

Abstract
The principle mitochondrial target where the respiratory inhibitors CO, CN- and NO act in the execution of their acute toxic effects is complex IV of the electron-transport chain, cytochrome c oxidase. However, there is a paucity of studies in the literature regarding the concerted effects of such poisons. Accordingly, the combined inhibitory effects of CO + CN-, NO + CN- and NO + CO on the activity of cytochrome c oxidase preparations are reported. Only in the case of CO + CN- do the effects of the two inhibitors seem to be additive as expected. NO appears to be antagonistic towards the effects of the other two inhibitors; that is, the effects of both CO and CN- on enzyme activity are ameliorated by NO when present. To further clarify these observations, the ligand-substitutions of heme-bound CN- by NO in cytochrome c oxidase and hemoglobin have also been briefly investigated. These results suggest that displacement of CN- from the ferric hemoproteins by NO is rate-limited by heme reduction - and in the case of the enzyme, the presence of non-ligand-binding electron-transfer centers facilitates the reaction. The findings are discussed in relation to the idea that NO does not behave as a classic reversible (by dissociation) inhibitor.
Keywords: Complex IV, hypoxia, mitochondria, respiratory poisons, smoke inhalation, supplemental oxygen, synergistic toxicity
Introduction
It is undoubtedly the case that, due to the widespread use of materials such as acrylonitrile copolymers and polyurethanes, the toxic effects of HCN in concert with those of CO have become of increasing concern worldwide in relation to victims of smoke inhalation (1-8). Furthermore, while the literature is not extensive, there are independent reports that the acute toxic effects of these two respiratory poisons are not additive, but synergistic (9-11). The observed synergistic lethality seems to involve the metabolic consequences of tissue hypoxia (9, 11) rather than changes in blood flow (9) or altered blood CO and CN- concentrations (10). It follows that the biochemistry underlying these findings is quite likely associated with inhibition of oxygen turnover at cytochrome c oxidase (complex IV of the mitochondrial electron-transport chain) since both CO and CN- are generally accepted to rapidly bind and inactivate the enzyme. Interestingly, it has been shown in rat brain, that one effect of CO in vivo is to elevate NO levels (12). Paradoxically, however, NO has been shown to either exacerbate (13, 14) or protect against (14, 15) the toxic effects of CN- depending upon the particular cell culture and/or conditions employed. As NO is yet another complex IV inhibitor, it is clearly to be anticipated that investigating the combined effects of these three inhibitory species on cytochrome c oxidase activity may very well provide some insight into the mechanism of the reported CO and CN- synergistic toxicity.
The active (O2-binding) site of cytochrome c oxidase is binuclear, consisting of haem a3 and CuB (16). The active-site pocket has long been known to be able to simultaneously accommodate more than one diatomic ligand, such as two NO molecules (17), or NO and CN- (18) or CO and CN- (19); hence, by implication, O2 together with another diatomic species. Therefore, studying the interactions of dual inhibitors with this particular enzyme is of some unusual importance. Here we report the results of a study concerning the combined effects of CO + CN-, NO + CN- and NO + CO on the activity of isolated bovine cytochrome c oxidase (ferrocytochrome c:oxygen oxidoreductase EC 1.9.3.1) and submitochondrial particles. The results appear to clarify some confounding observations reported in studies employing cultured cells and in vivo systems.
Experimental
Cytochrome c oxidase was prepared as previously described (20) from intact bovine heart mitochondria using a modified Harzell-Beinert procedure (without the preparation of Keilin-Hartree particles). The enzyme was determined to be spectroscopically pure if the 444 nm to 424 nm ratio for the reduced enzyme was 2.2 or higher (21). Derivatives were prepared in 50 mM potassium phosphate, 1 mM in sodium EDTA and 0.1% in lauryl maltoside, pH 7.4-7.8, to concentrations of 10-80 μ-M (in enzyme). Enzyme concentrations were determined as total heme a using the differential (absorption) extinction coefficient of Δε604 = 12 mM-1cm-1for the reduced minus oxidized spectra of the mammalian and bacterial enzymes, respectively (22). Concentrations throughout are given on a per enzyme concentration basis (NOT per [heme a]). When required, submitochondrial particles were prepared from intact mitochondria simply by dispersion in buffer, the resulting osmotic shock rupturing the mitochondrial membranes. Mild sonication of these preparations did not lead to any measurable changes in cytochrome c oxidase activity.
Ferrocytochrome c:O2 oxidoreductase activity was determined spectrophotometrically employing the high ionic strength method of Sinjorgo et al. (23). Using this assay, we routinely obtain a turnover number with respect to cytochrome c of 340 (± 30) s-1 (260 μM O2, 0.1 M sodium phosphate, 0.1% lauryl maltoside, pH 7.4, 22 °C) similar to that of the bovine enzyme isolated from a variety of tissues by others (23). Oxygen consumption kinetics were measured polarographically using a catalytic amount of cytochrome c (60 μM) and 5 mM sodium ascorbate as the reductant. Reactions were carried out at room temperature in 0.1 M potassium phosphate buffer, 0.1% lauryl maltoside, pH 7.4, 22 °C, at an initial oxygen concentration of ~130 μM. Nitric oxide decomposition is dependent upon oxygen concentration and governed by the equation -d[NO]/dt = 4k[NO]2[O2] with k = 2 x 106 M-2s-1 (24, 25). Consequently, starting with an oxygen concentration of ~130 μM, the initial rate of uncatalysed degradation of a 10 μM NO solution will be ~6 μM per minute at room temperature, but this slows dramatically as the reaction proceeds. All kinetic time courses for oxygen consumption (and ferrocytochrome c oxidation) were essentially linear in the range 10 - 60 s. Where required, rates were estimated from the linear-region slopes of the oxygen (or ferrocytochrome c) concentration versus time plots without applying corrections.
Hemoglobin A was obtained from Sigma, oxidized by K3Fe(CN)6 to metHb, further purified on a G-25 column and then dialyzed against high (100 mM) and low (10 mM) salt sequentially to rid the protein of excess [Fe(CN)6]3- and [Fe(CN)6]4- (26). MetHbCN was formed by titrating purified metHb with KCN until the fully formed species was detected spectrophotometrically at 540 nm (ε540 = 12.5 cm-1mM-1) (27).
All reagents were ACS grade, or better, used without further purification and unless stated to the contrary, were purchased from Aldrich or Sigma. Sodium dithionite, 87% minimum assay (+ H2O), was obtained from EM Science. Carbon monoxide and nitric oxide gases were obtained from Matheson Incorporated. Carbon monoxide saturated solutions were made by bubbling CO through buffer solution for 30 minutes. Nitric oxide was scrubbed with water and KOH pellets prior to use, bubbled through anaerobic buffer (prepared by bubbling argon through the solution) and added to enzyme samples volumetrically with gas-tight syringes. Buffered solutions never exhibited any significant change of pH (i.e. < 0.05 pH units) following NO additions.
Electronic absorption spectra were measured and photometric determinations made using Shimadzu UV-1650PC and UV-2501PC spectrophotometers. Rates of electron transfer from reduced cytochrome c to cytochrome c oxidase under saturating [O2] (260 μM at 22°C) were followed at 550 nm. A Clark-type electrode (Rank Brothers), calibrated using saturated sodium bisulphate (0% calibration) and air-saturated buffer (100% calibration), was employed to carry out the oxygen uptake experiments. The oxygen-depletion experiments performed under a closed-system configuration of the Clark-type electrode showed linearity from 100% to ~12% oxygen levels over a 3 minute period.
