

Summary
Missense mutations in human PLP1, the gene encoding myelin proteolipid protein (PLP), cause dysmyelinating Pelizaeus-Merzbacher disease of varying severity. Although disease pathology has been linked to retention of misfolded PLP in the endoplasmic reticulum (ER) and induction of the unfolded protein response (UPR), the molecular mechanisms that govern phenotypic heterogeneity remain poorly understood. To address this issue, we examined the cellular response to missense mutants of PLP that are associated with distinct disease phenotypes. We found that the mild-disease-associated mutants, W162L and G245A, were cleared from the ER comparatively quickly via proteasomal degradation and/or ER exit. By contrast, the more `aggressive' A242V mutant, which causes severe disease, was significantly more stable, accumulated at the ER and resulted in a specific activation of the UPR. On the basis of these findings, we propose that the rate at which mutant PLP proteins are cleared from the ER modulates disease severity by determining the extent to which the UPR is activated.
Keywords: ER-associated degradation, ER stress, Pelizaeus-Merzbacher disease, Protein misfolding, Unfolded protein response
Introduction
A number of clinically important human diseases are known to be caused by protein misfolding at the endoplasmic reticulum (ER). The ER is a major site for the synthesis of membrane and secretory proteins in eukaryotic cells, and nascent polypeptides are translocated across the ER membrane as extended chains, which then typically fold and oligomerise with the assistance of ER chaperones. Correctly folded and assembled proteins are packaged into transport vesicles for delivery to the Golgi complex, whereas those that have not acquired the proper conformation are normally retained in the ER by a quality-control system that helps to ensure that misfolded or misassembled proteins are not deployed within the cell. Persistently misfolded proteins are normally retrotranslocated into the cytosol and degraded by proteasomes via a process known as ER-associated degradation (ERAD) (Anelli and Sitia, 2008; Vembar and Brodsky, 2008). Situations that disrupt protein folding lead to the production of potentially toxic misfolded proteins in the ER; hence, eukaryotic cells employ a defence mechanism that maintains a balance between protein synthesis, folding and degradation, known as the unfolded protein response (UPR) (Ron and Walter, 2007; Schroder and Kaufman, 2005). The UPR can be initiated by three transmembrane stress sensors, IRE1, ATF6 and PERK, that are activated by the accumulation of misfolded proteins in the ER. These form distinct branches of the UPR that function to reduce the overall rate of protein synthesis and upregulate the folding and degradation capacity of the ER, thereby reducing levels of misfolded protein in the ER, and potentially restoring homeostasis (Friedlander et al., 2000; Ng et al., 2000). However, if the cell fails to re-establish ER and cellular homeostasis, prolonged UPR signalling can induce programmed cell death (Szegezdi et al., 2006).
A variety of pathological conditions are associated with the production of misfolded proteins at the ER. Some of these diseases, such as cystic fibrosis, are caused by a loss of function of the relevant protein, whereas others, such as retinitis pigmentosa, are due to a toxic gain of function of the misfolded mutant protein (Aridor and Balch, 1999; Rutishauser and Spiess, 2002). As a result, considerable progress has been made towards understanding the molecular mechanisms that mediate ER retention and ERAD of mutant proteins (Hebert and Molinari, 2007). Interestingly, there is a number of gain-of-function ER folding diseases in which different mutations in the same gene result in distinct disease phenotypes (Nelis et al., 1999). Little is known about whether variations in the ability of the ER quality-control machinery to deal with different mutant forms of the same protein contribute to such variations.
In order to address this issue, we examined the cellular response to different disease-associated mutants of proteolipid protein (PLP). PLP is a polytopic membrane protein that, together with its splice variant DM20, represents the major protein constituents of central nervous system (CNS) myelin. Mutations in the gene encoding PLP (PLP1) cause Pelizaeus-Merzbacher disease (PMD), an X-linked recessive leukodystrophy characterised by dysmyelination of the CNS (Koeppen and Robitaille, 2002; Yool et al., 2000). A variety of PLP missense mutations, duplications and deletions have been identified, and these cause a wide spectrum of disease phenotypes (Garbern, 2007). Because missense mutations usually cause more severe phenotypes than null mutations (Boison and Stoffel, 1994; Klugmann et al., 1997), it is believed that disease pathology is largely induced by the presence of the mutant protein, rather than by the absence of a functional polypeptide (or loss of function). Most of the missense mutations studied to date disrupt the trafficking of PLP to the cell surface (Gow et al., 1994b; Gow and Lazzarini, 1996; Woodward, 2008), and it is generally believed that the pathogenic mechanism underlying PMD is the accumulation of misfolded PLP in the ER leading to activation of the UPR and oligodendrocyte apoptosis (Gow and Lazzarini, 1996; Gow et al., 1998; Southwood et al., 2002). However, the disease caused by missense mutations can range in phenotypic severity from mild PMD and late-onset spastic paraplegia type 2 (SPG2), which is characterised by hypomyelination and thinning of the myelin sheath (Hodes et al., 1997; Kobayashi et al., 1994), to severe connatal PMD associated with widespread oligodendrocyte apoptosis and a virtual absence of compact myelin (Gencic et al., 1989; Hudson et al., 1989). The critical factors that determine whether a particular mutation of the gene encoding PLP gives rise to a mild disease with hypomyelination or a severe disease with oligodendrocyte apoptosis are currently poorly understood (Dhaunchak and Nave, 2007; Gow and Lazzarini, 1996; Gow et al., 1998; Kramer-Albers et al., 2006; Southwood and Gow, 2001).
One possibility that is consistent with the phenotypic variation observed in PMD patients is that different mutations might have distinct effects on the ability of the quality-control machinery to deal with the mutant proteins, thereby producing distinct cellular responses. To test this hypothesis, we examined the quality control and associated cellular responses to naturally occurring missense mutations in similar regions of PLP, which produce a markedly different phenotype. In particular, we focused on two neighbouring mutations within the fourth transmembrane (TM) domain of PLP, one that causes severe PMD and a second that is associated with mild disease. We find that, although both mutant proteins are strongly retained in the ER and subjected to ERAD, the efficiency with which they are degraded and the subsequent cellular response elicited are quite different. Thus, whereas the A242V mutant protein associated with severe PMD is relatively resistant to degradation and induces a robust ER-stress response, the mild-disease-associated G245A mutant is rapidly cleared from the ER and fails to activate the UPR. Hence, our studies reveal differential sensitivity of the ER-associated quality-control and degradation machineries to different mutant forms of the same protein. These findings have implications for our understanding of the mechanisms that determine the severity of PMD, and potentially other gain-of-function protein-folding diseases.
Results
Retention of disease-associated PLP mutants by the ER quality-control system
In order to examine how the ER quality-control machinery deals with different mutant forms of PLP associated with distinct disease phenotypes, we focused on missense mutations within the TM domains of the polypeptide, because these might be expected to induce comparable folding defects. Genotype-phenotype correlation studies have shown that about 80% of the TM-domain mutations in the PLP1 gene lead to severe PMD (Cailloux et al., 2000), with one well-studied example being the substitution of a valine for an alanine (A242V; msd mutation) in the fourth TM domain (Gencic and Hudson, 1990; Yamamoto et al., 1998). By marked contrast, mutation of a neighbouring glycine to alanine (G245A), and of a tryptophan to leucine (W162L) in the third TM domain, both produce a milder phenotype (Hubner et al., 2005) (see Fig. 1).
Fig. 1.

Locations of the PLP missense mutations under study. A two-dimensional model of PLP topology illustrating the four TM domains, cytosolic N- and C-termini, palmitoylation sites (squiggles), and disulphide bonds (straight lines). The relative positions of the missense mutations that have been examined in this study are indicated. The A242V (msd) mutation associated with severe PMD is in red, whereas the W162L and G245A mutations associated with mild PMD are in black. The Myc-His6 (mh) epitope tag at the C-terminus of the recombinant protein is also indicated.
