

Abstract
Here we use intravital imaging to demonstrate a reversible transition to a motile state as breast cancer cells spread. Imaging primary tumours reveals heterogeneity in cell morphology and motility. Two distinct modes of motility are observed: collective and single-celled. By monitoring the localisation of Smad2 and the activity of a TGFβ-dependent reporter gene during breast cancer cell dissemination we demonstrate that TGFβ signalling is transiently and locally activated in motile single cells. TGFβ1 switches cells from cohesive to single cell motility through a transcriptional programme involving Smad4, EGFR, Nedd9, M-RIP, FARP and RhoC. Blockade of TGFβ signalling prevents cells moving singly in vivo but does not inhibit cells moving collectively. Cells restricted to collective invasion are capable of lymphatic invasion but not blood-borne metastasis. Constitutive TGFβ signalling promotes single cell motility and intravasation but reduces subsequent growth in the lungs. Thus, transient TGFβ signalling is critical for blood-borne metastasis.
Introduction
Breast cancer metastasis starts with cell motility in the primary tumour leading to either local tissue invasion or entry into lymph or blood vessels1, 2. Analysis of fixed clinical material reveals that cancer cells can invade either cohesively or as single cells3. Metastases often retain many of the differentiated characteristics of the primary tumour including cell-cell contacts, but the behaviour and signalling that occurs as cells disseminate remains contentious. Epidermal Growth Factor (EGF) and Transforming Growth Factor β (TGFβ) signalling can promote tumour cell motility 4-6. Furthermore, these factors are up-regulated in breast cancer and correlate with adverse outcomes7-9. The TGFβ pathway is intriguing because it can promote growth arrest10, which seems incompatible with tumour progression. In some cases this paradox is resolved by loss of key mediators of the growth suppressive response to TGFβ in cancer cells11-13.
Alternatively, TGFβ signalling may only be active for limited periods as tumours disseminate and then return to low levels once metastases are established. Similarly, a reversible transition of cancer cells of epithelial origin to mesenchymal phenotypes as they metastasize has been suggested14, 15. This transition can be driven by TGFβ in experimental systems, however clinical data is less clear16. Signalling pathways may be activated in locally within tumours15 and live imaging studies have shown that tumour cell motility is unevenly distributed within primary tumours17, 18. However, heterogeneity in signalling within tumour micro-environments and cell motility have not been studied together.
TGFβ ligands bind to heterotetrameric complexes of receptors with serine-threonine kinase activity leading to an increase in their ability to phosphorylate Smad proteins. When Smad2 and Smad3 are phosphorylated they form complexes with Smad4 that accumulate in the nucleus and regulate transcription19. We use live imaging to investigate changes in TGFβ signalling as breast cancer cells become motile in primary tumours and subsequently colonize secondary sites. We demonstrate that TGFβ signalling is transiently and locally activated in disseminating single cells in vivo. Blockade of TGFβ signalling prevents cells moving singly in vivo but permits cells to move cohesively. Single cell motility is essential for blood-borne metastasis while cohesive invasion is capable of lymphatic spread.
Results
Intravital imaging of breast cancer cell dissemination
Rat mammary carcinoma cells (MTLn3E) were engineered to express either actin or a membrane localisation sequence fused to GFP to enable imaging of cell morphology before injection into the mammary fat pad. Figure1Ai shows that large areas of MTLn3E tumours contain closely packed cancer cells that retain significant localisation of β-catenin to cell junctions (Supplementary Figure1A&B). The majority of these cells were non-motile over periods of observation lasting up to two hours (Movie1, Figure1Aii and data not shown). Other areas of the tumour had more disorganised cell morphologies and motile cells were observed (second part of Movie1, Figure1Aiii-iv); these are apparent as adjacent red, green and blue images in Figure1Aiv (bottom right) and Figure1Bii&1Biii. On average 5% of cells were motile, but they were not homogeneously distributed. Many tumour areas monitored had no motile cells and other areas had >15% of motile cells (Figure1C and data not shown).
Figure 1. Transient acquisition of motile behaviour by breast cancer cells.