Cultured sheep pulmonary artery endotheial cells (SPAEC) were a gift from Bruce Pitt, Department of Environmental & Occupational Health, University of Pittsburgh. The SPAEC were grown in OptiMEM supplemented with 10% fetal bovine serum, 15 μg/mL endothelial cell growth supplement, 100 U/mL penicillin and 100 μg/mL streptomycin at 37 °C in a 5% CO2 atmosphere. Cells were plated into 24-well plates to ~95% confluence. Just prior to cyanide addition, media was removed and replaced with PBS (phosphate-buffered saline) after washing the cells once with PBS. Sodium cyanide (0 to 1.2 mM, final concentration) was added and cells were incubated for one hour at 37°C. Cells were then washed with PBS and 10% AlamarBlueTM added to assay for cell viability. Fluorescence changes (535 nm excitation, 590 nm emission) were monitored using a BGM Fluostar Galaxy plate reader. In those experiments where both L-NAME and sodium cyanide were added to cells, L-NAME was incubated with the SPAEC for 30 minutes in PBS before addition of cyanide as described above.
Results
Steady-state Inhibition Kinetics
To establish the effects of dual inhibitors on the activity of cytochrome c oxidase, the kinetics of both ferrocytochrome c oxidation and dioxygen depletion have been examined. It has been shown that incubation of cytochrome c oxidase with CO leads to deactivation of the enzyme by loss of subunit I (28). To avoid the possibility of such processes, unless explicitly stated to the contrary, samples of the enzyme were not pre-incubated with CO, CN-, or NO, turnover always being initiated before the addition of any inhibitors. To be sure that significant differences between inhibited rates and control (uninhibited) rates could routinely be obtained, we opted so far as was possible to work at single inhibitor concentrations sufficient to lower the measured activity by 50 - 60% of the control (maximal) rate (see Table I). This approach also ensured that when pairs of inhibitors were added together at these pre-established concentrations, either a net loss or gain in activity could be detected with roughly equal sensitivity. Since published Ki values can be laboratory and/or preparation dependent the precise quantities of inhibitor to be used were determined by a few trial measurements.
Table I.
Comparison of observed cytochrome c oxidase (complex IV) turnover numbers during single and dual inhibition by CO, KCN, NO, CO + KCN, NO + KCN, NO + CO.
| Inhibitor {concentration} | Turnover number (kcat, s-1)# |
|---|---|
| Uninhibited control* | 346 ± 28 |
| CO {0.5 mM} | 190 ± 21 |
| KCN {50 nM} | 136 ± 8 |
| NO {0.5 μM} | 184 ± 30 |
| CO {0.5 mM} & KCN {50 nM} | 109 ± 9 |
| KCN {50 nM} & NO {0.5 μM} | 176 ± 8 |
| NO {0.5 μM} & CO {0.5 mM} | 182 ± 11 |
1.2 nM enzyme, pH 7.4 in 0.1 M sodium phosphate buffer, 1 mM EDTA, 0.05% lauryl maltoside, 22 °C.
Calculated from fits in Figure 1A, quoted errors are standard deviations.
Within the limits of experimental uncertainty, the concerted inhibitory effects of CO + CN- on the rate of ferrocytochrome c oxidation by the isolated enzyme could reasonably be approximated as the sum of the effects of these two inhibitors acting separately (Figure 1A). It should be noted that, from a qualitative perspective, the addition of the two inhibitory effects gives essentially the same result as one could obtain by simply increasing the concentration of a single inhibitor. This, of course, is exactly the result expected if the two inhibitors both form inactive enzyme-inhibitor complexes, which upon dissociation lead to regeneration of the catalytically competent enzyme. Therefore, these findings are entirely in keeping with the widely held view (19, 29-31) that CO and CN- reversibly associate with the haem a3-CuB pair of cytochrome c oxidase, preventing reaction of the enzyme with substrate O2 while either inhibitory species remains bound in the binuclear site.
Figure 1. Dual inhibition of cytochrome c oxidase (complex IV) turnover (spectrophotometric measurements) by CO + CN-, NO + CN-, and NO + CO.

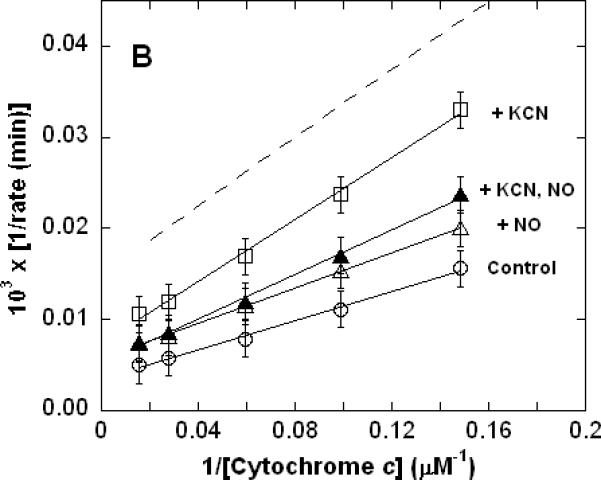

Lineweaver-Burk (double reciprocal) plots showing inhibition of ferrocytochrome c oxidation. Reaction conditions were 1.2 nM enzyme in 0.1 M aqueous potassium phosphate buffer, pH 7.4, 1.0 mM in EDTA, 0.05% in lauryl maltoside, 22 °C. A. Uninhibited control (○), 0.5 mM CO (▽), 50 nM KCN (□), 0.5 mM in CO and 50 nM in KCN (◆); B. uninhibited (○), 0.5 μM NO (Δ), 50 nM KCN (□), 50 nM in KCN and 0.5 μM in NO (▲); C. uninhibited (○), 0.5 mM CO (▽), 0.5 μM NO (Δ), 0.5 μM in NO and 0.5 mM in CO (▼). In each panel, the broken line represents the combined effect of the two relevant inhibitors predicted by simple summation of their individual measured effects.
Unlike the CO + CN- kinetics, the concerted inhibitory effects of NO + CN-(Figure 1B) and NO + CO (Figure 1C) were clearly not additive. Remarkably, with NO in excess, the inhibition observed for NO + CN- was actually less than that observed for the same concentration of CN- alone (Figure 1B). The kinetics of cytochrome c oxidase inhibition by NO + CO were indistinguishable from those of either NO, or CO, alone in this particular set of experiments (Figure 1C) - other data sets (e.g. see Figure 3C below) suggested that, in fact, the NO + CO kinetics were dominated by the effect of NO alone. It does not appear that these qualitatively very different findings to those obtained for CO + CN- (Figure 1A) can be explained by any straightforward considerations of relative affinities and mass action. Since the inhibition of cytochrome c oxidase by CO + CN- yields simple additive results, it must be the NO which is principally responsible for the interesting non-additive kinetics shown in Figure 1. More succinctly, the addition of NO (nominally `inhibitory') to the enzyme during turnover in the presence of either of the other two inhibitors did not lead to increased inhibition. It is to be stressed that in experiments where cytochrome c oxidase is inhibited by NO alone, there was no obvious indication in the kinetics of any unusual behavior. At [NO]:[O2] ratios upwards of 1:50, non-linear kinetics were evident; but in the NO concentration range employed to obtain the data of Figure 1, linearity of double-reciprocal plots, with a straightforward (additive) dependence of the slope on NO concentration, was reproducibly observed (Figure 2). This is also the case for double-reciprocal plots of CO (only) and CN- (only) at the concentrations employed here (not shown).
Figure 3. Dual inhibition of cytochrome c oxidase (complex IV) turnover (polarographic measurements) by CO + CN-, NO + CN-, and NO + CO.
