

Abstract
Nonribosomal peptides have a variety of medicinal activities including activity as antibiotics, antitumor drugs, immunosuppressives, and toxins. Their biosynthesis on multimodular assembly lines as a series of covalently tethered thioesters, in turn covalently attached on pantetheinyl arms on carrier protein way stations, reflects similar chemical logic and protein machinery to fatty acid and polyketide biosynthesis. While structural information on excised or isolated catalytic adenylation (A), condensation (C), peptidyl carrier protein (PCP) and thioesterase (TE) domains had been gathered over the past decade, little was known about how the NRPS catalytic and carrier domains interact with each other both within and across elongation or termination modules. This highlight reviews recent breakthrough achievements in both X-ray and NMR spectroscopic studies that illuminate the architecture of NRPS PCP domains, PCP-containing didomain-fragments and of a full termination module (C-A-PCP-TE).
1. Introduction
Two major classes of metabolites, polyketides (PK) and fatty acids (FA) on the one hand and nonribosomal peptides (NRP) on the other, are assembled in a series of iterative condensation steps while the reactants are tethered as thioesters on phosphopantetheinyl (4′-PP) arms of proteins.1-8 The covalent cofactor is post-translationally added to the 10-12 kDa carrier protein domains functioning within protein assemblies to accomplish the transport and presentation of substrates to diverse catalytic domains.9, 10
In each chain-elongating condensation, the growing FA, PK, or NRP acyl chain participates as the electrophilic partner, undergoing attack at the acyl thioester carbonyl while the monomer to be incorporated acts as nucleophile.11 For FA and PK chain growth, the iterative chemical step is C-C bond formation via a decarboxylative Claisen condensation from a (methyl)-malonyl-S-acyl carrier protein (ACP).12, 13 For NRP chain elongation, the downstream nucleophile is the amine of an aminoacyl-S-peptidyl carrier protein (PCP) to form the C-N amide (peptide) backbone linkage.14, 15 ACPs and PCPs are similar 3 or 4-helix bundles of 80-100 residue non-catalytic domains, one per biosynthetic module, that are the way stations for the growing PK or NRP chains.8, 16, 17
Fatty acid synthases (FAS) and polyketide synthases (PKS) can occur as distributed assemblies with each catalytic domain on a separate protein (type II FAS or PKS), e.g. in tetracycline and daunomycin biosynthesis.18, 19 Alternatively, the catalytic and carrier protein domains can be strung together into one or more functional modules within a single polypeptide chain. The erythromycin or 6-deoxyerythronolide B synthase (DEBS) has seven modules spread over three protein subunits and this multimodular assembly line is termed a type I PKS.20, 21 The most common architecture of NRP synthetase assembly lines is the type I pattern, exemplified by (a) aminoadipyl-cysteinyl-valine synthetase22, 23, a three module, ten domain 480 kDa single subunit enzyme that carries out the first committed step in penicillin biosynthesis, and (b) the seven module vancomycin synthetase, organized in a 3 module, 3 module, 1 module assembly of three subunit proteins.24, 25
Following two decades of intensive investigation of the chemical mechanisms of PKS and NRPS assembly lines, the chemical logic is fairly well deciphered.9, 11, 12, 26, 27 This includes the enzymatic priming of the apo forms of ACPs and PCPs by phosphopantetheinyl transferases (PPTases) to install the active thiol function of the pantetheinyl cofactor on the carrier proteins so that covalent acyl/peptidyl chain growth can proceed on the thiols.10, 28, 29 The selection and/or activation of monomeric building blocks in chain initiation involves acyl-CoAs by acyltransferase (AT) domains in PKS assembly lines and amino acids by adenylation (A) domains in NRPS assembly lines.6, 30-39 Chain elongation is mechanistically coupled to translocation, via a C-C bond forming step carried out by ketosynthase (KS) domains for polyketides and fatty acids and by C-N bond formation by condensation (C) domains in NRPS assemblies.40-46 The termination modules for PKS, FAS and NRPS typically contain a thioesterase (TE) domain as the most downstream domain and catalyze the disconnection of the full length acyl/peptidyl chain from the adjacent downstream ACP/PCP domain.2, 16, 17, 40, 47-51 TE domains can act as hydrolases, reductases, or regiospecific macrocyclization catalysts and can release the PK, FA or NRP nascent product.52-66 A variety of tailoring enzymes can work on the acyl or peptidyl chain. These tailoring reactions may occur in trans after the chain termination step on the nascent product but many (e.g. keto-reductases, dehydratases, enoyl-reductases in PKS modules and N-methyltransferases, epimerases, cyclases or FMN-dependent oxidases in NRPS modules) work in cis in a specific elongation module on the elongating acyl chains.44, 47, 49, 67-82
Comparable insights into how the structural architecture of type I FAS, PKS, and NRP assembly lines enable and constrain chain initiation, elongation, and termination events have lagged behind chemical insights. The first level of structural information has been obtained from X-ray crystallographic and NMR spectral analyses of separate constituent catalytic and carrier proteins from distributed type II assemblies or excised single domains from type I synthetases. Structural studies on the FAS and PKS catalytic subunits83-92 (KS, AT, KR, DH and, ER) first yielded to such structural information by protein crystallization and X-ray diffraction along with high-resolution liquid-state NMR spectra analysis of the 80-100 residue ACP proteins.84, 93-102 Structures of free standing or excised NRPS adenylation (A) domains were solved by X-ray crystallography103-106 as well as a C domain from the vibriobactin siderophore synthetase assembly line107 and tailoring enzymes such as the halogenases of the rebeccamycin or syringomycin synthetases.108, 109 The type I TE domain from the surfactin non-ribosomal peptide synthetase as well as the TEs from polyketide synthases such as erythromycin and picromycin have been also crystallized and the structures solved.59, 110-112 These structures provided insights into the general fold of isolated proteins and excised domains, their structural dynamic and high-resolution structural details helped to understand the interaction with their substrates, but the structures could not provide sufficient information on protein-protein or domain-domain interactions to elucidate the overall architectures the multienzyme assemblies of PKS, FAS or NRPS systems.
Most recently, structures of multidomainal fatty acid synthases of fungal (Thermomyces lanuginosus), yeast (Saccharomyces cerevisae) and mammalian (Sus scrofa) origin, free and in complex with NADP+ have been solved by X-ray crystallography, giving snap shots of the global orientation of catalytic domains relative to each other and giving hints as to the mobility of ACP and thioesterase domains. The invisibility of ACP and TE domains in the X-ray analyses of the mammalian and fungal FAS structures reflect the domains intrinsic flexibility.48, 113-118 These structures have been transformational accomplishments in delineating how the architectural arrays of the catalytic core (KS, AT/MPT) and tailoring (KR, DH, ER) domains interact to perform the chemical steps of fatty acid elongation. Further exciting details about the well-synchronized internal dynamics and domain-domain interactions have been discovered very recently for a metazoan fatty acid synthase by cryogenic electron microscopy and three-dimensional reconstruction methods.119 Analogously, pairs of PKS domains from the 6-deoxyerythronolide B synthase (DEBS) have been solved by efforts in X-ray crystallography83, 85, 120-124 to reveal how inter-domain linkers function and KS and AT domains to be oriented with respect to each other. Also, domains involved in the regioselective cyclization of polycyclic aromatic polyketide scaffolds by bacterial and fungal PKS have been studied to understand the control of ring size and connectivity.46, 56, 59, 125, 126
The canonical view of FAS and PKS organization in an elongation module of a type I assembly is now that AT-KS-ACP constitute the enzmyatic core assembly with defined interaction surfaces while the KR-DH-ER domains form an extension from which one, two, or three of the domains can be disabled or absent without affecting the core activity of the assembly line. The core AT-KS-ACP module enacts a chain elongation/chain translocation step to produce a β-ketoacyl-ACP product. Engagement of the loop catalytic domains before the growing acyl chain moves in the next condensation/translocation event to the ACP of the next downstream module controls the oxidation state at the Cβ position of the growing chain. Action of the KR alone leaves a β-OH-acyl-ACP, of KR plus DH yields an α,β-enoyl-ACP, while combined action of KR, DH and ER domains in that order yields a β-CH2-acyl-ACP at the end of a particular module's activity.11, 20
Recent studies on NRPS assembly lines have provided structural insight into NRPS architectural logic to a level comparable with that of PKS and FAS systems. These involve NMR studies on PCP domains127, 128, an external thioesterase (TEII)129 and on a PCP-TE didomain130, X-ray studies on a condensation (C) domain107, inter-subunit communication enabling COM-domains131, terminating type I thioesterases (TEI)110, 132 and adenylation (A) domains103-106, a dehydrolase domain involved in enterobactin synthesis (EntA)133, an excised PCP-C didomain134, the full length didomain protein (EntB) of a substrate delivering aryl carrier (ArCP) and a substrate-providing isochorismate lyase domain (ICL) from the enterobactin synthetase135, and on a full termination module C-A-PCP-TE.136 The structural studies have illuminated mechanistic issues still to be resolved in these antibiotic and antitumor agent assembly lines.137, 138
This highlight article examines recent conclusions from such NRPS structural work. We start with NMR studies on the peptidyl carrier proteins which must visit at least four catalytic partners. These essential catalytic partner proteins are priming PPTases, loading/initiating A domains, chain elongating upstream and downstream C domains and for some PCPs, chain terminating TE domains or tailoring enzymes. At least three of these carrier recognition processes are essential for each elongation cycle.17, 127, 139 Experiments focused on the protein dynamic and protein-protein interactions of the PCP have revealed that a defined conformational switch of this small shuttling domain is intimately linked to its ability to interact with multiple proteins.16, 128, 140 We then take up two different didomain combinations, a PCP-C and then a PCP-TE pair that function in cis in an elongation module in the tyrocidine synthetase and the termination module in enterobactin synthetase assembly lines, respectively.130, 134 The ‘pièce de résistance’ in the field is the recent X-ray structure of the termination module of the surfactin synthetase assembly line.136 This structure gives a snapshot of how the C-A-PCP-TE domains can interact at one moment in time. The complete termination module further reveals significant domain-domain interfaces and the positioning of linker regions between domains. This structural complex is a key starting point for subsequent efforts to chart possible pathways for how the PCP domain(s) in this termination module as well as in elongation and initiation modules must move and reorient to visit the C, A, downstream C, and the TE domains after initial interaction with the post-translational modifying 4′-phosphopantetheinyl transferases (PPTase). This termination module architecture also provides a building block for how multiple NRPS modules can come together to form full assembly lines.
2. A peptidyl carrier protein-centered view of NRPS assembly lines
The 80-100 residue peptidyl carrier proteins (PCPs) are at the center of the action in every module of NRPS assembly lines (as are the corresponding ACPs for FAS and PKS assemblies). A module is defined as the unit responsible for selection and addition of one substrate monomer into the growing, tethered peptidyl/acyl chain (Figure 1). A NRPS module is composed of a C, an A and a shuttling PCP domain. For a natural product enzymatic assembly line to function, the carrier proteins in each and every module must first be post-translationally modified on a conserved serine residue surrounded by a homologous pattern of amino acid residues, which together define a carrier protein-specific recognition site.50, 141-144 The 18 Å long prosthetic group, bearing the terminal thiol on which natural product chain growth subsequently proceeds, is covalently connected via a phosphodiester to the folded apo carrier proteins by action of a dedicated family of 4′-phosphopantetheinyl transferases, often encoded within the NRPS biosynthetic gene cluster.28, 29
Figure 1. Domain composition of some modular NRPS assembly lines.