A: i) An intravital image from a time series of an MTLn3E tumour with cells expressing GFP-actin (green) and collagen fibres (blue) shown (see Movie1). ii) Actin images from three different time points from panel i) are shown - overlay of images at different times in red, green and blue produces a white/grey image indicating no change in cell position (Figure 1Aii). iii) An intravital image from a second time series of a primary MTLn3E tumour with cells expressing GFP-actin (green) and collagen fibres (blue) shown (see Movie 1). iv) Actin images from three different time points from panel iii) are shown are shown over-laid – distinct areas of red, green and blue highlight motile cells. B: i) myr-GFP intravital primary tumour images from three different time points are shown overlaid in red, green and blue (see Movie 2). Panel ii) shows a region with rapidly moving disorganized cells, while panel iii) shows a region with slower moving cohesively organized cells. C: The number of motile cells observed per 20x field is shown. D: Left hand bar shows the proportion of non-motile and motile cells (74 movies analysed by two observers – error bar represents deviation between observers). Right hand bar shows the proportion of cells moving singly, singly but in the same path as another cell, cohesively in chains <20μm diameter and cohesively in groups >20μm diameter (74 movies analysed by two observers – error bars represent deviation between observers). E: The speed of motile cells either moving singly or cohesively is shown (>25 representative cells analysed for each category from 5 movies). F: i) An intravital image from a time series of a lymph node metastases with MTLn3E cells expressing fluorescently tagged actin (green) and collagen fibres (blue) shown. ii) Actin images from three different time points of the time-series shown in panel i) are shown overlaid in red, green and blue. G: Bars show the proportion of non-motile and motile MTLn3E cells in smaller (<100 cells) and larger (>100 cells) lymph node metastases (10 lymph nodes analysed in total).
Closer inspection revealed that some of the closely packed cells were moving, which are shown by the non-overlapping red, green and blue images of cell outlines in Figure1Biii. In some cases cells moved in groups several cells wide (Movie2), while in other instances cells were organised into chains only 2-3 cells wide (Movie3). We describe both these of movement as cohesive or collective. Cohesive motile cells accounted for about 20% of all motile cells (Figure1D). Cohesive cell movement was significantly slower than single cell motility (Figure1E). The different morphology and speed of cohesively moving cells leads suggest that this type of motility is distinct from single cell motility.
Previous studies have shown a correlation between single cell motility and metastasis 20-23. In agreement with this we occasionally observed single motile cells in the process of intravasation (Supplementary Figure1C and Movie4).
Motility is not maintained in lymph node metastases
We next investigated the behaviour of cells that had arrived in the inguinal lymph node. Figure 1F shows that almost all cells in lymph nodes were closely packed and non-motile (Figure1Fii and Movie5). Only 2% of cells were motile in larger more established metastases (Figure1G). In smaller metastases, which presumably contain more recently arrived cells, there was a slightly higher proportion of motile cells (Figure1G). Technical limits prevented imaging of the lungs of live mice. These data demonstrate that the acquisition of motile behaviour by cancer cells is both a localised and transient event.
Monitoring TGFβ signalling in vivo
TGFβ signalling can increase the invasive potential of cancer cells and promote epithelial to mesenchymal transitions implicated in cancer dissemination 14. Our data predict that the signalling events that promote cancer cell motility are locally and transiently activated. We therefore tested if activation of TGFβ signalling was associated with motile cancer cells in vivo. We used a TGFβ-dependent reporter (CAGA12-luciferase)24 to confirm that MTLn3E cells can respond to TGFβ (Figure 2A). Furthermore, TGFβ signalling leads to a reduced growth of MTLn3E cells in soft agar (Figure2B). Analysis of MTLn3E tumours revealed heterogeneous phosphorylation of Smad3 (indicative of active TGFβ signalling) in vivo with greatest levels apparent near the tumour margins and a subset of blood vessels (Figure2C & Supplementary Figure2). Analysis of human breast cancer samples and transgene-driven mouse tumours also revealed considerable heterogeneity in TGFβ signalling (Supplementary Figure2D&E). These data confirm that TGFβ signalling is non-uniformly active in tumours.