By using an established heterologous expression system (Swanton et al., 2003), we first tested whether the ER quality-control machinery is able to recognise and retain the mutant forms of PLP. Stable HeLa-cell lines expressing the different forms of PLP with a C-terminal Myc-His6 (mh) tag under the control of a tetracycline-inducible promoter were generated (Fig. 1) and the subcellular distribution of the proteins examined using immunofluorescence microscopy. Much of the wild-type (wt)-PLPmh colocalised with concanavalin A (ConA) bound to cell-surface glycoproteins (Fig. 2A, top row) and was visible on numerous microvilli protruding from the cell surface (Fig. 2A, insets). Very little colocalisation with the ER chaperone calnexin was observed (Fig. 2A, centre row), indicating that the vast majority of the wt protein was properly folded and had passed the ER quality control. A further pool of wt-PLPmh was seen in lysosome-associated membrane protein 1 (Lamp1)-containing late endosomal and/or lysosomal structures (Fig. 2A, bottom row), most probably representing protein that had been internalised from the cell surface (Gow et al., 1994a; Simons et al., 2002). Importantly, a very similar subcellular distribution of wt-PLP is seen in cultured oligodendrocytes (Dhaunchak and Nave, 2007; Kramer-Albers et al., 2006; Simons et al., 2002; Trajkovic et al., 2006), demonstrating that this HeLa-cell expression system provides a suitable model for studying the folding and quality control of PLP, consistent with previous studies, including our own (Sinoway et al., 1994; Swanton et al., 2003; Swanton et al., 2005). In contrast to the wt protein, A242V-PLPmh distribution was restricted to a reticular network that strongly colocalised with calnexin (Fig. 2B, centre row), consistent with this mutant being misfolded and retained by the ER quality-control machinery (Gow and Lazzarini, 1996; Kramer-Albers et al., 2006; Swanton et al., 2003). A characteristic feature of HeLa cells expressing A242V-PLPmh was the appearance of vacuolar-like structures (Fig. 2B, insets). The periphery of these structures contained both mutant PLPmh and calnexin, resembling the distended ER that has been observed under conditions of chronic ER stress (Alvarez et al., 1999; Dalal et al., 2004; Lass et al., 2008; Lin et al., 1999). The mild-disease-associated mutants W162L and G245A were also present in the ER, as evidenced by their colocalisation with calnexin (Fig. 2C,D, centre rows). In addition, a fraction of W162L-PLPmh seemed to be exported from the ER, and could be seen at the cell surface and also within Lamp1-positive structures (Fig. 2C, top and bottom rows). G245A-PLPmh, by contrast, showed no overlap with the late-endosomal and lysosomal marker (Fig. 2D, bottom row), and was not obvious at the plasma membrane (Fig. 2D, top row), suggesting that, similar to the severe-disease-associated A242V mutant, G245A-PLP is primarily retained in the ER.
Fig. 2.

Distinct subcellular distribution of wt and PMD-causing mutant PLP proteins. HeLa cells were induced to express (A) wt-, (B) A242V-, (C) W162L- or (D) G245A-PLPmh, fixed, permeabilised, and probed with Myc-specific antibodies (green in merge) and antibodies against either the ER-resident protein calnexin or the late-endosomal and lysosomal protein Lamp1 (red in merge). Alternatively, fixed wt-PLPmh- or mutant-PLPmh-expressing cells were stained with fluorescently modified ConA to selectively label cell-surface glycoproteins (red in merge) before permeabilisation and immunostaining with anti-Myc antibody. Colocalisation of PLPmh with specific organelle or plasma-membrane markers appears yellow. Insets (dashed boxes) show magnified areas. Scale bars: 10 μm.
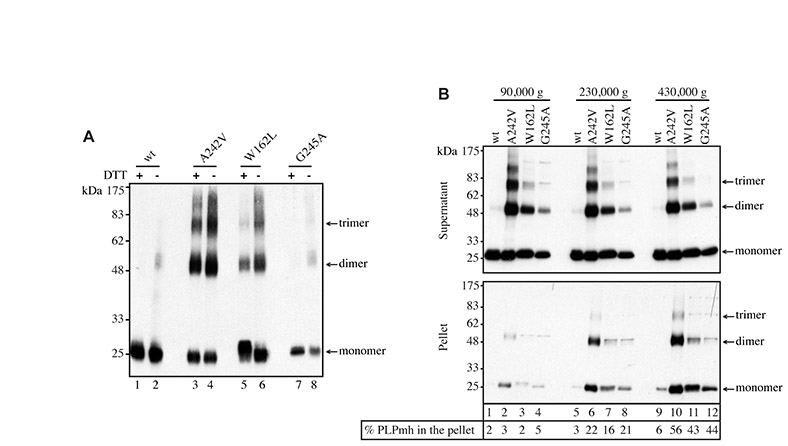
In order to provide a quantitative estimate of the amount of each PLPmh variant reaching the plasma membrane, cells were treated with a membrane-impermeable biotinylation reagent. Biotinylated (i.e. cell surface) PLPmh was isolated with NeutrAvidin beads, and detected by immunoblotting with anti-Myc antibody (Fig. 3A, top panel). Labelling of plasma-membrane-localised transferrin receptor (TfR) was used to control for loading and biotinylation efficiency in each experiment (Fig. 3A, bottom panel). A considerable fraction of wt-PLPmh was biotinylated and could be bound to NeutrAvidin beads following treatment of cells with sulpho-NHS-SS-biotin (Fig. 3A, cf. lanes 2 and 4). A significant amount of biotinylated W162L-PLPmh was also detected (Fig. 3A, lane 16), consistent with the subcellular distribution observed by immunofluorescence microscopy (Fig. 2C). By contrast, virtually no A242V- and even less G245A-PLPmh was detected in NeutrAvidin-bound fraction (Fig. 3A, lanes 8 and 12), providing further evidence that these mutants do not reach the cell surface efficiently (Fig. 3B). The ability of A242V-PLPmh, but not the wt or mild-disease-associated mutants, to form SDS-resistant oligomers (Swanton et al., 2005) was also clearly apparent (Fig. 3A, cf. lanes 1, 5, 9 and 13) and is discussed in more detail later.
Fig. 3.

Analysis of cell-surface expression of wt and mutant PLP. (A) HeLa cells expressing wt or mutant PLPmh were left untreated (–) or were treated (+) with membrane-impermeant sulpho-NHS-SS-biotin. Biotinylated proteins were precipitated using NeutrAvidin beads (beads, `B') and analysed by immunoblotting with anti-Myc or anti-TfR antibodies. The biotinylation levels of wt and mutant PLPmh were compared with 20% total cell lysates (total, `T'). (B) The levels of monomeric and oligomeric PLPmh on immunoblots were quantified by densitometry. Biotinylated PLPmh was expressed as a percentage of total PLPmh and normalised to the equivalent TfR levels. Histogram shows mean ± s.e.m. from three independent experiments (*P<0.05, **P<0.01, t-test).
Together, these results are consistent with the hypothesis that disease-associated mutations perturb the folding of PLP, thereby preventing authentic trafficking of the protein to the plasma membrane. Interestingly, the stringency with which the different mutants are retained by the ER quality-control machinery seems to vary. Hence, a fraction of W162L-PLPmh clearly exits the ER, progressing to the plasma membrane and lysosomal compartments. This suggests that the folding defect induced by this mutation might be comparatively subtle. By contrast, the A242V and G245A mutants are primarily retained in the ER. Because these two mutations produce very different disease phenotypes, they represent a particularly useful model that allows us to examine how the ability of the ER quality-control system to deal with different mutant forms of the same protein might contribute to disease severity.
Degradation of PLP mutants occurs at different rates and via distinct pathways
The underlying pathogenic mechanism of missense mutations in PLP is suggested to be the accumulation of the mutant protein in the ER, which leads to activation of the UPR and oligodendrocyte apoptosis (Gow and Lazzarini, 1996; Gow et al., 1998; Southwood and Gow, 2001; Southwood et al., 2002). The extent of PLP accumulation in the ER is likely to be controlled, at least in part, by the rate at which the polypeptide is degraded. Thus, the efficiency with which different mutant proteins are degraded could be an important factor determining disease pathology. Whereas plasma-membrane proteins can be endocytosed and degraded in lysosomes (Luzio et al., 2007), aberrant membrane proteins retained by the ER quality-control machinery are typically retrotranslocated and degraded by the ubiquitin-proteasome system in the cytosol (Vembar and Brodsky, 2008). In order to examine both the route and rate of degradation of the different forms of PLP, pulse-chase studies were performed in the presence of well-defined proteasome or lysosomal-protease inhibitors (Fig. 4A). These experiments revealed some striking differences in the pathways leading to degradation of the different PLPmh proteins.