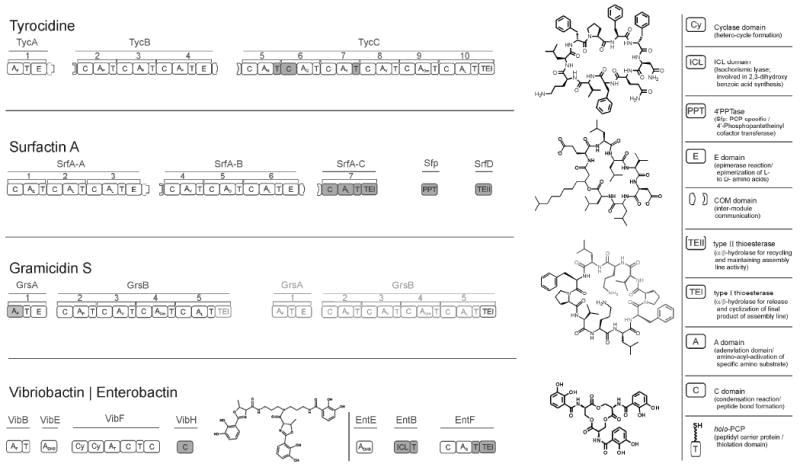
The multimodular assembly lines of the macrocyclic antibiotics Tyrocidine A, Surfactin A and Gramicidin S and the synthetases of the siderophores vibriobactin and enterobactin are shown in a graphical presentation beside the chemical structure of each assembly line. The assembly lines are represented in respective modules and domains for the synthesis of one full length natural product. The iteratively working Gramicidin assembly line is shown in two copies of its assembly line (black/gray). The chemical structure of Gramicidin S reflects this The definitions of domains: A = Adenylation, C = Condensation, T = Thiolation- or peptidyl carrier protein (PCP) are given in a figure legend. The filled domains within these assembly lines have their structures determined by X-ray crystallography or NMR spectroscopy.
Carrier proteins have been examined in the past using biochemical methods, by high resolution NMR spectroscopy and X-ray crystallography, as excised domains, as free standing single proteins, and during recognition by PPTases like Sfp from Bacillus subtilis and AcpS from Streptomyces coelicolor or Streptococcus pneumoniae.93-97, 99-102, 127, 128 The crystal structure of Sfp and the studied protein complex with PCP revealed that Sfp has a pseudo-homodimeric fold and provides one V-shaped interface for the 4′-PP cofactor modification of a PCP (Figure 2c); in comparison AcpS is crystallized as a symmetric homo-trimer and three catalytic sites for the post-translational modification of ACPs are located in the interface of AcpS monomers (Figure 2d).145-147 ACPs and PCPs had been shown to comprise of a three to four helix bundle, whereas the third helix is described as a short α- or 310- helix. However, X-ray studies did not deal with the structural flexibility and dynamics, essential for the function of carrier proteins to shuttle to, interact with, and present their tethered, growing acyl chain to every catalytic domain in any NRPS/PKS/FAS module. In comparison, early NMR analyses have given initial evidence that these small proteins may have some structural flexibility and conformational exchange also involving the 4′-PP arm.94, 95, 101
Figure 2. NMR-based structural information for the TycC PCP3 domain.
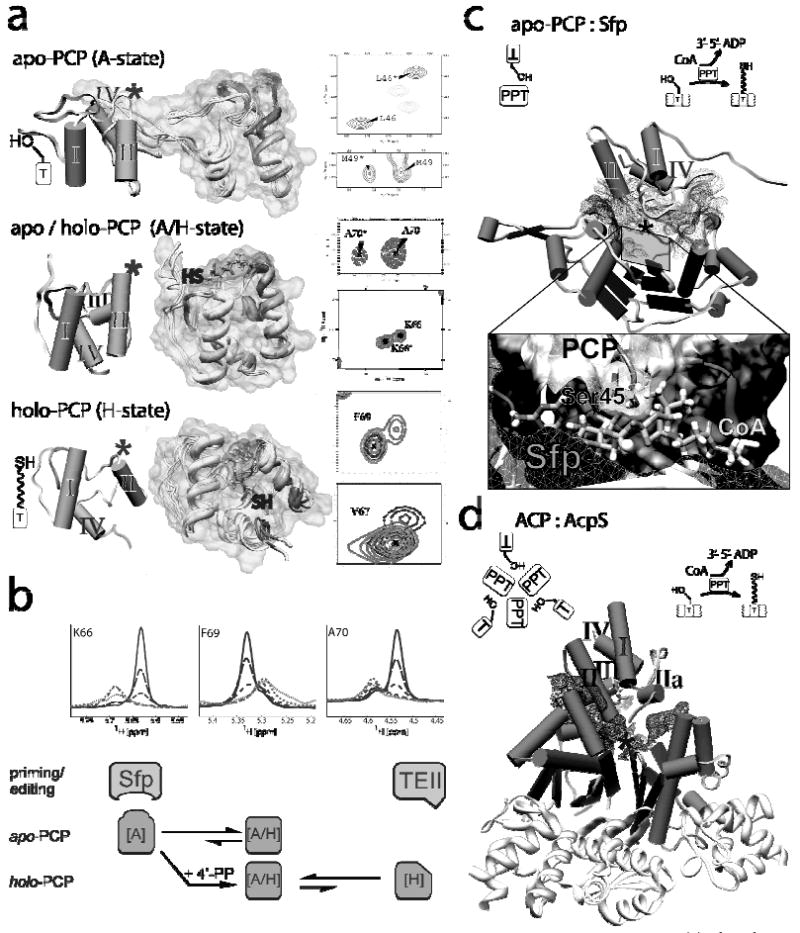
(a) the three conformational states observed for TycC3-PCP in NMR spectra of non-modified apo-PCP (A and A/H-states) and 4′-PP cofactor modified holo-PCP (A/H and H-states); The structures of all three states are shown with numbered helices; (*) marks the position of the active site serine for the post-translational modification. The chemical exchange between two conformers resulted in sets of two signals per amino acid residue in 15N-HSQC spectra. Examples of these double peaks are shown for apo-PCP (A-state) and holo-PCP (A/H-state). Substrate modification of the free HS-4′-PP cofactor arrests the structural dynamic and superposition of slow exchange-typical double peaks from apo- and holo-PCP and single peaks for acetyl-S-4′-PP-holo-PCP are shown (15N-HSQC of F69 and V67 for the H-state). HS marks the position of the 4′-PP thiol function in the A/H- and H-states and demonstrates its displacement on the surface of holo-TycC3-PCP. (b) The exchange ratio of apo-PCP is shifted toward the A-state in interaction with Sfp, while the editing TEII selects misacylated holo-PCP arrested in the H-state. The line-shapes analysis demonstrates the selection of the minor conformation. The diagram (below) shows the allosteric exchange model, dependent on the modification with the 4′-PP cofactor. (c, d) Comparison of the structural complexes of apo-TycC3-PCP : Sfp and ACP : AcpS. The interface of the protein complexes is shown by a mesh surface; illustrations demonstrate the assembly of the proteins in the complexes and the enzymatic function of the PPTases.
The apo and the holo forms of the peptidyl carrier protein, TycC3-PCP, excised from the third module of the third subunit (TycC) of the three protein, ten module tyrocidine A antibiotic synthetase (Figure 1), have been studied in detail by high resolution NMR spectroscopy.148-154 A subset of residues in apo-TycC3-PCP consistently showed two peaks in 15N-HSQC spectra for some backbone amides in two regions of the folded protein (Figure 2a). One region is around the active site residue Ser45 that will become phosphopantetheinylated in the holo form. The second region is located in the area of helix III including the linkers to helix αII and αIV. These NMR signals reflect slow chemical exchange, which could be described as slow conformational exchange between an A (for apo) state and a second, major conformer designated as A/H state, which is present for the apo- and holo protein conformers (Figure 2a). On post-translational conversion of apo to holo form of this PCP the A/H state predominates and a third H (holo) conformer is detected by NMR spectroscopy. In the equilibrating mixture of the two conformers of apo-PCP, the A state is the one that is selected to interact with the phosphopantetheinyl transferase Sfp to undergo post-translational modification. In the resulting holo form of PCP, as the A/H and H conformers exchange via loop/helix melting switch, the covalently attached 4′-PP cofactor rotates through approximately 100° over the surface of holo-TycC3-PCP and the thiol terminus of the pantetheinyl arm can move 30-35 Å (Figure 2a). The misacylated and arrested H state is the conformer detected by TEIIs (see section 3; (Figure 2b cartoon & lineshape analysis).128
This helix/loop switch may be a key swivel mechanism to move the pantetheinyl arm to allow the holo-PCP to visit C and A (and TE) domains within the cognate module and the C domain of adjacent modules in chain elongation steps. It is likely that different surface elements, loops, side chains, and charged residues of PCP domains may be recognized by partner C, A and TE domains.16, 139, 141 These effects on protein-protein recognition have been established by mutational studies with two PCP domains, from EntB and from EntF, in the E. coli enterobactin synthetase assembly line.155 These two PCPs, normally embedded as domains within the EntB and EntF proteins respectively, were recognized in the helix αII regions by their native PPTase EntD and in the loop II and helix III regions by the C-domain of EntF.139, 156
Establishing the structures of the major dynamic conformers of both apo and holo forms of PCP domains constitutes a foundation for evaluation of interactions with paired domains in more detail, taken up in the next section.
3. Recognition between pairs of domains in cis and in trans: PCP-TE interactions
The tethered natural product chains that grow as a series of elongating acyl thioesters attached to each carrier protein in each module of NRPS/PKS/FAS enzymatic machines are subject to chain disconnection chemistry by cleavage of the thermodynamically activated thioester bond in two distinct contexts.
The first is chain termination when a full length peptidyl/acyl chain has reached the most downstream carrier protein in an assembly line. The large majority of NRPS (and PKS and FAS) assembly lines terminate in a thioesterase (TE) domain which catalyzes the disconnection of the natural poduct from the assembly line. The TE domain in cis to the last PCP uses the side chain −OH of its active site serine residue to transfer the chain to form an O-peptidyl-TE acyl-enzyme intermediate.56, 59, 60, 157 The fate of the peptidyl-O-TE can vary, depending on the chemistry in the second step, from hydrolysis, to macrocyclization, to Dieckman cyclizations, thereby releasing the product as a carboxylic acid, a macrolactone or lactam, or a cyclic ketone, respectively.14, 55, 60, 158-160 Chain-terminating TEs are fascinating both for the chemical diversity of the second, product-determining step and for the recognition and transfer of the acyl chain from the immediately adjacent PCP in the PCP-TE didomain. The high resolution NMR structure of the EntF PCP-TE didomain revealed a well defined recognition site between both domains independent from the 4′-PP cofactor modification.130 This close interaction between the last carrier and the terminating thioesterase of an assembly line is defined by a remarkable specificity of the thioesterase.
The second context where TEs can catalyze hydrolytic disconnection of peptidyl-acyl-S-pantetheinyl-carrier protein linkages is in proof reading and editing functions.161-165 In several NRPS (and PKS) biosynthetic gene clusters, a second TE (TEII) is encoded, but produced in trans as a free-standing protein. This external thioesterase is observed to increase the throughput of an assembly line to generate mature, released product.161, 162, 164, 166-172 Mechanistic studies have revealed the TEIIs can patrol the assembly line modules and hydrolytically remove stalled non-native thioesters from the 4′-PP arm that would otherwise block the assembly line. While TEIIs can remove mis-aminoacylated substrates due to faulty activity of adenylation domains from several PCP domains, it may be their main function to hydrolyze acetyl-thioesters of acetyl-S-pantetheinyl-carrier proteins that block chain growth. Much of intracellular CoASH is present in the form of acetyl-CoA and the majority of PPTases do not discriminate well.29, 173 If they transfer not HS-pantetheinyl-P from CoASH but acetyl-S-pantetheinyl-P from the more common acetyl-CoA to apo carrier proteins, this mistake will shut down an assembly line. The editing TEIIs can rescue a dead assembly line by hydrolysis of the acetyl moiety to uncover the terminal SH on the 4′-PP arm for re-acylation with appropriate amino acid monomers.