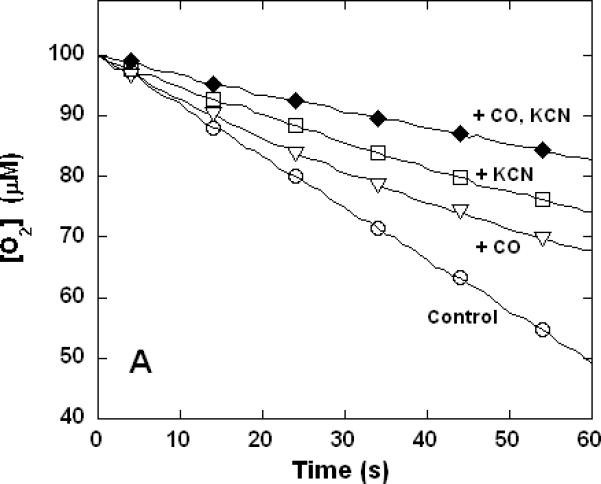
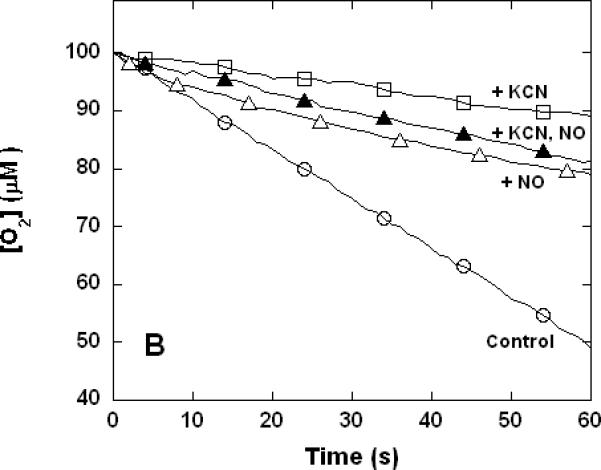
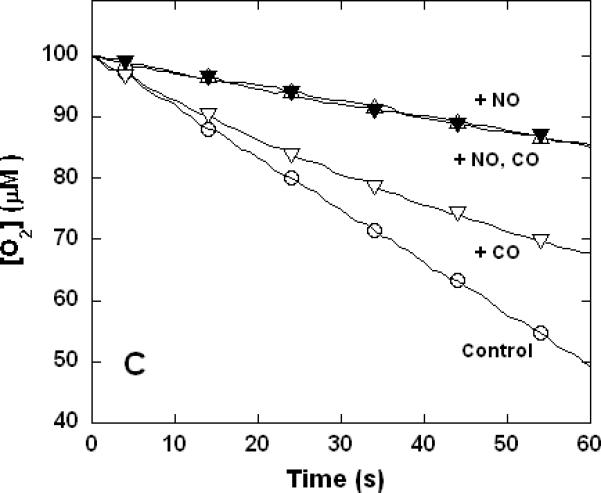
Clark-type oxygen electrode traces showing inhibition of O2 consumption, 500 nM enzyme, 60 μM ferrocytochrome c in 0.1 M aqueous potassium phosphate buffer, pH 7.4, 1.0 mM in EDTA, 0.05% in lauryl maltoside, 22 °C. Small adjustments to the locations of the traces relative to the ordinate axes have been made to assist with visual comparison. A. Uninhibited control (○), 1.0 mM CO (▽), 1.0 μM KCN (□), 1.0 mM in CO and 1.0 μM in KCN (◆); B. uninhibited (○), 8 μM NO (Δ), 2 μM KCN (□), 2 μM in KCN and 8 μM in NO (▲); C. uninhibited (○), 10 μM NO (Δ), 1.0 mM CO (▽), 10 μM in NO and 1.0 mM in CO (▼).
Figure 2. Lineweaver-Burk (double reciprocal) plot demonstrating inhibition of cytochrome c oxidase turnover by NO alone.
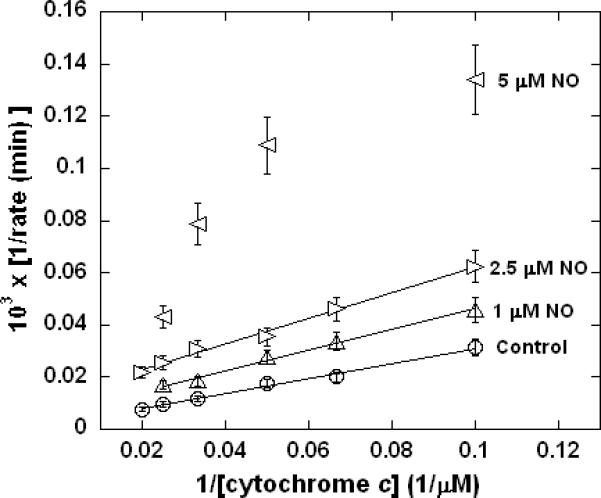
Measurements performed in 0.1 M aqueous potassium phosphate buffer, pH 7.4, 1.0 mM in EDTA, 0.05% in lauryl maltoside, using samples that were 5.0 nM in enzyme at 37 °C.
To verify that the unusual non-additive kinetic behavior of mixed NO + CN- and NO + CO inhibition were routinely to be observed irrespective of the procedure used, a more limited set of polarographic measurements was performed. Clarke-type oxygen electrodes are subject to drift and determinations made with these devices lack the sensitivity of the spectrophotometric method, which in practice requires that slightly different conditions be used in the two kinds of measurement. Here again, the inhibitory effects of CO and CN- combined to yield increased inhibition when the two were present together (Figure 3A). However, at the individual inhibitory levels used in these experiments, the combined inhibition of NO + CN- was less than that observed for CN- alone (Figure 3B) and the combined inhibition of NO + CO was the same as that observed for NO alone (Figure 3C) in good qualitative agreement with the spectrophotometrically determined results of Figure 1.
The ability of NO to counteract the inhibition of cytochrome c oxidase by CN- was further investigated by varying the CN- concentration at fixed [NO] (Figure 4A) and by varying the NO concentration at fixed [CN-] (Figure 4B). The most interesting feature of these data is that, at all ratios of [NO]:[CN-] examined, the inhibitory effects were never found to be quantitatively additive. That is, the combined inhibition observed for NO + CN- was always found to be significantly less than one would estimate by summation of the inhibition measured for NO alone and CN- alone at the same individual concentrations. NO appeared to dominate the inhibition kinetics only when present in greater than 4-fold (Figure 4B) to 7-fold (Figure 4A) excess. The difference between these estimates is not significant given the likely volumetric uncertainty involved in the assay procedures, where transfers of microliter quantities of gaseous reagents have been made with gas-tight syringes to reaction vessels closed with rubber septa. In measurements of cytochrome c oxidase activity in the presence of individual inhibitors, we typically find it necessary to add 5-10 times the concentration of NO compared with CN- to reaction mixtures in order to achieve the same degree of enzyme inhibition (e.g. see Table I). Thus, when both were present, the ratio of [NO]:[CN-] at which neither NO, nor CN-, inhibition was dominant (i.e. 4-7) corresponded well to the concentration ratio at which the degree of inhibition due to each inhibitor should be equal (i.e. 5-10). In situations where both inhibitors were present, the effect of NO was dominant when it was in greater than several-fold excess ([CN-] < 150 nM in Figure 4A) whereas the effect of CN- was dominant if NO were present at less than several-fold excess ([NO] < 400 nM in Figure 4B). At the higher inhibitor levels, where total inhibiton of the enzyme was > 60%, the difference between the results for NO + CN- and CN- only (Figure 4A) or NO + CN- and NO only (Figure 4B) were within the experimental uncertainty. In summary, so far as these data are able to discriminate, the mechanisms by which the two inhibitors function are apparently mutually exclusive (not additive). Consequently, under conditions where the inhibitory capacity of NO exceeds that of CN- (corresponding to a greater than ~4-fold concentration excess) NO inhibition will dominate and CN- inhibition will effectively be decreased.
Figure 4. Dependence of cytochrome c oxidase turnover on the relative concentrations of NO and CN- during mixed inhibition.
