-4, with an 18-kb deletion removing the 5' end of the DMI1 gene and upstream region (Ane et al., 2004). PCR templates were prepared to model the representation of a mutant within a genomic DNA pool. Genomic DNA of dmi1-4 and wild-type M. truncatula A17 were combined in ratios up to 1 mutant in 24,000 wild types to create a set of PCR templates useful for assessing the sensitivity of detection strategies (Fig. 1). Primers that spanned the deletion producing a 0.3-kb product were highly efficient, allowing detection of the deletion in a 1 in 8,000 dilution (Fig. 1A). A secondary PCR using nested primers allowed detection of the deletion up to a 1 in 24,000-fold dilution (Fig. 1A). However, when attempting to discover an unknown deletion, it is unlikely that primers would so closely span such a large deletion. Therefore, we tested the ability to detect the deletion using primers more distant to the deletion site. Primers spanning the deletion and producing 3- and 8-kb targets were far less efficient at detecting the mutation in a dilution series using a single PCR; however, in a secondary PCR, the 3-kb target could be detected in a 1:24,000 dilution, while the 8-kb target could be detected in a 1:8,000 dilution (Fig. 1, B and C).
Figure 1.

A reconstruction experiment showing the amplification of the M. truncatula DMI1 locus from genomic DNA of wild type (A17), the dmi1-4 mutant (D1), and pools containing dmi1-4 and wild-type DNA at ratios of 1:25 ng (1:25), 1:1,000 ng (1K), 1:4,000 ng (4K), 1:8,000 ng (8K), 1:12,000 ng (12K), 1:16,000 ng (16K), 1:20,000 ng (20K), and 1:24,000 ng (24K). Nested PCR primers flank the 18-kb deletion by 350 bp (A), 3.0 kb (B), and 8.0 kb (C). Amplification from the wild-type region is suppressed entirely under these conditions, allowing amplification of the mutant product in pools of over 1:24,000 for the 350-bp and 3.0-kb assays and 1:12,000 genomes for the 8.0-kb assay. 10, Primary PCR; 20, secondary PCR.
In spite of the success of target detection in the delete-a-gene mock, we were unable to discover deletions in target genes in a population of fast neutron-mutagenized M. truncatula using this strategy. We hypothesized that this may reflect the unlikely event of designing primers that sufficiently spanned these large deletions to allow discovery. It was thus desirable to establish a detection system that would allow discovery of smaller deletions. We set up a second recapitulation of delete-a-gene using a much smaller deletion, the nsp2-1 mutant, which possesses a 435-bp deletion (Oldroyd and Long, 2003). A secondary PCR using nested primers L7, R7, L5, and R5 (Fig. 2A) shows the 1,561-bp nsp2 to one amplicon barely able to compete for amplification with the 1,996-bp wild-type fragment at a ratio of one mutant in 25 wild types (Fig. 2A).
Figure 2.

A reconstruction experiment showing the amplification of the M. truncatula NSP2 locus from genomic DNA of wild-type M. truncatula (A17), the nsp2-1 mutant (N2), and pools containing nsp2-1 and wild-type DNA at ratios of 1:25 ng (1:25), 1:1,000 ng (1K), 1:4,000 ng (4K), 1:8,000 ng (8K), 1:12,000 ng (12K), 1:16,000 ng (16K), 1:20,000 ng (20K), and 1:24,000 ng (24K). Only the secondary PCR products are shown in each case. A, Nested PCR. Wild-type alleles are preferentially amplified. B, Poison primer suppression enhances detection of the mutant allele in pools of up to 1:1,000 genomes. C, Restriction suppression. EcoRV-treated templates allow reliable detection in pools of up to 4,000 plants. D, De-TILLING strategy. Combining poison primer and restriction suppression allows preferential amplification of the mutant allele in pools containing a 24,000-fold excess of wild-type sequences. 10, Primary PCR; 20, secondary PCR.