Fig. 4.

Pulse-chase analysis of wt- and mutant-PLP degradation. (A) HeLa cells were left uninduced (–) or induced to express wt-, W162L-, A242V- or G245A-PLPmh. Cells were treated for 1 hour with DMSO, Z-LLF-CHO (10 μM) or a mixture of leupeptin (100 μM) and pepstatin A (1 μg/ml), pulse-labelled for a further hour with [35S]methionine/cysteine, and then chased with unlabelled methionine/cysteine up to 8 hours in the continued presence of the compounds. [35S]-labelled PLPmh was recovered by immunoprecipitation with anti-Myc, resolved by SDS-PAGE and visualised by phosphorimaging. The radioactivity of the wt, W162L, A242V (monomer and dimer) and G245A-PLPmh bands at each time point was quantified by phosphorimaging and expressed as a percentage of the corresponding value at 0-hour chase. The graphs show the mean ± s.e.m. from at least three independent experiments (*P<0.05, **P<0.01, ***P<0.001, t-test). (B) HeLa cells were left uninduced (–) or induced to express A242V-PLPmh for 2 or 20 hours, and then subjected to pulse-chase analysis as described in Fig. 4A. [35S]-labelled monomeric and dimeric A242V-PLPmh was quantified by phosphorimaging as described in Fig. 4A. The graph shows the mean ± s.e.m. from at least three independent experiments.
As previously observed, wt-PLPmh is relatively stable in cultured cells (Kramer-Albers et al., 2006; Swanton et al., 2003), and was turned over with a half-life of ∼6.8 hours (Fig. 4A, wt). Treatment of cells with a combination of the lysosomal inhibitors leupeptin and pepstatin A blocked the degradation of the wt protein completely (Fig. 4A, wt). This suggests that the lysosome is the major site for degradation of wt-PLPmh, consistent with its lysosomal and plasma-membrane localisation observed by immunofluorescence microscopy (Fig. 2A). Indeed, accumulation of wt-PLPmh in Lamp1-positive structures was clearly visible in inhibitor-treated cells (supplementary material Fig. S1A). The proteasome inhibitor Z-LLF-CHO also caused some inhibition of degradation of wt-PLPmh (Fig. 4A, wt), reflecting either a minor fraction of the wt protein that misfolds and is degraded by ERAD or the inhibition of some lysosomal proteases by Z-LLF-CHO (Lee and Goldberg, 1998) (cf. supplementary material Fig. S1A).
We then examined the degradation of the different mutant forms of PLPmh. W162L-PLPmh was considerably less stable than the wt protein, having a half-life of ∼4.4 hours (Fig. 4A, W162L). Its degradation was reduced by the lysosomal-protease inhibitors leupeptin and pepstatin A, and even more strongly by Z-LLF-CHO (Fig. 4A, W162L). Lactacystin, a second proteasomal inhibitor, which is chemically unrelated to Z-LLF-CHO, also significantly stabilised W162L-PLPmh (supplementary material Fig. S2). Accumulation of the mutant protein was observed in the ER of Z-LLF-CHO-treated cells, and in the late endosomes and/or lysosomes of cells treated with leupeptin and pepstatin A (supplementary material Fig. S1B). These data suggest that both proteasomal and lysosomal pathways are involved in the degradation of W162L-PLPmh. By contrast, the severe-disease-associated A242V mutant was more slowly degraded, with a half-life of ∼5.6 hours (Fig. 4A, A242V). Degradation of A242V-PLPmh was almost entirely inhibited by Z-LLF-CHO, but unaffected by lysosomal-protease inhibitors (Fig. 4A, A242V). Together with the ER retention observed by immunofluorescence microscopy (Fig. 2B), this indicates that A242V-PLPmh is degraded via the ERAD pathway. Similarly, degradation of the mild-disease-associated G245A mutant was proteasome dependent but unaffected by inhibition of lysosomal proteases (Fig. 4A, G245A), suggesting that, similar to the A242V variant, it is also degraded by ERAD. We found that vacuolar-like structures surrounded by ER membrane were observed after treatment both of A242V-PLPmh- and G245A-PLPmh-expressing cells with Z-LLF-CHO (supplementary material Fig. S1C,D, insets), presumably in response to the inhibition of proteasomal degradation and increased accumulation of misfolded mutant PLPmh species in the ER. The role of the proteasome in the degradation of both of these PLP mutants was further supported by the accumulation of their polyubiquitylated forms in cells treated with proteasome inhibitor (supplementary material Fig. S3, square brackets).
Interestingly, the G245A mutant of PLPmh was degraded significantly more rapidly than the A242V mutant, with a half-life of ∼3.6 hours (Fig. 4A, G245A). This observation suggested that the severe-disease-associated A242V variant is more resistant to ERAD than is the G245A mutant that causes mild PMD. Consistent with this interpretation, in comparison with A242V-PLPmh, considerably less radiolabelled G245A-PLPmh was produced during the 1-hour pulse-labelling period (Fig. 4A, cf. A242V and G245A, DMSO panels, 0-hour chase). Furthermore, proteasome inhibition dramatically increased the amount of the radiolabelled G245A mutant generated during this time (Fig. 4A, G245A, cf. DMSO and Z-LLF-CHO panels, 0-hour chase), suggesting that rapid degradation of G245A-PLPmh limits the accumulation of this mutant protein. We considered the possibility that a simple accumulation of A242V-PLPmh in the ER could impede the ERAD pathway and repeated the pulse-chase assay using cells that contained different levels of total A242V mutant. The rate of degradation remained constant irrespective of whether cells were induced to express A242V-PLPmh for 2 hours or 20 hours prior to pulse-labelling (Fig. 4B). We therefore conclude that the difference in the rates of degradation of A242V and G245A-PLPmh reflects a difference in the ability of the ERAD machinery to dispose of these two mutant proteins.
When taken together, our results (summarised in Table 1) demonstrate that all three PLP mutants studied are degraded via the proteasome, reflecting their status as ERAD substrates. However, for the W162L mutant, ER retention is less stringent and lysosomal degradation also plays an important role in protein turnover. Significantly, the two PLP mutants that are associated with mild PMD are more efficiently cleared from the ER than the severe-disease-associated A242V mutant. Of particular interest is the observation that the A242V and G245A mutants, both of which are tightly ER-retained, are degraded at different rates, with the mild-disease-associated mutant being removed significantly more rapidly than the mutant linked to severe PMD.
Table 1.
Summary of the localisation and degradation characteristics of the PLPmh missense mutants
|
Localisation
|
Degradation
|
||||||
|---|---|---|---|---|---|---|---|
| PLPmh | Disease severity | ER | Late endosomes and/or lysosomes | % cell-surface PLPmh | Approximate half-life (hours) | Proteosomes | Lysosomes |
| wt | – | – | + | 14.9±2.4 | 6.8 | – | + |
| W162L | Mild | + | + | 7.5±0.4 | 4.4 | + | + |
| A242V | Severe | + | – | 0.8±0.1 | 5.6 | + | – |
| G245A | Mild | + | – | 4.3±2.1 | 3.6 | + | – |
Distinct physical properties of PLP mutants
In order to investigate the molecular basis for the difference in PLP degradation rates, we examined the physical properties of the mutant proteins. We have previously shown that a key feature of disease-associated PLP mutants is their tendency to rapidly assemble into SDS-resistant homo-oligomers (Swanton et al., 2005). This behaviour was apparent in the pulse-chase studies performed here (Fig. 4 and supplementary material Fig. S4), with SDS-resistant dimers of the severe-disease-associated mutant A242V clearly evident immediately after the 1-hour pulse-labelling period (Fig. 4A, A242V, DMSO panel, 0-hour chase). By contrast, the mild-disease-associated mutants, particularly the G245A variant, had a very modest tendency to form stable oligomers (Fig. 4A, W162L and G245A, DMSO panels). In both cases, dimer formation was accentuated in the presence of proteasome inhibitors (supplementary material Fig. S4, W162L and G245A, cf. DMSO and Z-LLF-CHO/Lactacystin panels), suggesting that these mutants can form oligomers if given sufficient opportunity. Hence, there seems to be a direct correlation between the rapid formation of SDS-resistant oligomers and the stability of the PLP mutants.
