

Abstract
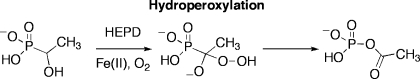
Hydroxyethylphosphonate dioxygenase (HEPD) catalyzes the O2-dependent cleavage of the carbon−carbon bond of 2-hydroxyethylphosphonate (2-HEP) to afford hydroxymethylphosphonate (HMP) and formate without input of electrons or use of any organic cofactors. Two mechanisms have been proposed to account for this reaction. One involves initial hydroxylation of substrate to an acetal intermediate and its subsequent attack onto an Fe(IV)-oxo species. The second mechanism features initial hydroperoxylation of substrate followed by a Criegee rearrangement. To distinguish between the two mechanisms, substrate analogues were synthesized and presented to the enzyme. Hydroxymethylphosphonate was converted into phosphate and formate, and 1-hydroxyethylphosphonate was converted to acetylphosphate, which is an inhibitor of the enzyme. These results provide strong support for a Criegee rearrangement with a phosphorus-based migrating group and require that the O−O bond of molecular oxygen is not cleaved prior to substrate activation. (2R)-Hydroxypropylphosphonate partitioned between conversion to 2-oxopropylphosphonate and hydroxymethylphosphonate, with the latter in turn converted to phosphate and formate. Collectively, these results support a mechanism that proceeds by hydroperoxylation followed by a Criegee rearrangement.
Introduction
Natural product phosphonates are in wide use in medicine and agriculture.(1) One member of this class of natural products, phosphinothricin (PT), is an unusual amino acid with a phosphinate side chain (Scheme 1). Synthetic PT (glufosinate) is the active component of the commercially important herbicides Liberty, Ignite, and Basta, which are used in conjunction with transgenic crops such as corn, soybean, cotton, and canola. The mode of action of PT is due to the unique carbon−phosphorus−carbon moiety that mimics the δ-carboxylate of glutamate thereby inhibiting glutamine synthetase resulting in a toxic buildup of ammonia and glutamine starvation in the plant.(2) Recent genetic and biochemical studies identified 2-hydroxyethylphosphonate (2-HEP) and hydroxymethylphosphonate (HMP) as intermediates in the PT biosynthetic pathway.(3) During PT biosynthesis, the carbon−carbon bond of 2-HEP is cleaved to afford HMP and formate by the product of the phpD gene, hydroxyethylphosphonate dioxygenase (HEPD, Scheme 1). Initial characterization has revealed that HEPD is a member of the cupin superfamily of proteins and that it only requires molecular oxygen and ferrous iron for activity.(4) 2-HEP analogues deuterium-labeled at the C1 or C2 positions showed that the hydrogen atoms at C1 are retained in HMP and that one of the hydrogen atoms at C2 is present in formate. In addition, labeling studies with 18O2 and H218O demonstrated that HEPD is a dioxygenase. The active site of HEPD contains a 2-His/1-Glu facial triad(5) characteristic of a large class of nonheme iron dependent enzymes.(6) A cocrystal structure, containing 2-HEP coordinated to a Cd2+-ion in the active site, demonstrated bidentate metal coordination of the substrate.(4) Although HEPD lacks significant sequence homology to other previously characterized enzymes, the crystal structure revealed that the active site architecture and tertiary structure of HEPD are similar to 2-hydroxypropylphosphonate epoxidase (HppE),(7) an enzyme involved in the biosynthesis of the phosphonate antibiotic fosfomycin.(8) HEPD and HppE not only share similar structures but also act on similar substrates, 2-HEP and (2S)-hydroxypropylphosphonate ((2S)-HPP), respectively.
Scheme 1. 2-HEP is Converted to HMP and Formate by HEPD during the Biosynthesis of PT.

On the basis of the initial characterization, two mechanisms were proposed for the transformation of 2-HEP into formate and HMP (Scheme 2). In both mechanisms, catalysis is initiated by bidentate coordination of substrate to the ferrous iron followed by binding of molecular oxygen to form an Fe(III)−O2•− species, which is postulated to abstract a hydrogen atom from C2 of 2-HEP to form Fe(III)−O−OH (I) and a carbon centered radical. Although these initial steps are not uniformly accepted for nonheme iron dependent enzymes, elegant studies by the laboratories of Bollinger and Krebs have provided spectroscopic and kinetic support for the feasibility of these steps for myo-inositol oxygenase (MIOX).9−11 The proposed mechanistic pathways for HEPD then bifurcate. In one mechanism, the ferric hydroperoxide and carbon-based radical proceed to form an alkylhydroperoxide and a ferrous iron center (Scheme 2). The peroxy hemiacetal intermediate formed can then undergo a Criegee rearrangement to afford O-formyl-HMP (IV). Hydrolysis of this intermediate at C1 (necessary to account for observed isotope washout during the transformation(4)) would afford the products. Alternatively, if the substrate radical in intermediate I reacts with the terminal oxygen atom of the ferric-hydroperoxide, the substrate is hydroxylated and an Fe(IV)=O intermediate (II) is generated. The hemiacetal in intermediate II can then undergo a retro-Claisen-like carbon−carbon bond scission affording formate and a Horner−Wadsworth−Emmons-like carbanion (III) that is stabilized by metal coordination. This electron rich intermediate can then attack the oxygen atom of the electrophilic ferryl species to provide the observed products.(4)
Scheme 2. Proposed Mechanisms for Conversion of 2-HEP to HMP and Formate by HEPD.