We therefore attempted to develop a system that could detect these small deletions in large pools of wild-type plants. Poison primer suppression was first described for detecting deletions induced by the mutagen trimethylpsoralen in mutant populations of Caenorhabditis elegans (Edgley et al., 2002). In addition to the nested PCR assay described above, a third poison primer is included in the first round of PCR (Fig. 2B). A small PCR product, known as the suppressor fragment, is produced between the poison primer and one of the external primers. The suppressor fragment is amplified more efficiently than the full-length wild-type product due to its smaller size. Amplification from a mutant template present within the DNA pool, in which the poison primer binding site has been deleted, produces a single amplicon from the external primers. During the second round of nested PCR, the suppressor fragment, lacking one of the external primer binding sites, cannot act as a template. Only the deletion allele and wild-type allele will now be amplified. Because the production of the wild-type amplicon has been limited by competition from the suppressor fragment in the first round, the mutant amplicon is able to successfully compete for amplification. A poison primer strategy that included the L7B poison primer in the first round of PCR allowed detection of the nsp2-1 mutant at a pooling level of 1:1,000 in the second round of PCR (Fig. 2B). This is consistent with the pooling depth used in the original C. elegans study (1:1,200), although for a small target of 701 bp, a detection sensitivity of 1:5,000 was demonstrated (Edgley et al., 2002).
To further enhance the sensitivity of deletion detection, we assessed the capability of restriction enzymes to suppress the production of wild-type amplicons. In this strategy, a nested PCR assay is designed centered upon restriction sites unique within the amplified region. Predigesting the DNA pool with this restriction enzyme will destroy a majority of the wild-type template, allowing the mutant allele, in which this restriction site has been deleted, to successfully compete for amplification. Predigesting the nsp2-1 pooled templates with EcoRV and amplifying using the standard nested PCR protocol increased the detection sensitivity to 1:4,000 (Fig. 2C). Amplification from the wild-type allele is not completely suppressed, and the mutant allele is not reliably amplified in the more highly pooled templates.
We assessed how integrating both restriction suppression and poison primers impacted on deletion detection sensitivity. Amplifying from a predigested template and including the poison primer, designed to bind within 30 bp of the restriction site, we achieved much greater detection sensitivities. The nsp2 deletion removes only 20% of the amplified region, yet using the poison primer and restriction suppression we were able to detect the deletion mutant in pools containing a 24,000-fold excess of wild-type sequences (Fig. 2D).
Mutagenesis and Population Structure
To create a population for De-TILLING, wild-type M. truncatula seeds were mutagenized by exposure to fast neutron radiation. The most effective mutagenic dose of fast neutron radiation was determined to provide a maximum number of deletions per line while retaining a practical level of plant survival and fertility. A 50% survival in the treated M1 plants represents a reasonable balance between mutagenesis and fertility. The segregation of albino phenotypes in the M2 progeny of mutagenized seed has also been used as an indicator of mutagenic rate. For a fast neutron-mutagenized population of Arabidopsis, an albino frequency of 2% has been equated with around 10 induced deletions per line (Koornneef et al., 1982). The extrapolation of this to other species must be made with caution. Since the effective fast neutron dose, usually expressed in Gray (Gy), varies markedly between species (Koornneef et al., 1982), and the genetic basis of this phenotype may vary. The effective dosage is also influenced by the water content of seeds and must be adjusted for each new batch. In our experience, the mutagenic effect of fast neutron bombardment, as measured by M1 survival and albino frequency, varies markedly between seed batches. These measures should therefore be made for treated seed batches for every new population. A pilot experiment indicated that a fast neutron dose of 32.5 Gy resulted in between 10% and 30% survival and represented a maximum usable dose. A later experiment assessed mutagenic effect following replicated fast neutron doses of 32.5 Gy. M1 survival varied between 35% and 77% (Supplemental Data S1). The M1 survival for the seed batches selected for the fast neutron-mutagenized population varied between 21% and 50%. An assessment of 1440 M1 lines (288 families) revealed an albino frequency of 2.57% in the M2 progeny (Supplemental Data S1). A more thorough analysis of the mutagenic effect of fast neutron bombardment could be made through analysis of tiling arrays, which would enable us to describe both the size distribution and number of induced deletions per line. However, this has not been carried out for this study.
The M1 seeds were grown to maturity in groups of five plants in a single container and seed pooled from each container. DNA was isolated from 25 seedlings representing each pool, and this DNA was normalized in a 96-well format. A stack of five 96-well plates was considered a tower and represented 12,000 M2 seedlings from 2,400 M1 plants. Thirteen towers were produced to give a total population of 156,000 M2 plants, derived from an original population of 31,200 mutagenized M1 plants. Each tower was pooled in rows, columns, and plates to create 25 3D pools per tower (Fig. 3).
Figure 3.

