

Abstract
Increased levels of reactive oxygen species (ROS) by hyperglycemia can induce apoptosis of renal cells and diabetic nephropathy. The redox balance in the renal cell seems, therefore, of the utmost importance. ROS-mediated apoptosis may be further aggravated by an inadequate cytoprotective response against ROS. When there are insufficient cytoprotective and ROS scavenging molecules, ROS lead to considerable cellular damage and to a point of no return in apoptosis. Induction of cytoprotective proteins may prevent or attenuate apoptosis, renal cell injury, and finally diabetic nephropathy. Here, we discuss some mechanisms of apoptosis and several strategies that have been probed to ameliorate, or to prevent apoptosis in the diabetic kidney.
Keywords: Diabetes, Apoptosis, Kidney, Diabetic nephropathy, Cytoprotective proteins, Anti-oxidants
Introduction
Diabetes mellitus forms an enormous and still increasing health burden worldwide [1]. Mortality and morbidity are caused in particular by the occurrence of complications [2]. One of the microvascular complications is diabetic nephropathy in both T1DM and T2DM, which is the leading cause of adult renal failure at the moment [3]. A key role in the pathogenesis of diabetic nephropathy has been attributed to hyperglycemia as it leads to the formation of advanced glycation end products (AGEs) and the mitochondrial production of reactive oxygen species (ROS), and, subsequently, to cell death and kidney dysfunction [4]. In this review, we will focus on the pathogenic role of oxidative stress and apoptosis in diabetic nephropathy. Potential strategies to prevent or treat the renal damage inflicted by ROS-induced apoptosis are discussed.
Diabetic nephropathy
Diabetic nephropathy can be characterized by both glomerular and tubulointerstitial injury, each, however, to a hugely variable extent [5]. An important histological hallmark of diabetic nephropathy is proliferation of mesangial cells, and, as a result, the expansion of extracellular matrix (ECM) in the mesangium [6]. Thickening of the glomerular basement membrane (GBM) is another feature of diabetic nephropathy [7]. One of the clinical manifestations and main predictor of diabetic nephropathy is emerging microalbuminuria in the early phase of the disease that further develops to massive macroalbuminuria during overt diabetic nephropathy and end stage renal disease. Damage to the capillary filter, existing of podocytes, GBM and glomerular endothelium, may be responsible for the development of albuminuria. We have recently shown that heparanase-mediated loss of heparan sulfate in the GBM is a possible key mediator in the development of proteinuria and overt diabetic nephropathy. However, we could also observe changes in tubular heparan sulfate [8–12]. Notably, an aberrant expression of heparanase and heparan sulfate in certain cancer cells has been implicated to play a role in apoptosis [13, 14]. However, the possible role of renal heparanase expression in apoptosis during the development of diabetic nephropathy remains to be addressed. Historically, mesangial cells have been attributed a central role in glomerular-associated diabetic nephropathy. However, also other glomerular cell types, like endothelial cells, and podocytes have been implicated in the genesis of diabetic nephropathy, while inflammatory cells such as macrophages can further exacerbate the oxidative stress and inflammatory processes leading to apoptosis.
Alternatively, renal deterioration may also be related to chronic hypoxia, that can for example occur following vasoconstriction by decreased levels of nitric oxide (NO) or increased levels of angiotensin II [15]. The occurrence of interstitial fibrosis may additionally deteriorate the diffusion of oxygen to the renal cells. During hypoxia ROS formation can occur via increased electron flow at complex III of the mitochondrial electron transport chain and via NADPH oxidase [16, 17]. Experimental research has shown that hyperglycemia interferes with the beneficial VEGF response to hypoxia in proximal tubular cells [18]. As a result, these and numerous other factors may cause hypoxia, the formation of ROS, cellular injury and finally apoptosis of renal cells. In summary, both the glomerular and the tubular compartment may be involved in the initiation of diabetic nephropathy.
Redox balance in the kidney
Within renal cells a balance between pro- and anti-oxidants is kept under physiological conditions. Although ROS have important functions in normal cellular signaling and regulation, in excess, they may result in oxidative damage to proteins, lipids and DNA, ultimately leading to apoptosis and renal injury [19]. ROS can activate several pro-inflammatory transcription factors, resulting in the production of cytokines and chemokines and vascular adhesion molecules, and, subsequently, the influx of inflammatory cells into the kidney. This formation of renal inflammation initiated by ROS further prolongs and exacerbates ROS-mediated cell injury, apoptosis and kidney dysfunction. To keep the redox balance under control a complex network of pro- and anti-oxidant enzymes exists. However, it can be envisioned that in case the cytoprotective anti-oxidant factors, as exemplified in Fig. 1, get overwhelmed as may occur following hyperglycemia, the balance gets skewed towards oxidative stress, and may lead to apoptosis and kidney injury.
Fig. 1.