Initial support for the hydroxylation mechanism came from the observation that HEPD oxidizes 2-HPP to 2-oxopropylphosphonate (OPP), with the enzyme being inactivated in the process.(4) Both proposed mechanisms can account for the oxidation of 2-HPP and the inactivation of HEPD, but the mechanisms of enzyme inactivation are different (Scheme 3). The hydroperoxylation mechanism predicts that the enzyme is inactivated via oxidation of ferrous iron by hydrogen peroxide produced in the Criegee rearrangement whereas in the hydroxylation mechanism the metal is trapped in the inactive Fe(IV) state. Since hydrogen peroxide was not observed in the previous study, the latter mechanism was favored.(4) To further investigate the mechanism of catalysis, in this study additional substrate analogues, putative catalytic intermediates, and labeled substrates were synthesized and presented to HEPD.
Scheme 3. Two Proposed Mechanisms That Can Account for Inactivation of HEPD during Oxidation of 2-HPP.

Results
HMP is Converted to Phosphate and Formate by HEPD
Incubation of 2-HEP (2 mM) with HEPD (10 μM) resulted in the expected appearance of a peak in the 31P NMR spectrum corresponding to HMP. Unexpectedly, after full conversion of 2-HEP to HMP, the peak corresponding to HMP slowly decreased in intensity with the concomitant appearance of a new peak at δ = 2.3 ppm that was shown to correspond to phosphate by spiking with authentic material. When HMP (2 mM) was incubated with HEPD (10 μM) the recorded 31P NMR spectrum showed appearance of the same new peak (Figure 1a). To identify the carbon-containing product, synthetic 2H2−HMP was incubated with HEPD, and aliquots of the reaction were quenched at various time intervals. Organic acids were derivatized to the 2-nitrophenylhydrazide and analyzed via LC-MS, demonstrating time-dependent production of 2H-formate (Figure 1b). Leaving out any component from the standard assay conditions (HEPD, HMP, or Fe(II)) did not result in production of formate. Additionally, when 2-HEP was incubated for extended periods of time with stoichiometric amounts of HEPD, all substrate was consumed and two equivalents of formate were produced, one from conversion of 2-HEP to HMP and formate, and one from the subsequent oxidation of HMP to phosphate and formate.
Figure 1.

HEPD catalyzes oxidation of HMP to afford (a) phosphate (Pi), as evidenced by 31P NMR spectroscopy, and (b) 2H-formate from 2H2−HMP as the carbon containing product in a time dependent fashion as evidenced by LC-MS.
O-Formyl-HMP (OFHMP) is Not Accepted as a Substrate
OFHMP (IV, Scheme 2) is a proposed intermediate on the hydroperoxylation mechanistic pathway. Therefore, OFHMP was synthesized and presented as a potential substrate to ferrous HEPD, the oxidation state of the enzyme that OFHMP would interact with as an intermediate (Scheme 2). OFHMP is not particularly stable and was partially hydrolyzed to HMP and formate during workup of the synthetic procedure. The sample presented to HEPD therefore contained both OFHMP and HMP in a 1:1 ratio. A control experiment showed that further hydrolysis is very slow under the HEPD assay conditions without HEPD. Thus, a sample of OFHMP and HMP (1:1) was incubated with HEPD and the reaction was monitored by 31P NMR spectroscopy revealing peaks corresponding to phosphate and OFHMP, and a HMP peak that had strongly decreased in intensity compared to OFHMP (Figure S1). The ratio of the intensities of the peaks corresponding to phosphate and OFHMP was 1.0:0.9 indicating that most of the HMP present in the original mixture had been oxidized to phosphate, consistent with the data presented in the previous section. However, the experiment also showed that no enzyme catalyzed hydrolysis of OFHMP had occurred.
1-Hydroxyethylphosphonate and 1-Hydroxy-2,2,2-trifluoroethylphosphonate Are Substrates
Given that HMP is a substrate for HEPD, 1-hydroxyethylphosphonate (1-HEP) and 1-hydroxy-2,2,2-trifluoroethylphosphonate (1-HEP-CF3) were tested as substrate analogues. Under multiple turnover conditions (10 μM HEPD and 2 mM substrate analogue), neither compound underwent chemistry discernible by 31P NMR spectroscopy. Each substrate analogue (1.0 mM) was then incubated with 0.5 equiv of HEPD and, after acidic workup, two peaks were observed in both experiments. One peak corresponded to the starting material and the other peak at δ = 2.3 ppm corresponded to phosphate.
To determine the other products generated from 1-HEP, the substrate analogue (0.3 mM) was incubated with HEPD (0.1 mM) for 2 h, followed by analysis of organic acids by LC-MS after derivatization as described in the experimental section. A peak was observed in the chromatogram with a retention time and mass consistent with derivatized acetate. The produced acetate was quantified (0.11 mM), closely corresponding to the amount of HEPD in the assay. The phosphorus-containing product before acidic workup was then determined by incubating the analogue with HEPD and analysis by 31P NMR spectroscopy (Figure 2a). The spectrum showed three peaks; one for unreacted 1-HEP, a small peak for phosphate and an additional peak at δ = −2.2 ppm. Sulphuric acid was then added to the assay, the precipitated enzyme was removed by centrifugation, and another 31P NMR spectrum was recorded. The spectrum showed a peak for 1-HEP and a peak for phosphate suggesting the third peak at δ = −2.2 ppm was associated with an acid labile intermediate that can be converted to phosphate under acidic conditions. This intermediate was then identified as acetyl phosphate (AP) by spiking with authentic material. Because 1-HEP was only converted when incubated with high concentrations of HEPD, the enzyme is likely inactivated during the conversion of 1-HEP to acetylphosphate. Indeed, preincubation of HEPD (2 μM) with acetylphosphate (100 μM) inhibited oxidation of its native substrate 2-HEP. Similarly, incubation of HEPD (100 μM) with 1 equiv of AP decreased the rate of formate production by 67% compared to an assay without AP.