Misfolded proteins, including some mutant PLP, can form aberrant disulphide bonds (Dhaunchak and Nave, 2007) and might also be prone to aggregation, either of which could potentially impede ERAD (Hosokawa et al., 2006). However, no obvious correlation between the extent of disulphide bonding and the degradation rates of the different mutants was seen (supplementary material Fig. S5A), and the sedimentation behaviour of the different mutants was similar (supplementary material Fig. S5B). These results suggest that neither variable formation of aberrant oxidation products nor variable aggregation of the mutant proteins underlies the differences in the rates of degradation observed.
Differential activation of the UPR by PLP mutants
The UPR is believed to modulate disease severity in PMD (Southwood et al., 2002), and we therefore compared the magnitude of the UPR induced by expression of the disease-associated mutant forms of PLP. In this context, we focussed on the two primarily ER-retained mutants, A242V and G245A, because they are degraded via ERAD at different rates and are associated with diseases of varying severity. For such comparative analyses, it was important to ensure that the rate of synthesis of the two proteins was similar. This was achieved by lowering the concentration of doxycycline used to induce expression of A242V-PLPmh (Fig. 5A).
Fig. 5.

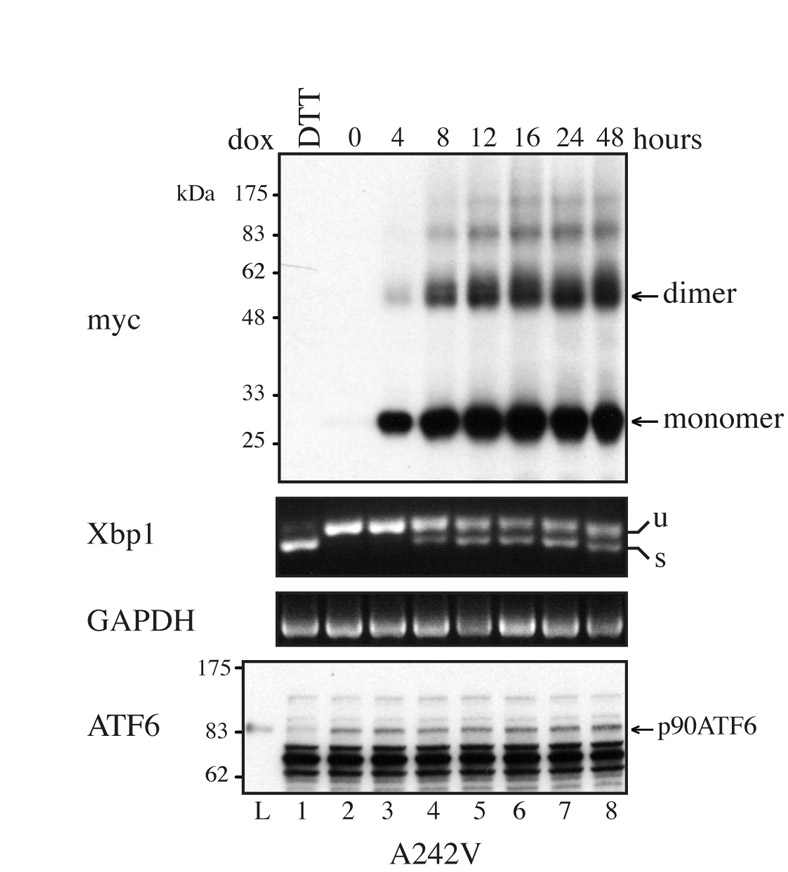
Expression of A242V-PLP activates IRE1 signalling. (A) HeLa cells were induced to express G245A-PLPmh by using 1 μg/ml doxycycline, whereas expression of A242V-PLPmh was induced with 0.05 μg/ml doxycycline (A242VlowDOX). Cells were treated with Z-LLF-CHO (10 μM) for 1 hour, pulse-labelled with [35S]methionine/cysteine in the presence of Z-LLF-CHO for the times indicated, lysed and lysates immunoprecipitated with anti-Myc. Immunoprecipitated [35S]-labelled PLPmh was quantified by phosphorimaging and expressed as a percentage of the amount of protein synthesised after 10 minutes in order to normalise differences in cell number. The graph shows the mean from two independent experiments. (B) HeLa cells were induced to express wt-, A242VlowDOX-, G245A- or W162L-PLPmh for increasing periods of time (0-48 hours). Treatment of uninduced cells with 10 mM DTT for 2 hours served as a positive control to trigger Xbp1-mRNA splicing. PLPmh was detected by immunoblotting RIPA-solubilised cell lysates with anti-Myc. Tubulin was assessed as a protein loading control. Alternatively, total RNA was isolated from cells and Xbp1-mRNA splicing was determined by RT-PCR. Unspliced (u) and spliced (s) Xbp1 mRNA are indicated. GAPDH served as a cDNA loading control. ATF6 immunoblots were performed by analysing RIPA lysates prepared from a second independent experiment. Treatment of uninduced cells with 2 mM DTT for 2 hours served as a positive control to trigger cleavage of the full-length ATF6 protein (p90 ATF6). Specificity of the anti-ATF6 antibody was confirmed using a commercial ATF6-transfected cell lysate (Lysate, `L'; Imgenex). (C) A242V-PLPmh-expressing HeLa cells were induced with increasing concentrations of doxycycline (0.01-1 μg/ml) for 24 hours. Cell lysates were analysed by western blotting with anti-Myc or anti-tubulin, and Xbp1-mRNA splicing was determined by RT-PCR.
We first examined the ER-stress response to the synthesis of these mutant forms of PLP by monitoring the IRE1-XBP1 branch of the UPR (Yoshida et al., 2001). Treatment of cells with the reducing agent dithiothreitol (DTT), a potent inducer of the UPR, resulted in the appearance of spliced Xbp1 mRNA (Fig. 5B, Xbp1 panel; cf. lanes 1 and 2, 9 and 10, 17 and 18, 25 and 26). By contrast, Xbp1-mRNA splicing was not visible in cells expressing wt-PLPmh, demonstrating that synthesis of the properly folded protein did not induce the UPR over a 48-hour period (Fig. 5B, Xbp1 panel, lanes 2-8). Xbp1-mRNA splicing was measured at several different times following induction of mutant PLPmh expression, because UPR signalling can diminish with time (Lin et al., 2007; Rutkowski et al., 2006). The severe-disease-associated A242V-PLPmh accumulated rapidly in cells following addition of doxycycline (Fig. 5B, Myc panel, lanes 10-16). Spliced Xbp1 mRNA was clearly visible after 8 hours (Fig. 5B, Xbp1 panel, lane 12, see arrow), demonstrating that expression of this mutant protein induces a rapid ER-stress response. Interestingly, prolonged expression of A242V-PLPmh at low levels of induction resulted in attenuation of Xbp1-mRNA splicing (Fig. 5B, Xbp1 panel, lanes 14-16), consistent with observations that IRE1 activation is reduced during persistent ER stress (Lin et al., 2007; Rutkowski et al., 2006). Despite having a similar rate of synthesis to A242V-PLPmh (Fig. 5A), less G245A-PLPmh accumulated in doxycycline-induced cells (Fig. 5B, Myc panel, cf. lanes 10-16 and 18-24), reflecting the more efficient degradation of this mild-disease-associated mutant. No spliced Xbp1 mRNA could be detected in cells expressing G245A-PLPmh, even after 48 hours of continual synthesis of the mutant protein (Fig. 5B, Xbp1 panel, lanes 18-24). This is an important finding because it suggests that efficient removal of G245A from the ER by ERAD protects the cells by preventing the activation of the UPR. Likewise, expression of W162L-PLPmh, which is effectively cleared from the ER by a combination of ERAD and export, failed to induce Xbp1-mRNA splicing (Fig. 5B, Xbp1 panel, lanes 26-32). Thus, there is a clear correlation between the stability, and hence load, of the different mutant forms of PLP in the ER and their ability to induce the IRE1-XBP1 arm of the ER-stress response. The dependence of IRE1 activation on misfolded protein load was confirmed by varying the steady-state level of A242V-PLPmh using different doxycycline concentrations (Fig. 5C). As predicted, the extent of Xbp1-mRNA splicing increased in proportion to total levels of the mutant protein (Fig. 5C), and this splicing was maintained when the mutant was expressed at high levels (supplementary material Fig. S6).