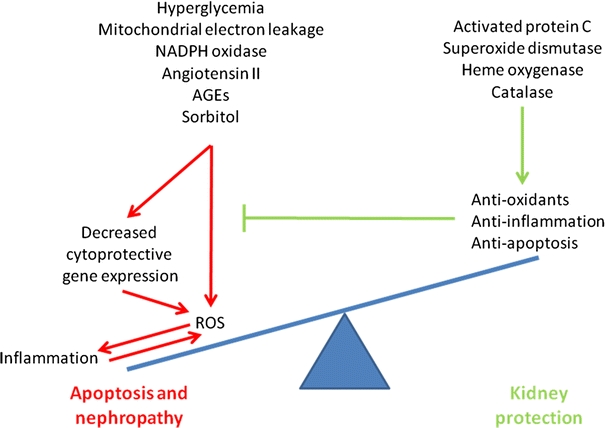
Excess ROS leads to apoptosis and diabetic nephropathy. Model demonstrating the different causes of ROS formation that result in the induction of inflammation by pro-inflammatory genes, leukocyte infiltration, and aggravation of ROS formation. Ultimately, this may lead to apoptosis and kidney dysfunction. We postulate that in humans induction of cytoprotective proteins, such as HO-1, SOD or catalase, will restore this ROS-mediated skewing of the redox balance by generating products that protect against oxidative stress, and/or inflammation and apoptosis, or a combination
Mitochondria-derived ROS constitute the main source of intracellular ROS that are responsible for apoptosis and diabetic nephropathy [20, 21]. As described above, both the glomeruli and the tubulointerstitial compartments can be subjected to the hyperglycemia-induced increase in mitochondrial ROS production. During periods of hyperglycemia, renal cells fail to attenuate the entrance of glucose into the cell, resulting in such increased substrate levels for the mitochondrial electron transport chain that this leads to an overflow of the respiratory chain. Normally, a small amount of ROS are formed as a result of electron carriers reacting directly with oxygen, but during this overflow backed up electrons result in even more reactive electron carriers and thus enhanced production of ROS. It has been demonstrated that high glucose levels result in increased levels of ROS, AGEs and sorbitol, and higher activity of protein kinase C that have been demonstrated to contribute to cellular injury and apoptosis [22].
Other strong contributors to ROS formation in the diabetic kidney are the superoxide producing enzyme NADPH oxidase (NOX), and xanthine oxidase, which catalyzes oxidation of hypoxanthine (the occurrence of different NOX isoforms in different cells of the diabetic kidney is reviewed in [4]). Nitric oxide synthase produces the free radical nitric oxide, which, under normal conditions acts protective, but under oxidative circumstances, can be converted into other reactive nitrogen species (RNS), including peroxynitrite [23].
On the other hand, there are intrinsic renal cytoprotective anti-oxidant factors and protein responses that keep the ROS and RNS in balance. The cytoprotective anti-oxidant proteins include for example superoxide dismutases (SOD) that convert superoxide to hydrogen peroxide, which, in turn, is rendered inactive by catalase and glutathione peroxidase. The heme oxygenase (HO) system is another crucial anti-oxidant system in the kidney [24, 25]. HO degrades the pro-oxidant heme into iron, carbon monoxide and biliverdin [23]. Biliverdin is than directly converted into the strong anti-oxidant bilirubin by biliverdin reductase. HO-derived iron can, like heme, catalyze the Fenton reaction resulting in the production of reactive hydroxyl groups, but is scavenged by co-induced ferritin [23]. The strongly inducible HO-1 isoform has been shown to protect against oxidative stress, inflammation and apoptosis in the kidney [23, 26–28], and also the constitutively expressed HO-2 isoform is getting recognized for its protection against oxidative stress and inflammatory processes [29]. In addition to an increased formation of ROS, a decreased anti-oxidant capacity results in skewing of the redox balance. It is likely that in diabetes patients a combination of increased ROS formation and diminished anti-oxidant defense results in apoptosis of renal cells. This is exemplified by the findings of Quan et al., who demonstrated that hyperglycemia decreases HO-activity in the aortic endothelium and increases the production of ROS [30], suggesting that patients with hyperglycemia are deprived from the protective effects of the HO-effector molecules CO, bilirubin and ferritin. Moreover, hyperglycemia was shown to decrease the protective effect of the HO-system in the vascular system of animal models [30, 31], resulting in an impaired defense against oxidative stress and inflammation [30, 32]. In addition, it has been shown that HO-1 and the cytoprotective heat shock protein 72 expression is decreased in skeletal muscles of patients with type 2 diabetes (T2DM) [33]. In contrast, Hayashi demonstrated that during streptozotocin (STZ)-induced diabetes in rats, which is an established model for mainly T1DM, HO-1 levels were up-regulated in glomeruli, whereas the expression of other cytoprotective enzymes such as catalase, GSH peroxidase and Cu-, Zn- superoxide dismutase were unchanged compared to control rats [34, 35]. Several studies have shown a protective role of HO-1 against oxidants in the kidney, as reviewed in [36, 37]. In summary, when hyperglycemia leads to the sudden production of ROS, and the anti-oxidant capacity is not sufficient, induction of apoptosis leads to renal injury and diabetic nephropathy.
Mechanisms of apoptosis
The apoptotic process is regulated by certain proteins, the so-called cysteine-dependent specific aspartate proteases or caspases. Caspases have a role in the induction of apoptosis as well as in the execution phase of apoptosis, and play a central role in diabetic nephropathy. Caspase-dependent apoptosis can be induced by either the extrinsic or the intrinsic pathway.