Figure 2.

31P NMR spectra after incubation of HEPD with (a) 1-HEP, (b) 1-HEP-CF3. Pi = inorganic phosphate.
The 31P NMR spectrum for the reaction of 1-HEP-CF3 with HEPD also showed a peak for the starting material and a peak for phosphate (Figure 2b). To determine the identity of any nonphosphorus containing products, 1-HEP-CF3 (0.3 mM) was incubated with HEPD (0.1 mM) and the 19F NMR spectrum was recorded. Two peaks were observed, with one peak corresponding to unreacted 1-HEP-CF3 and a second signal corresponding to trifluoroacetate, as demonstrated with an authentic sample. Integration of the peaks in the spectrum showed a ratio of 1:2 for trifluoroacetate:1-HEP-CF3. The amount of trifluoroacetate and phosphate formed was approximately equal to the amount of HEPD in the reaction, suggesting that HEPD was inactivated after converting 1-HEP-CF3 to trifluoroacetate and phosphate. However, no reduction of formate production from 2-HEP was observed after preincubation of HEPD with trifluoroacetate by itself or with trifluoroacetate and phosphate.
Stoichiometry of Conversion of 2-HPP to OPP
We previously reported that 2-HPP is accepted as a substrate for HEPD and converted to OPP.(4) To determine the stereochemistry of this transformation (2S)- and (2R)-HPP were prepared(12) and presented to HEPD and a 31P NMR spectrum was recorded after 2 h. The spectrum showed oxidation for the R but not the S isomer. The stoichiometry of (2R)-HPP consumed to OPP produced was determined by incubation of HEPD with a 3-fold excess of (2R)-HPP and quantification of the OPP generated via LC-MS. Reaction of 0.1 mM, 0.25 mM, or 0.5 mM HEPD with 0.3 mM, 0.75 mM, and 1.5 mM (2R)-HPP produced OPP concentrations of 0.052 ± 0.011 mM, 0.122 ± 0.005 mM and 0.220 ± 0.016 mM, respectively. These three experiments demonstrate stoichiometries of OPP produced to HEPD of 0.52:1, 0.49:1 and 0.44:1, respectively. In the hydroperoxylation mechanism, each conversion of (2R)-HPP to OPP would produce one equivalent of H2O2. We previously showed that one equivalent of H2O2 rapidly inactivates approximately two ferrous HEPD molecules,(4) and hence the observed stoichiometries at first glance appear to agree very well with the hydroperoxylation mechanism (i.e., each turnover of HEPD results in one OPP and ∼2 oxidized, inactive enzymes). One caveat is that complete conversion of (2R)-HPP to OPP is assumed in this model.
To probe if (2R)-HPP was cleanly converted to OPP, HEPD (0.5 mM) was incubated with (2R)-HPP (1.5 mM). EDTA and dithionite were added to improve signal quality through sharpening of the peaks in the 31P NMR spectrum (Figure 3) that were very broad at the high enzyme concentrations used. The spectrum after 2 h showed peaks corresponding to (2R)-HPP, OPP, and phosphate in a 1:0.4:0.33 ratio; this ratio did not change after the initial enzymatic conversion. Since all phosphorus-containing products originated from (2R)-HPP, the concentrations of (2R)-HPP, OPP and phosphate are approximately 867, 346, and 286 μM, respectively. To account for the presence of phosphate, OPP was incubated under identical conditions with stoichiometric amounts of HEPD and the 31P NMR spectrum was recorded, but no conversion of OPP to phosphate was observed. Then (2R)-HPP (1.5 mM) was incubated with HEPD (0.5 mM) for 2 h and the sample was analyzed for organic acids. Both acetate and formate (364 and 216 μM, respectively) were observed in significantly higher concentrations than in control reactions (62 and 16 μM of acetate and formate, respectively). Collectively, these results are consistent with partitioning of (2R)-HPP to generate OPP, which is unreactive toward further enzymatic oxidation, in addition to acetate and HMP (Scheme 4). Subsequent oxidation of HMP produces the observed formate and phosphate products, as discussed above.
Figure 3.

31P NMR spectrum after incubation of HEPD with (2R)-HPP demonstrating formation of OPP as well as inorganic phosphate (Pi).
Scheme 4. Reaction of (2R)-HPP Partitions between Formation of OPP and Phosphate (presumably via HMP).

Fate of the Hydroxyl Oxygen of 2-HEP
The previously proposed mechanisms(4) were restricted by the outcome of studies using either 18O2 or H218O, which provided complementary evidence that the hydroxyl oxygen in the HMP product was derived in part from molecular oxygen and in part from solvent. A third possibility that was not considered involves partial incorporation of the hydroxyl oxygen from 2-HEP into HMP. A possible mechanism for such a process is shown in Scheme 5 and in Scheme S1 in the Supporting Information. After generation of the peroxy hemiacetal (V), this species could be converted into a dioxirane intermediate (VI). Such a mechanism has precedent in reactions in organic solvents(13) and usually involves an activated oxygen leaving group, a role that might be played by an active site acid in the enzyme (Scheme 5). The carbonyloxide resonance structures VII can be drawn for the dioxirane that then illustrate a mechanism for exchange analogous to that proposed for the observed exchange in organic solvents. For HEPD, exchange of the hydroxide originating from the peroxy group with solvent and reformation of a peroxy hemiacetal could result in incorporation of oxygen from substrate and/or molecular oxygen (Scheme 5), and/or solvent (Scheme S1) into the hydroxyl of the HMP product. To probe the feasibility of such a mechanism, the hydroxyl group in 2-HEP was synthetically labeled with 18O,(14) and the labeled substrate was presented to HEPD. Analysis of HMP by LC-MS and of formate via derivatization to the TBDMS ester and analysis by GCMS revealed that the 18O label was present exclusively in formate and not HMP (Figure 4). In combination with the observed 40% wash-in of oxygen derived from solvent into the HMP product,(4) the mechanism in Scheme 5 is unlikely (see also Scheme S1).