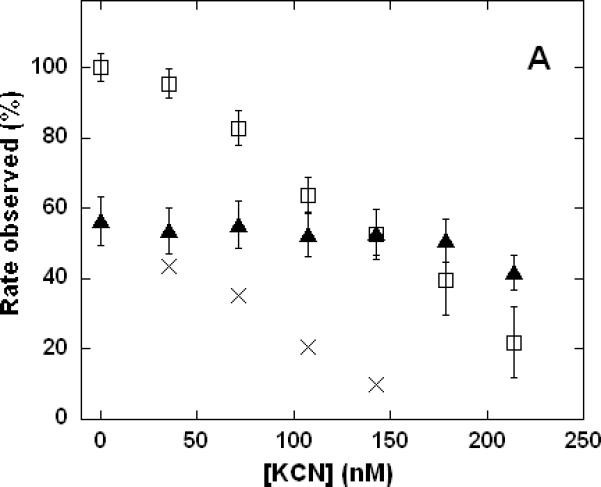
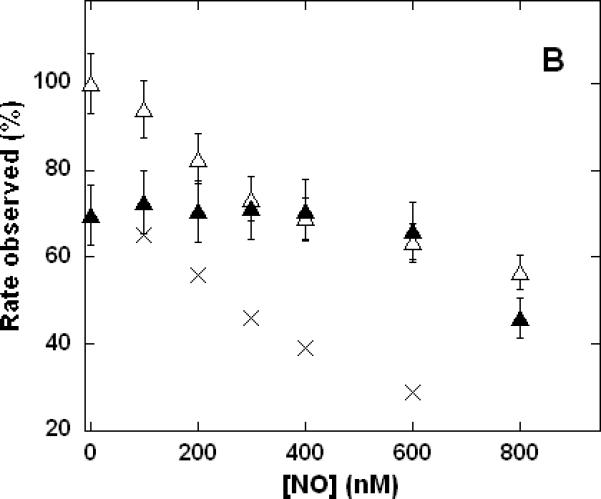
Measurements performed in 0.1 M aqueous potassium phosphate buffer, pH 7.4, 1.0 mM in EDTA, 0.05% in lauryl maltoside, 22 °C, 2.4 nM enzyme concentration. A. Variable [KCN], without NO (□), NO added to 1.0 μM (▲); B. variable [NO], without KCN (Δ), KCN added to 80 nM (▲). Each point is the mean of 3-6 measurements, error bars represent standard deviations. Total inhibition predicted (assuming no interaction) by simple summation of the individual effects due to NO plus CN- (×).
Characterization of the Inhibited Forms
In the presence of an electron source, the cyanide-inhibited cytochrome c oxidase exists as a partially-reduced derivative in which CN- is bound to heme a3 in its ferric form, CuB (and the other centres) being reduced (18, 29). This species, which exhibits a well-known broad EPR spectrum (Figure 5, broken trace) attributed to cyanoferriheme a3, has also been detected during enzyme turnover in the presence of CN-, although it was apparently not the major inhibited form (30). Upon exposure to NO, the broad EPR signal was rapidly converted to a much sharper spectrum exhibiting a three-line hyperfine structure around g = 2 (Figure 5, solid trace). This signal has been unambiguously shown to derive from a five-coordinate nitrosylferroheme species (20). That is, NO displaces CN- from heme a3 with near-concomitant reduction of the heme from the ferric to ferrous oxidation level. Given the similarity in the ligand properties of NO and CO, it was to be expected that the latter would also undergo an analogous substitution reaction with the partially-reduced cyanide-inhibited cytochrome c oxidase, but this cannot be so convincingly demonstrated by EPR spectroscopy as the carbonmonoxyferroheme a3 final product is EPR silent.
Figure 5. X-band electron paramagnetic resonance (EPR) spectra showing displacement of CN- by NO at heme a3 of cytochrome c oxidase.
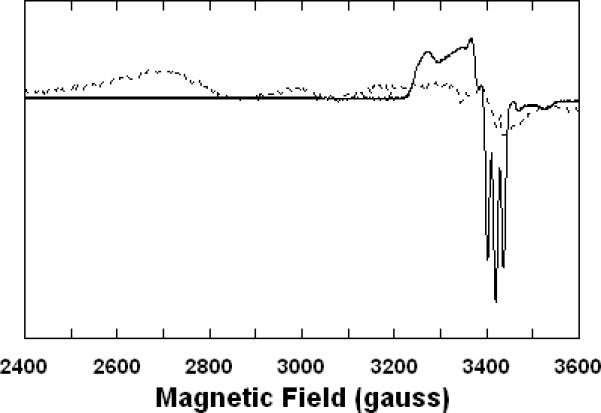
Sample preparations were carried out in 0.1 M aqueous potassium phosphate buffer, pH 7.4, 1.0 mM in EDTA, 0.05% in lauryl maltoside, 22 °C, prior to freezing in EPR tubes. Recording conditions: 0.2 mW microwave power, 4 G modulation amplitude, 1 × 104 amplifier gain, 15 K sample temperature. Broken trace: partially-reduced cyanide adduct, 60 μM in enzyme, 1.0 mM in KCN, ~1 mM in Na2S2O4. Solid trace: partially-reduced cyanide adduct plus NO, 60 μM in enzyme, 1.0 mM in KCN, ~1 mM in Na2S2O4, 1.9 mM (1.0 atm) NO.
The displacement of CN- from heme a3 in cytochrome c oxidase by CO can be conveniently followed by electronic absorption spectroscopy. The partially-reduced cyanide-inhibited enzyme exhibited a 428 nm Soret absorption band (Figure 6, broken trace) that was shifted to 433 nm following the introduction of CO (Figure 6, dotted trace) with an accompanying increase in the intensity of the visible-region (α) band at 604 nm (not shown). These electronic absorption changes, which were complete by the time the sample could be returned to the spectrophotometer following addition of CO, are strong indicators of CO binding to heme a3 in the ferrous state, confirming the expected reaction to have taken place. Interestingly, following NO addition to the partially-reduced cyanide-inhibited enzyme a more complicated envelope with two maxima at 430 nm and 442 nm was obtained (Figure 6, solid trace). In fact, these two features were slightly variable in relative intensity between samples (not shown) demonstrating that they represent two distinct chemical species rather than being two bands in the spectrum of a single derivative. Since Soret features at > 440 nm are associated with exogenousligand-free ferrous heme a3, this spectrum provides evidence that once NO has displaced CN-, the NO itself may then be lost from the ferrous heme. This does not necessarily mean, however, that the NO dissociates - it may be lost by conversion to some other species that is not a good ligand. In summary, both CO and NO, are seemingly able to rapidly displace CN- from ferric haem a3 to form, respectively, ferrous CO and ferrous NO adducts, presumably with the acquisition of the necessary electron from a reduced center nearby, such as CuB. In the case of the NO adduct, there is some indication in the absorption spectrum for subsequent partial loss of bound NO.
Figure 6. Electronic absorption spectra of cytochrome c oxidase derivatives showing displacement of CN- by both NO and CO.
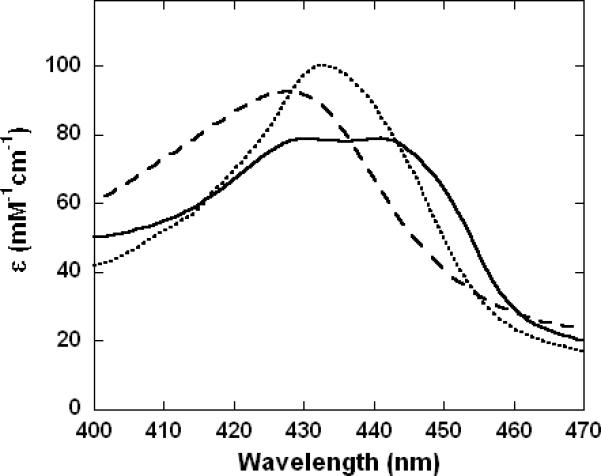
Samples were prepared in 100 mM aqueous potassium phosphate buffer, pH 7.4, 0.05 % lauryl maltoside, 22°C, 1.00 cm pathlengths. Broken trace: partially-reduced cyanide adduct, 12 μM in enzyme, 0.2 mM in KCN, ~1 mM in Na2S2O4; dotted trace: partially-reduced cyanide adduct plus CO, 12 μM in enzyme, 0.2 mM in KCN, ~1 mM in Na2S2O4, ~1 mM (1.0 atm) CO; solid trace: partially-reduced cyanide adduct plus NO, 12 μM in enzyme, 0.2 mM in KCN, ~1 mM in Na2S2O4, 1.9 mM (1.0 atm) NO.