The extrinsic pathway involves a death-inducing ligand such as for example Fas ligand (FasL) or TNFα interacting with their corresponding death receptor on the plasma membrane, for this example Fas receptor and TNF receptor, respectively. After binding of these ligands to the death receptors Fas-associated death domain (FADD) or TNF-receptor-associated death domain (TRADD), caspase-8 is recruited and activated to form a death-inducing signaling complex (DISC). DISC can propagate the death signal directly by activating the effector caspases-3, -6 and -7 [38, 39], or indirectly via cleavage of Bid, which upon translocation to the mitochondria leads to mitochondrial outer membrane permeabilization (MOMP) and subsequent release of pro-apoptotic proteins, such as cytochrome c [40]. Cytochrome c is part of the caspase-dependent intrinsic pathway discussed below and can interact with apoptotic protease activating factor-1 (Apaf-1) and procaspase 9 to form an apoptosome. This complex is able to activate caspase 9, which in turn activates the effector caspases 3, 6 and 7, leading to apoptosis [41].
The second apoptotic pathway is the intrinsic pathway, in which the mitochondrion is the central regulator [42]. The point-of-no-return for the intrinsic pathway is MOMP, which is controlled by the family of Bcl-2 proteins [43]. Bcl-2 proteins are either anti-apoptotic (Bcl-2, Bcl-XL, Bcl-w) or pro-apoptotic (Bax, Bak, Bad, Bim) and the balance between these two groups ultimately determines cell survival or cell death. Especially the pro-apoptotic family member Bax seems important since it can oligomerize or form complexes with other proteins to form channels such as the permeability transition pore complex (PTPC) and mitochondrial apoptosis-induced channel (MAC) in the mitochondrial outer membrane, which can facilitate the release of pro-apoptotic molecules such as cytochrome c [44–46].
Diabetes and apoptosis in the kidney
As outlined above, both acute and chronic hyperglycemia leads to oxidative stress and this is the major trigger for tubular and glomerular cells to go into apoptosis as determined in animal models and in vitro cell culture systems [20, 22, 47]. High glucose levels cause ROS-dependent apoptosis of mesangial cells via Bax-mediated mitochondrial permeability and subsequent cytochrome c release [48]. In murine and human renal mesangial cells high glucose levels caused an increased Bax/Bcl-2 ratio, associated with cytochrome c release from mitochondria and subsequent the pro-apoptotic caspase-3 activation [49]. Enhanced levels of glucose generated NADPH oxidase-mediated and mitochondrial-mediated ROS formation that activated pro-apoptotic p38 MAPK and caspase-3 activation in podocytes in vitro, whereas inhibition of NADPH oxidase-induced ROS formation prevented podocyte apoptosis in vivo [50]. Inhibition of p38 MAPK inhibited the diabetes-mediated decrease in the anti-apoptotic Bcl-2 expression and activation of caspase-3 in mesangial cells [51].
In renal tubular epithelial cells high glucose-mediated oxidative stress induced an increased Bax protein expression, which was accompanied by a reduced Bcl-2 expression [52]. These data are in line with earlier findings that during diabetes gene expression of pro-apoptotic Bax was increased, whereas anti-apoptotic Bcl-2 and Bcl-XL expression was down-regulated [53]. Different caspases, in particular caspase-3 and -9 play a crucial role in high glucose-induced apoptosis of proximal tubular epithelial cells [54].
In a gene profiling study of the tubulointerstitium from T1DM and T2DM nephropathy biopsy specimens it was shown that two death receptors (osteoprotegerin and Fas), and the death ligand TRAIL were strongly upregulated [55]. Glomerular and proximal tubular expression of TRAIL was confirmed by immunohistochemistry and was shown to be higher in diabetic kidneys than controls.
It was further found that protein C (PC) modulates mitochondrial apoptosis in diabetic nephropathy [56]. In glomerular endothelial cells, a complex of thrombomodulin and thrombin activates PC, that subsequently activates the protease-activated receptor-1 (PAR-1) in the proximity of the endothelial PC-receptor (EPCR), resulting in cytoprotective signaling. Hyperglycemia interferes with thrombomodulin-dependent PC activation, resulting in less protection and even promotion of release of apoptosis-inducing factors like cytochrome c and Smac/Diablo leading to apoptosis of endothelial cells and podocytes [19, 56].
In summary, data on renal apoptosis in diabetic nephropathy are scarce and scattered, but both the intrinsic and extrinsic apoptotic pathway seem to be involved.
Anti-oxidants and cytoprotective proteins in the prevention of ROS-induced apoptosis of renal cells and diabetic nephropathy
Several studies have targeted oxidative and nitrosative stress, and thus apoptosis of renal cells caused by hyperglycemia, through administration of anti-oxidants or overexpression of cytoprotective proteins.
Anti-oxidant administration to animals has demonstrated protective effects against the development of diabetic nephropathy. In addition to endogenous anti-oxidant compounds like glutathione, thioredoxin, and biliverdin/bilirubin also dietary anti-oxidants, such as vitamin C and E and beta-carotene may restore the redox balance. The number of apoptotic proximal tubular epithelial cells, proteinuria, glomerular and tubulointerstitial sclerosis, and renal malondialdehyde, as index of oxidative stress, were significantly decreased after anti-oxidant treatment with vitamin C to T2DM rats when compared with untreated T2DM rats [57]. Like vitamin C, vitamin E normalized diabetes-induced renal dysfunction such as glomerular volume and TGF-beta production in STZ-induced diabetic rats [58]. High glucose-induced ROS formation and mesangial and proximal tubular epithelial cell apoptosis could be inhibited in in vitro models, using the anti-oxidant taurine [49, 52].