Scheme 5. Alternative Mechanism That Would Result in Retention of 18O Label from 2-18OH-HEP in HMP.

Blue circles indicate oxygen from substrate, orange circles indicate oxygen from O2, and half filled circles represent oxygens with potential solvent wash-out/wash-in.
Figure 4.

(a) Extracted ion chromatograms for HMP (black line) and 18O HMP (gray line) from reaction of 2-18OH-HEP. (b) Mass spectrum of 18O formate (105 m/z), the peak at m/z 103 is spurious formate.
Studies with Additional Substrate Analogues
Other analogues of HEP tested as potential substrates of HEPD are shown in Figure 5. The compounds were incubated with HEPD under both catalytic and single turnover conditions and the assays were analyzed via 31P NMR spectroscopy and LC-MS. Both methods showed that none of the tested analogues gave rise to new products when compared to authentic standards.
Figure 5.

Additional analogues tested as substrates for HEPD.
Discussion
The studies with structural analogues of 2-HEP presented here provide new information toward a better understanding of the mechanism of HEPD. At present, we do not have direct evidence for the early steps in the proposed mechanistic cycle, but because external reducing equivalents are not required for the overall transformation, and because the substrate does not contain low energy electrons, there appears to be no pathway for formation of more reactive species than a formally ferric-superoxo intermediate. In addition, the results of the current study also strongly suggest the O−O bond is not broken prior to hydrogen atom abstraction from substrate. Two other nonheme iron dependent enzymes also achieve substrate oxidation without input of exogenous electrons. For myo-inositol oxygenase (MIOX), Bollinger and Krebs and co-workers have provided strong spectroscopic and kinetic evidence that a ferric-superoxo species is formed reversibly in the confines of an enzyme active site and that it is capable of abstracting a hydrogen atom from a C−H bond that is activated by a geminal hydroxyl group.9−11,15,16 DFT studies on isopenicillin N-synthase (IPNS) concluded that the formation of a ferric superoxo form of the enzyme is favorable only because of charge donation from a thiolate ligand from substrate; similar calculations on phenylalanine hydroxylase, another facial triad, mononuclear nonheme iron dependent enzyme, demonstrated that oxygen binding to the ferrous enzyme was highly unfavorable.(17) In the latter computations, two water molecules occupied the sites that in HEPD are occupied by alkoxide and phosphonate ligands of the 2-HEP substrate. Although the oxygen ligands of 2-HEP may not provide the same charge donation as the thiolate ligand of the δ-(l-α-aminoadipoyl)-l-cysteinyl-d-valine (ACV) substrate of IPNS, substrate coordination in HEPD, as well as in HppE,(7) may also enhance oxygen binding and activation.
With respect to the steps after hydrogen atom abstraction, this study with substrate analogs strongly supports a hydroperoxylation pathway for HEPD. Most insightful are the experiments with 1-HEP resulting in the formation of acetylphosphate. This transformation is most readily explained as a Criegee-like rearrangement (Scheme 6) in which a phosphorus atom migrates. Acetylphosphate is shown to be a potent inhibitor of HEPD explaining why only a single turnover occurs, with the inhibition likely due to the resemblance of acetylphosphate to HMP-formyl ester. The proposed phosphorus migration has precedent in Baeyer−Villiger oxidation of dialkyl acylphosphonates in organic solvents that have been shown to proceed through a Criegee intermediate.(18) The hydroperoxylation mechanism can also explain the observed conversion of HMP into phosphate and formate with formylphosphate a likely intermediate (Scheme 6). Unlike acetyl phosphate, which has been shown to be stable at neutral pH for months,(19) formylphosphate is unstable and readily hydrolyzes nonenzymatically to phosphate and formate as observed by Benkovic and co-workers for the reaction catalyzed by PurT GAR formyltransferase.(20) HEPD is regenerated in its Fe(II) state in this model and, unlike with 1-HEP, the enzyme is not inactivated due to the rapid hydrolysis of formylphosphate, accounting for the catalytic turnover of HMP to phosphate and formate. Elucidating the details of the mechanism by which the alkylhydroperoxides are formed from a ferric hydroperoxide and a carbon based radical requires further studies.
Scheme 6. Mechanism for Reaction of 1-Hydroxyethylphosphonates with HEPD.

The results with 1-HEP-CF3 can also be rationalized by the hydroperoxylation pathway. After initial hydroperoxylation, Criegee-like rearrangement would result in trifluoroacetylphosphate, which is more reactive toward hydrolysis than acetylphosphate and is not detected directly. Instead, the hydrolysis products trifluoroacetate and phosphate were observed. Why HEPD is inactivated during this process resulting in a single turnover is not clear as trifluoroacetate is not an inhibitor. It is possible that trifluoroacetyl phosphate remains bound to the iron center after its formation in the active site and that hydrolysis only occurs after protein denaturation associated with product analysis.