Structure of a De-TILLING tower. Each tower consists of 480 pools, each of which is genomic DNA of 25 seedlings taken from the pooled M2 progeny of five mutagenized M1 plants. Each tower is initially pooled into 25 row, column, and plate pools. These are used to create a pair of reciprocal HTPs. The De-TILLING population is initially screened using the 54 HTP representing the 13 towers twice. When a deletion mutant is detected, 25 3D pools are screened to locate the mutant to an individual well. The mutant is then recovered from the identified pool.
A problem intrinsic to any PCR-based screening strategy is the generation of false positives due to production of spurious PCR products. In many circumstances, sequencing the spurious product would be sufficient to separate genuine from spurious products. Characterization of 31 spurious products produced using the De-TILLING screening strategy (e.g. Fig. 4B) demonstrated that these products invariably originated from the target sequence and in addition were structurally identical to deletion alleles. These possessed deletions ranging from 249 bp to 1.7 kb with an average internal deletion size of 1,261 bp representing 55% of the amplified region. These amplicons were not reproduced in subsequent PCR reactions. This phenomenon was also noted in C. elegans deletion detection platforms by Jansen et al. (1997) and Liu et al. (1999). Lui noted a similar number of false positive amplicons of this type using unmutagenized genomic DNA and suggested they may arise from polymerase slippage across gaps formed by secondary loops in the DNA template. To address this problem, the 3D pools were finally combined to create four reciprocal half tower pools (HTPs) for each tower (Fig. 3). The entire population consists of 54 HTPs in which each of the 13 towers is represented twice. Detection of a mutation therefore results in an identical pair of PCR fragments from two of the HTPs. When a mutant is detected within the HTPs, both PCR amplicons are sequenced to validate and characterize the deletion allele. Subsequent PCR screening of 25 3D pools for that tower allows the identification of a single seed packet from which the mutant originated. This packet of seeds can then be screened for individual plants harboring the characterized deletion.
Figure 4.

Recovering a mutant for a LysM receptor-like kinase. A, Assays were designed around five restriction sites unique within 2- to 2.3-kb regions. The StyI assay (red) detected a 422-bp deletion within the population. B, Two identical PCR products indicate the presence of a deletion allele within tower 4. Note the spurious PCR products in towers 6 and 7 that are not real detection events. C, Amplification from the 3D pools of tower 4 locates a single M2 pool containing the mutant. D, PCR screening of 29 seedlings grown from this pools allow the lysM1-1 mutant to be recovered.
Detecting Novel Deletion Mutants in the Population
We searched for deletions in a LysM receptor-like kinase. Five restriction sites, unique within regions of approximately 2.4 kb, were identified within and closely adjacent to the 1.9-kb coding region (Fig. 4A). De-TILLING assays were designed centered upon each restriction site, resulting in nested PCR amplicon sizes of 2.0 to 2.3 kb. A single deletion allele was detected possessing a 422-bp deletion (Fig. 4B). This was uniquely identified by the StyI-based assay as none of the other targeted restriction sites were removed by the deletion. Following detection, the pool from which the mutation originated was identified by screening the 3D pools of tower 4 (Fig. 4C). We then recovered the mutant from a screen of 29 M2 plants from the identified pool (Fig. 4D). Despite the deletion only removing 18.1% of this 2.3-kb nested product, we were able to detect this mutant using the De-TILLING method.
To demonstrate the utility of the De-TILLING strategy for recovering mutations in small genes, we targeted the transcription factor EFD (for ethylene response factor required for nodule differentiation; Vernie et al., 2008) initially identified as being up-regulated in M. truncatula nodules using microarray analyses (El Yahyaoui et al., 2004) and suppression subtractive hybridization approaches (Godiard et al., 2007). The coding region of this gene consisted of two exons of 85 and 503 bp. Because no sequence downstream of the second exon was available, we were limited to designing targets around the first exon giving us a very small target of only 85 bp. To increase the number of De-TILLING assays we could design, we searched for restriction sites up to 250 bp outside the coding region. We identified three targetable restriction sites around which De-TILLING assays of 2.1, 2.4, and 2.9 kb were designed. Screening the population revealed a mutant in tower 4 for the EcoRI-based assay (Fig. 5A). Sequencing of these amplicons revealed a 1,570-bp deletion entirely removing the exon 1 and the 3' end of exon 2. This deletion completely abolishes the production of the EFD transcript, as verified by quantitative reverse transcription PCR analysis (Vernie et al., 2008). Following screening of the 3D pools, we were able to recover two mutant plants from 50 seeds taken from the identified family, one heterozygous and one homozygous for the efd1-1 mutation.
Figure 5.