We also examined activation of a second UPR sensor, ATF6, a 90-kDa transmembrane protein that is proteolytically activated under conditions of ER stress. The ATF6 cleavage product is unstable, and therefore ATF6 activation is often measured by following loss of the full-length protein (DuRose et al., 2006; Haze et al., 1999; Rutkowski et al., 2006). Induction of the UPR with DTT resulted in the disappearance of virtually all full-length ATF6 (Fig. 5B, ATF6 panel, cf. lanes 1 and 2, 9 and 10, 17 and 18, 25 and 26), demonstrating efficient ATF6 cleavage. By contrast, no loss of p90 ATF6 was observed upon induction of wt-, G245A- or W162L-PLPmh expression (Fig. 5B, ATF6 panel, lanes 2-8, 18-24, 26-32). This suggests that synthesis of these forms of PLP did not stimulate ATF6 cleavage, and is consistent with their inability to activate IRE1. Expression of A242V-PLPmh also failed to induce observable cleavage of p90 ATF6 (Fig. 5B, ATF6 panel, lanes 10-16), despite causing activation of IRE1-XBP1 (Fig. 5B, Xbp1 panel, lanes 12 and 13). Because measuring the disappearance of full-length ATF6 might not be sufficiently sensitive to detect a small degree of ATF6 cleavage, we increased A242V-PLPmh expression using a higher doxycycline concentration. However, no evidence of ATF6 cleavage was seen even upon prolonged high-level expression of the mutant protein, which induced robust Xbp1 splicing (supplementary material Fig. S6, cf. Xbp1 and ATF6 panels). Therefore, we conclude that expression of A242V-PLPmh does not efficiently activate ATF6 in this system.
In order to examine the consequences of UPR activation by mutant PLP, we focused on the transcription factor CHOP, which is upregulated by ER stress and is a key mediator of ER-stress-induced apoptosis (Oyadomari and Mori, 2004). Levels of CHOP mRNA, assessed by reverse-transcriptase PCR (RT-PCR), were low under basal conditions and increased markedly upon treatment of cells with DTT (Fig. 6A, cf. lanes 1 and 2, 9 and 10, 17 and 18, 25 and 26, 33 and 34). The abundance of CHOP mRNA was not altered by expression of wt-PLPmh, or of the G245A and W162L variants (Fig. 6A, lanes 2-8, 18-24 and 26-32), providing further evidence that the rapidly cleared mutant proteins do not induce the UPR. By contrast, synthesis of the severe-disease-associated A242V mutant at a similar rate caused a dramatic increase in CHOP mRNA within 8 hours (Fig. 6A, lanes 10-16). As observed for IRE1-XBP1, an even greater upregulation of CHOP mRNA was provoked by increased levels of A242V-PLPmh (Fig. 6A, lanes 34-40). Because CHOP is generally associated with proapoptotic UPR signalling (Oyadomari and Mori, 2004), we next examined the effect of the different mutant proteins on cell viability. In line with their inability to induce any of the UPR markers tested, wt-PLPmh and the mild-disease-associated W162L and G245A mutants had little effect on cell viability (Fig. 6B). Cells also tolerated similar rates of A242V-PLPmh synthesis, showing only a very marginal decrease in viability after 48 hours (Fig. 6B). However, inducing higher expression levels of this mutant resulted in a clear loss of cell viability (Fig. 6B), consistent with the idea that a threshold of CHOP expression is required to promote apoptosis (Rutkowski et al., 2006). These findings further highlight the correlation between load of misfolded protein in the ER, UPR signalling and cell fate.
Fig. 6.

Expression of A242V-PLP upregulates CHOP expression and decreases cell viability. (A) HeLa cells were induced to express wt-, A242V-, G245A- or W162L-PLPmh for increasing periods of time (0-48 hours). Total RNA was isolated and levels of CHOP mRNA determined by RT-PCR. Treatment of uninduced cells with 5 mM DTT for 4 hours served as a positive control to induce CHOP mRNA levels. GAPDH served as a cDNA loading control. (B) HeLa cells were induced to express wt-, A242V-, G245A- or W162L-PLPmh for the indicated periods of time. Cell viability was measured via the MTT assay, and expressed relative to uninduced cells. The graph shows the mean ± s.e.m. from three experiments.
Together, these results suggest that the rate at which mutant proteins are cleared from the ER is an important factor in determining their ability to induce the UPR. Consistent with a role of the UPR in determining PMD pathology, the two mutant forms of PLP that cause mild PMD are degraded comparatively quickly and fail to activate the UPR, whereas the more stable severe-disease-associated variant rapidly induces an ER-stress response, resulting in upregulation of the UPR target gene CHOP.
Discussion
It is believed that the pathology of PMD, and many other gain-of-function ER folding diseases, is caused by activation of the UPR, which leads to programmed cell death. Thus, factors that govern the extent and duration of UPR activation in response to expression of different mutant proteins are likely to play an important role in determining disease pathology. However, little is known about the nature of these factors, and the molecular basis for the variation in the severity of PMD remains unclear. In order to address this issue, we used a stable inducible HeLa-cell system to describe the fundamental cellular events associated with expression of three different mutant forms of PLP associated with diseases of varying severity. Together, our results suggest that the rate at which different mutant proteins are cleared from the ER determines their ability to activate an ER-stress response and might thus play an important role in determining disease pathology. Hence, whereas the more aggressive A242V mutant has an inherently slower turnover and induces ER stress, the mild-disease-associated W162L and G245A variants are more rapidly degraded and fail to activate the UPR.
Immunofluorescence microscopy and quantitative cell-surface biotinylation revealed that all three mutant forms of PLP were subjected to some degree of ER retention. Of particular interest was the observation that both the A242V and G245A mutants were almost exclusively ER localised. These mutations are associated with severe and mild forms of the disease, respectively, and therefore ER retention alone cannot explain the differences in phenotypic severity. A fraction of the W162L mutant was able to pass the ER quality control, reaching the cell surface and late endosomes and/or lysosomes. By contrast, a GFP-tagged version of the W162L mutant transiently expressed in COS-7 cells was found to accumulate in a perinuclear compartment (Koizume et al., 2006). This apparent discrepancy might be due to the general tendency of PLP to be retained intracellularly in COS-7 cells (Kramer-Albers et al., 2006; Thomson et al., 1997). Importantly, trafficking of wt-PLP in the inducible HeLa cells used here mimics that seen in oligodendroglial cells (Dhaunchak and Nave, 2007; Kramer-Albers et al., 2006), with very little, if any, wt-PLP detectable in the ER. Thus, we are confident that inducible expression of PLP in HeLa cells provides a valuable system with which to analyse the cellular processing of mutant forms of this protein, and that our results reveal fundamental principles about this process.
Significantly, the A242V and G245A mutants were both retained in the ER and degraded via ERAD, yet they produce distinct disease phenotypes. This observation provided us with a useful model with which to examine whether variations in the ability of the ER quality-control machinery to deal with different mutant forms of the same protein could underlie differences in cellular pathology. Strikingly, the two mutant proteins were turned over with very different kinetics. Although both were degraded via ERAD, G245A-PLP was degraded significantly faster than the A242V mutant. As a result, A242V-PLP accumulated at the ER to a greater extent. Importantly, when synthesised at comparable rates, only the more stable A242V mutant induced the UPR. These results suggest that the efficiency of ERAD is an important factor in determining the extent of UPR activation caused by expression of different mutant forms of the same protein (see model in Fig. 7).
Fig. 7.

Schematic for the postulated molecular mechanism underlying mild and severe PMD resulting from different PLP missense mutations.