Two recent complementary NMR studies have illuminated an in cis and an in trans interaction of PCP-TE pairs.129, 130, 138 The in cis 38 kDa PCP-TE pair was derived from the EntF termination module (C-A-T-TE) of the enterobactin synthetase assembly line, responsible for cyclotrimerization of tethered N-2,3-dihydroxyl-benzoylserine to release the enterobactin trilactone siderophore. The in trans pair involved the TycC3-PCP noted above with the editing TEII from surfactin synthetase. These studies required the development of new methodologies in NMR spectroscopy and structure calculation for sorting out high resonance overlaps in the NMR spectra of the protein complexes.129, 174, 175
The apo form of the EntF PCP-TE was solved after mutation of the active site serine Ser 48 of the PCP domain to alanine to obtain homogeneous protein samples.130 The apo PCP domain exhibited the anticipated fold with inter-domain linker mobility. The TE domain adopts the overall fold of α/β hydrolases, previously seen in X-ray structures of the surfactin and fengycin synthetase TEIs.110, 132 A well-defined interface between the PCP and TE domains was observed and the active sites of PCP and TE were separated by a distance traversable by an acylated pantetheine arm, consistent with a functional conformation (Figure 3a, b). Titration of the PCP-TE didomain with different phosphopantetheinyl transferases (Sfp, EntD, and AcpS) suggested capture of an open conformer of the structurally exchanging PCP-TE system by the PCP-modifying enzymes Sfp and EntD. Both PPTases recognize the EntF PCP domain. Analogous titration of the PCP-TE with the EntF C domain revealed a specific interaction surface on the PCP domain distinct from that recognized by the TE domain, consistent with dynamics of each domain within an NRPS module and selected subsets of PCP conformers interacting with A, C, TE and PPTase partners selectively. These parallel, non-overlaping interaction sites on the PCP surface further supports a limited internal structural flexibility as the driving force to shuttle substrates in an elongation or termination module.
Figure 3. NMR-based structures of an in cis PCP-TE pair from the EntF NRPS module of the enterobactin synthetase and an in trans complex of TycC3-PCP and the editing SrfTEII.
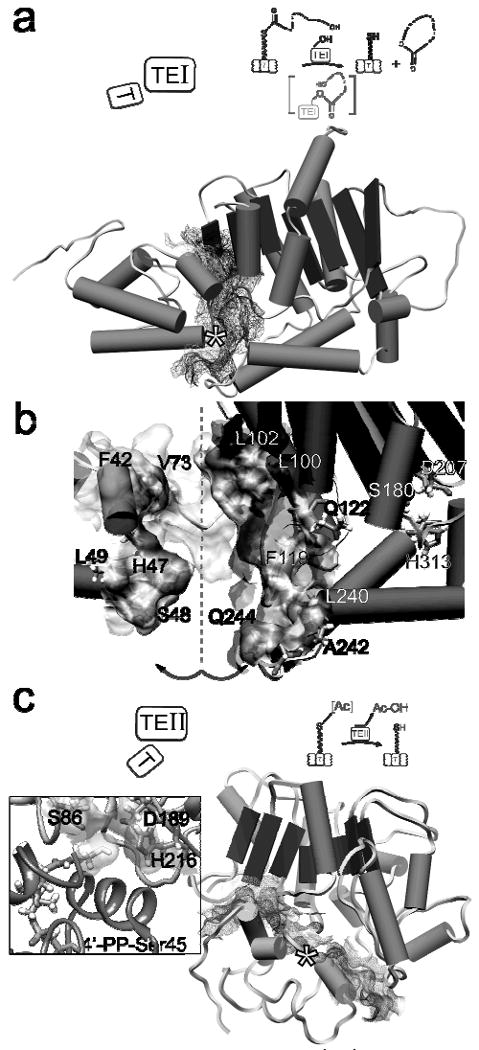
(a) Structure of the EntF PCP-TE didomain; (*) indicates position of the active site serine of the PCP. (b) Opening the interface by a rotation along the dashed line allows identification of residues involved in domain-domain recognition. The active site residues of the TE (S180, D207, and H313) are indicated. (c) The TycC3-PCP : SrfTEII complex demonstrates a more compact interface compared to the EntF PCP-TE didomain (a). The inset interface of PCP and TEII shows the position of the modelled 4′-PP thiol relative to the active site residues of the TEII. Illustrations demonstrate the orientation of the domains and the enzymatic functions of the EntF PCP-TE didomain and the PCP : TEII complex.
The TycC3PCP : SrfTEII structural studies129 provided several kinds of complementary and additional information. The first was the structure of the editing TEII, allowing comparison with the chain-terminating TEIs found in the NRPS and PKS assembly lines, and establishing structural flexibility of the TEII. The active site of SrfTEII was shallow and more accessible compared to TEIs, consistent with hydrolytic editing of small acyl groups, but not the large peptidyl chains of normal carrier protein tethered intermediates. Titration with the misacylated acetyl-S-4′-PP-PCP domain revealed the PCP to be in the H conformational state (see previous section 2) and shifts the structural exchange equilibrium of the SrfTEII, for specific complex formation, towards one conformation. Finally, as shown in Figure 3b & 3c the recognition interfaces of the PCP-TEI pair are distinct from that of the PCP : TEII, enabling the TEII to access PCP-bound intermediates while the most downstream PCP and the TEI are in an active conformation.
4. PCP-C didomain X-ray information
A consideration of the alignment of the core C-A-PCP triad of domains across multiple elongation modules in NRPS assembly lines suggests the PCPn in modulen must interface with the Cn+1 in the immediate downstream modulen+1 during peptidyl chain elongation. This interaction can be transient, lasting only long enough for the growing peptidyl chain on the 4′-PP arm of PCPn to be captured by the free amino group of the aminoacyl-S-4′-PP-PCPn+1 in the Cn+1 domain active site. Alternatively, it could be a longer-lived, stable interface that helps position upstream and downstream modules in functional orientations, both when two modules are in cis in the same protein subunit and when they act in trans across subunits via defined interfaces.
Intensive efforts by the Marahiel and Essen groups at the Philipps-University in Marburg, Germany to crystallize a variety of multi-domain fragments of NRPS assembly lines led in 2007134 to the X-ray structure determination at 1.8 Å resolution of a PCP-C didomain (Figure 4), again from the third subunit of the tyrocidine synthetase assembly line of Bacillus brevis. Tyrocidine is a cyclic decapeptide macrolactam, in which D-Phe1 and L-Leu10 are joined head to tail in a TE-mediated macrolactamization release step. The ten modules of this assembly line are distributed over three separate subunits TycA, -B and -C in a one, three, six module organization, respectively, such that the steps that form the peptide links between D-Phe1 and L-Pro2 and D-Phe4 and L-Asn5 are in trans while the other seven condensations occur in cis (Figure 1). The didomain construct that resulted in useful protein crystallization was derived from the third subunit TycC and involved the fifth PCP and the sixth C domain. This protein contains the ninth PCP (PCP9) in the overall assembly line and the last C domain (C10) (Figures 1, 4). The C domain uses L-Leu-S-4′-PP-PCP10 as a nucleophilic donor and the nonapeptidyl-S-4′-PP-PCP9 as the electrophilic acceptor in the last peptide bond forming step of the assembly line. This final condensation step occurs just prior to transfer of the decapeptidyl-S-4′-PP-PCP10 product chain to the TE domain for off-loading and subsequent head-to-tail macro-lactamization. Thus, as for any elongation module, PCP9 must be able to interact specifically and sequentially, with the adjacent A domain (A9) to load ornithine from activated ornithinyl-AMP, as ornithinyl-S-4′-PP-PCP9, and to transfer this substrate to the C9 active site for peptide bond formation to form the nonapeptidyl-S-4′-PP-PCP9. PCP9 must sub-sequently present the nonapeptidyl chain to C10 to fulfill this shuttling function; all three catalytic domains (A9, C9, and C10) should be able to achieve proximity to the carrier domain PCP9 at some point in the loading/elongation cycle.
Figure 4. X-ray structure of the Tyc PCP9-C10 didomain of the tyrocidine assembly line.
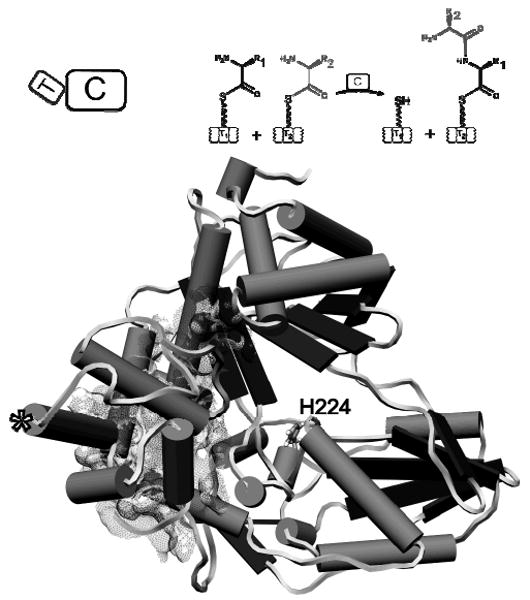
The PCP domain [(*) indicates the active site serine] faces the opposite direction to the active site histidine (H224) of the Tyc C10 domain. Illustrations indicate the orientation of domains and the enzymatic function of the PCP-C didomain.
The TycC PCP-C didomain crystal structure should give insight into both the fold of the PCP and C domains and the relative orientation of upstream donor PCP to immediately adjacent downstream partner C domain. Residues 1-82 comprise PCP9 and residues 101-522 make up C10. The PCP domain with the active site serine residue replaced by an alanine represents the apo form of the carrier domain in a fold similar to the A/H state described in the apo-PCP NMR studies noted in an earlier section. The C domain has the V-shaped architecture reminiscent of the previously solved free-standing C domain, VibH107 in the vibriobactin synthetase assembly line2, 176, with a presumed active site His 224 residue in the cleft of the V-shaped protein.134 The N-terminal and C-terminal pseudo-domains of the V-shaped C10 look to have a swivel point around Ser 268 and potential motion around this swivel point may be important for peptide bond formation by C domains.
While one might have anticipated this PCP-C donor/acceptor pair would be oriented such that Ser 43, the active site serine in the PCP domain that must normally become post-translationally phosphopantetheinylated (if the protein were not a mutant) with a 18 Å long 4′-PP cofactor, would be within 20-25 Å of the C domain active site, this was not the case. As noted in figure 4, Ser 43 in the PCP domain and His 224 in the C domain are 47 Å apart.
The authors proposed several possibilities to explain this unproductive orientation of the PCP and C domains. One is that this orientation could be imposed by crystal packing forces: in essence this could then be a nonphysiological didomain architecture, and therefore this X-ray snapshot could represent some trapped inactive state. Or this snapshot could be a state visited by the PCP9-C10 in cis pair perhaps between catalytic cycles. If so, dramatic domain movements/reorientations would be required to yield a conformer where the 4′-PP arm (absent from the apo form of PCP9) could bring the peptidyl chain into the C10 domain active site. A third alternative is that this PCP-C didomain orientation reflects a state in which the PCP9 is oriented not to the downstream C10 domain but back towards the C9 domain (not present in this excised fragment). In this context, the 4′-PP arm on the holo-form of PCP9 would be carrying Orn9 as the nucleophile for the C9 domain mediated condensation cycle with the octapeptidyl chain tethered as a thioester on the 4′-PP arm of PCP8. To validate this supposition it would, of course, be nice to be able to crystallize and structurally analyze an excised, native Cn-An-PCPn-Cn+1 multidomain construct.