In an effort to better understand the general features of a reaction where CN- attached to a ferric heme is displaced by ligands having a marked preference for ferrous heme (namely, NO and CO) we performed a limited set of experiments with hemoglobin. Following the admission of NO gas to an anaerobic sample of cyanomethemoglobin, the visible region absorption spectrum slowly (kobs ~0.5 hr-1) changed from that of the ferric heme-CN- derivative to that of a mixture (32) of 5-coordinate (minor component) and 6-coordinate ferrous heme-NO adducts (Figure 7A). Significantly, when the analogous substitution was attempted with CO (not shown) there was no detectable reaction after several hours. The rate of the overall reaction with NO was found to be independent of pH (one pH unit either side of 7.4, not shown) but linearly dependent upon [NO] (Figure 7B). The obvious reasonable interpretation of these observations is that reduction of the heme is rate limiting and also, while NO may act as the necessary reducing agent, CO cannot. This supports the idea that reaction of the partially-reduced cyanide-adduct of cytochrome c oxidase is possible with CO and faster in the case of NO because of the proximity of reduced centers in the enzyme that are able to act as electron sources.
Figure 7. Reaction of NO with cyanomethemoglobin (metHbCN).
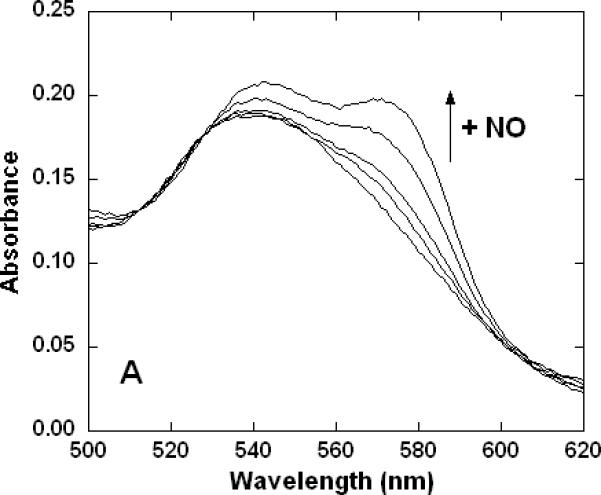
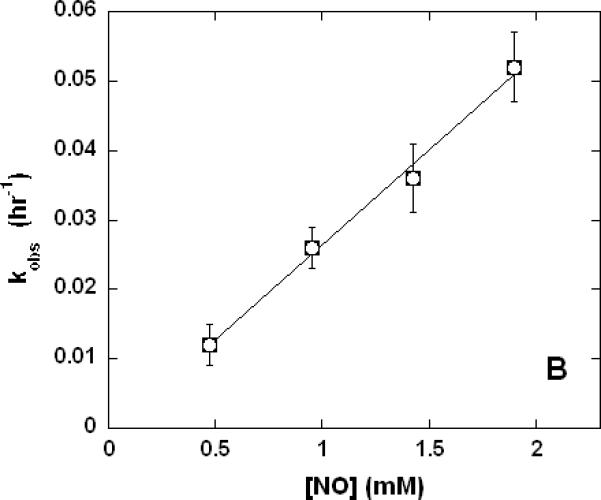
Samples were prepared in 100 mM aqueous potassium phosphate buffer, pH 7.4, 22°C, 1.00 cm pathlengths. A. Electronic absorption spectral changes (500-620 nm range) over time (~1 hr) of 14 μM metHbCN plus 0.95 mM (0.5 atm) NO. B. Linear dependence of the rate of reaction on the NO concentration.
Biological Significance
To evaluate the possible biological significance of the present findings, we undertook a brief investigation of the effects of added KCN on endothelial cells. It is most convenient to investigate the effects of varying near-physiological NO levels by addition of nitric oxide synthase (NOS) inhibitors to endothelial cells since these are net producers of NO. The addition of the NOS inhibitor Nω-nitro-L-arginine methyl ester (L-NAME) to sheep pulmonary artery endothelial cells (SPAEC) has no measurable effect on metabolic activity (Figure 8, broken line) for several hours. However, the SPAEC undergo a significantly increased loss of metabolic activity in the presence of L-NAME + KCN (Figure 8, ∎) compared to KCN alone (Figure 8, ∎).These findings clearly indicate that NO, at endogenously generated levels in SPAEC, is an antagonist of KCN toxicity. There is also a limited amount of supporting data from other laboratories. Baskin et al. (33) have reported a protective effect of NO donors against the effects of NaCN in mice. Leavesley et al. (14) similarly found a protective effect of NO donors in cultured cells exposed to KCN, but surprisingly, these same authors report apparent enhancement of CN- toxicity by low level NO production (see Discussion).
Figure 8. Resistance of sheep pulmonary artery endothelial cells (SPAEC) to CN- is suppressed in the presence of a nitric oxide synthase (NOS) inhibitor.
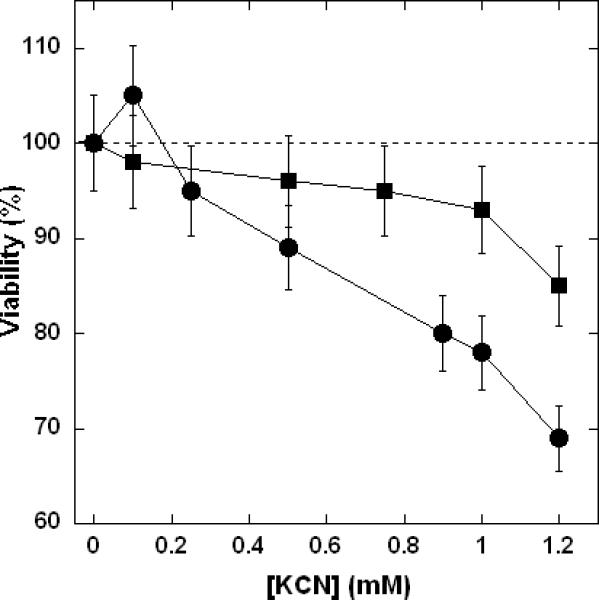
Viability was determined 2 hours after exposures to KCN by AlamarBlue™ (see Experimental for details). Variable KCN (■), variable KCN plus 0.5 mM L-NAME (●). The broken line indicates the effect of the NOS inhibitor L-NAME on the SPAEC in the absence of KCN (by extrapolation from the point where [KCN] = 0).
Discussion
Functional Reserve of Cytochrome c Oxidase Activity?
On a per molar basis, the rate of ferrocytochrome c oxidation by its oxidase is expected to be four times the rate of oxygen removal, since four electrons are required for the conversion of one dioxygen to two water molecules during the enzyme-catalyzed reaction. However, even using samples of the purified enzyme, it can be very difficult in practice to straightforwardly compare kinetic determinations performed spectrophotometrically and polarographically. The two sets of measurements are optimally made under rather different sets of conditions (see Experimental) where the principal sources of error are not the same. Furthermore, there is typically some variation in the specific activity of different enzyme preparations and a loss in activity of individual preparations upon storage - which are important considerations given that the two procedures can seldom be performed at the same time. Thus, if a spectrophotometrically determined specific activity for cytochrome c oxidase is found to differ somewhat from four times the polarographically determined rate on a per molar basis, this need not necessarily be significant. The situation is even more problematic if all the components of the electron-transport chain (ETC) are present under partially-inhibitory conditions. For example, inhibition of the cytochrome c oxidase (complex IV) by CN- will result in accumulation of electrons in complexes I, II and III, this in turn leading to an increase in superoxide (O -2) generation. Under spectrophotometric assay conditions, some of the electron accumulation in complexes I, II and III could, conceivably, be due to back-flow from ferrocytochrome c and, conversely, O -2 reacts with ferricytochrome c to regenerate the starting form of the cytochrome. That is, there are complex IV-independent reactions that can both oxidize and reduce cytochrome c. Consequently, any spectrophotometric measurement of cytochrome c oxidase activity under cyanide-inhibited conditions where other ETC components are present could be erroneously elevated or lowered depending upon the relative rates of such interfering reactions. When working with purified preparations of the enzyme, back-flow of electrons is of no concern, but we did verify that interference from O -2 generation was insignificant in the present studies by observing that the addition of cyanide-insensitive manganese superoxide dismutase had no effect on the measured activity (not shown).