The results of the detailed investigation of the conversion of (2R)-HPP to OPP are also consistent with initial hydroperoxylation followed by a Criegee rearrangement. Previously, we did not observe hydrogen peroxide production,(4) which appeared to be incompatible with the hydroperoxylation mechanism because stoichiometric conversion of (2R)-HPP to OPP should produce 1 equivalent of H2O2. Half an equivalent of H2O2 has been shown to inactivate approximately 1 equiv of Fe(II)-HEPD,(4) and thus ∼0.5 equivalents of H2O2 should remain upon oxidation of (2R)-HPP and should have been detected. However, as noted in the Supporting Information of our previous study, a caveat in this model was that the conversion of (2R)-HPP to OPP could not be quantitated at the time. In this study, we were able to analyze the products by 31P NMR spectroscopy after solving previous problems with peak broadening. The results show that conversion of (2R)-HPP to OPP is not quantitative and that (2R)-HPP oxidation partitions between formation of OPP and HMP along with acetate (Scheme 7). The latter reaction is similar to the physiological reaction, conversion of 2-HEP to HMP and formate. Presumably, an intermediate in this process is O-acetyl-hydroxymethylphosphonate (VIII) that is hydrolyzed to acetate and HMP. HMP is then converted to formate and phosphate as discussed above. The partitioning also explains why H2O2 was not observed in the previous study since only ∼0.6 equivalents of OPP (and hence H2O2) are formed with respect to HEPD. This amount is sufficient to completely oxidize the enzyme, but very little H2O2 would remain after HEPD oxidation. The partitioning between carbon migration and peroxide elimination suggests that adding an additional methyl group to C2 results in less favorable orbital overlap for a Criegee rearrangement such that peroxide elimination competes. The observed conversion of (2R)-HPP to OPP is reminiscent of the conversion by HppE of (2R)-HPP, the enantiomer of the physiological substrate, to OPP. Thus, when a hydrogen atom is available for abstraction at C2, both HEPD and HppE carry out the same transformation with (2R)-HPP. On the other hand, unlike HppE, HEPD cannot be coerced to abstract a hydrogen atom from C1 of (2S)-HPP. We note that the observed oxidation of (2R)-HPP and not (2S)-HPP by HEPD is consistent with the active site geometry and bidentate binding mode of 2-HEP observed in the cocrystal structure of Cd2+-HEPD with 2-HEP.(4)
Scheme 7. Mechanism for Oxidation of (2R)-HPP to OPP as well as Acetate, Formate, and Phosphate.

Unfortunately, attempts to obtain direct evidence for the hydroperoxylation/Criegee rearrangement mechanism with 2-HEP as substrate have so far been inconclusive. OFHMP, the penultimate intermediate in the hydroperoxylation pathway with 2-HEP, was synthesized and presented as a substrate for HEPD but enzymatic hydrolysis was not observed. This result may be rationalized by inability of the product to bind to the reactive form of the enzyme, which may only be accessed during turnover. Alternative direct support for the hydroperoxylation followed by a Criegee rearrangement may be obtained with 2-HEP stereospecifically labeled at C1. Net inversion of configuration is predicted by this mechanism (retention in the Criegee rearrangement step and inversion for subsequent hydrolysis at C1), whereas net retention of configuration is most likely for the hydroxylation mechanism; experiments to examine these predictions are ongoing.
In summary, the studies presented here with substrate analogues provide evidence for a hydroperoxylation mechanism for HEPD that is followed by a Criegee-like rearrangement. As such, the enzyme has similarities with flavin dependent Bayer-Villigerases,21−24 except that the substrate for HEPD is at a lower oxidation level than the carbonyl-containing substrates of these enzymes. The corollary of the hydroperoxylation mechanism of HEPD is that the O−O bond of molecular oxygen is not broken prior to substrate activation, as has been proposed for the aforementioned nonheme iron enzymes MIOX and IPNS, for a diiron(II/III) superoxide model complex,(25) and for the copper-dependent enzymes dopamine β monooxygenase, peptidyl glycine α-hydroxylating monooxygenase, and galactose oxidase.26−30 We note that Seto and co-workers in pioneering studies had proposed that phosphonoacetaldehyde was converted to HMP via a Baeyer−Villiger like oxidation to OFHMP followed by nonenzymatic hydrolysis.(31) Although the molecular details turn out to be different, the overall biosynthetic logic is remarkably similar.
Experimental Procedures
General
HEPD was overexpressed in E. coli as an N-terminal His6-fusion protein and purified via affinity chromatography as previously described.(4) Protein concentrations were determined by the method of Bradford using bovine serum albumin as the standard. In addition, a theoretical extinction coefficient of 79 995 M−1 cm−1 was calculated using Expert Protein Analysis System (ExPASy, http://www.expasy.org) and concentrations determined using this value were within ∼10% of the concentrations determined by the Bradford assay. Thus, while small deviations from the reaction stoichiometries reported herein are possible, they would not change the interpretation of these studies. All activity assays were performed in HEPES buffer (25 mM, pH = 7.5) unless otherwise noted. LC-MS was performed on an Agilent 1200 series quad pump system equipped with a diode array detector and a G1956B mass spectrometer with a multimode-electrospray/atmospheric pressure chemical ionization (MM-ES+APCI) source. The column used for separation was a Synergi 4 μ Fusion-RP 80A column (150 × 4.6 mm, 4 μm, Phenomenex Torrance, CA) using a flow rate of 0.5 mL/min. UV−visible absorption spectra were recorded on a Varian Cary 4000 UV−vis spectrophotometer. GC-MS was performed on an Agilent 6890N gas chromatograph equipped with an electron impact (EI) ionization source.
NMR spectra were recorded on a Varian Unity 500 or Varian Unity Inova 600 spectrometer. Proton and carbon chemical shifts are reported in δ values relative to an external standard of 0.1% tetramethylsilane in CDCl3 (0.00 ppm). Phosphorus shifts are reported in δ values relative to an external standard of 85% phosphoric acid (0.00 ppm). Fluorine shifts are reported in δ values relative to an external standard of CFCl3 (0.00 ppm). Fast atom bombardment (FAB) mass spectrometry for characterization of synthetic compounds was performed by the University of Illinois Mass Spectrometry Center using a Waters 70-SE-4F mass spectrometer.