This first X-ray structure of a PCP-C didomain emphasizes both: (1) the value of a high resolution X-ray structure and (2) its limitations in that the species crystallized lacked the 4′-PP business arm of the PCP domain and represents only one snapshot in time. Further the “nonproductive” orientation of PCP9 to C10 emphasizes the ambiguity of one static structure as a possible unproductive conformer vs. a conformer that represents the orientation of the two domains at an earlier step in which PCP9 interacts with the (missing) upstream C9 domain rather than the (present) downstream C10 domain.
To sort out this and other ambiguities of architecture and mechanism will require many X-ray-based snapshots of different intermediates to reconstruct the whole picture of assembly line domain-domain and module-module interactions. This structure of an unfunctional didomain also emphasizes the complementary need to study functional structural dynamic of protein-protein and inter-domain interactions during the many steps of peptidyl chain growth, e.g. by NMR spectroscopy. For example, for the ten module tyrocidine synthetase, there are at least thirty discrete covalent chemical steps (not to mention the many non-covalent changes of protein scaffolds) required for the biosynthesis of each cyclic tyrocidine antibiotic molecule that rolls off the TE domain of the assembly line.
5. Architecture of a NRPS termination module
The crystal structure of the four-domain C-A-PCP-TE termination module of the surfactin A synthetase from Bacillus subtilis has recently been determined at 2.6 Å resolution (Figure 5a), again by combined efforts of the Marahiel and Essen groups.136 Surfactin synthetase has seven modules and generates an N-acyl-heptapeptido-lactone biosurfactant.177, 178 The native termination module has 1274 amino acids and a mass of 144 kDa. To obtain crystals suitable for structure determination, two conditions were necessary. One was the point mutation of the active site serine Ser 1003 in the PCP domain to alanine so that the mutant carrier mimicked the apo-form of the PCP. The second feature was that the construct contained a C-terminal myc-His6-tag comprising an additional 16 amino acid residues, employed to allow affinity purification. Eventually, the C-terminal tag formed an α-helix that acted as a docking element with the C domain of a second C-A-PCP-TE molecule in the crystal packing; both modifications may have enabled crystallization of the observed conformer.
Figure 5. X-ray structure of the full length termination module of the surfactin synthetase (SrfA-C).
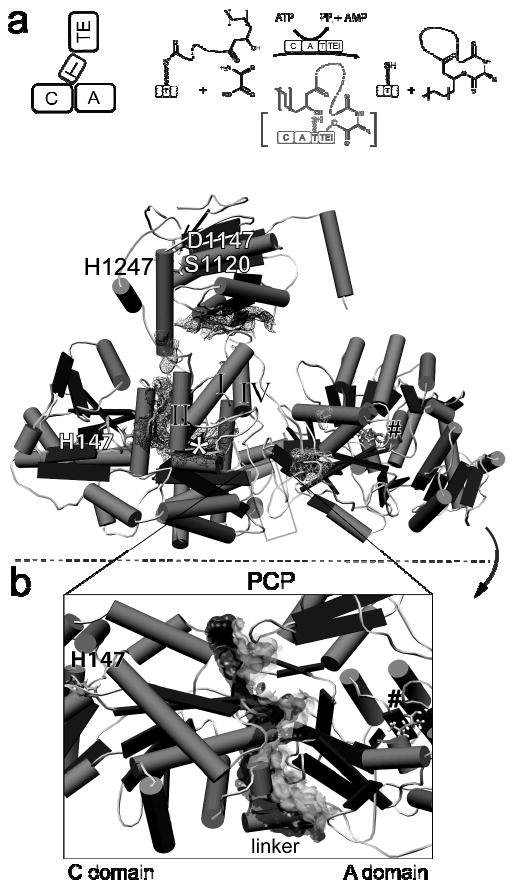
(a) The structure of the C-A-PCP-TE module shows the peptidyl carrier domain oriented towards the C domain. The positions of the active site residues of the C domain (His 147), of PCP (*), and of the TE domain (Ser 1120, Asp 1147, His 1247) are indicated. A substrate leucine is bound inside the A domain (#). The interaction surfaces of the peptidyl carrier domain with surrounding domains are shown by mesh surfaces. Illustrations indicate the orientation of domains of the termination module and the enzymatic function of SrfA-C. (b) Rotating the structure reveals a large stable interface between the C and A domains (joint surface). This rigid interface defines the module C-A-PCP as a stable unit in NRPS assembly lines. The positions of the adjacent domains are indicated.
The structure of the Srf TE domain in the complete termination module aligned well with the previously reported structure of the excised Srf TE domain at a RMSD 0.72 Å for the Cα traces, suggesting little effect of the C-A-PCP domains on the TE fold. The C-A-PCP tridomain assembly represents the core unit of all NRPS elongation modules so the molecular snapshot of this crystal structure will be precedent for domain structures and orientations accessible to elongation modules as well as termination modules.
The structure of the A domain in this termination module reveals two subdomains: the core in the N-terminal region and a C-terminal catalytic subdomain, with the catalytic loop displaced some 16 Å from positions in previously solved single A domain structures.103-106 On the basis of this observation, the authors proposed that A domains must (1) open to bind the cognate amino acid substrate (in this case leucine, observed in the A domain active site), (2) close down to hold onto the aminoacyl-AMP intermediate, and then (3) open back up to allow access of the HS-4′-PP arm of the holo-PCP domain to capture the activated amino acyl group held in the A domain active site. The C domain in this module is similar in architecture to the two C domains previously solved by X-ray crystallography, the stand alone VibH and the C domain described above in the PCP9-C10 didomain of the tyrocidine synthetase. The canyon in the C domain is proposed to be accessible to a lipohexa-peptidyl chain from a (missing) upstream PCP and also to the L-leucyl7-S-4′-PP-PCP within the termination module.
Of particular note is the interaction between the C domain, the A domain, and the intervening 32 residue linker which is well ordered with a structurally rigid 10 residue α-helical stretch that packs along the C-A didomain interface (Figure 5b). Tanovic et al. argue that these extensive interactions suggest the C-domain-A-domain orientation does not change and that the C-A didomain forms a structurally rigid platform that remains invariant during the several steps of the catalytic chain elongation/termination process.136, 179 In contrast, the 15 residue and 9 residue linkers between the A-PCP and the PCP-TE domains, respectively, make fewer interactions and are therefore proposed to be flexible, consistent with the domain movements/rotations required during one or more catalytic steps.
It remains unclear which stage in an NRPS catalytic cycle has been captured by this crystal structure. The active site His 147 of the C domain and the substrate amino acid leucine coordinated in the A domain are 63 Å apart, too far to be bridged by a 4′-PP arm which at full extension would reach 18 Å. The absence of the 4′-PP arm, (recall that the mutant apo-PCP domain in the crystallized module has the Ser 1003 mutated to Ala) also precludes knowing where the 4′-PP chain would be located in this module conformer, i.e. whether it would be pointed toward the C domain, the A domain, or the TE domain, or would be invisible due to conformational flexibility.
Given the interaction in the crystal of the α-helical C-terminal myc-His6-tag with the N-terminus of the C domain of an adjacent C-A-PCP-TE molecule, Tanovic et al. speculate that this helix may mimic the C-terminus of the upstream module and may presage the docking of the prior subunit SrfA-B, containing module 6, on this termination module 7 (SrfA-C). This coordination of a single helix may inform how to assemble seven modules in tandem array to build the full multimodular three protein assembly line for surfactin biosynthesis. If the speculation of Tanovic et al. is a valid prediction, the coordination of an adjacent module may have general impact for other multi-module assembly line architectures of NRP synthetases.
This pioneering X-ray structure of an NRPS termination module gives the first insights into the architecture of elongation and termination modules and the first snapshot along a multi snapshot trajectory of an NRPS assembly line. It is the starting point for further structural investigations with different reaction intermediates or stable analogs, particularly with the 4′-PP arm installed on the native PCP domain. From a PCP-centric perspective, the presence of the 4′-PP cofactor in the holo form of PCP domains in either X-ray- or NMR-based further analysis of full modules and natively interacting domains will be crucially important for advancing to the next level of understanding. It is likely that the conformational switching and distinct orientations of the 4′-PP arm, as determined for isolated PCP domains and in PCP-TE didomains, may likewise affect the PCP conformations and consequent orientations towards the C, A or TE domains in an enzymatic active module.
6. Conclusions: next steps and remaining questions for NRPS (and PKS) assembly lines
The dramatic progress in structure elucidation of NRPS domains, didomain complexes and a termination module, the analysis of dynamic processes and recognition interfaces of isolated domain and in complexes over the past few years, now allows speculation on additional structural and catalytic issues of assembly line function to be raised.
Orientation of carriers in PCP-containing complexes in NMR and X-ray structures
The dramatic progress in structure elucidation of NRPS domains, didomain complexes and a termination module, the analysis of dynamic processes and recognition interfaces of isolated domain and in complexes over the past few years, now allows speculation on additional structural and catalytic issues of assembly line functions.
Orientation of carriers in PCP- and ACP-containing complexes in NMR and X-ray structures
The gallery of NMR and X-ray structures now available of PCP domains in combination with one or more adjacent domains (ICL-PCP, PCP-C, PCP-TE, C-A-PCP-TE) gives initial insights into several orientations that carrier protein domains may occupy with respect to precursor-generating domains, and upstream and downstream catalytic partner domains.130, 134-136 Undoubtedly, future efforts will bring more examples of conformational exchange processes into focus and might reveal the role of the phosphopantetheine arms and the thiol-bound growing product chains on specific partner recognition events. The corresponding orientations and flexibility of ACP domains in FAS, PKS and hybrid PKS-NRPS assemblies will also be informative on the spectrum of interactions that these 10 - 12 kDa domains are involved in biosynthetic cycles and the role of surface patches and individual side chains for interactions with partner proteins.
The ACP-TE didomains of the homo-dimeric fatty acid synthetase from Sus scrofa are so mobile within the FAS active site cleft that even in the recent 3.2 Å maps of this mammalian FAS-I these domains are invisible.115, 118 An artificial disulphide bridge between the carrier-attached 4′-PP cofactor thiol group and a cysteine residue, trapping the ACP in the fungal FAS, freezes out the rotational freedom of the carrier domain inside the otherwise dome-like enclosed fungal fatty acid synthetase and provides hints for the interactions between carrier domain and catalytic domains in fungal FAS systems.48, 116 A sequential interaction of the carrier-tethered substrate with catalytic domains is necessary for the successful synthesis of secondary metabolites in the PKS, FAS and NRPS assemblies. E. J. Brignole et al. could demonstrate how global protein dynamics of an entire metazoan fatty acid synthase can easily accomplish selective protein-protein interactions.119 Interestingly, protein density corresponding to the ACP-TE didomains of the dimeric metazoan FAS was likely observed during these cryogenic EM studies.