It has been reported that exposure to KCN of mouse brain in vivo (34) and rat mesencephalic cells in culture (14) requires > 50% inhibition of cytochrome c oxidase before the rate of O2 consumption is decreased. If not an experimental artifact, this is a potentially very interesting observation indeed because it suggests the presence of some additional critical site(s) other than the terminal oxidase where the acute effects of CN- toxicity are expressed. However, the “threshold effect” analysis (35, 36) adopted to generate these results, is subject to the interference and consequent interpretational difficulties we have just described. So, at the level of the intact respiratory chain, there may be some threshold of cytochrome c oxidase inhibition by CN- below which O2 consumption is not decreased - but this remains to be verified unambiguously.
Inhibition by Nitric Oxide
We have shown that under appropriate turnover conditions, where NO is one of a pair of dual inhibitors, the measured cytochrome c oxidase inhibition kinetics essentially represent the effects of NO alone (Figures 1B & 1C and 3B & 3C). That is, intriguingly, NO appears able to act as an antagonist of both CO and CN- inhibition of the enzyme. As the inhibition kinetics of CO + CN- (in the absence of NO) are additive in the expected manner (Figures 1A and 3A) one is drawn to the conclusion that it is the behavior of NO which is in some way significantly different to that of these other two inhibitors. More specifically, the mechanism of NO inhibition cannot simply involve the reversible formation of an inactive enzyme-inhibitor complex; because if it did, the measured kinetic effects of NO + CN- would be qualitatively similar to those exhibited by CO + CN-, which is clearly not the case. Further to this point, the affinity of CN- for cytochrome c oxidase (KD = 20 nM, k2 ~106 M-1s-1) (14, 37) is about two orders of magnitude less than that of NO (KD = 0.1 nM, k2 ~108 M-1s-1) (14, 38) when these parameters are determined for each inhibitory species in the absence of the other. Consequently, when both inhibitors are present, from consideration of mass action alone it appears as though NO should influence the measured rate of cytochrome c oxidase turnover even if CN- is in 100-fold excess. That this is clearly not the case in turnover experiments (Figure 4B) casts additional doubt on the notion that NO behaves as a classic reversible (by dissociation) inhibitor and argues that the reported affinity parameters cannot all be physically meaningful in the present context of mixed (NO + CN-) inhibition.
It has been suggested that NO be considered a substrate of cytochrome c oxidase (20, 39) as the enzyme is known to catalyze the oxidation of NO to NO -2 under a variety of conditions (40, 41). This observation conveniently facilitates an explanation of the antagonistic effect of NO on the CO and CN- inhibition of the enzyme. The conventional reaction catalyzed by cytochrome c oxidase is the four-electron reduction of molecular oxygen to water:
| (Eq. 1) |
With CO or CN- bound in the active site, this reaction cannot take place and, apparently, neither can any other O2-consuming reaction. However, with NO occupying the active site, we propose that an alternative reaction is possible, in which NO can be considered an auxiliary substrate:
| (Eq. 2) |
The conditions and mechanism by which the conversion of NO to NO -2 might occur have been controversial, most other authors favoring CuB as the site of NO oxidation. However, as NO has recently been shown to undergo what seems to be the same reaction with a bacterial terminal oxidase lacking CuB (42), the favored view is probably erroneous. Fortunately, the only features of the reaction of cytochrome c oxidase with NO needed to explain the present results are that (i) NO becomes oxidized to nitrite in a process consuming O2 (i.e. NO does not behave as a reversibly bound competitive inhibitor) (41, 42); (ii) the rate of the reaction expressed in Equation 2 must be slower than the conventional reaction of Equation 1 as NO certainly does have a readily detected inhibitory effect on the enzyme (Figure 2); (iii) the affinity of NO (taking into account mass action) for the enzyme must be greater than those of both CO and CN-, so that NO effectively out-competes the other two inhibitors for occupation of the active site, rendering their contributions to the overall kinetics negligible (Figures 1B & 1C and 3B & 3C). Given the above three stipulations, NO can dominate the observed inhibition when CO or CN- are also present because displacement of CO or CN- by NO is facile and the subsequent conversion of NO to NO -2 is slow in comparison. In other words, the observed kinetics are rate-limited by the NO-consuming reaction in Equation 2 (or some similar process) irrespective of whether or not CO and CN- are also present.
Intriguingly, while finding exogenously administered NO-donors to antagonize CN - inhibition, the same group of authors also report that endogenous (low level) NO production enhanced CN- inhibition in mesencephalic cells (14). No plausible biochemical explanation for these findings was offered. Mechanistically, it is not at all obvious how the action of NO could change from being toxic at low concentrations to protective at higher concentrations - and this is contrary to the current results where endogenous levels of NO were found to ameliorate CN- toxicity in endothelial cells (Figure 8). Conflicting views concerning the effect of low NO levels towards cyanide toxicity could reflect differences in behavior of particular cell lines, properties of end points observed, or other experimental variables between laboratories. However, resolution of this paradox is beyond the scope of the present work and must await further studies.
Supplemental Oxygen Therapy
An unexpected beneficial effect of supplemental O2 delivery in the amelioration of cyanide toxicity has been the subject of dozens of experimental investigations and clinical case reports (43). There has been no rational basis for this particular treatment as cyanide-inhibited cytochrome c oxidase cannot be reactivated by O2 (44). However, O2 is also a substrate for nitric oxide synthase (NOS) and so, we can at least now postulate that supplemental O2 leads to increased NO which, as we show here, is an antagonist of CN- inhibition of cytochrome c oxidase. Unfortunately, this explanation cannot be complete as supplemental O2 is most effective when delivered in addition to nitrite and thiosulphate (45). Nitrite is another potential source of NO (46), suggesting that constituent NOS alone might not be able to generate enough NO. Thiosulfate, a substrate for the enzyme rhodanese, promotes the conversion of CN- to the less toxic thiocyanate ion (SCN-). Conceivably, some portion of the supplemental O2 may become diverted into O2- production. Subsequently generated derivatives of O -2 (e.g. hydrogen peroxide and peroxynitrite) could then, perhaps, start oxidizing CN- to less toxic species by means of reactions catalyzed by the numerous peroxidatic hemoproteins present in biological systems (47, 48). Again, however, examining such possibilities is outside the focus of the present investigation.
Inhibition by Carbon Monoxide
So far as the isolated enzyme is concerned, compared with NO and CN-, CO is a relatively poor inhibitor of cytochrome c oxidase (Figures 1 and 3). There are those who suggest that the observed toxicity can stem from direct inhibition of cytochrome c oxidase by CO (49, 50) and others who have argued that all the cellular toxic effects of CO are likely due to the elevation of the steady-state NO levels mediated by competition between CO and endogenous NO for the same preferred locations (51, 52). However, the ~50% inhibition of cytochrome c oxidase reported (50) following exposure of mitochondrial preparations to 0.05% CO cannot be readily explained by either mechanism. The matter has recently been clarified by Iheagwara et al. (28) who showed that exposure of mice to low levels of CO led to substantial loss of cytochrome c oxidase activity in conjunction with decreased mitochondrial content of total heme a and the associated subunit I. This recently identified signaling process almost certainly provides a much more significant contribution to the net toxicity observed (in all experiments with viable mitochondria, cultured cells, excised tissues and in vivo) than competitive binding of CO to the active site of the enzyme. Finally, we note that the reported synergism in the toxicity of CO + HCN, is thought to be primarily associated with hypoxia at the cellular level (9-11). Cleary, however, the present study suggests that this does not arise through any interaction at the oxygen-binding site of cytochrome c oxidase, since inhibition by CO + CN- is the one simple additive case we observed (Figures 1A and 3A).