Unfortunately, translational studies to the administration of solely anti-oxidants did not yet result in potent protection against apoptosis in human diabetic nephropathy as reviewed in [4]. Possibly induction of endogenous cytoprotective proteins (see Table 1) may be more efficiently translated to the clinical situation than administered anti-oxidants. Endogenous cytoprotective proteins may be targeted to the kidney or generated in the kidney, and, therefore, function more efficiently than exogenous added anti-oxidants, since they act within the cell or cellular compartment where ROS are actually generated. Recently, it was shown that ROS production was reduced in a T1DM and T2DM mouse models with transgenic overexpression of the anti-oxidant enzyme catalase in proximal tubular cells [59, 60]. This resulted in reduced pro-apoptotic Bax and active caspase-3 levels. Overexpression of catalase in the proximal tubular cells protected diabetic mice from increased blood pressure, albuminuria, glomerulosclerosis, interstitial fibrosis and tubular apoptosis. These findings are in line with the observations that catalase deficiency in mice renders kidneys more prone to oxidative stress [61], while humans deficient in catalase are predisposed to cumulative oxidant damage leading to T1DM and T2DM [62].
Table 1.
Targeted cytoprotective genes and effects on renal cell apoptosis and nephropathy
| Targeted genes in diabetic animal models | Effects on diabetic renal cell apoptosis and nephropathy |
|---|---|
| Transgenic catalase mice | Reduced albuminuria, glomerulosclerosis, interstitial fibrosis and tubular apoptosis [59, 60] |
| Catalase-deficient mice | Kidneys are more prone to oxidative stress [61] |
| HO-1 gene transfer in mice or rats | Reduced apoptosis and kidney injury [63] |
| HO-2-deficient mice | Enhanced STZ-induced diabetic nephropathy, which could be restored by HO-1 gene transfer [64] |
| SOD transgenic mice | Protection against diabetic nephropathy [68] |
| Thioredoxin-1 transgenic mice | Diminished albuminuria and tubular apoptosis [67] |
| Peroxiredoxin-3 transgenic mice | Protection against hyperglycemia and glucose intolerance and reduced fibroblast apoptosis by reducing mitochondrial H2O2 production [69] |
| Glutathione peroxidase-1-deficient mice | No effects on T1DM-induced kidney injury [70] |
| Mice with a reduction of thrombo-modulin-dependent protein C activation | Aggravated glomerular apoptosis and diabetic nephropathy [19, 56] |
| Mice expressing a hyper-activatable protein C mutation | Protein C activation prevents glomerular apoptosis and diabetic nephropathy [19, 56] |
Pharmacological induction or gene transfer of HO-1 in diabetic rats reduced ROS formation and caused an increase in anti-oxidants biliverdin/bilirubin, which resulted in an improvement of vascular and renal function [63]. Induction of HO-1 led to increased levels of the anti-apoptotic phosphorylated AKT (pAkt) and BcL-XL. Additionally, HO-2 deficiency enhanced STZ-induced diabetic nephropathy, possibly via increased levels of superoxide anions [64]. Pharmacological up-regulation of HO-1 by heme or cobalt protoporphyrin in HO-2-deficient T1DM mice resulted in reduced superoxide anions and improved diabetic kidney morphology and function [64]. Selective overexpression of the cytoprotective enzyme HO-1 in the endothelial cell line HMEC-1 was shown to prevent hyperglycemia-mediated O2 − formation and thus blocked ROS-induced DNA damage and caspase activation [25]. This study indicates that endothelial cell survival after oxidative injury may be enhanced by increased HO-1 expression, thus blocking inflammation and apoptosis.
In isolated lymphocytes of patients affected by T2DM complicated with nephropathy evidence was found of systemic oxidative stress, and the observed induction of HO-1 and thioredoxin reductase-1 was thought to reflect a response in counteracting the intracellular pro-oxidant status [65].
In mice overexpressing SOD, diabetes-induced renal injury was also abrogated, most likely due to reduced superoxide–NO interactions [66–68]. SOD overexpression increased anti-apoptotic glomerular Wnt5a/beta-catenin signaling and abrogated diabetes-induced caspase-3 cleavage, DNA damage and subsequent mesangial apoptosis[68]. Overexpression of manganese (Mn)SOD that exclusively is expressed in mitochondria appeared strongly protective against glucose-induced ROS, AGEs and sorbitol formation in endothelial cells [22], corroborating the prominent role of mitochondria in ROS-induced renal cell apoptosis.
Transgenic mice overexpressing thioredoxin-1 were protected against diabetic nephropathy as shown by reduced albuminuria and tubular apoptosis [67]. Furthermore, markers of systemic and renal oxidative stress were attenuated in this model, again indicating a significant role for oxidative stress in the development of diabetic nephropathy. Transgenic mice overexpressing peroxiredoxin-3, a mitochondria-specific thioredoxin peroxidase, caused a reduction of mitochondrial-produced H2O2 and an increased resistance to stress-induced fibroblast cell death and apoptosis [69], whereas, surprisingly, glutathione peroxidase-1-deficient animals showed similar levels of diabetes-induced kidney injury as wild type controls [70]. Inhibition of mitochondrial NADPH-dependent isocitrate dehydrogenase activity led to an increase of apoptosis mediated by active caspase-3, PARP and a decreased Bcl-2 expression, whereas overexpression of the enzyme resulted in decreased apoptosis of HEK293 cells associated with a decreased expression in PARP and Bax [71]. Also in other studies it was shown that reduction of oxidative stress by anti-oxidant enzyme activity led to improved renal function after hyperglycemic injury because of a reduction in tubular cell apoptosis [72, 73].