In the well-characterized two module enterobactin synthetase, several questions remain open at the moment: (1) Assuming the two Ent modules along with the adenylation domain EntE assemble completely into one multi-protein complex before becoming fully enzymatically active, as described for PKS assembly lines such as DEBS20, 30, 83, 85, 120-122, 180, 181 or the fungal and mammalian FAS systems114, 116, can it be assumed that the full assembly state is operant for all assembly lines, including NRPS systems? The recently discovered, biochemically and structurally analyzed COM-domains on NRPS assemblies131, 137, 182 support the idea that natural product synthetases assemble completely prior to the enzymatic activity. But e.g. the andrimid assembly line45, 183 is in many discrete subunits of single enzymes or didomainal proteins and for a full assembly prior enzymatic activity, without obvious COM or docking domains, additional mechanisms must be available for inter-domain recognition and protein-protein interactions. (2) The vast product diversity of NRPS assembly lines in general represents an extensive variety of tailoring functions. How is the action of the wide variety of in trans tailoring enzymes70 including halogenases108, 184, hydroxylases74, 185, oxidases, methyl-transferases69, 186, 187, and glycosyltransferases82, 188-190 coordinated in the continuing biosynthetic process in either pre-assembled or randomly interacting assembly lines? (3) The PPTase of the enterobactin assembly line (EntD), responsible for the post-translational modification of the EntF and EntB carrier domains, is a separately expressed and membrane associated protein155, 156, 191-193: an immediate post-translational cytosolic modification with the 4′-PP cofactor of the not-yet entirely folded EntF NRPS module with properly arranged domains seems unlikely. Considering the wide interface described between bulky PPTases and apo-carrier domains, sufficient conformational freedom and structural space is essential for the 4′-PP cofactor modification in every C-A-PCP-(TE) NRPS module. More detailed structural information on domains, multi-domain proteins and their relative orientation to adjacent domains is essential for modelling of full assembly lines and for understanding how PPTases gain access to the PCP domains.
By combining a series of structural snapshots of A domains utilizing the recently solved crystal structures of the D-alanyl adenylation (A) domain DltA103, 104, Yonus et al. demonstrate a defined structural dynamic of a small lid-like domain dependent on the loading state of the A domain. The observation of structural exchange processes on modules of NRPS assembly lines, encourages speculations about rather small and well coordinated structural exchange processes implemented in every NRPS domain of an assembly line, as opposed to large displacing movements of a single domain.
Surface recognition and structural flexibility of carrier protein domains
Equivalent structural snapshots of ACPs enable a similar picture of structural exchange for ACP domains analogous to those described for TycC3-PCP in section 2: NMR studies show structural exchange between two conformations94, 95, 99-101, 128, 194 and dynamics involving helix αII and helix III of holo-ACP domains, with an exchange rate of 200 sec-1.95 X-ray crystallographic and NMR spectroscopic analyses describe free holo-carrier domains with the 4′-PP cofactor either buried in the core between helix αI and αII94, 95, 128, 146 or with the cofactor exposed to the solvent94, 128, 146, 195, while substrate-modified carriers are shown with a shifted structural exchange equilibrium to one conformational state95, 194, 196 and with the substrate-S-4′-PP cofactor embedded in the carrier core. The disordering flexibility of helices αII and III might allow access of the substrate-S-4′-PP cofactor to the protective and often hydrophobic environment of the carrier protein core, while the H-state might reflect this opening movement of the three-to-four helix bundle. This observation is consistent with biochemical evidence of chemically or isomeric labile substrates being stabilized while tethered as thioesters to their native holo-carriers.40, 197, 198
Because of the assumed high mobility of carrier protein domains in any functional NRPS or PKS module, the ongoing studies of PCP domains interacting with their cognate catalytic domains from isolated assembly lines may be of great use in defining surface areas for selective interaction partners that may be generalizable. Thus, the titrations of the PCPs, e.g. in EntB and EntF as apo-, holo- and substrates-modified holo-forms, in the enterobactin synthetase assembly line155 with PPTases, C domains, A domains, and the chain-terminating TE domain should give baseline information testable subsequently in multiple systems, both by mutagenesis and structural analysis including NMR spectroscopy, X-ray crystallography and cryogenic electron microscopy.
Intra- and inter-module architectural connections
The recent structural insights into the architecture of the homo-dimeric, multi-domain iterative mammalian FAS-I indicate some similarities to PKS modules, such as those in the erythromycin synthase assembly line.114, 115 Coupled with the structure of the NRPS termination module, implementing the structural basis for standard NRPS elongation modules, these architectures form the basis set for subsequent predictions for how modules can be strung together to form the multimodular type I PKS and NRPS assembly lines. They also should reveal how the subunits can be mixed and matched in hybrid NRPS-PKS assembly lines to produce such therapeutic molecules as rapamycin, bleomycin, and epothilones.199-203 Two additional features are of note: the first is how PKS and NRPS optional subunits can be genetically shuffled in and out of any module to control the tailoring of a growing chain in any modules; the second is how different subunits in multi-subunit assembly lines find only the correct partners for chain elongation.121, 122, 131, 182, 204
In this connection the mammalian FAS structure indicates that the KS-AT di-domain is the central platform for fatty acid and polyketide chain elongation chemistry. The modifying domains for the PKS and FAS chain elongation, keto reduction (KR), dehydration (DH) and enoyl reduction (ER) are on a separate loop so their presence (as active or inactive forms) or absence does not interrupt the C-C bond forming decarboxylative Claisen chain elongation chemistry. The location of the ACP domain is not fully understood other than it must be highly mobile and be able to visit both the central platform domains and the optional tailoring domains within the time regime before chain transfer to the next downstream module.
This architectural principle of a core KS-AT platform may be carried over in the NRPS module architecture with the C-A didomain as the analogous central platform.136 The PCP domains visualized to date in the PCP-C, PCP-TE, and C-A-PCP-TE structures must be mobile to visit the other catalytic domains. There is no comparable high resolution data on where optional in cis tailoring domains are placed in NRPS modules but sequence data suggests methyltransferases (MT) and other optional domains can be inserted at a particular loop region of A domains which would be notionally similar to the KR-DH-ER loops of FAS and PKS modules.
Architectural interfaces between NRPS and PKS modules
In NRP-PKS hybrid molecules the NRPS and PKS modules of the biosynthetic enzymatic assembly lines can be organized both in cis within hybrid subunits and in trans as separate subunits. In each setting there is a chain transfer recognition issue at two levels: (1) at the small molecule or substrate recognition level for an NRPS-PKS interface a KS domain must accept an upstream peptidyl chain on a PCP rather than an ACP domain while at a PKS-NRPS interface it is the C domain that must condense a ketide chain growing on an upstream ACP domain. (2) at the protein-protein recognition level presumably the ACP vs. PCP distinctions must be ignored/accommodated by the heterologous C/KS domain. Thus, structural study of hybrid module interactions may reveal the PCP/ACP-partner protein recognition codes that can be overridden by Nature as it evolves/constructs a new PK-NRP hybrid as in the biogenesis of the antibiotic andrimid and in the coronatine assembly line.45, 198
Subunit docking codes
One of the recent advances in defining protein-protein recognition elements between subunits is deciphering the code for docking domain interactions at the C-terminus of one subunit (often an ACP or PCP) and the N-terminus of the next subunit (often a C or KS domain).121, 122, 131, 137, 201, 205 However, there are many noncanonical subunit interfaces where different domains are found at the breakpoints and suggest additional layers of protein recognition codes. Recent NMR insights into the in trans structures of PKS N- and C-terminal docking domains in the erythromycin assembly line121, 206, 207 and of NRPS-NRPS N-terminal docking or communication domain in the tubulysin assembly line122 provide insight into how subunits are selectively paired to ensure desired chain growth and elongation across the four possible pairs of homologous NRPS-NRPS, PKS-PKS, and heterologous NRPS-PKS, and PKS-NRPS subunit/domain interfaces. The future ability to engineer this inter-subunit and domain-domain comunication will be a key feature of combinatorial biosynthesis strategies.
Acknowledgments
We thank Tanja Mittag, Hospital for Sick Children, Toronto, Canada for technical support with the lineshape analysis and critical reading of the manuscript. We gratefully acknowledge the financial support in part by the National Institutes of Health Grant GM20011 and GM49338 (C.T.W.) and the Human Frontier Science Program (A.K.).
Footnotes