Figure 2. TGFβ signalling in MTLn3E cells.
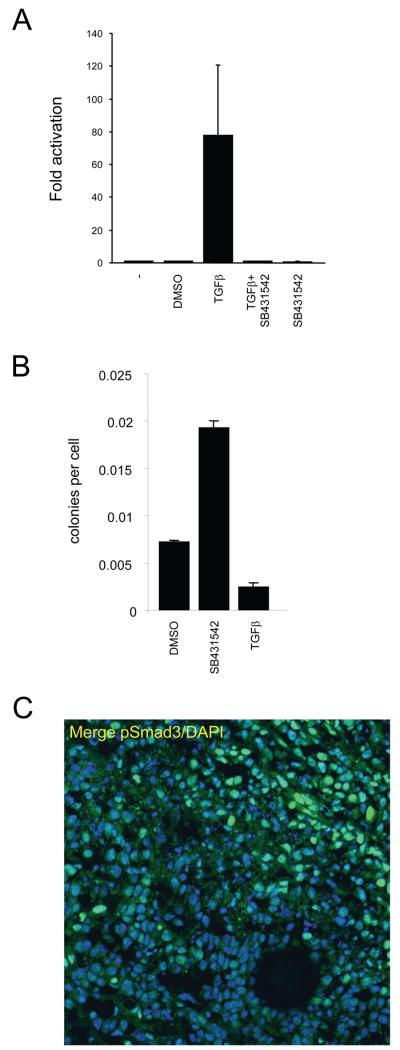
A: Assay showing changes in CAGA12-luciferase reporter activity in MTLn3E cells in response to 2ng/ml TGFβ1 +/−10μM SB431542 (an inhibitor of the TGFβ type I receptor Alk 5 - also inhibits Alk4&7). B: Soft agar assays were set up using MTLn3E cells that had been cultured in control media (+ vehicle) or with 2ng/ml TGFβ1 or 10μM SB431542 for 24 hours. Average of two experiments (error bars represent half range). C: MTLn3E tumour stained for pSmad3 (green) and DAPI (blue), tumour margin is in top right corner.
Nuclear localisation of Smad2 in single motile cells in vivo
To monitor TGFβ signalling with cellular resolution in vivo we adopted two approaches. We engineered MTLn3E cells to express Smad2 fused to GFP, which accumulates in the nucleus in response to TGFβ signalling25. To aid visualisation of the nucleus cells were also made to express Orange Fluorescent Protein26 fused to an NLS (Supplementary Figure3A&B). Imaging of tumours expressing GFP-Smad2 and OFP-NLS was performed and GFP-Smad2 localisation was correlated with cell behaviour. GFP-Smad2 localisation was heterogeneous in vivo; Figure 3A shows predominantly cytoplasmic Smad2 on the left compared to more even distribution between nucleus and cytoplasm with occasional cells exhibiting nuclear accumulation on the right (compare also Figure 3Bi&3Biii). Timelapse analysis revealed that all singly-moving cells had Smad2 in the nucleus (Figure3Bi - note co-localisation of GFP-Smad2 with Orange-NLS marker in motile cell, Figure 3C and Movie6). In contrast, cells moving collectively had Smad2 in the cytoplasm (Figure3Bii and Movie7). Cytoplasmic localisation of Smad2 was also common in non-motile cells (Figure 3Biii & 3C and Movie8) although some stationary cells had similar levels of Smad2 in the nucleus as in the cytoplasm. The nuclear accumulation of Smad2 seen in singly-moving cells was not found in cells that had disseminated to local lymph nodes or larger lung metastases. However, we occasionally noted nuclear Smad2 in isolated cells in the lungs (Figure3D). These data imply a transient nuclear accumulation of Smad2 during metastasis.
Figure 3. Nuclear accumulation of Smad2 in singly-moving cells.