Activated PC mediates protection against mitochondrial apoptosis in the glomerulus, while high glucose levels abrogate activation of PC and its down-stream protective pathways. Interestingly, it has recently been found that increasing activated PC during hyperglycemia avoids oxidative stress and restores activated PC-mediated cytoprotection, and prevents mitochondria-dependent apoptosis of endothelial cells and podocytes, and diabetic nephropathy in T1DM [19, 56].
In summary, anti-oxidants and cytoprotective proteins can counteract ROS-induced apoptosis of renal cells and the development of diabetic nephropathy (see also Table 1, Fig. 1).
Concluding remarks
In the diabetic kidney ROS are produced via different pathways. When cytoprotective and anti-oxidant capacity is insufficient, ROS will induce apoptosis, which may finally result in diabetic nephropathy. Therapeutic options may include restoration of the redox balance by anti-oxidant compounds such as vitamin C and E. Unfortunately, routine anti-oxidants have demonstrated minimal renoprotection in humans with diabetic nephropathy, despite many positive observations in animal models [4]. However, for the treatment or prevention of diabetic nephropathy, the induction of cytoprotective proteins with anti-oxidant activities, such as PC, catalase, SOD and HO-1, constitute a valid alternative strategy. In fact, manipulating cytoprotective proteins and their effector molecules seem to reduce apoptosis in the diabetic kidney, and may therefore be beneficial, as demonstrated in animal models for diabetic nephropathy. These observations warrant further translational approaches addressing cytoprotective proteins in humans with diabetic nephropathy.
Acknowledgment
D. Dekker, MSc is supported by the Dutch Diabetes Research Foundation (Grant No. 2006.00.055).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- AGE
Advanced glycation end products
- Apaf-1
Apoptotic protease activating factor-1
- Bcl2
B-cell lymphoma 2
- CO
Carbon monoxide
- DISC
Death-inducing signaling complex
- DNA
Deoxyribonucleic acid
- ECM
Extracellular matrix
- EPCR
Endothelial PC-receptor
- FADD
Fas-associated death domain
- GBM
Glomerular basement membrane
- GSH
Glutathione
- HO
Heme oxygenase
- MAC
Mitochondrial apoptosis-induced channel
- MAPK
Mitogen-activated protein kinases
- MOMP
Mitochondrial outer membrane permeabilization
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NO
Nitric oxide
- PAR-2
Protease-activated receptor-1
- PARP
Poly (ADP-ribose) polymerase
- PC
Protein C
- PTPC
Permeability transition pore complex
- ROS
Reactive oxygen species
- RNS
Reactive nitrogen species
- Smac
Second mitochondria-derived activator of caspases
- SOD
Superoxide dismutase
- STZ
Streptozotocin
- TNFα
Tumor necrosis factor-α
- TRADD
TNF-receptor-associated death domain
- TRAIL
TNF-related apoptosis-inducing ligand
- VEGF
Vascular endothelial growth factor
- Wnt
Wingless-type MMTV integration site family
Contributor Information
F. A. D. T. G. Wagener, Phone: +31-24-3616539, FAX: +31-24-3540339, Email: f.wagener@ncmls.ru.nl
J. van der Vlag, Phone: +31-24-3616539, FAX: +31-24-3540339, Email: j.vandervlag@nier.umcn.nl
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes—estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Morrish NJ, Wang SL, Stevens LK, Fuller JH, Keen H. Mortality and causes of death in the WHO multinational study of vascular disease in diabetes. Diabetologia. 2001;44:S14–S21. doi: 10.1007/PL00002934. [DOI] [PubMed] [Google Scholar]
- 3.Kiberd B. The chronic kidney disease epidemic: stepping back and looking forward. J Am Soc Nephrol. 2006;17:2967–2973. doi: 10.1681/ASN.2006020123. [DOI] [PubMed] [Google Scholar]
- 4.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 5.Fioretto P, Stehouwer CDA, Mauer M, et al. Heterogeneous nature of microalbuminuria in NIDDM: studies of endothelial function and renal structure. Diabetologia. 1998;41:233–236. doi: 10.1007/s001250050895. [DOI] [PubMed] [Google Scholar]
- 6.Osterby R, Gundersen HJG, Horlyck A, Kroustrup JP, Nyberg G, Westberg G. Diabetic glomerulopathy—structural characteristics of the early and advanced stages. Diabetes. 1983;32:79–82. doi: 10.2337/diab.32.2.s79. [DOI] [PubMed] [Google Scholar]
- 7.Dalla VM, Saller A, Mauer M, Fioretto P. Role of mesangial expansion in the pathogenesis of diabetic nephropathy. J Nephrol. 2001;14(Suppl 4):S51–S57. [PubMed] [Google Scholar]
- 8.van den Hoven MJ, Rops AL, Bakker MA, et al. Increased expression of heparanase in overt diabetic nephropathy. Kidney Int. 2006;70:2100–2108. doi: 10.1038/sj.ki.5001985. [DOI] [PubMed] [Google Scholar]