- 1f7l AcpS : Coenzyme A complex
- 1f80 AcpS : ACP complex
- 1qr0 Sfp : Coenzyme A complex
- 2gdw TycC3-PCP (A/H-state)
- 2gdx TycC3-PCP (H-state)
- 2gdy TycC3-PCP (A-state)
- 2ge0 Sfp : CoA complex (model)
- 2ge1 Sfp : Mg2+: CoA : TycC3-PCP (model)
- 2jbz AcpS : Coenzyme A complex
- 2jgp TycC PCP5-C6 didomain
- 2ron SrfTEII
- 2roq EntF PCP-TE
- 2k2q SrfTEII : TycC3-PCP complex
- 2vsq SrfA-C module
References
- 1.Hori K, Yamamoto Y, Minetoki T, Kurotsu T, Kanda M, Miura S, Okamura K, Furuyama J, Saito Y. J Biochem. 1989;106:639–645. doi: 10.1093/oxfordjournals.jbchem.a122909. [DOI] [PubMed] [Google Scholar]
- 2.Keating TA, Marshall CG, Walsh CT. Biochemistry. 2000;39:15522–15530. doi: 10.1021/bi0016523. [DOI] [PubMed] [Google Scholar]
- 3.Kessler N, Schuhmann H, Morneweg S, Linne U, Marahiel MA. J Biol Chem. 2004;279:7413–7419. doi: 10.1074/jbc.M309658200. [DOI] [PubMed] [Google Scholar]
- 4.Kleinkauf H, Roskoski R, Jr, Lipmann F. Proc Natl Acad Sci U S A. 1971;68:2069–2072. doi: 10.1073/pnas.68.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crosa JH, Walsh CT. Microbiol Mol Biol Rev. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee SG, Lipmann F. Proc Natl Acad Sci U S A. 1977;74:2343–2347. doi: 10.1073/pnas.74.6.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pfeifer E, Pavela-Vrancic M, von Dohren H, Kleinkauf H. Biochemistry. 1995;34:7450–7459. doi: 10.1021/bi00022a019. [DOI] [PubMed] [Google Scholar]
- 8.Stein T, Vater J, Kruft V, Otto A, Wittmann-Liebold B, Franke P, Panico M, McDowell R, Morris HR. J Biol Chem. 1996;271:15428–15435. doi: 10.1074/jbc.271.26.15428. [DOI] [PubMed] [Google Scholar]
- 9.Finking R, Marahiel MA. Annu Rev Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 10.Lipmann F. Adv Microb Physiol. 1980;21:227–266. doi: 10.1016/s0065-2911(08)60357-4. [DOI] [PubMed] [Google Scholar]
- 11.Cane DE, Walsh CT. Chem Biol. 1999;6:R319–25. doi: 10.1016/s1074-5521(00)80001-0. [DOI] [PubMed] [Google Scholar]
- 12.Fischbach MA, Walsh CT. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 13.Khosla C, Kapur S, Cane DE. Curr Opin Chem Biol. 2009 doi: 10.1016/j.cbpa.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh CT. Acc Chem Res. 2008;41:4–10. doi: 10.1021/ar7000414. [DOI] [PubMed] [Google Scholar]
- 15.Walsh CT. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 16.Lai JR, Koglin A, Walsh CT. Biochemistry. 2006;45:14869–14879. doi: 10.1021/bi061979p. [DOI] [PubMed] [Google Scholar]
- 17.Stachelhaus T, Huser A, Marahiel MA. Chem Biol. 1996;3:913–921. doi: 10.1016/s1074-5521(96)90180-5. [DOI] [PubMed] [Google Scholar]
- 18.Pickens LB, Tang Y. Metab Eng. 2008 doi: 10.1016/j.ymben.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye J, Dickens ML, Plater R, Li Y, Lawrence J, Strohl WR. J Bacteriol. 1994;176:6270–6280. doi: 10.1128/jb.176.20.6270-6280.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 21.Tang L, Yoon YJ, Choi CY, Hutchinson CR. Gene. 1998;216:255–265. doi: 10.1016/s0378-1119(98)00338-2. [DOI] [PubMed] [Google Scholar]
- 22.MacCabe AP, van Liempt H, Palissa H, Unkles SE, Riach MB, Pfeifer E, von Dohren H, Kinghorn JR. J Biol Chem. 1991;266:12646–12654. [PubMed] [Google Scholar]
- 23.Diez B, Gutierrez S, Barredo JL, van Solingen P, van der Voort LH, Martin JF. J Biol Chem. 1990;265:16358–16365. [PubMed] [Google Scholar]
- 24.Walsh C. Science. 1999;284:442–443. doi: 10.1126/science.284.5413.442. [DOI] [PubMed] [Google Scholar]
- 25.Hubbard BK, Walsh CT. Angew Chem Int Ed Engl. 2003;42:730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 26.Clardy J, Fischbach MA, Walsh CT. Nat Biotechnol. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- 27.Grunewald J, Marahiel MA. Microbiol Mol Biol Rev. 2006;70:121–146. doi: 10.1128/MMBR.70.1.121-146.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lambalot RH, Gehring AM, Flugel RS, Zuber P, LaCelle M, Marahiel MA, Reid R, Khosla C, Walsh CT. Chem Biol. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 29.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, Walsh CT. Biochemistry. 1998;37:1585–1595. doi: 10.1021/bi9719861. [DOI] [PubMed] [Google Scholar]
- 30.Lau J, Cane DE, Khosla C. Biochemistry. 2000;39:10514–10520. doi: 10.1021/bi000602v. [DOI] [PubMed] [Google Scholar]
- 31.Hans M, Hornung A, Dziarnowski A, Cane DE, Khosla C. J Am Chem Soc. 2003;125:5366–5374. doi: 10.1021/ja029539i. [DOI] [PubMed] [Google Scholar]
- 32.Rusnak F, Faraci WS, Walsh CT. Biochemistry. 1989;28:6827–6835. doi: 10.1021/bi00443a008. [DOI] [PubMed] [Google Scholar]
- 33.Heaton MP, Neuhaus FC. J Bacteriol. 1992;174:4707–4717. doi: 10.1128/jb.174.14.4707-4717.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reichert J, Sakaitani M, Walsh CT. Protein Sci. 1992;1:549–556. doi: 10.1002/pro.5560010410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gocht M, Marahiel MA. J Bacteriol. 1994;176:2654–2662. doi: 10.1128/jb.176.9.2654-2662.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dieckmann R, Lee YO, van Liempt H, von Dohren H, Kleinkauf H. FEBS Lett. 1995;357:212–216. doi: 10.1016/0014-5793(94)01342-x. [DOI] [PubMed] [Google Scholar]
- 37.Ehmann DE, Shaw-Reid CA, Losey HC, Walsh CT. Proc Natl Acad Sci U S A. 2000;97:2509–2514. doi: 10.1073/pnas.040572897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roche ED, Walsh CT. Biochemistry. 2003;42:1334–1344. doi: 10.1021/bi026867m. [DOI] [PubMed] [Google Scholar]
- 39.Villiers BR, Hollfelder F. Chembiochem. 2009;10:671–682. doi: 10.1002/cbic.200800553. [DOI] [PubMed] [Google Scholar]
- 40.Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. J Am Chem Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergendahl V, Linne U, Marahiel MA. Eur J Biochem. 2002;269:620–629. doi: 10.1046/j.0014-2956.2001.02691.x. [DOI] [PubMed] [Google Scholar]
- 42.Belshaw PJ, Walsh CT, Stachelhaus T. Science. 1999;284:486–489. doi: 10.1126/science.284.5413.486. [DOI] [PubMed] [Google Scholar]
- 43.Clugston SL, Sieber SA, Marahiel MA, Walsh CT. Biochemistry. 2003;42:12095–12104. doi: 10.1021/bi035090+. [DOI] [PubMed] [Google Scholar]
- 44.Luo L, Kohli RM, Onishi M, Linne U, Marahiel MA, Walsh CT. Biochemistry. 2002;41:9184–9196. doi: 10.1021/bi026047+. [DOI] [PubMed] [Google Scholar]
- 45.Fortin PD, Walsh CT, Magarvey NA. Nature. 2007;448:824–827. doi: 10.1038/nature06068. [DOI] [PubMed] [Google Scholar]
- 46.Kelly WL, Hillson NJ, Walsh CT. Biochemistry. 2005;44:13385–13393. doi: 10.1021/bi051124x. [DOI] [PubMed] [Google Scholar]
- 47.Kopp F, Linne U, Oberthur M, Marahiel MA. J Am Chem Soc. 2008;130:2656–2666. doi: 10.1021/ja078081n. [DOI] [PubMed] [Google Scholar]
- 48.Leibundgut M, Jenni S, Frick C, Ban N. Science. 2007;316:288–290. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]
- 49.Vaillancourt FH, Vosburg DA, Walsh CT. Chembiochem. 2006;7:748–752. doi: 10.1002/cbic.200500480. [DOI] [PubMed] [Google Scholar]
- 50.Finking R, Mofid MR, Marahiel MA. Biochemistry. 2004;43:8946–8956. doi: 10.1021/bi0496891. [DOI] [PubMed] [Google Scholar]
- 51.Marshall CG, Burkart MD, Meray RK, Walsh CT. Biochemistry. 2002;41:8429–8437. doi: 10.1021/bi0202575. [DOI] [PubMed] [Google Scholar]
- 52.Gerber R, Lou L, Du L. J Am Chem Soc. 2009 doi: 10.1021/ja8091054. [DOI] [PubMed] [Google Scholar]
- 53.Robbel L, Hoyer KM, Marahiel MA. FEBS J. 2009 doi: 10.1111/j.1742-4658.2009.06897.x. [DOI] [PubMed] [Google Scholar]
- 54.Sims JW, Schmidt EW. J Am Chem Soc. 2008;130:11149–11155. doi: 10.1021/ja803078z. [DOI] [PubMed] [Google Scholar]
- 55.Hoyer KM, Mahlert C, Marahiel MA. Chem Biol. 2007;14:13–22. doi: 10.1016/j.chembiol.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He W, Wu J, Khosla C, Cane DE. Bioorg Med Chem Lett. 2006;16:391–394. doi: 10.1016/j.bmcl.2005.09.077. [DOI] [PubMed] [Google Scholar]
- 57.Lin H, Thayer DA, Wong CH, Walsh CT. Chem Biol. 2004;11:1635–1642. doi: 10.1016/j.chembiol.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 58.Steller S, Sokoll A, Wilde C, Bernhard F, Franke P, Vater J. Biochemistry. 2004;43:11331–11343. doi: 10.1021/bi0493416. [DOI] [PubMed] [Google Scholar]
- 59.Boddy CN, Schneider TL, Hotta K, Walsh CT, Khosla C. J Am Chem Soc. 2003;125:3428–3429. doi: 10.1021/ja0298646. [DOI] [PubMed] [Google Scholar]
- 60.Kohli RM, Takagi J, Walsh CT. Proc Natl Acad Sci U S A. 2002;99:1247–1252. doi: 10.1073/pnas.251668398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kohli RM, Trauger JW, Schwarzer D, Marahiel MA, Walsh CT. Biochemistry. 2001;40:7099–7108. doi: 10.1021/bi010036j. [DOI] [PubMed] [Google Scholar]
- 62.Schwarzer D, Mootz HD, Marahiel MA. Chem Biol. 2001;8:997–1010. doi: 10.1016/s1074-5521(01)00068-0. [DOI] [PubMed] [Google Scholar]
- 63.Trauger JW, Kohli RM, Walsh CT. Biochemistry. 2001;40:7092–7098. doi: 10.1021/bi010035r. [DOI] [PubMed] [Google Scholar]
- 64.Gokhale RS, Hunziker D, Cane DE, Khosla C. Chem Biol. 1999;6:117–125. doi: 10.1016/S1074-5521(99)80008-8. [DOI] [PubMed] [Google Scholar]
- 65.Shaw-Reid CA, Kelleher NL, Losey HC, Gehring AM, Berg C, Walsh CT. Chem Biol. 1999;6:385–400. doi: 10.1016/S1074-5521(99)80050-7. [DOI] [PubMed] [Google Scholar]
- 66.Larsen NA, Lin H, Wei R, Fischbach MA, Walsh CT. Biochemistry. 2006;45:10184–10190. doi: 10.1021/bi060950i. [DOI] [PubMed] [Google Scholar]
- 67.Palaniappan N, Alhamadsheh MM, Reynolds KA. J Am Chem Soc. 2008;130:12236–12237. doi: 10.1021/ja8044162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neary JM, Powell A, Gordon L, Milne C, Flett F, Wilkinson B, Smith CP, Micklefield J. Microbiology. 2007;153:768–776. doi: 10.1099/mic.0.2006/002725-0. [DOI] [PubMed] [Google Scholar]
- 69.Pelzer S, Wohlert SE, Vente A. Ernst Schering Res Found Workshop. 2005;51:233–259. doi: 10.1007/3-540-27055-8_11. [DOI] [PubMed] [Google Scholar]
- 70.Walsh CT, Chen H, Keating TA, Hubbard BK, Losey HC, Luo L, Marshall CG, Miller DA, Patel HM. Curr Opin Chem Biol. 2001;5:525–534. doi: 10.1016/s1367-5931(00)00235-0. [DOI] [PubMed] [Google Scholar]