Detection of the efd-1 mutant. A, Identical PCR products occurring in tower 4 reveal the presence of efd-1, an ERF transcription factor mutant possessing a 1,571-bp deletion. B, Amplification from the 3D pools reveals the row, column, and plate location of the efd-1 mutant containing M2 pool within tower 4.
We attempted to recover deletions in 14 genes using a minimum of five well-distributed targets per locus. Target sizes ranged from 1.2 to 3.2 kb with an average target amplicon size of 2,335 bp. The sizes of deletions that we detected using the De-TILLING method were 422, 1,270, 1,570, and 1,723 bp with target amplicons sizes of 2.3 kb (18.2% deletion), 1.9 kb (67.9%), 2.9 kb (54.1%), and 2.6 kb (67.5%), respectively (Table I). All mutations completely removed the targeted restriction site and the poison primer binding site.
Table I.
Loci targeted by De-TILLING indicating the range of target sizes and the size of deletions identified
| Gene Name | Exons (Length, bp) | Assays | Deletions (% of Amplicon) |
|---|---|---|---|
| ERF1 | 1 (83) | 3 | 1,570 (54%) |
| HAP2-1 | 6 (1,599) | 6 | – |
| HK4 | 1 (3,366) | 5 | 1,723 (68%) |
| LysMorf | 1 (1,914) | 5 | 422 (18%) |
| MADS1 | 4 (725) | 10 | – |
| COI1-1 | 3 (1,806) | 5 | – |
| Cyc | 11 (1,334) | 6 | – |
| B3 | 11 (946) | 7 | – |
| CLE13 | 1 (255) | 5 | – |
| Della1 | 1 (1,785) | 5 | – |
| ENOD40-1 | 1 (720) | 7 | – |
| MCA8 | 8 (3,246) | 7 | 1,270 (68%) |
| GRAS1 | 2 (2,161) | 6 | – |
| GRAS2 |
1 (1,515) |
5 |
– |
DISCUSSION
Here, we describe a combination of detection strategies that greatly enhances the utility of deletion detection in mutant populations to that previously described. De-TILLING can be used to recover deletion mutants for the majority of plant species and offers several advantages over conventional TILLING. A standard TILLING population of 4,000 lines requires amplification, CelI digestion, and analysis of fluorescently labeled PCR products for 500 samples. To reach acceptable levels of cost and efficiency, small, heavily mutagenized populations are essential. Mutants recovered from these populations will possess a very large number of nontarget mutations. For an Arabidopsis TILLING population, conservative estimates suggest the density of mutations in exons to be approximately three per megabase (Colbert et al., 2001). The impact of this is around 20 to 25 profoundly affected genes per EMS-mutagenized genome (Henikoff and Comai, 2003). This can affect the viability of mutants and confound and delay downstream analyses. To achieve saturation, fast neutron-mutagenized populations need to be larger than those created using EMS; however, the mutants recovered will be knockouts and will possess far fewer nontarget mutations. The large population sizes in De-TILLING are offset by the high level of pooling employed, which allows great efficiencies of time and cost in comparison to the standard 8-fold pooling of TILLING.
The size of the population of plants needed to saturate a genome depends firstly on the rate at which loci are deleted from the genome and secondly on the number of deletions that can be detected using the screening method. For Arabidopsis lines exposed to fast neutron radiation at a standard dose of 60 Gy, Koornneef et al. (1982) estimated an average of approximately 2,500 lines are required to inactivate a gene once. With around 27,000 coding sequences in the Arabidopsis genome (TAIR8 release 2008), this implies that around 10 to 11 genes are randomly deleted per line. The albino rate measured for this Arabidopsis population (approximately 2%) is similar to that measured for the M. truncatula De-TILLING population (2.57%). This suggests a similar number of deleted loci per line in the M. truncatula population. However, we must make this comparison with caution, as the basis of the albino phenotype in M. truncatula is not well defined.