The precise mechanism through which the proximal ER-stress sensors are activated is not yet clear, but for IRE1 might involve direct binding of misfolded proteins to the lumenal domain of the protein (Credle et al., 2005; Zhou et al., 2006). Thus, it is formally possible that the difference in IRE1 induction by the two ER-retained mutant proteins reflects variations in their ability to bind to IRE1. Nevertheless, when the steady-state level of A242V-PLP was decreased sufficiently, this mutant also failed to induce Xbp1-mRNA splicing (Fig. 5B,C). This supports our suggestion that the extent of UPR activation reflects differences in the stability of mutant polypeptides, and thus the level to which they accumulate in the ER, and not some other, as-yet poorly defined, property of particular mutant proteins. Recent work indicates that the three stress sensors in mammalian cells (IRE1, ATF6 and PERK) possess distinct sensitivity to different forms of ER stress (DuRose et al., 2006). Such a model would be consistent with our observation that expression of A242V-PLPmh failed to induce efficient cleavage of ATF6, despite causing robust activation of IRE1 and induction of CHOP mRNA (Fig. 6A and supplementary material Fig. S6). Because the PERK pathway plays a dominant role in CHOP upregulation during ER stress (Harding et al., 2000; Oyadomari and Mori, 2004), the increase in CHOP seen here most likely reflects activation of PERK, suggesting that expression of A242V-PLPmh activates both IRE1 and PERK.
The half-life of A242V-PLP reported here (5.5 hours) is longer than that observed in immortalized oligodendrocytes (3 hours) (Kramer-Albers et al., 2006). This might be due to differences in the expression systems used, the timecourse over which degradation was followed and/or intrinsic differences in the ERAD capacity of the cells. Nevertheless, regardless of the absolute rate of A242V-PLP degradation, our findings clearly support a model in which variation in the relative stability of different mutant forms of PLP in the ER underlies their ability to induce ER stress. This correlation between mutant-protein stability and activation of the UPR extended to the W162L mutant, which was also degraded comparatively rapidly and failed to induce UPR signalling. In this case, a considerable fraction of the mutant protein escaped the ER quality-control system and was directed to lysosomes, either via post-ER quality-control systems (Arvan et al., 2002) or following the normal trafficking pathway of the wt protein (Simons et al., 2002). In either case, forward transport of W162L-PLP probably contributed to the effective clearance of this mutant protein from the ER and its failure to induce ER stress. Similarly, a recent study showed that a proportion of the I186T (rsh) mutant of PLP, which also causes mild disease, exits the ER in immortalized oligodendrocytes and is degraded by proteasome- and lysosome-dependent pathways (Kramer-Albers et al., 2006). In contrast to the W162L mutant studied here, Kramer-Albers et al. found that the half-life of I186T-PLP was comparable to that of the severe-disease-associated mutant A242V-PLP. This difference in stability of the two `mild' mutants relative to A242V-PLP probably reflects variations in their trafficking efficiency and/or partitioning between proteasomal and lysosomal degradation pathways. In any event, the results of Kramer-Albers et al. support our hypothesis that the ability of certain mutant proteins to partially escape ER quality control reduces their capacity to activate the UPR, therefore disrupting cell function (Fig. 7).
The `set point' that determines whether cells survive or die when faced with ER stress varies between cell types, and there is some evidence that myelinating cells might have a distinct response to UPR activation (Lin and Popko, 2009; Southwood et al., 2002). For example, CHOP, which is generally considered to promote apoptosis, seems to have a protective effect in oligodendrocytes (Southwood et al., 2002). As a result, the ultimate consequences of UPR activation in HeLa cells might not be directly comparable to those in oligodendrocytes. However, expression of A242V-PLPmh in HeLa cells was accompanied by induction of CHOP, as seen in oligodendrocytes from mice and humans harbouring PLP1 mutations (Southwood et al., 2002), suggesting that this early response to expression of mutant PLP is conserved. In the case of HeLa cells, high-level A242V-PLPmh expression was also correlated with an increase in cell death, although further studies using oligodendrocytes will be needed to fully elucidate the consequences of UPR activation in these specialised cells.
Because the UPR is believed to modulate the pathology of PMD, these results provide insight into the molecular mechanisms that govern the severity of this disease. Our data suggest that the extent of UPR activation is determined, at least in part, by the rate at which the mutant protein is cleared from the ER. Removal from the ER might occur via forward trafficking, or through effective ERAD, either of which seems to minimise UPR signalling. These findings might also be relevant to other ER folding diseases whose pathology is dictated by the UPR.
We previously established a direct correlation between the extent of mutant PLP oligomerisation and the severity of PMD (Swanton et al., 2005), and our current data support the proposal that there is a link between the stability of PLP mutants and the rate of homo-oligomerisation (Fig. 7). The observation that the A242V and G245A mutant proteins are degraded with different efficiencies and exhibit distinct oligomeric properties suggests that these missense mutations produce distinct effects on the protein structure, despite being located in the same regions of the polypeptide. Although the high-resolution structure of PLP has not yet been resolved, modelling the fourth TM domain of PLP predicts that the two mutations produce similar structural alterations (supplementary material Fig. S7). Because the presence of β-branched valine residues in TM helices can facilitate helix-helix interactions by providing a conformationally restricted interface (Curran and Engelman, 2003), we speculate that the A242V substitution might specifically increase the dimerisation interface of this mutant, thus increasing the propensity of A242V-PLP to form dimers.
In conclusion, our studies provide evidence for a link between the stability of different mutant forms of PLP, their ability to induce the UPR and the severity of the disease that they cause. Thus, we suggest that the efficiency of ERAD is an important factor governing the extent of UPR activation in response to expression of different mutant proteins. These findings have implications for other gain-of-function ER folding diseases in which the UPR modulates disease pathology, and suggest that strategies aimed at facilitating the removal of mutant proteins from the ER continue to have considerable therapeutic potential (Powers et al., 2009).
Materials and Methods
Reagents and antibodies
Media and reagents were from Lonza (Wokingham, UK), Z-LLF-CHO from Calbiochem, protease-inhibitor cocktail from Sigma, and leupeptin, pepstatin A and lactacystin from BIOMOL (Exeter, UK). Antibodies were from Upstate (Myc epitope), Sigma (Myc epitope and calnexin), Developmental Studies Hybridoma Bank, University of Iowa (Lamp1), BIOMOL (polyubiquitin conjugates, clone FK2), Zymed (TfR) and Imgenex (ATF6). Anti-α-tubulin was kindly provided by Keith Gull (University of Oxford, UK). Fluorophore- and HRP-conjugated secondary antibodies were from Molecular Probes (Paisley, UK) and Sigma, respectively.
Molecular cloning and DNA manipulations
The cDNA encoding wt-PLPmh (Swanton et al., 2003) was subcloned into the NotI and EcoRV sites of the tetracycline-responsive expression vector pTRE2hyg (Clontech), and QuikChange site-directed mutagenesis (Stratagene) was used to generate the W162L or G245A point mutations.
Cell culture
Inducible HeLa-cell lines expressing W162L and G245A-PLPmh were generated by transfection of HeLa Tet-On cells (Clontech) with the relevant pTRE2hyg plasmids using Lipofectamine 2000 (Invitrogen). Stable transfectants were selected by resistance to 400 μg/ml hygromycin B. Clonal cell lines were chosen based on the level of inducible expression, and maintained in DMEM containing 10% foetal bovine serum and 2 mM L-glutamine, plus 100 μg/ml geneticin and 100 μg/ml hygromycin B at alternate passages. Expression was induced by 1 μg/ml doxycycline and analyses were conducted after 20-24 hours, unless stated otherwise.
Immunofluorescence microscopy
Cells grown on coverslips were fixed with 3% formaldehyde and 0.2% glutaraldehyde in PBS for 30 minutes. After unreacted fixative was quenched, cells were permeabilised with 0.1% Triton X-100 and 0.05% SDS in PBS for 4 minutes, then incubated for 60 minutes with primary antibodies diluted into PBS. Cells were washed with PBS and incubated with the appropriate fluorophore-conjugated secondary antibodies diluted in PBS for another 60 minutes. After washing with PBS, coverslips were mounted with Mowiol and analysed using an Olympus BX60 upright microscope driven by MetaMorph software (Universal Imaging Corporation).