2-HEP and HMP were synthesized as previously reported.(4) The following substrate analogues were synthesized according to literature procedures: 2-18OH-HEP,(14) (2R)- and (2S)-HPP,(12) 3-hydroxypropylphosphonate,(32) 2-hydroxybutylphosphonate,(33) 3-fluoro-2-hydroxypropylphosphonate,(34) 2-fluoroethylphosphonate,(35) phosphonoacetaldehyde,(36) and 1-hydroxyethylphosphonate.(37) Ethylphosphonate and aminoethylphosphonate were purchased from Sigma-Aldrich. 2-Nitrophenylhydrazine (NPH) was purchased from Acros Organics. N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) was purchased from Chem-Impex (Wood Dale, IL). Dichloromethane was distilled over CaH2. All other chemicals were purchased from Sigma Aldrich at the highest purity and used without further purification.
Synthesis of O-Formyl-HMP
A round-bottom flask equipped with a magnetic stir bar and reflux condenser was charged with dibenzyl HMP(38) (500 mg, 1.7 mmol), which was dissolved in formic acid (3 mL) and heated under reflux while stirring overnight. The solution was allowed to cool to 25 °C, and Et2O (20 mL) was added. A solution of aqueous NaOH (1 M) was added dropwise until a second layer formed. The layers were separated and water was removed from the aqueous (bottom) layer under reduced pressure to afford product as a white solid. This material was a 1:1 mixture of the desired product and HMP. OFHMP was relatively stable in buffered HEPES solution (25 mM, pH = 7.5). 1H NMR (500 MHz, D2O) δ 4.09 (d, J = 8.5 Hz, 2H, CH2), 7.99 (s, 1H CHO); 13C NMR (125 MHz, D2O) δ 59.4 (d, J = 156.9 Hz), 163.5; 31P NMR (202 MHz, D2O) δ 13.7.
Synthesis of 1-Hydroxy-2,2,2-trifluoroethylphosphonate
Under a nitrogen atmosphere a round-bottom flask equipped with a magnetic stir bar and reflux condenser was charged with commercially available diethyl 1-hydroxy-2,2,2-trifluoroethylphosphonate (1 g, 4.2 mmol, 1 equiv), which was dissolved in dry DCM (10 mL). To the solution was added bromotrimethylsilane (2 mL, 15.1 mmol, 3.6 equiv) and the orange solution was heated under reflux while stirring for 2 h. The solution was allowed to cool to 25 °C and solvent was removed under reduced pressure. The resulting residue was taken up in 1:1 H2O:EtOH (10 mL) and solvent was removed under reduced pressure to afford product (663 mg, 3.7 mmol, 88%) as a tan oil. 1H NMR (500 MHz, D2O) δ 4.09 (m); 13C NMR (125 MHz, D2O) δ 27.76 (d, J = 8.5), 66.01 (m); 31P NMR (202 MHz, D2O) δ 11.13 (bs); 19F NMR (215 MHz, D2O) δ 17.34 (at); HRMS (FAB+) calcd for (C2H4F3O4P+H+) 180.9878 m/z found 180.9880 m/z.
Identification of Assay Products via LC-MS
Enzymatic assays were quenched by adding H2SO4 (30 mM) and the protein was pelleted via microcentrifugation (1 min at 16.1g). The supernatant was analyzed via LC-MS (positive mode) using aqueous 0.1% formic acid as an isocratic mobile phase and scanning masses of 50−500 m/z.
Identification of Assay Products via 31P NMR Spectroscopy
HEPD was separated from the assay via centrifugal filtration (Millipore Micron YM-30, 30 kDa nominal molecular weight limit). D2O (20% v/v) was added to the flow-through and the 31P NMR spectrum was recorded on a Varian UI600 (frequency for phosphorus: 242 MHz).
Aqueous Derivatization and Quantification of Organic Acids
Following a modified literature procedure,(39) to a 100 μL sample was added propionic acid as an internal standard (10 μL of a 1 mM stock solution in 1:1 pyridine:HCl), followed by EDC (10 μL of a 0.29 M stock solution), and subsequently NPH (10 μL of a 0.12 M stock solution in 250 mM aqueous HCl). The orange solution was heated to 60 °C in a water bath for 15 min and precipitated protein was pelleted via microcentrifugation (1 min at 16.1g). The supernatant (20 μL) was analyzed via LC-MS using an isocratic mobile phase (50:50 MeOH:H2O + 0.1% formic acid) for separation. Authentic standards of sodium 13C-formate, sodium acetate, and propionic acid were also derivatized and analyzed by monitoring the absorbance at 400 nm and by MM-ES+APCI (negative mode) and found to have retention times of 5.81, 5.93, and 6.93 min, respectively. The observed m/z values for each derivatized compound corresponded to the monodeprotonated hydrazide and were: 181 (13C-formate), 194 (acetate), and 208 (propionic acid).
Solutions of varying concentrations of each of these acids were made in HEPES buffer (25 mM, pH = 7.5), derivatized, and analyzed via LC-MS as described previously using single ion monitoring (SIM) for the derivatized acid of interest. The area of the peak corresponding to derivatized acid was divided by the area of the derivatized propionic acid peak to afford a response factor, which was plotted against the acid concentration to afford a linear relationship. This equation was used to quantify the organic acid concentration in the enzymatic assays.