A: Low magnification image of a primary MTLn3E tumour expressing GFP-Smad2 and NLS-Orange. B: Time series of higher magnification images of GFP-Smad2 and NLS-Orange expressing primary tumours are shown. i), ii) and iii) show areas including singly-moving (see also Movie 6), cohesively moving (see also Movie 7), and stationary cells (see also Movie 8), respectively. C: Quantification of Smad2 localisation in non-motile, singly-moving, and collectively moving cells in primary tumours, lymph node and lung metastases. 26 movies from 6 mice were analysed D: GFP-Smad2 localisation in a lymph node metastasis and different size lung metastases.
Activation of a TGFβ reporter in single motile cells in vivo
Nuclear accumulation of Smad2 does not necessarily indicate activation of a transcriptional response. We therefore used a CAGA12-CFP reporter to determine which cells had activated a TGFβ transcriptional response (Supplementary Figure3C&D). Figure 4A shows that expression of the CAGA12-CFP reporter was heterogeneous. Strikingly, almost all cells moving singly were CFP positive whereas collectively moving cells were CFP negative (Figure4A&B, Movie9). CAGA12-CFP expression was low in lymph node and larger lung metastases, although it was sometimes observed in smaller lung metastases (Figure4A&B). These observations support the idea of a transient activation of TGFβ signalling in singly-moving cells during metastasis. The presence of non-motile cells with nuclear Smad2 and CAGA12-CFP expression indicates that TGFβ signalling is not sufficient to drive cancer cell motility.
Figure 4. Activation of Smad-dependent transcription in singly-moving cells.

A: i) Low magnification image of a primary MTLn3E tumour constitutively expressing myristoylated-Cherry (red) and expressing CFP (cyan) from a Smad-dependent promoter; collagen second harmonic signal is in blue. Arrows indicate motile cells – see also right-hand panels and Movie 9. ii) Image of an MTLn3E lymph node metastasis constitutively expressing myr-Cherry and expressing CFP from a Smad-dependent promoter; collagen second harmonic signal is in blue. iii-iv) Images of different size MTLn3E lung metastases constitutively expressing myr-Cherry and expressing CFP from a Smad-dependent promoter. B: Quantification of the proportion of CAGA12::CFP positive cells in non-motile, singly-moving, and collectively moving cells in primary tumours, lymph node and lung metastases. Average data from four independent clones is shown with 24 from 13 mice were analysed, standard error is shown. C: Quantification of the proportion of vimentin promoter::GFP positive cells in non-motile, singly-moving, and collectively moving cells in primary tumours and lymph node metastases. Average of two independent clones is shown.
We confirmed these results using a second breast cancer model. Orthotopic tumours were generated using 410.4 mouse mammary carcinoma cells 27. Cell motility was only observed in a subset of 410.4 cells (~1%) and the majority of these cells moved as single cells (data not shown). Importantly, Supplementary Figure4 shows that there was an increase in TGFβ signalling in the single cells as judged by either Smad2 localisation or CAGA12-CFP expression that was not maintained in lymph nodes (see also Movies10&11). Thus transient and reversible changes in TGFβ signalling occur in multiple models of breast cancer metastasis.
TGFβ signalling is implicated in promoting the mesenchymal characteristics during cancer invasion. MTLn3E cells express intermediate levels of several mesenchymal markeres including Twist, Snail and vimentin. Of these, only the levels of vimentin increased in response to TGFβ treatment (Supplementary Figure3E). We therefore investigated if vimentin expression was altered in motile cells in vivo. MTLn3E cells were engineered to contain the vimentin promoter controlling GFP expression and a constitutive promoter driving expression of mRFP-actin (Supplementary Figure3F). Intravital imaging revealed a heterogeneous pattern of vimentin expression. Similar to the CAGA12 reporter, a greater proportion of cells moving singly were positive for vimentin expression (Figure4C and Movie12), which probably reflects elevated TGFβ signalling. However, unlike activation of TGFβ signalling, vimentin expression was also compatible with cohesive movement (Figure4C). In cohesively-moving cells the basal level of vimentin expression is not TGFβ regulated because GFP-Smad2 and CAGA12-CFP cell lines show that TGFβ signalling is very low in these cells.