PCR-based methods do not recover deletions of all sizes. Only a subset of the induced deletions will be detected by any screening method. Deletions must be small enough to be flanked by the nested PCR primers and large enough to produce mutant amplicons whose amplification can be separated from the wild type. Our screening was carried out using an average target size of 2.3 kb and ranged from 1.2 to 3.2 kb. Detected deletions removed from 18% to 68% of the target region, although we were able to model detection of a 14% deletion using the nsp2-1 mutant. We can therefore estimate that we would be targeting deletions within the range of 0.2 to 2.2 kb using this method. The estimate of 10 deleted coding regions per line would include many deletions outside this range. While limiting detection to small deletions is highly desirable, this increases the population size needed to achieve saturation. The number of lines (n) needed to increase the probability of recovering a mutant (F) to any level is related to the observed frequency of detectable deletions (P) through the formula:
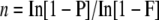 |
Screening 13 towers (31,200 M1 plants) enabled us to recover mutants in four out of the 14 genes we targeted. A population of 125,000 M1 would therefore give an 80% probability of recovery. Given that the relationship of diminishing returns exists for any reverse genetics screening platform, a combination of approaches will always be the most effective strategy. Increasing the size range of detectable deletions to 4 kb may improve the recovery of mutants. Extending this strategy to include large deletions to increase recovery would, however, lead to the problems highlighted by Li et al. (2001) of routinely disrupting more than one gene. While deletion of single genes is required in the majority of cases, such an approach can be advantageous for removing tandemly duplicated genes, which is discussed below.
Recovering a deletion removing an 85-bp exon of the EFD transcription factor demonstrated the utility of De-TILLING for the targeting of small genes. Mutagenizing small genes is problematic for reverse genetic strategies based on insertional and point mutation inducing mutagens. The probability of identifying an insertion is dependent upon the size and structure of the targeted gene. The probability of finding a mutant possessing an insertion in a particular Arabidopsis gene can be calculated using the formula:
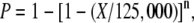 |
where P is the probability of recovering the desired mutant, X is the size of the gene in kilobase pairs, 125,000 is the approximate size in kilobases of the Arabidopsis genome, and n is the number of inserts in the mutant library (Krysan et al., 1999). Therefore, for an Arabidopsis collection containing 100,000 insertions, there is only a 55% probability of identifying an insertion mutant for a 1-kb target. For a similar Medicago collection, with a genome size of 400 Mb, the probability is approximately 22%. Using deletion mutagenesis, the probability of recovering a mutant is entirely independent of the size of the target sequence. It is far easier to hit a small target with a large deletion than an insertion or point mutation. The structure of the gene is also significant. Genes possessing small exons and large introns will be more difficult to mutagenize using TILLING or insertional techniques. Insertions are unlikely to be found within small exons, and those that fall within introns or intergenic regions are likely to have no effect on protein function. TILLING relies on the identification of approximately 1-kb regions with a high probability of introducing deleterious point mutations. Where a gene is structured as small exons and large introns, it is difficult to identify useful target regions for TILLING. Deletion-based reverse genetic platforms do not carry these limitations.
Deletion-based reverse genetic systems have the ability to inactivate multiple genes. Plant genomes are highly redundant, and it is estimated that <10% of the genes tagged in Arabidopsis are likely to generate a phenotypic change (Meinke et al., 2003). For a mutant phenotype to become apparent, it is sometimes necessary for multiple members of a gene family to be inactivated. For unlinked loci, it is possible to stack insertions or point mutations within a single line to investigate gene function. However, where homologous genes are present in tightly linked tandem arrays, recombination becomes extremely improbable and therefore difficult to achieve. In general, over 15% of the identified genes in sequenced plant genomes are members of tandem-arrayed gene families (Jander and Barth, 2007). This is slightly higher in Arabidopsis, where about 4,000 genes are tandemly repeated as two or more copies (Arabidopsis Genome Initiative, 2000). In Arabidopsis, the recombination rate is estimated to be around 200 kb/centimorgan. For two mutations in Arabidopsis separated by 5 kb, a homozygous double mutant would be recovered only once in every 64 million F2 progeny (Jander and Barth, 2007). Alternative strategies for recovering double mutants of tandemly homologous genes are therefore very attractive. Deletions introduced by fast neutron mutagenesis, unlike point mutations and DNA insertions, have the capacity to remove multiple adjacent genes; however the delete-a-gene detection procedures are more suited to this than the De-TILLING methods described here.
In addition to its use as a research tool, deletion mutagenesis has the potential to find application in crop improvement programs. The use of fast neutron mutagenesis is applicable to any plant species. It is conducted on large batches of dry seed at very low cost and is therefore ideally suited to applications in crop improvement. Fast neutron-mutagenized lines with lower levels of nontarget mutations and an absence of any foreign DNA sequences may be more acceptable to consumers concerned with the perceived dangers of genetic modification.