Acknowlegements
Supported by the National Institutes of Health (HL61411 to J.P and L.L.P.)
Bibliography
- (1).Ferrari LA, Arado MG, Giannuzzi L, Mastrantonio G, Guatelli MA. Hydrogen cyanide and carbon monoxide in blood of convicted dead in a polyurethane combustion: a proposition for the data analysis. Forensic Sci Int. 2001;121:140–143. doi: 10.1016/s0379-0738(01)00464-9. [DOI] [PubMed] [Google Scholar]
- (2).Alarie Y. Toxicity of fire smoke. Crit Rev Toxicol. 2002;32:259–289. doi: 10.1080/20024091064246. [DOI] [PubMed] [Google Scholar]
- (3).Alcorta R. Smoke inhalation & acute cyanide poisoning. Hydrogen cyanide poisoning proves increasingly common in smoke-inhalation victims. Jems. 2004;29(suppl 615):16–17. quiz suppl. [PubMed] [Google Scholar]
- (4).Riddle K. Hydrogen cyanide: fire smoke's silent killer. Jems. 2004;29(suppl 5) [PubMed] [Google Scholar]
- (5).Walsh DW, Eckstein M. Hydrogen cyanide in fire smoke: an underappreciated threat. Emerg Med Serv. 2004;33:160–163. [PubMed] [Google Scholar]
- (6).Yeoh MJ, Braitberg G. Carbon monoxide and cyanide poisoning in fire related deaths in Victoria, Australia. J Toxicol Clin Toxicol. 2004;42:855–863. doi: 10.1081/clt-200035211. [DOI] [PubMed] [Google Scholar]
- (7).Wardaszka Z, Niemcunowicz-Janica A, Janica J, Koc-Zorawska E. [Levels of carbon monoxide and hydrogen cyanide in blood of fire victims in the autopsy material of the Department of Forensic Medicine, Medical University of Bialystok] Arch Med Sadowej Kryminol. 2005;55:130–133. [PubMed] [Google Scholar]
- (8).Eckstein M, Maniscalco PM. Focus on smoke inhalation--the most common cause of acute cyanide poisoning. Prehosp Disaster Med. 2006;21:s49–55. doi: 10.1017/s1049023x00015909. [DOI] [PubMed] [Google Scholar]
- (9).Pitt BR, Radford EP, Gurtner GH, Traystman RJ. Interaction of carbon monoxide and cyanide on cerebral circulation and metabolism. Arch Environ Health. 1979;34:345–349. [PubMed] [Google Scholar]
- (10).Norris JC, Moore SJ, Hume AS. Synergistic lethality induced by the combination of carbon monoxide and cyanide. Toxicology. 1986;40:121–129. doi: 10.1016/0300-483x(86)90073-9. [DOI] [PubMed] [Google Scholar]
- (11).Moore SJ, Ho IK, Hume AS. Severe hypoxia produced by concomitant intoxication with sublethal doses of carbon monoxide and cyanide. Toxicol Appl Pharmacol. 1991;109:412–420. doi: 10.1016/0041-008x(91)90004-x. [DOI] [PubMed] [Google Scholar]
- (12).Mendelman A, Zarchin N, Meilin S, Guggenheimer-Furman E, Thom SR, Mayevsky A. Blood flow and ionic responses in the awake brain due to carbon monoxide. Neurol Res. 2002;24:765–772. doi: 10.1179/016164102101200861. [DOI] [PubMed] [Google Scholar]
- (13).Gunasekar PG, Borowitz JL, Isom GE. Cyanide-induced generation of oxidative species: involvement of nitric oxide synthase and cyclooxygenase-2. J Pharmacol Exp Ther. 1998;285:236–241. [PubMed] [Google Scholar]
- (14).Leavesley HB, Li L, Prabhakaran K, Borowitz JL, Isom GE. Interaction of cyanide and nitric oxide with cytochrome c oxidase: implications for acute cyanide toxicity. Toxicol Sci. 2008;101:101–111. doi: 10.1093/toxsci/kfm254. [DOI] [PubMed] [Google Scholar]
- (15).Jensen MS, Nyborg NC, Thomsen ES. Various nitric oxide donors protect chick embryonic neurons from cyanide-induced apoptosis. Toxicol Sci. 2000;58:127–134. doi: 10.1093/toxsci/58.1.127. [DOI] [PubMed] [Google Scholar]
- (16).Yoshikawa S, Shinzawa-Itoh K, Tsukihara T. Crystal structure of bovine heart cytochrome c oxidase at 2.8 A resolution. J Bioenerg Biomembr. 1998;30:7–14. doi: 10.1023/a:1020595108560. [DOI] [PubMed] [Google Scholar]
- (17).Brudvig GW, Stevens TH, Chan SI. Reactions of nitric oxide with cytochrome c oxidase. Biochemistry. 1980;19:5275–5285. doi: 10.1021/bi00564a020. [DOI] [PubMed] [Google Scholar]
- (18).Hill BC, Brittain T, Eglinton DG, Gadsby PM, Greenwood C, Nicholls P, Peterson J, Thomson AJ, Woon TC. Low-spin ferric forms of cytochrome a3 in mixed-ligand and partially reduced cyanide-bound derivatives of cytochrome c oxidase. Biochem J. 1983;215:57–66. doi: 10.1042/bj2150057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hill BC. The pathway of CO binding to cytochrome c oxidase. Can the gateway be closed? FEBS Lett. 1994;354:284–288. doi: 10.1016/0014-5793(94)01140-0. [DOI] [PubMed] [Google Scholar]
- (20).Pearce LL, Bominaar EL, Hill BC, Peterson J. Reversal of cyanide inhibition of cytochrome c oxidase by the auxiliary substrate nitric oxide: an endogenous antidote to cyanide poisoning? J Biol Chem. 2003;278:52139–52145. doi: 10.1074/jbc.M310359200. [DOI] [PubMed] [Google Scholar]
- (21).Gibson Q, Palmer G, Wharton D. J Biol Chem. 1965;240:915–920. [PubMed] [Google Scholar]
- (22).van Gelder BF. On cytochrome c oxidase: I. The extinction coefficients of cytochrome a and cytochrome a3. Biochimica et Biophysica Acta. 1966;118:36–46. doi: 10.1016/s0926-6593(66)80142-x. [DOI] [PubMed] [Google Scholar]
- (23).Sinjorgo KM, Durak I, Dekker HL, Edel CM, Hakvoort TB, van Gelder BF, Muijsers AO. Bovine cytochrome c oxidases, purified from heart, skeletal muscle, liver and kidney, differ in the small subunits but show the same reaction kinetics with cytochrome c. Biochim Biophys Acta. 1987;893:251–258. doi: 10.1016/0005-2728(87)90046-6. [DOI] [PubMed] [Google Scholar]
- (24).Ford PC, Wink DA, Stanbury DM. Autoxidation kinetics of aqueous nitric oxide. FEBS Lett. 1993;326:1–3. doi: 10.1016/0014-5793(93)81748-o. [DOI] [PubMed] [Google Scholar]
- (25).Lewis RS, Deen WM. Kinetics of the reaction of nitric oxide with oxygen in aqueous solutions. Chem Res Toxicol. 1994;7:568–574. doi: 10.1021/tx00040a013. [DOI] [PubMed] [Google Scholar]
- (26).Bonaventura C, Godette G, Tesh S, Holm DE, Bonaventura J, Crumbliss AL, Pearce LL, Peterson J. Internal electron transfer between hemes and Cu(II) bound at cysteine beta93 promotes methemoglobin reduction by carbon monoxide. J Biol Chem. 1999;274:5499–5507. doi: 10.1074/jbc.274.9.5499. [DOI] [PubMed] [Google Scholar]
- (27).Antonini E, Brunori M. Hemoglobin and myoglobin in their reactions with ligands. North-Holland; Amsterdam: 1971. Chapter 3: the derivatives of ferric hemoglobin and myoglobin; pp. 40–54. [Google Scholar]