TGFβ switches cells to single cell motility in vitro
The data presented above show a striking correlation between the mode of migration used and TGFβ signalling in vivo; however, they do not demonstrate that TGFβ causally determines the mode of migration. To test this we investigated how TGFβ affected the motility of MTLn3E cells. When seeded at low density MTLn3 cells grow as distinct colonies. Cells within these colonies are constantly in motion but almost never move as single cells away from the colony (Movie13). When cells are cultured in the presence of TGFβ1 they no longer grow as discrete colonies but instead move as single cells (Figure5A&B). This response could be simply quantified by calculating the average area occupied by a cell before a neighbouring cell is present. The kinetics of the switch to single cell motility were slow (Figure5A). Prolonged exposure to TGFβ1 promoted an increase in actin stress fibres and a loss of β-catenin localisation from cell-cell contacts (Figure5B). Consistent with this we found that tumour areas with high pSmad3 levels had lost β-catenin localisation at cell contacts (Supplementary Figure5A). 410.4 cells underwent similar changes in motility and morphology following TGFβ1 treatment (Supplementary Figure6A). Unlike TGFβ, EGF did not promote cell scattering but increased the speed at which cells moved in cohesive groups (Supplementary Figure6B&C).
Figure 5. TGFβ requires Smad4 to switch cells to single cell motility.

A: Time course of switch to single cell motility induced by 2ng/ml TGFβ. Scattering of colonies was used as a readout of single cell motility. B: F-actin and β-catenin staining of MTLn3E cells seeded at low density in the presence or absence of TGFβ 2ng/ml for 40 hours (see also Movie 9). C: Graph shows the cell scattering 24hr after addition of 2ng/ml TGFβ in control and Smad1, 2, 3, 4, 5, 1&5 or 2&3 siRNA smartpool transfected cells (average of two or three experiments, * p<0.05, ** p<0.01 student t-test). D: Western blot showing the efficiency of Smad4 depletion using 4 different siRNA sequences. Graph shows the cell scattering 24hr after addition of 2ng/ml TGFβ following transfection of three different Smad4 siRNA sequences (averages and standard deviations from one representative experiment of three are shown, * p<0.05). E: β-catenin staining of MTLn3E cells in control and Smad4 siRNA transfected cells 24hr after addition of 2ng/ml TGFβ.
The slow kinetics of the response to TGFβ suggest that the switch to single cell motility may be driven by transcriptional responses mediated by Smad transcription factors. In both MTLn3E and 410.4 cells transfection of siRNA targeting Smad4 profoundly reduced cell scattering in response to TGFβ, while depletion of Smad3 led to a more modest reduction in scattering (Figure5C-E and Supplementary Figure6B). Having established a key role for TGFβ-mediated transcription we performed microarray analysis to identify TGFβ target genes in MTLn3E cells. A large number of genes were regulated by TGFβ signalling including many well established targets (CTGF & PAI-1). Several poorly-characterised TGFβ regulated genes were also identified (Supplementary Table 1 with qRT-PCR confirmation in Supplementary Figure7). Many of these genes could potentially be implicated in the switch away from cohesive cell motility. EGFR, AP-1 family members (c-jun and junB), various proteins involved in Rho signalling (RhoC, MPRIP – a Rho interacting regulator of myosin phosphatase activity, Farp1 - a FERM domain containing Rho exchange factor, Nedd9 - an atypical Rac activator, RhoQ/TC10), and a range of molecules implicated in cell-cell adhesion could all affect the mode of cell motility (Supplementary Table 1). We tested if these genes were required for the switch from cohesive to single cell motility. Figure6A shows depletion of MPRIP, Farp1, Nedd9, c-jun, EGFR and CTGF all reduced TGFβ-induced cell scattering although none completely abrogated the response. Depletion of RhoC alone had little effect but combined depletion of RhoC and its close homologue RhoA significantly reduced cell scattering. Similar results were obtained in 410.4 cells (Supplementary Figure6C). We additionally used multiple siRNA sequences to confirm the effect of one of the less well-studied target genes, MPRIP (Supplementary Figure6E&F). We further demonstrated the importance of Rho-mediated regulation of contractility, EGFR signalling and JNK/c-jun signalling using the pharmacological inhibitors Y27632 (targets Rho-ROCK signalling), AG1478 (targets EGFR), and SP600125 (targets JNK signalling), respectively (Figure6 and Supplementary Figure6D).