In comparison with the well-established TILLING method, De-TILLING can be used to isolate mutants at a fraction of the time and cost. Fast neutron mutagenesis generates complete knockout mutants that do not possess the very high number of background mutations that are typical for TILLING mutants. De-TILLING can also address the problems of targeting small genes, a problem that is intrinsic to all methods based on insertion and point mutation. As the cost of sequencing continues to fall, the low cost, scalability, and technical simplicity of De-TILLING has the potential to become a valuable tool in a wide variety of plant species.
MATERIALS AND METHODS
Fast Neutron Population
Wild-type Medicago truncatula seed (A17, Jemalong) was exposed to fast neutron radiation at a dose of 32.5 to 35 Gy at the Atomic Energy Research Institute (Budapest). The rate of mutagenesis was assessed by M1 survival (37–48%) and albino rate (2.57%) as calculated as a percentage of M1 treated lines displaying albino phenotypes in the M2 plants. Five M1 plants were grown in soil in a single pot and the seed pods collected in a single pool. These were mechanically threshed and the seeds archived at 4°C. Twenty-five seeds from each M2 family were germinated. The seedlings were freeze dried and the tissue ground.
DNA Extraction and Pooling
A 30-mg sample of the tissue from each M2 family was aliquoted for DNA extraction. Extractions were carried out in 96-well format using the DNeasy 96 plant kit (Qiagen) and eluted in 200 μL. DNA was spectrophotometrically quantified and normalized to 50 ng μL−1. The population was structured into towers consisting of five 96-well plates. Samples from each row, column, and plate of each tower were combined to create a 3D pooling structure. The 3D pools were then combined to create four HTPs per tower, which were directly screened using the De-TILLING method. HTPs from each tower were digested with a range of restriction enzymes and stored at −20°C.
Automated De-TILLING Assay Design
A PERL script, known as MtMutDetect.pl, was designed to align a coding and a genomic sequence and, using a user-modifiable list of enzymes, identify restriction sites within and adjacent to exon sequences unique within PCR amplicons of a defined size range. The program then performs automated primer design and returns a five-primer De-TILLING assay centered upon the targeted restriction sites. Poison primers were designed within 30 bp of the targeted restriction site. Parameters can be set to determine the amplicon size, the number of enzymes, and the amount of intron sequence to be included in the identification of targetable restriction sites.
PCR Conditions
Nested PCR reactions were performed in a total of 50 μL using Taq Polymerase Master Mix (Qiagen), 240 ng of digested genomic HTP DNA, and 10 pmols of each primer. PCR was carried out using an MJ Research tetrad PTC-225 peltier thermocyler over 40 cycles of 30 s at 94°C, 30 s at 55°C, and 2 min and 30 s at 74°C.
dmi1-4 and nsp2-1 Detection Reconstruction Experiments
Genomic DNAs of A17, dmi1-4, and nsp2-1 were prepared, quantified, and normalized as described above. These were pooled at mutant to wild-type ratios of 1:25, 1:1,000, 1:4,000, 1:8,000, 1:12,000, 1:16,000, 1:20,000, and 1:24,000. Six microliters of each pool (240 ng) was used as template in all amplifications. The following primers were used to amplify dmi1-4 deletion borders: dmi1-4-F (5'-TCTTCTTAATTTCATGTGCATAATTGTCG-3'), dmi1-4 (0.3kb)-R1 (5'-TCAATTTGATGGTGCATAATAGCA-3'), dmi1-4 (0.3kb)-R2 (5'-AGGCAGTAATATGGAATGGACA-3'), dmi1-4 (3kb)-R1 (5'-TCTTCTGTCATTCACCTGAGGCT-3'), dmi1-4 (3kb)-R2 (5'-GTTGATAATAGCACGCTCTTGGT-3'), dmi1-4 (8kb)-R1 (5'-CTGTCACCATTTCTGTATGTGCT-3'), and dmi1-4 (8kb)-R2 (5'-TCTGTATGTGCTGCGATTTTCAC-3'). F and R1 primers were used in the first round PCR, and F and R2 were used in the second round PCR.
A De-TILLING assay was designed to detect the nsp2-1 deletion (Fig. 2B). This consisted of two external primer L7 (5'-TTGCATTCACATCAGGTAGGA-3') and R7 (5'-GAGCAATTTGAACCTCTCACG-3'), a poison primer L7B (5'-AATCAAGCCATCATCGAAGC-3') and nested primers L5 (5'-TGACAACAGCGCACATAACA-3') and R5 (5'-AAACCAAAACGCACACACAA-3'; Fig. 2). Amplifications were carried out identical to those described below for De-TILLING screening.