Analysis of HMP as a Substrate for HEPD
In a typical assay HEPD (10 μM) and HMP (2 mM) were added to oxygenated buffer (25 mM HEPES, pH 7.5) and incubated at 25 °C for 2 h. The sample was then analyzed via 31P NMR spectroscopy (proton coupled and decoupled). The carbon-containing product was identified by running an assay similar to the one described above except using 2H2−HMP as the substrate and subsequent derivatization and quantification of organic acids as described above.
Test of O-Formyl-HMP as a Substrate for HEPD
In a typical assay, O-formyl-2-hydroxymethylphosphonate (OFHMP, 2 mM) was incubated with HEPD (10 μM) for 2 h and samples were analyzed via 31P NMR spectroscopy. A control sample without HEPD was also analyzed in a similar fashion.
Oxidation of HPP to OPP
To determine which enantiomer of HPP is oxidized, HEPD (500 μM) and either (2S)- or (2R)-HPP (1.5 mM) were incubated in oxygenated HEPES buffer (25 mM, pH 7.5) for 2 h at 25 °C. Then the products were analyzed by 31P NMR spectroscopy and LC-MS. The masses for protonated 2-HPP (141) and OPP (139) were extracted from the total ion chromatogram (TIC) to afford a single peak in each extracted ion chromatogram (EIC) with retention times of 6 and 7 min, respectively, that were identical to authentic standards of 2-HPP and OPP.
To determine the stoichiometry of the oxidation of (2R)-HPP to OPP via LC-MS, first a linear relationship between OPP concentration and area of the peak in the EIC was established by analyzing varying concentrations of synthetic OPP in buffer (50, 100, 250, 500, and 1000 μM). Similar assays as previously described were then carried out with varying concentrations of HEPD (100, 250, and 500 μM) and (2R)-HPP (3-fold excess over HEPD), which were analyzed via LC-MS.
To determine the stoichiometry of the oxidation of (2R)-HPP to OPP by NMR spectroscopy, (2R)-HPP (1.5 mM) was incubated with HEPD (500 μM) for 2 h. EDTA (50 mM) then dithionite (10 mM) and D2O (20% v/v) were added and the 31P NMR spectrum was recorded. The EDTA and dithionite were necessary to abrogate the line broadening effect of a high concentration of paramagnetic Fe(III) in the sample. Additionally, (2R)-HPP (1.5 mM) was incubated with HEPD (500 μM) for 2 h and the sample was analyzed for organic acids as described above.
Analysis of 1-Hydroxyethyl Phosphonates as Substrates for HEPD
To identify the carbon-containing product of 1-hydroxyethylphosphonate, HEPD (100 μM) was incubated in oxygenated buffer with 1-HEP (300 μM) for 2 h at 25 °C. The organic acids were then derivatized and analyzed via LC-MS as described above. To identify the product of 1-HEP-CF3, HEPD (100 μM) was incubated in oxygenated HEPES buffer with 1-HEP-CF3 (300 μM) for 2 h at 25 °C, then D2O (20% v/v) was added and the 19F NMR spectrum recorded. Standards of 1-HEP-CF3 and sodium trifluoroacetate showed peaks at −72.5 ppm and −76.0 ppm, respectively. To identify the phosphorus containing product, 1-hydroxy ethylphosphonates (1.5 mM) were incubated with HEPD (500 μM) for 2 h. EDTA (50 mM) then dithionite (10 mM) and D2O (20% v/v) were added and the 31P NMR spectrum was recorded. Standards of 1-HEP and 1-HEP-CF3 under similar conditions had chemical shifts of 19.2 ppm and 7.3 ppm, respectively. Phosphate, phosphite, and acetyl phosphate under these conditions had chemical shifts of 2.3 ppm, 4 ppm, and −2.2 ppm respectively.
Tracking the Fate of the Hydroxyl Group from 2-18OH-HEP
HEPD (2 μM) and 2-18OH-HEP (2 mM) were added to oxygenated buffer, incubated at 25 °C for 2 h, and then analyzed via LCMS as described above. The masses for protonated HMP and 18OH-HMP (115 and 117, respectively) were extracted to afford a single peak in each EIC with a retention time of 5.4 min. Each monoisotopic product peak was separately integrated to afford the isotopic composition. Additionally, formate was analyzed via GC-MS as previously reported(4) to determine the isotopic composition.
Acknowledgments
This work was supported by the National Institutes of Health (PO1 GM077596 to W.A.V.). We thank Scott Denmark (UIUC) for helpful discussions.