Biotinylation of cell-surface proteins
Cell-surface proteins were biotinylated using 0.25 mg/ml sulpho-NHS-SS-biotin (Pierce) in PBS for 30 minutes at 4°C. Unreacted biotin reagent was quenched by adding ice-cold Tris-HCl, pH 7.6, to 50 mM. Cells were washed with PBS, lysed in RIPA buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.2% deoxycholate) containing protease-inhibitor cocktail and incubated at 4°C for 1 hour. Insoluble material was removed by centrifugation at 5000 g for 10 minutes at 4°C. Biotinylated conjugates were affinity-purified with NeutrAvidin resin (Pierce) at room temperature for 60 minutes, resolved by SDS-PAGE and detected by immunoblotting.
Pulse-labelling/chase analysis
Cells were incubated in methionine/cysteine-free DMEM for 20 minutes, then pulse-labelled in the same medium containing 20 μCi/ml EasyTag [35S]methionine/cysteine (PerkinElmer) for the indicated periods of time. After pulse-labelling, cells were rinsed twice with PBS, then incubated with normal growth medium containing 4 mM non-radioactive methionine and cysteine for up to 8 hours (chase). At the indicated times of pulse (pulse-labelling analysis) or chase (pulse-chase analysis), cells were rinsed twice with ice-cold PBS and lysed in immunoprecipitation (IP) buffer (10 mM Tris-HCl, pH 7.6, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100) containing protease-inhibitor cocktail at 4°C for 1 hour. After centrifugation at 5000 g for 10 minutes at 4°C, clarified cell lysates were processed for denaturing IP as described (Swanton et al., 2003). Radiolabelled proteins were visualised by SDS-PAGE and phosphorimaging. [35S]-labelled PLPmh was quantified using AIDA software, and GraphPad Prism 4 software was used for data and statistical analysis. The mean values from the pulse-labelling studies were fitted with one-phase-exponential-association curves according to the formula y = ymax × [1–exp(–kx)] (where k=rate constant and x=time), whereas the averaged data from the pulse-chase studies were fitted with one-phase-exponential-decay curves according to the formula y = 100 × exp(–kx). The decay rate constant (k), which is representative of the rate of protein degradation, was determined for each curve and the half-life calculated (t1/2=0.69/k).
RT-PCR analysis of Xbp1-mRNA splicing and CHOP-mRNA induction
Cells were harvested in Trizol (Invitrogen) and total RNA was extracted according to the manufacturer's instructions. RNA (1 μg) was reverse-transcribed into single-stranded cDNA using the AMV-RT system (Roche), and the cDNA used as template for PCR amplification of Xbp1, CHOP or GAPDH mRNA. Primers used for RT-PCR analysis included: Xbp1, 5′-ACAGCGCTTGGGGATGGATG-3′ and 5′-TGACTGGGTCCAAGTTGTCC-3′; CHOP, 5′-CCCTGCTTCTCTGGCTTGGCTGAC-3′ and 5′-GCTCCCAATTGTTCATGCTTGGTGC-3′; and GAPDH, 5′-GACCCCTTCATTGACCTCAACTACATG-3′ and 5′-GTCCACCACCCTGTTGCTGTAGCC-3′. PCR products were resolved on 2% agarose gels.
Cell-viability assay
Cell viability was assessed by measuring their ability to metabolise 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma). 500 μl of 2.5 mg/ml MTT was added to each well of a 24-well plate and cells incubated at 37°C for 2 hours. The media was discarded and reduced crystals of MTT solubilised with 500 μl DMSO. 150 μl of DMSO solution was transferred to a well of a 96-well plate so as to create triplicates of each sample, and absorbance at 570 nm was measured with an automatic plate reader.
Supplementary Material
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/122/21/3942/DC1
This work was supported by The Wellcome Trust. We thank Martin Lowe, Craig McKibbin and Jim Warwicker for critical comments on this manuscript and helpful discussions. Deposited in PMC for release after 6 months.
References
- Alvarez, A. I., Arostegui, E., Martin, R., Molina, M., Duran, M. and Tejada, M. I. (1999). Role of the Pro23Leu mutation in a family affected by retinitis pigmentosa in the Basque Country. Clin. Genet. 56, 407-408. [DOI] [PubMed] [Google Scholar]
- Anelli, T. and Sitia, R. (2008). Protein quality control in the early secretory pathway. EMBO J. 27, 315-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor, M. and Balch, W. E. (1999). Integration of endoplasmic reticulum signaling in health and disease. Nat. Med. 5, 745-751. [DOI] [PubMed] [Google Scholar]
- Arvan, P., Zhao, X., Ramos-Castaneda, J. and Chang, A. (2002). Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic 3, 771-780. [DOI] [PubMed] [Google Scholar]
- Boison, D. and Stoffel, W. (1994). Disruption of the compacted myelin sheath of axons of the central nervous system in proteolipid protein-deficient mice. Proc. Natl. Acad. Sci. USA 91, 11709-11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cailloux, F., Gauthier-Barichard, F., Mimault, C., Isabelle, V., Courtois, V., Giraud, G., Dastugue, B. and Boespflug-Tanguy, O. (2000). Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur. J. Hum. Genet. 8, 837-845. [DOI] [PubMed] [Google Scholar]
- Credle, J. J., Finer-Moore, J. S., Papa, F. R., Stroud, R. M. and Walter, P. (2005). On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 102, 18773-18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran, A. R. and Engelman, D. M. (2003). Sequence motifs, polar interactions and conformational changes in helical membrane proteins. Curr. Opin. Struct. Biol. 13, 412-417. [DOI] [PubMed] [Google Scholar]
- Dalal, S., Rosser, M. F., Cyr, D. M. and Hanson, P. I. (2004). Distinct roles for the AAA ATPases NSF and p97 in the secretory pathway. Mol. Biol. Cell 15, 637-648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaunchak, A. S. and Nave, K. A. (2007). A common mechanism of PLP/DM20 misfolding causes cysteine-mediated endoplasmic reticulum retention in oligodendrocytes and Pelizaeus-Merzbacher disease. Proc. Natl. Acad. Sci. USA 104, 17813-17818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhe-Paganon, S., Werner, E. D., Nishi, M., Hansen, L., Chi, Y. I. and Shoelson, S. E. (2004). A phenylalanine zipper mediates APS dimerization. Nat. Struct. Mol. Biol. 11, 968-974. [DOI] [PubMed] [Google Scholar]
- DuRose, J. B., Tam, A. B. and Niwa, M. (2006). Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol. Biol. Cell 17, 3095-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander, R., Jarosch, E., Urban, J., Volkwein, C. and Sommer, T. (2000). A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2, 379-384. [DOI] [PubMed] [Google Scholar]
- Garbern, J. Y. (2007). Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell Mol. Life Sci. 64, 50-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gencic, S. and Hudson, L. D. (1990). Conservative amino acid substitution in the myelin proteolipid protein of jimpymsd mice. J. Neurosci. 10, 117-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gencic, S., Abuelo, D., Ambler, M. and Hudson, L. D. (1989). Pelizaeus-Merzbacher disease: an X-linked neurologic disorder of myelin metabolism with a novel mutation in the gene encoding proteolipid protein. Am. J. Hum. Genet. 45, 435-442. [PMC free article] [PubMed] [Google Scholar]
- Gow, A. and Lazzarini, R. A. (1996). A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat. Genet. 13, 422-428. [DOI] [PubMed] [Google Scholar]
- Gow, A., Friedrich, V. L., Jr and Lazzarini, R. A. (1994a). Intracellular transport and sorting of the oligodendrocyte transmembrane proteolipid protein. J. Neurosci. Res. 37, 563-573. [DOI] [PubMed] [Google Scholar]
- Gow, A., Friedrich, V. L., Jr and Lazzarini, R. A. (1994b). Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J. Neurosci. Res. 37, 574-583. [DOI] [PubMed] [Google Scholar]
- Gow, A., Southwood, C. M. and Lazzarini, R. A. (1998). Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J. Cell Biol. 140, 925-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M. and Ron, D. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099-1108. [DOI] [PubMed] [Google Scholar]