- 9.van den Hoven MJ, Rops AL, Vlodavsky I, Levidiotis V, Berden JH, van der Vlag J. Heparanase in glomerular diseases. Kidney Int. 2007;72:543–548. doi: 10.1038/sj.ki.5002337. [DOI] [PubMed] [Google Scholar]
- 10.van den Hoven MJ, Wijnhoven TJ, Li JP, et al. Reduction of anionic sites in the glomerular basement membrane by heparanase does not lead to proteinuria. Kidney Int. 2008;73:278–287. doi: 10.1038/sj.ki.5002706. [DOI] [PubMed] [Google Scholar]
- 11.Wijnhoven TJM, Lensen JFM, Rops ALWM, et al. Aberrant heparan sulfate profile in the human diabetic kidney offers new clues for therapeutic glycomimetics. Am J Kidney Dis. 2006;48:250–261. doi: 10.1053/j.ajkd.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Wijnhoven TJM, van den Hoven MJW, Ding H, et al. Heparanase induces a differential loss of heparan sulphate domains in overt diabetic nephropathy. Diabetologia. 2008;51:372–382. doi: 10.1007/s00125-007-0879-6. [DOI] [PubMed] [Google Scholar]
- 13.Ginath S, Menczer J, Friedmann Y, et al. Expression of heparanase, Mdm2, and erbB2 in ovarian cancer. Int J Oncol. 2001;18:1133–1144. doi: 10.3892/ijo.18.6.1133. [DOI] [PubMed] [Google Scholar]
- 14.Ikeguchi M, Hirooka Y, Kaibara N. Heparanase gene expression and its correlation with spontaneous apoptosis in hepatocytes of cirrhotic liver and carcinoma. Eur J Cancer. 2003;39:86–90. doi: 10.1016/S0959-8049(02)00558-0. [DOI] [PubMed] [Google Scholar]
- 15.Singh DK, Winocour P, Farrington K. Mechanisms of disease: the hypoxic tubular hypothesis of diabetic nephropathy. Nat Clin Pract Nephrol. 2008;4:216–226. doi: 10.1038/ncpneph0757. [DOI] [PubMed] [Google Scholar]
- 16.Goyal P, Weissmann N, Grimminger F, et al. Upregulation of NAD(P)H oxidase 1 in hypoxia activates hypoxiainducible factor 1 via increase in reactive oxygen species. Free Radic Biol Med. 2004;36:1279–1288. doi: 10.1016/j.freeradbiomed.2004.02.071. [DOI] [PubMed] [Google Scholar]
- 17.Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Katavetin P, Miyata T, Inagi R, et al. High glucose blunts vascular endothelial growth factor response to hypoxia via the oxidative stress-regulated hypoxia-inducible factor/hypoxia-responsible element pathway. J Am Soc Nephrol. 2006;17:1405–1413. doi: 10.1681/ASN.2005090918. [DOI] [PubMed] [Google Scholar]
- 19.Brownlee M. Preventing kidney cell suicide. Nat Med. 2007;13:1284–1285. doi: 10.1038/nm1107-1284. [DOI] [PubMed] [Google Scholar]
- 20.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 21.Brownlee M. The pathobiology of diabetic complications—a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 22.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 23.Wagener FADT, Volk HD, Willis D, et al. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev. 2003;55:551–571. doi: 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- 24.Abraham NG, Kappas A. Heme oxygenase and the cardiovascular-renal system. Free Radic Biol Med. 2005;39:1–25. doi: 10.1016/j.freeradbiomed.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Asija A, Peterson SJ, Stec DE, Abraham NG. Forum original research communication—targeting endothelial cells with heme oxygenase-1 gene using VE-cadherin promoter attenuates hyperglycemia-mediated cell injury and apoptosis. Antioxid Redox Signal. 2007;9:2065–2074. doi: 10.1089/ars.2007.1804. [DOI] [PubMed] [Google Scholar]
- 26.Scharstuhl A, Mutsaers R, Pennings B, Zizi M, Russel F, Wagener F. Curcumin-induced apoptosis of human dermal fibroblasts is mediated by apoptosis inducing factor (Aif) and not by caspases. Wound Repair Regen. 2009;17:A18. [Google Scholar]
- 27.Wagener FADT, Eggert A, Boerman OC, et al. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. 2001;98:1802–1811. doi: 10.1182/blood.V98.6.1802. [DOI] [PubMed] [Google Scholar]
- 28.Wagener FADT, Toonen EJM, Wigman L, et al. HMOX1 promoter polymorphism modulates the relationship between disease activity and joint damage in rheumatoid arthritis. Arthritis Rheum. 2008;58:3388–3393. doi: 10.1002/art.23970. [DOI] [PubMed] [Google Scholar]
- 29.Seta F, Bellner L, Rezzani R, et al. Heme oxygenase-2 is a critical determinant for execution of an acute inflammatory and reparative response. Am J Pathol. 2006;169:1612–1623. doi: 10.2353/ajpath.2006.060555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quan S, Kaminski PM, Yang LM, et al. Heme oxygenase-1 prevents superoxide anion-associated endothelial cell sloughing in diabetic rats. Biochem Biophys Res Commun. 2004;315:509–516. doi: 10.1016/j.bbrc.2004.01.086. [DOI] [PubMed] [Google Scholar]