Supporting Information Available
Supplementary Figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Metcalf W. W.; van der Donk W. A. Annu. Rev. Biochem. 2009, 78, 65–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderscheid R.; Wild A. J. Plant Physiol. 1986, 123, 135–142. [Google Scholar]
- Blodgett J. A.; Thomas P. M.; Li G.; Velasquez J. E.; van der Donk W. A.; Kelleher N. L.; Metcalf W. W. Nat. Chem. Biol. 2007, 3, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchillo R. M.; Zhang H.; Blodgett J. A. V.; Whitteck J. T.; Li G.; Nair S. K.; van der Donk W. A.; Metcalf W. W. Nature 2009, 459, 871–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehntop K. D.; Emerson J. P.; Que L. Jr. J. Biol. Inorg. Chem 2005, 10, 87–93. [DOI] [PubMed] [Google Scholar]
- Ryle M. J.; Hausinger R. P. Curr. Opin. Chem. Biol. 2002, 6, 193–201. [DOI] [PubMed] [Google Scholar]
- Higgins L. J.; Yan F.; Liu P.; Liu H. W.; Drennan C. L. Nature 2005, 437, 838–844. [DOI] [PubMed] [Google Scholar]
- Liu P.; Murakami K.; Seki T.; He X.; Yeung S. M.; Kuzuyama T.; Seto H.; Liu H. W. J. Am. Chem. Soc. 2001, 123, 4619–4620. [DOI] [PubMed] [Google Scholar]
- Xing G.; Barr E. W.; Diao Y.; Hoffart L. M.; Prabhu K. S.; Arner R. J.; Reddy C. C.; Krebs C.; Bollinger J. M. Jr. Biochemistry 2006, 45, 5402–5412. [DOI] [PubMed] [Google Scholar]
- Xing G.; Diao Y.; Hoffart L. M.; Barr E. W.; Prabhu K. S.; Arner R. J.; Reddy C. C.; Krebs C.; Bollinger J. M. Jr. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 6130–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollinger J. M. Jr.; Diao Y.; Matthews M. L.; Xing G.; Krebs C. Dalton Trans. 2009, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z. B.; Liu P. H.; Murakami K.; Kuzuyama T.; Seto H.; Liu H. W. Angew. Chem., Int. Ed. 2002, 41, 4529. [DOI] [PubMed] [Google Scholar]
- Edwards J. O.; Pater R. H.; Curclf R.; Furia F. D. Photochem. Photobiol. 1979, 30, 63–70. [Google Scholar]
- Hammerschmidt F. Angew. Chem., Int. Ed. Engl. 1994, 33, 341–342. [Google Scholar]
- Kim S. H.; Xing G.; Bollinger J. M. Jr.; Krebs C.; Hoffman B. M. J. Am. Chem. Soc. 2006, 128, 10374–10375. [DOI] [PubMed] [Google Scholar]
- Xing G.; Hoffart L. M.; Diao Y.; Prabhu K. S.; Arner R. J.; Reddy C. C.; Krebs C.; Bollinger J. M. Jr. Biochemistry 2006, 45, 5393–5401. [DOI] [PubMed] [Google Scholar]
- Brown C. D.; Neidig M. L.; Neibergall M. B.; Lipscomb J. D.; Solomon E. I. J. Am. Chem. Soc. 2007, 129, 7427–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon N. J.; Evans S. A. Jr. J. Org. Chem. 1993, 58, 4516–4519. [Google Scholar]
- Lipmann F.; Tuttle L. C. J. Biol. Chem. 1944, 153, 571–582. [Google Scholar]
- Marolewski A. E.; Mattia K. M.; Warren M. S.; Benkovic S. J. Biochemistry 1997, 36, 6709–6716. [DOI] [PubMed] [Google Scholar]
- Ryerson C. C.; Ballou D. P.; Walsh C. Biochemistry 1982, 21, 2644–2655. [DOI] [PubMed] [Google Scholar]
- Kamerbeek N. M.; Moonen M. J.; Van Der Ven J. G.; Van Berkel W. J.; Fraaije M. W.; Janssen D. B. Eur. J. Biochem. 2001, 268, 2547–2557. [DOI] [PubMed] [Google Scholar]
- Sheng D.; Ballou D. P.; Massey V. Biochemistry 2001, 40, 11156–11167. [DOI] [PubMed] [Google Scholar]
- Malito E.; Alfieri A.; Fraaije M. W.; Mattevi A. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 13157–13162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X.; Que L. Jr. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 5340–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. P.; Blackburn N. J.; Klinman J. P. Biochemistry 2006, 45, 15419–15429. [DOI] [PubMed] [Google Scholar]
- Evans J. P.; Ahn K.; Klinman J. P. J. Biol. Chem. 2003, 278, 49691–49698. [DOI] [PubMed] [Google Scholar]
- Chen P.; Solomon E. I. J. Am. Chem. Soc. 2004, 126, 4991–5000. [DOI] [PubMed] [Google Scholar]
- Chen P.; Bell J.; Eipper B. A.; Solomon E. I. Biochemistry 2004, 43, 5735–5747. [DOI] [PubMed] [Google Scholar]
- Humphreys K. J.; Mirica L. M.; Wang Y.; Klinman J. P. J. Am. Chem. Soc. 2009, 131, 4657–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto H.; Kuzuyama T. Nat. Prod. Rep. 1999, 16, 589–596. [DOI] [PubMed] [Google Scholar]
- Capson T. L.; Thompson M. D.; Dixit V. M.; Gaughan R. G.; Poulter C. D. J. Org. Chem. 1988, 53, 5903–5908. [Google Scholar]
- Schweifer A.; Hammerschmidt F. Bioorg. Med. Chem. Lett. 2008, 18, 3056–3059. [DOI] [PubMed] [Google Scholar]
- Ridge J. A.; Roberts M. F.; Schaffer M. H.; Stark G. R. J. Biol. Chem. 1976, 251, 5966–5975. [PubMed] [Google Scholar]
- Saunders B. C.; Stacey G. J. J. Chem. Soc. 1948, 1773–1779. [DOI] [PubMed] [Google Scholar]
- Isbell A. F.; Englert L. F.; Rosenberg H. J. Org. Chem. 1969, 34, 755–756. [Google Scholar]
- Baraldi P. G.; Guarneri M.; Moroder F.; Simoni D.; Vicentini C. B. Synthesis 1982, 70–71. [Google Scholar]
- Haemers T.; Wiesner J.; Giessmann D.; Verbrugghen T.; Hillaert U.; Ortmann R.; Jomaa H.; Link A.; Schlitzer M.; Van Calenbergh S. Bioorg. Med. Chem. 2008, 16, 3361–3371. [DOI] [PubMed] [Google Scholar]
- Albert D. B.; Martens C. S. Marine Chem. 1997, 56, 27–37. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.