- 71.Li Y, Weissman KJ, Muller R. J Am Chem Soc. 2008;130:7554–7555. doi: 10.1021/ja8025278. [DOI] [PubMed] [Google Scholar]
- 72.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 73.Schneider TL, Walsh CT. Biochemistry. 2004;43:15946–15955. doi: 10.1021/bi0481139. [DOI] [PubMed] [Google Scholar]
- 74.Strieker M, Kopp F, Mahlert C, Essen LO, Marahiel MA. ACS Chem Biol. 2007;2:187–196. doi: 10.1021/cb700012y. [DOI] [PubMed] [Google Scholar]
- 75.Balibar CJ, Vaillancourt FH, Walsh CT. Chem Biol. 2005;12:1189–1200. doi: 10.1016/j.chembiol.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 76.Patel HM, Tao J, Walsh CT. Biochemistry. 2003;42:10514–10527. doi: 10.1021/bi034840c. [DOI] [PubMed] [Google Scholar]
- 77.Schauwecker F, Pfennig F, Grammel N, Keller U. Chem Biol. 2000;7:287–297. doi: 10.1016/s1074-5521(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 78.Luo Y, Lin S, Zhang J, Cooke HA, Bruner SD, Shen B. J Biol Chem. 2008;283:14694–14702. doi: 10.1074/jbc.M802206200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fu H, Alvarez MA, Khosla C, Bailey JE. Biochemistry. 1996;35:6527–6532. doi: 10.1021/bi952957y. [DOI] [PubMed] [Google Scholar]
- 80.Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. J Am Chem Soc. 2005;127:17393–17404. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schoenafinger G, Schracke N, Linne U, Marahiel MA. J Am Chem Soc. 2006;128:7406–7407. doi: 10.1021/ja0611240. [DOI] [PubMed] [Google Scholar]
- 82.Fischbach MA, Lin H, Liu DR, Walsh CT. Proc Natl Acad Sci U S A. 2005;102:571–576. doi: 10.1073/pnas.0408463102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Keatinge-Clay A. J Mol Biol. 2008;384:941–953. doi: 10.1016/j.jmb.2008.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tang Y, Lee HY, Tang Y, Kim CY, Mathews I, Khosla C. Biochemistry. 2006;45:14085–14093. doi: 10.1021/bi061187v. [DOI] [PubMed] [Google Scholar]
- 85.Chen AY, Cane DE, Khosla C. Chem Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pan H, Tsai S, Meadows ES, Miercke LJ, Keatinge-Clay AT, O'Connell J, Khosla C, Stroud RM. Structure. 2002;10:1559–1568. doi: 10.1016/s0969-2126(02)00889-4. [DOI] [PubMed] [Google Scholar]
- 87.Keatinge-Clay AT, Stroud RM. Structure. 2006;14:737–748. doi: 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 88.Keatinge-Clay AT, Maltby DA, Medzihradszky KF, Khosla C, Stroud RM. Nat Struct Mol Biol. 2004;11:888–893. doi: 10.1038/nsmb808. [DOI] [PubMed] [Google Scholar]
- 89.Korman TP, Tan YH, Wong J, Luo R, Tsai SC. Biochemistry. 2008;47:1837–1847. doi: 10.1021/bi7016427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Korman TP, Hill JA, Vu TN, Tsai SC. Biochemistry. 2004;43:14529–14538. doi: 10.1021/bi048133a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Keatinge-Clay AT. Chem Biol. 2007;14:898–908. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 92.Hadfield AT, Limpkin C, Teartasin W, Simpson TJ, Crosby J, Crump MP. Structure. 2004;12:1865–1875. doi: 10.1016/j.str.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 93.Alekseyev VY, Liu CW, Cane DE, Puglisi JD, Khosla C. Protein Sci. 2007;16:2093–2107. doi: 10.1110/ps.073011407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma AK, Sharma SK, Surolia A, Surolia N, Sarma SP. Biochemistry. 2006;45:6904–6916. doi: 10.1021/bi060368u. [DOI] [PubMed] [Google Scholar]
- 95.Zornetzer GA, Fox BG, Markley JL. Biochemistry. 2006;45:5217–5227. doi: 10.1021/bi052062d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Findlow SC, Winsor C, Simpson TJ, Crosby J, Crump MP. Biochemistry. 2003;42:8423–8433. doi: 10.1021/bi0342259. [DOI] [PubMed] [Google Scholar]
- 97.Crump MP, Crosby J, Dempsey CE, Parkinson JA, Murray M, Hopwood DA, Simpson TJ. Biochemistry. 1997;36:6000–6008. doi: 10.1021/bi970006+. [DOI] [PubMed] [Google Scholar]
- 98.Kim Y, Prestegard JH. Proteins. 1990;8:377–385. doi: 10.1002/prot.340080411. [DOI] [PubMed] [Google Scholar]
- 99.Johnson MA, Peti W, Herrmann T, Wilson IA, Wuthrich K. Protein Sci. 2006;15:1030–1041. doi: 10.1110/ps.051964606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim Y, Ohlrogge JB, Prestegard JH. Biochem Pharmacol. 1990;40:7–13. doi: 10.1016/0006-2952(90)90171-g. [DOI] [PubMed] [Google Scholar]
- 101.Kim Y, Prestegard JH. Biochemistry. 1989;28:8792–8797. doi: 10.1021/bi00448a017. [DOI] [PubMed] [Google Scholar]
- 102.Holak TA, Kearsley SK, Kim Y, Prestegard JH. Biochemistry. 1988;27:6135–6142. doi: 10.1021/bi00416a046. [DOI] [PubMed] [Google Scholar]
- 103.Du L, He Y, Luo Y. Biochemistry. 2008;47:11473–11480. doi: 10.1021/bi801363b. [DOI] [PubMed] [Google Scholar]
- 104.Yonus H, Neumann P, Zimmermann S, May JJ, Marahiel MA, Stubbs MT. J Biol Chem. 2008;283:32484–32491. doi: 10.1074/jbc.M800557200. [DOI] [PubMed] [Google Scholar]
- 105.May JJ, Kessler N, Marahiel MA, Stubbs MT. Proc Natl Acad Sci U S A. 2002;99:12120–12125. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Conti E, Stachelhaus T, Marahiel MA, Brick P. EMBO J. 1997;16:4174–4183. doi: 10.1093/emboj/16.14.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Keating TA, Marshall CG, Walsh CT, Keating AE. Nat Struct Biol. 2002;9:522–526. doi: 10.1038/nsb810. [DOI] [PubMed] [Google Scholar]
- 108.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Biochemistry. 2007;46:1284–1292. doi: 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- 109.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 110.Bruner SD, Weber T, Kohli RM, Schwarzer D, Marahiel MA, Walsh CT, Stubbs MT. Structure. 2002;10:301–310. doi: 10.1016/s0969-2126(02)00716-5. [DOI] [PubMed] [Google Scholar]
- 111.Tsai SC, Miercke LJ, Krucinski J, Gokhale R, Chen JC, Foster PG, Cane DE, Khosla C, Stroud RM. Proc Natl Acad Sci U S A. 2001;98:14808–14813. doi: 10.1073/pnas.011399198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tsai SC, Lu H, Cane DE, Khosla C, Stroud RM. Biochemistry. 2002;41:12598–12606. doi: 10.1021/bi0260177. [DOI] [PubMed] [Google Scholar]
- 113.Jenni S, Ban N. Acta Crystallogr D Biol Crystallogr. 2009;65:101–111. doi: 10.1107/S0907444909000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leibundgut M, Maier T, Jenni S, Ban N. Curr Opin Struct Biol. 2008;18:714–725. doi: 10.1016/j.sbi.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 115.Maier T, Leibundgut M, Ban N. Science. 2008;321:1315–1322. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- 116.Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Science. 2007;316:254–261. doi: 10.1126/science.1138248. [DOI] [PubMed] [Google Scholar]
- 117.Jenni S, Leibundgut M, Maier T, Ban N. Science. 2006;311:1263–1267. doi: 10.1126/science.1123251. [DOI] [PubMed] [Google Scholar]
- 118.Maier T, Jenni S, Ban N. Science. 2006;311:1258–1262. doi: 10.1126/science.1123248. [DOI] [PubMed] [Google Scholar]
- 119.Brignole EJ, Smith S, Asturias FJ. Nat Struct Mol Biol. 2009;16:190–197. doi: 10.1038/nsmb.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tang Y, Kim CY, Mathews II, Cane DE, Khosla C. Proc Natl Acad Sci U S A. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Broadhurst RW, Nietlispach D, Wheatcroft MP, Leadlay PF, Weissman KJ. Chem Biol. 2003;10:723–731. doi: 10.1016/s1074-5521(03)00156-x. [DOI] [PubMed] [Google Scholar]
- 122.Richter CD, Nietlispach D, Broadhurst RW, Weissman KJ. Nat Chem Biol. 2008;4:75–81. doi: 10.1038/nchembio.2007.61. [DOI] [PubMed] [Google Scholar]
- 123.Tsuji SY, Cane DE, Khosla C. Biochemistry. 2001;40:2326–2331. doi: 10.1021/bi002463n. [DOI] [PubMed] [Google Scholar]
- 124.Gokhale RS, Lau J, Cane DE, Khosla C. Biochemistry. 1998;37:2524–2528. doi: 10.1021/bi971887n. [DOI] [PubMed] [Google Scholar]
- 125.Lesburg CA, Zhai G, Cane DE, Christianson DW. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 126.Ames BD, Korman TP, Zhang W, Smith P, Vu T, Tang Y, Tsai SC. Proc Natl Acad Sci U S A. 2008;105:5349–5354. doi: 10.1073/pnas.0709223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Weber T, Baumgartner R, Renner C, Marahiel MA, Holak TA. Structure. 2000;8:407–418. doi: 10.1016/s0969-2126(00)00120-9. [DOI] [PubMed] [Google Scholar]
- 128.Koglin A, Mofid MR, Lohr F, Schafer B, Rogov VV, Blum MM, Mittag T, Marahiel MA, Bernhard F, Dotsch V. Science. 2006;312:273–276. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]
- 129.Koglin A, Lohr F, Bernhard F, Rogov VV, Frueh DP, Strieter ER, Mofid MR, Guntert P, Wagner G, Walsh CT, Marahiel MA, Dotsch V. Nature. 2008;454:907–911. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Frueh DP, Arthanari H, Koglin A, Vosburg DA, Bennett AE, Walsh CT, Wagner G. Nature. 2008;454:903–906. doi: 10.1038/nature07162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hahn M, Stachelhaus T. Proc Natl Acad Sci U S A. 2004;101:15585–15590. doi: 10.1073/pnas.0404932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Samel SA, Wagner B, Marahiel MA, Essen LO. J Mol Biol. 2006;359:876–889. doi: 10.1016/j.jmb.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 133.Sundlov JA, Garringer JA, Carney JM, Reger AS, Drake EJ, Duax WL, Gulick AM. Acta Crystallogr D Biol Crystallogr. 2006;62:734–740. doi: 10.1107/S0907444906015824. [DOI] [PubMed] [Google Scholar]
- 134.Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen LO. Structure. 2007;15:781–792. doi: 10.1016/j.str.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 135.Drake EJ, Nicolai DA, Gulick AM. Chem Biol. 2006;13:409–419. doi: 10.1016/j.chembiol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 136.Tanovic A, Samel SA, Essen LO, Marahiel MA. Science. 2008;321:659–663. doi: 10.1126/science.1159850. [DOI] [PubMed] [Google Scholar]
- 137.Weissman KJ, Muller R. Chembiochem. 2008;9:826–848. doi: 10.1002/cbic.200700751. [DOI] [PubMed] [Google Scholar]
- 138.Kapur S, Khosla C. Nature. 2008;454:832–833. doi: 10.1038/454832a. [DOI] [PubMed] [Google Scholar]