- Haze, K., Yoshida, H., Yanagi, H., Yura, T. and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert, D. N. and Molinari, M. (2007). In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 87, 1377-1408. [DOI] [PubMed] [Google Scholar]
- Hodes, M. E., Blank, C. A., Pratt, V. M., Morales, J., Napier, J. and Dlouhy, S. R. (1997). Nonsense mutation in exon 3 of the proteolipid protein gene (PLP) in a family with an unusual form of Pelizaeus-Merzbacher disease. Am. J. Med. Genet. 69, 121-125. [PubMed] [Google Scholar]
- Hosokawa, N., Wada, I., Natsuka, Y. and Nagata, K. (2006). EDEM accelerates ERAD by preventing aberrant dimer formation of misfolded alpha1-antitrypsin. Genes. Cells 11, 465-476. [DOI] [PubMed] [Google Scholar]
- Hubner, C. A., Orth, U., Senning, A., Steglich, C., Kohlschutter, A., Korinthenberg, R. and Gal, A. (2005). Seventeen novel PLP1 mutations in patients with Pelizaeus-Merzbacher disease. Hum. Mutat. 25, 321-322. [DOI] [PubMed] [Google Scholar]
- Hudson, L. D., Puckett, C., Berndt, J., Chan, J. and Gencic, S. (1989). Mutation of the proteolipid protein gene PLP in a human X chromosome-linked myelin disorder. Proc. Natl. Acad. Sci. USA 86, 8128-8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugmann, M., Schwab, M. H., Puhlhofer, A., Schneider, A., Zimmermann, F., Griffiths, I. R. and Nave, K. A. (1997). Assembly of CNS myelin in the absence of proteolipid protein. Neuron 18, 59-70. [DOI] [PubMed] [Google Scholar]
- Kobayashi, H., Hoffman, E. P. and Marks, H. G. (1994). The rumpshaker mutation in spastic paraplegia. Nat. Genet. 7, 351-352. [DOI] [PubMed] [Google Scholar]
- Koeppen, A. H. and Robitaille, Y. (2002). Pelizaeus-Merzbacher disease. J. Neuropathol. Exp. Neurol. 61, 747-759. [DOI] [PubMed] [Google Scholar]
- Koizume, S., Takizawa, S., Fujita, K., Aida, N., Yamashita, S., Miyagi, Y. and Osaka, H. (2006). Aberrant trafficking of a proteolipid protein in a mild Pelizaeus-Merzbacher disease. Neuroscience 141, 1861-1869. [DOI] [PubMed] [Google Scholar]
- Kramer-Albers, E. M., Gehrig-Burger, K., Thiele, C., Trotter, J. and Nave, K. A. (2006). Perturbed interactions of mutant proteolipid protein/DM20 with cholesterol and lipid rafts in oligodendroglia: implications for dysmyelination in spastic paraplegia. J. Neurosci. 26, 11743-11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass, A., Kujawa, M., McConnell, E., Paton, A. W., Paton, J. C. and Wojcik, C. (2008). Decreased ER-associated degradation of alpha-TCR induced by Grp78 depletion with the SubAB cytotoxin. Int. J. Biochem. Cell Biol. 40, 2865-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, D. H. and Goldberg, A. L. (1998). Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 8, 397-403. [DOI] [PubMed] [Google Scholar]
- Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., Shokat, K. M., Lavail, M. M. and Walter, P. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T. Y., Wang, S. M., Fu, W. M., Chen, Y. H. and Yin, H. S. (1999). Toxicity of tunicamycin to cultured brain neurons: ultrastructure of the degenerating neurons. J. Cell Biochem. 74, 638-647. [DOI] [PubMed] [Google Scholar]
- Lin, W. and Popko, B. (2009). Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci. 12, 379-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio, J. P., Pryor, P. R. and Bright, N. A. (2007). Lysosomes: fusion and function. Nat. Rev. Mol. Cell. Biol. 8, 622-632. [DOI] [PubMed] [Google Scholar]
- Nelis, E., Haites, N. and Van Broeckhoven, C. (1999). Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum. Mutat. 13, 11-28. [DOI] [PubMed] [Google Scholar]
- Ng, D. T., Spear, E. D. and Walter, P. (2000). The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 150, 77-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari, S. and Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381-389. [DOI] [PubMed] [Google Scholar]
- Powers, E. T., Morimoto, R. I., Dillin, A., Kelly, J. W. and Balch, W. E. (2009). Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Annu. Rev. Biochem. 78, 959-991. [DOI] [PubMed] [Google Scholar]
- Ron, D. and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 8, 519-529. [DOI] [PubMed] [Google Scholar]
- Rutishauser, J. and Spiess, M. (2002). Endoplasmic reticulum storage diseases. Swiss Med. Wkly. 132, 211-222. [DOI] [PubMed] [Google Scholar]
- Rutkowski, D. T., Arnold, S. M., Miller, C. N., Wu, J., Li, J., Gunnison, K. M., Mori, K., Sadighi Akha, A. A., Raden, D. and Kaufman, R. J. (2006). Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder, M. and Kaufman, R. J. (2005). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739-789. [DOI] [PubMed] [Google Scholar]
- Simons, M., Kramer, E. M., Macchi, P., Rathke-Hartlieb, S., Trotter, J., Nave, K. A. and Schulz, J. B. (2002). Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus-Merzbacher disease. J. Cell Biol. 157, 327-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinoway, M. P., Kitagawa, K., Timsit, S., Hashim, G. A. and Colman, D. R. (1994). Proteolipid protein interactions in transfectants: implications for myelin assembly. J. Neurosci. Res. 37, 551-562. [DOI] [PubMed] [Google Scholar]
- Southwood, C. and Gow, A. (2001). Molecular pathways of oligodendrocyte apoptosis revealed by mutations in the proteolipid protein gene. Microsc. Res. Tech. 52, 700-708. [DOI] [PubMed] [Google Scholar]
- Southwood, C. M., Garbern, J., Jiang, W. and Gow, A. (2002). The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 36, 585-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton, E., High, S. and Woodman, P. (2003). Role of calnexin in the glycan-independent quality control of proteolipid protein. EMBO J. 22, 2948-2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton, E., Holland, A., High, S. and Woodman, P. (2005). Disease-associated mutations cause premature oligomerization of myelin proteolipid protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 102, 4342-4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi, E., Logue, S. E., Gorman, A. M. and Samali, A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880-885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, C. E., Montague, P., Jung, M., Nave, K. A. and Griffiths, I. R. (1997). Phenotypic severity of murine Plp mutants reflects in vivo and in vitro variations in transport of PLP isoproteins. Glia 20, 322-332. [DOI] [PubMed] [Google Scholar]
- Trajkovic, K., Dhaunchak, A. S., Goncalves, J. T., Wenzel, D., Schneider, A., Bunt, G., Nave, K. A. and Simons, M. (2006). Neuron to glia signaling triggers myelin membrane exocytosis from endosomal storage sites. J. Cell Biol. 172, 937-948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar, S. S. and Brodsky, J. L. (2008). One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944-957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward, K. J. (2008). The molecular and cellular defects underlying Pelizaeus-Merzbacher disease. Expert Rev. Mol. Med. 10, e14. [DOI] [PubMed] [Google Scholar]
- Yamamoto, T., Nanba, E., Zhang, H., Sasaki, M., Komaki, H. and Takeshita, K. (1998). Jimpy(msd) mouse mutation and connatal Pelizaeus-Merzbacher disease. Am. J. Med. Genet. 75, 439-440. [PubMed] [Google Scholar]
- Yool, D. A., Edgar, J. M., Montague, P. and Malcolm, S. (2000). The proteolipid protein gene and myelin disorders in man and animal models. Hum. Mol. Genet. 9, 987-992. [DOI] [PubMed] [Google Scholar]
- Yoshida, H., Matsui, T., Yamamoto, A., Okada, T. and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881-891. [DOI] [PubMed] [Google Scholar]
- Zhou, J., Liu, C. Y., Back, S. H., Clark, R. L., Peisach, D., Xu, Z. and Kaufman, R. J. (2006). The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc. Natl. Acad. Sci. USA 103, 14343-14348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}