De-TILLING Screening and Sequencing Analysis
The first-round PCR was analyzed using an e-Gel (Invitrogen) to check for the production of the suppressor fragment. Second-round PCR was identical but scaled to 20 μL, using 2 μL of a 10−2 dilution of the first round PCR products as template and nested primers. The second round products were then analyzed on a 1.2% TBE agarose gel and putative deletion containing fragments recovered using QIAquick Gel extraction kits (Qiagen). The products were then sequenced using the second-round primers and an ABI3730 automated sequencer. The sequences were then compared to the wild type to determine the deletion junction.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Data S1. M1 survival and M2 albino frequencies in the Medicago De-TILLING population.
Supplementary Material
Acknowledgments
We thank Prof. Virginia Walbot for helpful discussions; Paul Bailey for compiling the script of the MtMutDetect.pl De-TILLING assay design software; Joe Palfalvi for providing fast neutron irradiation and useful discussions; and Jonathan Clarke, David Baker, and Bethany McCullagh for providing DNA extraction and sequencing services. We also thank Katy Owen and Gemma Lynes for their extensive contribution to the daily laboratory work in establishing the De-TILLING seed and DNA archives along with glasshouse assistants Ruth Pothecary, Emma Thompson, Paul Ward, Kate Bowdrey, Catherine French, Megan Murray, Richard Birkinshaw, Clare Harden, and Lucy Foulston.
This work was supported by the European Union as part of the Grain Legume Integrated Project, by a grant in aid for the Biotechnology and Biological Sciences Research Council, by the Samuel Roberts Noble Foundation, and by the National Science Foundation (grant no. DBI0703285).
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Christian Rogers (christian.rogers@bbsrc.ac.uk).
The online version of this article contains Web-only data.
Open Access articles can be viewed online without a subscription.
References
- Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen HM, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R, et al (2003) Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301 653–657 [DOI] [PubMed] [Google Scholar]
- Ane JM, Kiss GB, Riely BK, Penmetsa RV, Oldroyd GED, Ayax C, Levy J, Debelle F, Baek JM, Kalo P, et al (2004) Medicago truncatula DMI1 required for bacterial and fungal symbioses in legumes. Science 303 1364–1367 [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408 796–815 [DOI] [PubMed] [Google Scholar]
- Azpiroz-Leehan R, Feldmann KA (1997) T-DNA insertion mutagenesis in Arabidopsis: going back and forth. Trends Genet 13 152–156 [DOI] [PubMed] [Google Scholar]
- Bruggemann E, Handwerger K, Essex C, Storz G (1996) Analysis of fast neutron-generated mutants at the Arabidopsis thaliana HY4 locus. Plant J 10 755–760 [DOI] [PubMed] [Google Scholar]
- Colbert T, Till BJ, Tompa R, Reynolds S, Steine MN, Yeung AT, McCallum CM, Comai L, Henikoff S (2001) High-throughput screening for induced point mutations. Plant Physiol 126 480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgley M, D'Souza A, Moulder G, McKay S, Shen B, Gilchrist E, Moerman D, Barstead R (2002) Improved detection of small deletions in complex pools of DNA. Nucleic Acids Res 30 e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yahyaoui F, Küster H, Ben Amor B, Hohnjec N, Puhler A, Becker A, Gouzy J, Vernie T, Gough C, Niebel A, et al (2004) Expression profiling in Medicago truncatula identifies more than 750 genes differentially expressed during nodulation, including many potential regulators of the symbiotic program. Plant Physiol 136 3159–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godiard L, Niebel A, Micheli F, Gouzy J, Ott T, Gamas P (2007) Identification of new potential regulators of the Medicago truncatula-Sinorhizobium meliloti symbiosis using a large-scale suppression subtractive hybridization approach. Mol Plant Microbe Interact 20 321–332 [DOI] [PubMed] [Google Scholar]
- Haas BJ, Wortman JR, Ronning CM, Hannick LI, Smith RK, Maiti R, Chan AP, Yu CH, Farzad M, Wu DY, et al (2005) Complete reannotation of the Arabidopsis genome: methods, tools, protocols and the final release. BMC Biol 3 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Comai L (2003) Single-nucleotide mutations for plant functional genomics. Annu Rev Plant Biol 54 375–401 [DOI] [PubMed] [Google Scholar]