- 139.Zhou Z, Lai JR, Walsh CT. Chem Biol. 2006;13:869–879. doi: 10.1016/j.chembiol.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 140.Lai JR, Fischbach MA, Liu DR, Walsh CT. Proc Natl Acad Sci U S A. 2006;103:5314–5319. doi: 10.1073/pnas.0601038103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhou Z, Lai JR, Walsh CT. Proc Natl Acad Sci U S A. 2007;104:11621–11626. doi: 10.1073/pnas.0705122104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lai JR, Fischbach MA, Liu DR, Walsh CT. J Am Chem Soc. 2006;128:11002–11003. doi: 10.1021/ja063238h. [DOI] [PubMed] [Google Scholar]
- 143.Mofid MR, Finking R, Essen LO, Marahiel MA. Biochemistry. 2004;43:4128–4136. doi: 10.1021/bi036013h. [DOI] [PubMed] [Google Scholar]
- 144.Mofid MR, Finking R, Marahiel MA. J Biol Chem. 2002;277:17023–17031. doi: 10.1074/jbc.M200120200. [DOI] [PubMed] [Google Scholar]
- 145.Dall'Aglio P, Arthur C, Law CKE, Crump MP, Crosby J, Hadfield AT. 2006 doi: 10.2210/pdb2jbz/pdb. [DOI] [Google Scholar]
- 146.Parris KD, Lin L, Tam A, Mathew R, Hixon J, Stahl M, Fritz CC, Seehra J, Somers WS. Structure. 2000;8:883–895. doi: 10.1016/s0969-2126(00)00178-7. [DOI] [PubMed] [Google Scholar]
- 147.Reuter K, Mofid MR, Marahiel MA, Ficner R. EMBO J. 1999;18:6823–6831. doi: 10.1093/emboj/18.23.6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Guntert P, Mumenthaler C, Wuthrich K. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 149.Otting G, Wuthrich K. Q Rev Biophys. 1990;23:39–96. doi: 10.1017/s0033583500005412. [DOI] [PubMed] [Google Scholar]
- 150.Wider G, Wuthrich K. Curr Opin Struct Biol. 1999;9:594–601. doi: 10.1016/s0959-440x(99)00011-1. [DOI] [PubMed] [Google Scholar]
- 151.Wuthrich K. J Biol Chem. 1990;265:22059–22062. [PubMed] [Google Scholar]
- 152.Riek R, Wider G, Pervushin K, Wuthrich K. Proc Natl Acad Sci U S A. 1999;96:4918–4923. doi: 10.1073/pnas.96.9.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. Proc Natl Acad Sci U S A. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pervushin K, Riek R, Wider G, Wuthrich K. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gehring AM, Mori I, Walsh CT. Biochemistry. 1998;37:2648–2659. doi: 10.1021/bi9726584. [DOI] [PubMed] [Google Scholar]
- 156.Gehring AM, Bradley KA, Walsh CT. Biochemistry. 1997;36:8495–8503. doi: 10.1021/bi970453p. [DOI] [PubMed] [Google Scholar]
- 157.Tseng CC, Bruner SD, Kohli RM, Marahiel MA, Walsh CT, Sieber SA. Biochemistry. 2002;41:13350–13359. doi: 10.1021/bi026592a. [DOI] [PubMed] [Google Scholar]
- 158.Kopp F, Marahiel MA. Nat Prod Rep. 2007;24:735–749. doi: 10.1039/b613652b. [DOI] [PubMed] [Google Scholar]
- 159.Strieker M, Marahiel MA. Chembiochem. 2009;10:607–616. doi: 10.1002/cbic.200800546. [DOI] [PubMed] [Google Scholar]
- 160.Trauger JW, Kohli RM, Mootz HD, Marahiel MA, Walsh CT. Nature. 2000;407:215–218. doi: 10.1038/35025116. [DOI] [PubMed] [Google Scholar]
- 161.Schwarzer D, Mootz HD, Linne U, Marahiel MA. Proc Natl Acad Sci U S A. 2002;99:14083–14088. doi: 10.1073/pnas.212382199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yeh E, Kohli RM, Bruner SD, Walsh CT. Chembiochem. 2004;5:1290–1293. doi: 10.1002/cbic.200400077. [DOI] [PubMed] [Google Scholar]
- 163.Guo ZF, Sun Y, Zheng S, Guo Z. Biochemistry. 2009 doi: 10.1021/bi802165x. [DOI] [PubMed] [Google Scholar]
- 164.Chen D, Wu R, Bryan TL, Dunaway-Mariano D. Biochemistry. 2009;48:511–513. doi: 10.1021/bi802207t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Leduc D, Battesti A, Bouveret E. J Bacteriol. 2007;189:7112–7126. doi: 10.1128/JB.00755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Linne U, Schwarzer D, Schroeder GN, Marahiel MA. Eur J Biochem. 2004;271:1536–1545. doi: 10.1111/j.1432-1033.2004.04063.x. [DOI] [PubMed] [Google Scholar]
- 167.Kotowska M, Pawlik K, Smulczyk-Krawczyszyn A, Bartosz-Bechowski H, Kuczek K. Appl Environ Microbiol. 2009;75:887–896. doi: 10.1128/AEM.01371-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhou Y, Meng Q, You D, Li J, Chen S, Ding D, Zhou X, Zhou H, Bai L, Deng Z. Appl Environ Microbiol. 2008;74:7235–7242. doi: 10.1128/AEM.01012-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hu Z, Pfeifer BA, Chao E, Murli S, Kealey J, Carney JR, Ashley G, Khosla C, Hutchinson CR. Microbiology. 2003;149:2213–2225. doi: 10.1099/mic.0.26015-0. [DOI] [PubMed] [Google Scholar]
- 170.Kim BS, Cropp TA, Beck BJ, Sherman DH, Reynolds KA. J Biol Chem. 2002;277:48028–48034. doi: 10.1074/jbc.M207770200. [DOI] [PubMed] [Google Scholar]
- 171.Kotowska M, Pawlik K, Butler AR, Cundliffe E, Takano E, Kuczek K. Microbiology. 2002;148:1777–1783. doi: 10.1099/00221287-148-6-1777. [DOI] [PubMed] [Google Scholar]
- 172.Heathcote ML, Staunton J, Leadlay PF. Chem Biol. 2001;8:207–220. doi: 10.1016/s1074-5521(01)00002-3. [DOI] [PubMed] [Google Scholar]
- 173.Zhou Z, Koglin A, Wang Y, McMahon AP, Walsh CT. J Am Chem Soc. 2008;130:9925–9930. doi: 10.1021/ja802657n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Frueh DP, Vosburg DA, Walsh CT, Wagner G. J Biomol NMR. 2006;34:31–40. doi: 10.1007/s10858-005-5338-4. [DOI] [PubMed] [Google Scholar]
- 175.Frueh DP, Sun ZY, Vosburg DA, Walsh CT, Hoch JC, Wagner G. J Am Chem Soc. 2006;128:5757–5763. doi: 10.1021/ja0584222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wyckoff EE, Smith SL, Payne SM. J Bacteriol. 2001;183:1830–1834. doi: 10.1128/JB.183.5.1830-1834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cosmina P, Rodriguez F, de Ferra F, Grandi G, Perego M, Venema G, van Sinderen D. Mol Microbiol. 1993;8:821–831. doi: 10.1111/j.1365-2958.1993.tb01629.x. [DOI] [PubMed] [Google Scholar]
- 178.Nakano MM, Magnuson R, Myers A, Curry J, Grossman AD, Zuber P. J Bacteriol. 1991;173:1770–1778. doi: 10.1128/jb.173.5.1770-1778.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dieckmann R, Pavela-Vrancic M, von Dohren H, Kleinkauf H. J Mol Biol. 1999;288:129–140. doi: 10.1006/jmbi.1999.2671. [DOI] [PubMed] [Google Scholar]
- 180.Tang Y, Chen AY, Kim CY, Cane DE, Khosla C. Chem Biol. 2007;14:931–943. doi: 10.1016/j.chembiol.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Richter CD, Stanmore DA, Miguel RN, Moncrieffe MC, Tran L, Brewerton S, Meersman F, Broadhurst RW, Weissman KJ. FEBS J. 2007;274:2196–2209. doi: 10.1111/j.1742-4658.2007.05757.x. [DOI] [PubMed] [Google Scholar]
- 182.Kumar P, Li Q, Cane DE, Khosla C. J Am Chem Soc. 2003;125:4097–4102. doi: 10.1021/ja0297537. [DOI] [PubMed] [Google Scholar]
- 183.Jin M, Fischbach MA, Clardy J. J Am Chem Soc. 2006;128:10660–10661. doi: 10.1021/ja063194c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 185.Singh GM, Fortin PD, Koglin A, Walsh CT. Biochemistry. 2008;47:11310–11320. doi: 10.1021/bi801322z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Simunovic V, Zapp J, Rachid S, Krug D, Meiser P, Muller R. Chembiochem. 2006;7:1206–1220. doi: 10.1002/cbic.200600075. [DOI] [PubMed] [Google Scholar]
- 187.Weinig S, Hecht HJ, Mahmud T, Muller R. Chem Biol. 2003;10:939–952. doi: 10.1016/j.chembiol.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 188.Losey HC, Peczuh MW, Chen Z, Eggert US, Dong SD, Pelczer I, Kahne D, Walsh CT. Biochemistry. 2001;40:4745–4755. doi: 10.1021/bi010050w. [DOI] [PubMed] [Google Scholar]
- 189.Losey HC, Jiang J, Biggins JB, Oberthur M, Ye XY, Dong SD, Kahne D, Thorson JS, Walsh CT. Chem Biol. 2002;9:1305–1314. doi: 10.1016/s1074-5521(02)00270-3. [DOI] [PubMed] [Google Scholar]
- 190.Mulichak AM, Losey HC, Walsh CT, Garavito RM. Structure. 2001;9:547–557. doi: 10.1016/s0969-2126(01)00616-5. [DOI] [PubMed] [Google Scholar]
- 191.Armstrong SK, Pettis GS, Forrester LJ, McIntosh MA. Mol Microbiol. 1989;3:757–766. doi: 10.1111/j.1365-2958.1989.tb00224.x. [DOI] [PubMed] [Google Scholar]
- 192.Woodrow GC, Young IG, Gibson F. Biochim Biophys Acta. 1979;582:145–153. doi: 10.1016/0304-4165(79)90297-6. [DOI] [PubMed] [Google Scholar]
- 193.Hantash FM, Earhart CF. J Bacteriol. 2000;182:1768–1773. doi: 10.1128/jb.182.6.1768-1773.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wu BN, Zhang YM, Rock CO, Zheng JJ. Protein Sci. 2009;18:240–246. doi: 10.1002/pro.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Evans SE, Williams C, Arthur CJ, Burston SG, Simpson TJ, Crosby J, Crump MP. Chembiochem. 2008;9:2424–2432. doi: 10.1002/cbic.200800180. [DOI] [PubMed] [Google Scholar]
- 196.Roujeinikova A, Simon WJ, Gilroy J, Rice DW, Rafferty JB, Slabas AR. J Mol Biol. 2007;365:135–145. doi: 10.1016/j.jmb.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 197.Castonguay R, Valenzano CR, Chen AY, Keatinge-Clay A, Khosla C, Cane DE. J Am Chem Soc. 2008;130:11598–11599. doi: 10.1021/ja804453p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Strieter ER, Koglin A, Aron ZD, Walsh CT. J Am Chem Soc. 2009;131:2113–2115. doi: 10.1021/ja8077945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gatto GJ, Jr, McLoughlin SM, Kelleher NL, Walsh CT. Biochemistry. 2005;44:5993–6002. doi: 10.1021/bi050230w. [DOI] [PubMed] [Google Scholar]
- 200.Schneider TL, Shen B, Walsh CT. Biochemistry. 2003;42:9722–9730. doi: 10.1021/bi034792w. [DOI] [PubMed] [Google Scholar]
- 201.O'Connor SE, Chen H, Walsh CT. Biochemistry. 2002;41:5685–5694. doi: 10.1021/bi020006w. [DOI] [PubMed] [Google Scholar]
- 202.Julien B, Shah S, Ziermann R, Goldman R, Katz L, Khosla C. Gene. 2000;249:153–160. doi: 10.1016/s0378-1119(00)00149-9. [DOI] [PubMed] [Google Scholar]
- 203.Liu F, Garneau S, Walsh CT. Chem Biol. 2004;11:1533–1542. doi: 10.1016/j.chembiol.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 204.Gokhale RS, Tsuji SY, Cane DE, Khosla C. Science. 1999;284:482–485. doi: 10.1126/science.284.5413.482. [DOI] [PubMed] [Google Scholar]
- 205.Stein DB, Linne U, Hahn M, Marahiel MA. Chembiochem. 2006;7:1807–1814. doi: 10.1002/cbic.200600192. [DOI] [PubMed] [Google Scholar]
- 206.Wu N, Tsuji SY, Cane DE, Khosla C. J Am Chem Soc. 2001;123:6465–6474. doi: 10.1021/ja010219t. [DOI] [PubMed] [Google Scholar]
- 207.Wu N, Cane DE, Khosla C. Biochemistry. 2002;41:5056–5066. doi: 10.1021/bi012086u. [DOI] [PubMed] [Google Scholar]