- 31.Abraham NG, Rezzani R, Rodella L, et al. Overexpression of human heme oxygenase-1 attenuates endothelial cell sloughing in experimental diabetes. Am J Physiol Heart Circ Physiol. 2004;287:H2468–H2477. doi: 10.1152/ajpheart.01187.2003. [DOI] [PubMed] [Google Scholar]
- 32.Di Pascoli M, Rodella L, Sacerdoti D, Bolognesi M, Turkseven S, Abraham NG. Chronic CO levels has a beneficial effect on vascular relaxation in diabetes. Biochem Biophys Res Commun. 2006;340:935–943. doi: 10.1016/j.bbrc.2005.12.082. [DOI] [PubMed] [Google Scholar]
- 33.Bruce CR, Carey AL, Hawley JA, Febbraio MA. Intramuscular heat shock protein 72 and heme oxygenase-1 mRNA are reduced in patients with type 2 diabetes—evidence that insulin resistance is associated with a disturbed antioxidant defense mechanism. Diabetes. 2003;52:2338–2345. doi: 10.2337/diabetes.52.9.2338. [DOI] [PubMed] [Google Scholar]
- 34.Hayashi K, Haneda M, Koya D, Maeda S, Isshiki K, Kikkawa R. Enhancement of glomerular heme oxygenase-1 expression in diabetic rats. Diabetes Res Clin Pract. 2001;52:85–96. doi: 10.1016/S0168-8227(01)00218-2. [DOI] [PubMed] [Google Scholar]
- 35.Koya D, Hayashi K, Kitada M, Kashiwagi A, Kikkawa R, Haneda M. Effects of antioxidants in diabetes-induced oxidative stress in the glomeruli of diabetic rats. J Am Soc Nephrol. 2003;14:S250–S253. doi: 10.1097/01.ASN.0000077412.07578.44. [DOI] [PubMed] [Google Scholar]
- 36.Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 gene ablation and expression. J Am Soc Nephrol. 2000;11:965–973. doi: 10.1681/ASN.V115965. [DOI] [PubMed] [Google Scholar]
- 37.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70:432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 38.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 39.Walczak H, Krammer PH. The Cd95 (Apo-1/Fas) and the trail (Apo-2L) apoptosis systems. Exp Cell Res. 2000;256:58–66. doi: 10.1006/excr.2000.4840. [DOI] [PubMed] [Google Scholar]
- 40.Li HL, Zhu H, Xu CJ, Yuan JY. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/S0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 41.Zou H, Henzel WJ, Liu XS, Lutschg A, Wang XD. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/S0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 42.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4:139–163. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 43.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 44.Crompton M. Bax, Bid and the permeabilization of the mitochondrial outer membrane in apoptosis. Curr Opin Cell Biol. 2000;12:414–419. doi: 10.1016/S0955-0674(00)00110-1. [DOI] [PubMed] [Google Scholar]
- 45.Kinnally KW, Antonsson B. A tale of two mitochondrial channels, MAC and PTP, in apoptosis. Apoptosis. 2007;12:857–868. doi: 10.1007/s10495-007-0722-z. [DOI] [PubMed] [Google Scholar]
- 46.Liu XS, Kim CN, Yang J, Jemmerson R, Wang XD. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/S0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 47.Ha H, Hwang IA, Park JH, Lee HB. Role of reactive oxygen species in the pathogenesis of diabetic nephropathy. Diabetes Res Clin Pract. 2008;82:S42–S45. doi: 10.1016/j.diabres.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 48.Kang BPS, Urbonas A, Baddoo A, Baskin S, Malhotra A, Meggs LG. IGF-1 inhibits the mitochondrial apoptosis program in mesangial cells exposed to high glucose. Am J Physiol Renal Physiol. 2003;285:F1013–F1024. doi: 10.1152/ajprenal.00209.2003. [DOI] [PubMed] [Google Scholar]
- 49.Kang BPS, Frencher S, Reddy V, Kessler A, Malhotra A, Meggs LG. High glucose promotes mesangial cell apoptosis by oxidant-dependent mechanism. Am J Physiol Renal Physiol. 2003;284:F455–F466. doi: 10.1152/ajprenal.00137.2002. [DOI] [PubMed] [Google Scholar]
- 50.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. doi: 10.2337/diabetes.55.01.06.db05-0894. [DOI] [PubMed] [Google Scholar]
- 51.Jung DS, Li JJ, Kwak SJ, et al. FR167653 inhibits fibronectin expression and apoptosis in diabetic glomeruli and in high-glucose-stimulated mesangial cells. Am J Physiol Renal Physiol. 2008;295:F595–F604. doi: 10.1152/ajprenal.00624.2007. [DOI] [PubMed] [Google Scholar]
- 52.Verzola D, Bertolotto MB, Villaggio B, et al. Taurine prevents apoptosis induced by high ambient glucose in human tubule renal cells. J Investig Med. 2002;50:443–451. doi: 10.2310/6650.2002.32504. [DOI] [PubMed] [Google Scholar]
- 53.Ortiz A, Ziyadeh FN, Neilson EG. Expression of apoptosis-regulatory genes in renal proximal tubular epithelial cells exposed to high ambient glucose and in diabetic kidneys. J Investig Med. 1997;45:50–56. [PubMed] [Google Scholar]
- 54.Allen DA, Harwood SM, Varagunam M, Raftery MJ, Yaqoob MM. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J. 2003;17:908. doi: 10.1096/fj.02-0130fje. [DOI] [PubMed] [Google Scholar]