- Jander G, Barth C (2007) Tandem gene arrays: a challenge for functional genomics. Trends Plant Sci 12 203–210 [DOI] [PubMed] [Google Scholar]
- Jansen G, Hazendonk E, Thijssen KL, Plasterk RHA (1997) Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nat Genet 17 119–121 [DOI] [PubMed] [Google Scholar]
- Koornneef M, Dellaert LWM, Vanderveen JH (1982) EMS-induced and radiation-induced mutation frequencies at individual loci in Arabidopsis thaliana (L.) Heynh. Mutat Res 93 109–123 [DOI] [PubMed] [Google Scholar]
- Krysan PJ, Young JC, Sussman MR (1999) T-DNA as an insertional mutagen in Arabidopsis. Plant Cell 11 2283–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lassner M, Zhang YL (2002) Deleteagene: a fast neutron deletion mutagenesis-based gene knockout system for plants. Comp Funct Genomics 3 158–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Song YJ, Century K, Straight S, Ronald P, Dong XN, Lassner M, Zhang YL (2001) A fast neutron deletion mutagenesis-based reverse genetics system for plants. Plant J 27 235–242 [DOI] [PubMed] [Google Scholar]
- Liu LX, Spoerke JM, Mulligan EL, Chen J, Reardon B, Westlund B, Sun L, Abel K, Armstrong B, Hardiman G, et al (1999) High-throughput isolation of Caenorhabditis elegans deletion mutants. Genome Res 9 859–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martienssen RA (1998) Functional genomics: probing plant gene function and expression with transposons. Proc Natl Acad Sci USA 95 2021–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum CM, Comai L, Greene EA, Henikoff S (2000. a) Targeted screening for induced mutations. Nat Biotechnol 18 455–457 [DOI] [PubMed] [Google Scholar]
- McCallum CM, Comai L, Greene EA, Henikoff S (2000. b) Targeting induced local lesions in genomes (TILLING) for plant functional genomics. Plant Physiol 123 439–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinke DW, Meinke LK, Showalter TC, Schissel AM, Mueller LA, Tzafrir I (2003) A sequence-based map of Arabidopsis genes with mutant phenotypes. Plant Physiol 131 409–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldroyd GED, Long SR (2003) Identification and characterization of nodulation-signaling pathway 2, a gene of Medicago truncatula involved in Nod factor signaling. Plant Physiol 131 1027–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parinov S, Sevugan M, Ye D, Yang WC, Kumaran M, Sundaresan V (1999) Analysis of flanking sequences from dissociation insertion lines: a database for reverse genetics in Arabidopsis. Plant Cell 11 2263–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parinov S, Sundaresan V (2000) Functional genomics in Arabidopsis: large-scale insertional mutagenesis complements the genome sequencing project. Curr Opin Biotechnol 11 157–161 [DOI] [PubMed] [Google Scholar]
- Roberts MA, Reader SM, Dalgliesh C, Miller TE, Foote TN, Fish LJ, Snape JW, Moore G (1999) Induction and characterization of Ph1 wheat mutants. Genetics 153 1909–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessions A, Burke E, Presting G, Aux G, McElver J, Patton D, Dietrich B, Ho P, Bacwaden J, Ko C, et al (2002) A high-throughput Arabidopsis reverse genetics system. Plant Cell 14 2985–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speulman E, Metz PLJ, van Arkel G, Hekkert PTL, Stiekema WJ, Pereira A (1999) A two-component Enhancer-Inhibitor transposon mutagenesis system for functional analysis of the Arabidopsis genome. Plant Cell 11 1853–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman MR, Amasino RM, Young JC, Krysan PJ, Austin-Phillips S (2000) The Arabidopsis knockout facility at the University of Wisconsin-Madison. Plant Physiol 124 1465–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadege M, Wen JQ, He J, Tu HD, Kwak Y, Eschstruth A, Cayrel A, Endre G, Zhao PX, Chabaud M, et al (2008) Large-scale insertional mutagenesis using the Tnt1 retrotransposon in the model legume Medicago truncatula. Plant J 54 335–347 [DOI] [PubMed] [Google Scholar]
- Vernie T, Moreau S, de Billy F, Plet J, Combier JP, Rogers C, Oldroyd G, Frugier F, Niebel A, Gamas P (2008) EFD is an ERF transcription factor involved in the control of nodule number and differentiation in Medicago truncatula. Plant Cell 20 2696–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Lim J, Dale JM, Chen HM, Shinn P, Palm CJ, Southwick AM, Wu HC, Kim C, Nguyen M, et al (2003) Empirical analysis of transcriptional activity in the Arabidopsis genome. Science 302 842–846 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.