- (28).Iheagwara KN, Thom SR, Deutschman CS, Levy RJ. Myocardial cytochrome oxidase activity is decreased following carbon monoxide exposure. Biochim Biophys Acta. 2007;1772:1112–1116. doi: 10.1016/j.bbadis.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Eglinton DG, Johnson MK, Thomson AJ, Gooding PE, Greenwood C. Near-infrared magnetic and natural circular dichroism of cytochrome c oxidase. Biochem J. 1980;191:319–331. doi: 10.1042/bj1910319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jensen P, Wilson MT, Aasa R, Malmstrom BG. Cyanide inhibition of cytochrome c oxidase. A rapid-freeze e.p.r. investigation. Biochem J. 1984;224:829–837. doi: 10.1042/bj2240829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Einarsdottir O, Dyer RB, Lemon DD, Killough PM, Hubig SM, Atherton SJ, Lopez-Garriga JJ, Palmer G, Woodruff WH. Photodissociation and recombination of carbonmonoxy cytochrome oxidase: dynamics from picoseconds to kiloseconds. Biochemistry. 1993;32:12013–12024. doi: 10.1021/bi00096a011. [DOI] [PubMed] [Google Scholar]
- (32).Fago A, Crumbliss AL, Peterson J, Pearce LL, Bonaventura C. The case of the missing NO-hemoglobin: spectral changes suggestive of heme redox reactions reflect changes in NO-heme geometry. Proc Natl Acad Sci U S A. 2003;100:12087–12092. doi: 10.1073/pnas.2032603100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Baskin SI, Nealley EW, Lempka JC. Cyanide toxicity in mice pretreated with diethylamine nitric oxide complex. Human & experimental toxicology. 1996;15:13–18. doi: 10.1177/096032719601500103. [DOI] [PubMed] [Google Scholar]
- (34).Pettersen JC, Cohen SD. The effects of cyanide on brain mitochondrial cytochrome oxidase and respiratory activities. J Appl Toxicol. 1993;13:9–14. doi: 10.1002/jat.2550130104. [DOI] [PubMed] [Google Scholar]
- (35).Davey GP, Peuchen S, Clark JB. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem. 1998;273:12753–12757. doi: 10.1074/jbc.273.21.12753. [DOI] [PubMed] [Google Scholar]
- (36).Mazat JP, Letellier T, Bedes F, Malgat M, Korzeniewski B, Jouaville LS, Morkuniene R. Metabolic control analysis and threshold effect in oxidative phosphorylation: implications for mitochondrial pathologies. Molecular and cellular biochemistry. 1997;174:143–148. [PubMed] [Google Scholar]
- (37).Jones MG, Bickar D, Wilson MT, Brunori M, Colosimo A, Sarti P. A re-examination of the reactions of cyanide with cytochrome c oxidase. Biochem J. 1984;220:57–66. doi: 10.1042/bj2200057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Giuffre A, Barone MC, Mastronicola D, D'Itri E, Sarti P, Brunori M. Reaction of nitric oxide with the turnover intermediates of cytochrome c oxidase: reaction pathway and functional effects. Biochemistry. 2000;39:15446–15453. doi: 10.1021/bi000447k. [DOI] [PubMed] [Google Scholar]
- (39).Cooper CE. Nitric oxide and cytochrome oxidase: substrate, inhibitor or effector? Trends Biochem Sci. 2002;27:33–39. doi: 10.1016/s0968-0004(01)02035-7. [DOI] [PubMed] [Google Scholar]
- (40).Torres J, Sharpe MA, Rosquist A, Cooper CE, Wilson MT. Cytochrome c oxidase rapidly metabolises nitric oxide to nitrite. FEBS Lett. 2000;475:263–266. doi: 10.1016/s0014-5793(00)01682-3. [DOI] [PubMed] [Google Scholar]
- (41).Pearce LL, Kanai AJ, Birder LA, Pitt BR, Peterson J. The catabolic fate of nitric oxide: the nitric oxide oxidase and peroxynitrite reductase activities of cytochrome oxidase. J Biol Chem. 2002 doi: 10.1074/jbc.M109838200. [DOI] [PubMed] [Google Scholar]
- (42).Borisov VB, Forte E, Sarti P, Brunori M, Konstantinov AA, Giuffre A. Nitric oxide reacts with the ferryl-oxo catalytic intermediate of the CuB-lacking cytochrome bd terminal oxidase. FEBS Lett. 2006;580:4823–4826. doi: 10.1016/j.febslet.2006.07.072. [DOI] [PubMed] [Google Scholar]
- (43).Litovitz T. The use of oxygen in the treatment of acute cyanide poisoning. In: Ballantyne B, Marrs TC, editors. Clinical and Experimental Toxicology of Cyanides. Wright Ltd.; Bristol UK: 1987. pp. 467–472. [Google Scholar]
- (44).Isom GE, Way JL. Effects of oxygen on the antagonism of cyanide intoxication: cytochrome oxidase, in vitro. Toxicol Appl Pharmacol. 1984;74:57–62. doi: 10.1016/0041-008x(84)90269-2. [DOI] [PubMed] [Google Scholar]
- (45).Way JL, Leung P, Cannon E, Morgan R, Tamulinas C, Leong-Way J, Baxter L, Nagi A, Chui C. The mechanism of cyanide intoxication and its antagonism. Ciba Found Symp. 1988;140:232–243. doi: 10.1002/9780470513712.ch14. [DOI] [PubMed] [Google Scholar]
- (46).Dejam A, Hunter CJ, Schechter AN, Gladwin MT. Emerging role of nitrite in human biology. Blood Cells Mol Dis. 2004;32:423–429. doi: 10.1016/j.bcmd.2004.02.002. [DOI] [PubMed] [Google Scholar]
- (47).Moreno SN, Stolze K, Janzen EG, Mason RP. Oxidation of cyanide to the cyanyl radical by peroxidase/H2O2 systems as determined by spin trapping. Arch Biochem Biophys. 1988;265:267–271. doi: 10.1016/0003-9861(88)90127-0. [DOI] [PubMed] [Google Scholar]
- (48).Stolze K, Moreno SN, Mason RP. Free radical intermediates formed during the oxidation of cyanide by horseradish peroxidase/H2O2 as detected with nitroso spin traps. J Inorg Biochem. 1989;37:45–53. doi: 10.1016/0162-0134(89)80028-5. [DOI] [PubMed] [Google Scholar]
- (49).Miro O, Casademont J, Barrientos A, Urbano-Marquez A, Cardellach F. Mitochondrial cytochrome c oxidase inhibition during acute carbon monoxide poisoning. Pharmacol Toxicol. 1998;82:199–202. doi: 10.1111/j.1600-0773.1998.tb01425.x. [DOI] [PubMed] [Google Scholar]
- (50).Alonso JR, Cardellach F, Lopez S, Casademont J, Miro O. Carbon monoxide specifically inhibits cytochrome c oxidase of human mitochondrial respiratory chain. Pharmacol Toxicol. 2003;93:142–146. doi: 10.1034/j.1600-0773.2003.930306.x. [DOI] [PubMed] [Google Scholar]
- (51).Thom SR, Ischiropoulos H. Mechanism of oxidative stress from low levels of carbon monoxide. Res Rep Health Eff Inst. 1997:1–19. discussion 21-17. [PubMed] [Google Scholar]
- (52).Thom SR, Fisher D, Xu YA, Garner S, Ischiropoulos H. Role of nitric oxide-derived oxidants in vascular injury from carbon monoxide in the rat. Am J Physiol. 1999;276:H984–992. doi: 10.1152/ajpheart.1999.276.3.H984. [DOI] [PubMed] [Google Scholar]