- 55.Lorz C, Benito-Martin A, Boucherot A, et al. The death ligand TRAIL in diabetic nephropathy. J Am Soc Nephrol. 2008;19:904–914. doi: 10.1681/ASN.2007050581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Isermann B, Vinnikov IA, Madhusudhan T, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 57.Lee EY, Lee NY, Hong SW, Chung CH, Hong SY. Blockade of oxidative stress by vitamin C ameliorates albuminuria and renal sclerosis in experimental diabetic rats. Yonsei Med J. 2007;48:847–855. doi: 10.3349/ymj.2007.48.5.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Craven PA, DeRubertis FR, Kagan VE, Melhem M, Studer RK. Effects of supplementation with vitamin C or E on albuminuria, glomerular TGF-beta, and glomerular size in diabetes. J Am Soc Nephrol. 1997;8:1405–1414. doi: 10.1681/ASN.V891405. [DOI] [PubMed] [Google Scholar]
- 59.Brezniceanu ML, Liu F, Wei CC, et al. Catalase overexpression attenuates angiotensinogen expression and apoptosis in diabetic mice. Kidney Int. 2007;71:912–923. doi: 10.1038/sj.ki.5002188. [DOI] [PubMed] [Google Scholar]
- 60.Brezniceanu ML, Liu F, Wei CC, et al. Attenuation of interstitial fibrosis and tubular apoptosis in db/db transgenic mice overexpressing catalase in renal proximal tubular cells. Diabetes. 2008;57:451–459. doi: 10.2337/db07-0013. [DOI] [PubMed] [Google Scholar]
- 61.Kobayashi M, Sugiyama H, Wang DH, et al. Catalase deficiency renders remnant kidneys more susceptible to oxidant tissue injury and renal fibrosis in mice. Kidney Int. 2005;68:1018–1031. doi: 10.1111/j.1523-1755.2005.00494.x. [DOI] [PubMed] [Google Scholar]
- 62.Goth L, Eaton JW. Hereditary catalase deficiencies and increased risk of diabetes. Lancet. 2000;356:1820–1821. doi: 10.1016/S0140-6736(00)03238-4. [DOI] [PubMed] [Google Scholar]
- 63.Di Noia MA, Van Driesche S, Palmieri F, et al. Heme oxygenase-1 enhances renal mitochondrial transport carriers and cytochrome c oxidase activity in experimental diabetes. J Biol Chem. 2006;281:15687–15693. doi: 10.1074/jbc.M510595200. [DOI] [PubMed] [Google Scholar]
- 64.Goodman AI, Chander PN, Rezzani R, et al. Heme oxygenase-2 deficiency contributes to diabetes-mediated increase in superoxide anion and renal dysfunction. J Am Soc Nephrol. 2006;17:1073–1081. doi: 10.1681/ASN.2004121082. [DOI] [PubMed] [Google Scholar]
- 65.Calabrese V, Mancuso C, Sapienza M, et al. Oxidative stress and cellular stress response in diabetic nephropathy. Cell Stress Chaperones. 2007;12:299–306. doi: 10.1379/CSC-270.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeRubertis FR, Craven PA, Melhem MF, Salah E. Attenuation of renal injury in db/db mice overexpressing superoxide dismutase—evidence for reduced superoxide-nitric oxide interaction. Diabetes. 2004;53:762–768. doi: 10.2337/diabetes.53.3.762. [DOI] [PubMed] [Google Scholar]
- 67.Hamada Y, Miyata S, Nii-Kono T, et al. Overexpression of thioredoxin1 in transgenic mice suppresses development of diabetic nephropathy. Nephrol Dial Transplant. 2007;22:1547–1557. doi: 10.1093/ndt/gfm099. [DOI] [PubMed] [Google Scholar]
- 68.Lin CL, Wang JY, Ko JY, et al. Superoxide destabilization of beta-catenin augments apoptosis of high-glucose-stressed mesangial cells. Endocrinology. 2008;149:2934–2942. doi: 10.1210/en.2007-1372. [DOI] [PubMed] [Google Scholar]
- 69.Chen LJ, Na R, Gu MJ, et al. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell. 2008;7:866–878. doi: 10.1111/j.1474-9726.2008.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Haan JB, Stefanovic N, Nikolic-Paterson D, et al. Kidney expression of glutathione peroxidase-1 is not protective against streptozotocin-induced diabetic nephropathy. Am J Physiol Renal Physiol. 2005;289:F544–F551. doi: 10.1152/ajprenal.00088.2005. [DOI] [PubMed] [Google Scholar]
- 71.Shin AH, Kil IS, Yang ES, Huh TL, Yang CH, Park JW. Regulation of high glucose-induced apoptosis by mitochondrial NADP(+)-dependent isocitrate dehydrogenase. Biochem Biophys Res Commun. 2004;325:32–38. doi: 10.1016/j.bbrc.2004.09.218. [DOI] [PubMed] [Google Scholar]
- 72.Kanwar YS, Akagi S, Nayak B, et al. Renal-specific oxidoreductase biphasic expression under high glucose ambience during fetal versus neonatal development. Kidney Int. 2005;68:1670–1683. doi: 10.1111/j.1523-1755.2005.00611.x. [DOI] [PubMed] [Google Scholar]
- 73.Zhang YL, Wada J, Hashimoto I, et al. Therapeutic approach for diabetic nephropathy using gene delivery of translocase of inner mitochondrial membrane 44 by reducing mitochondrial superoxide production. J Am Soc Nephrol. 2006;17:1090–1101. doi: 10.1681/ASN.2005111148. [DOI] [PubMed] [Google Scholar]