Figure 6. Roles of multiple TGFβ target genes in the switch to single cell motility.

A: Graph shows the cell scattering 24hr after addition of 2ng/ml TGFβ in mock, control, RhoA, RhoC, RhoA+C, MPRIP, FARP1, RhoQ, NEDD9, FXYD5, CTGF, EGFR, PAI-1, c-Jun and JunB siRNA smartpool transfected cells (average of 2-4 experiments, * p<0.05, ** p<0.01 student t-test). B: Graph shows the cell scattering 24hr after addition of 2ng/ml TGFβ in control, 10 μM SB431542, 10 μM AG1478, 10 μM SP600125, and 10 μM Y27632 treated cells. C: β-catenin staining of MTLn3E cells in mock, control, RhoA+C, MPRIP, FARP1, NEDD9, CTGF, EGFR, and c-Jun siRNA transfected cells 24hr after addition of 2ng/ml TGFβ. D: Analysis of single ‘amoeboid’ cell motility: mock, control, RhoA+C, MPRIP, FARP1, NEDD9, CTGF, EGFR, and c-Jun siRNA transfected MTLn3E cells were plated as single cells on deformable collagen gels and the distance moved over 90 minutes was measured. Box and whisker plots show mean, quartiles and 10 and 90 percentiles (average of 2-4 experiments, * p<0.05, ** p<0.01 student t-test) E: F-actin staining of MTLn3E cells in mock, control, RhoA+C, MPRIP, FARP1, NEDD9, CTGF, EGFR, and c-Jun siRNA transfected cells plated as single cells on deformable collagen gels.
To understand how TGFβ target genes promote single cell motility we investigated their role in adherens junction organisation, cell motility and F-actin organisation. Figure6C shows that depletion of RhoA&C together, Farp1, MPRIP, and c-jun reduced adherens junction disruption following TGFβ treatment; whereas EGFR, Nedd9, and CTGF depletion had little effect. Although some TGFβ targets were not required for adherens junction disassembly they may be needed for other aspects of single cell motility. We tested this by culturing cells in conditions that favour single ‘amoeboid-type’ cell motility28 and analysing their movement and morphology (Movie14). EGFR, Nedd9 and c-jun were all required for efficient amoeboid motility and this corresponded with defects in the formation of F-actin rich protrusions at the front of the cell (Figure6D&E, protrusions marked with arrows). The effects of targeting Rho signalling through depletion of RhoA&C, MPRIP or Farp1 were less pronounced although in all cases elongated cells with tail retraction defects were observed. Together these data indicate that TGFβ promotes single cell motility by regulating a transcriptional programme with different genes playing distinct roles in the switch.
TGFβ signalling switches cells from cohesive to single cell movement in vivo
We further tested the role of TGFβ in cohesive and single cell motility by blocking signalling in a cell autonomous manner. MTLn3E cells were generated that expressed a ‘dominant-negative’ TGFβ type-II receptor fused toGFP (TGFβRDN-GFP) (Supplementary Figure8A-C). The behaviour of these cell lines was compared to control cell lines in vivo. Figure7A shows intravital imaging of a ‘mosiac’ tumour containing CFP-expressing control cells and TGFβRDN-GFP expressing cells. Strikingly, while numerous control cells are observed moving as single cells none of the TGFβRDN-GFP expressing cells are motile (Movie15). Interestingly, analysis of numerous tumours revealed that cells expressing TGFβRDN-GFP could still move cohesively (shown by imperfect overlay of red, green and blue images in Figure7B and Movie16). In fact this type of motility was observed more frequently (Figure7C). Comparable numbers of motile control and TGFβRDN-GFP cells were observed, but there was a striking switch in the type of motility.
Figure 7. TGFβ signalling is required for single cell motility in vivo.

A: An image from a time-series of a primary MTLn3E tumour containing a mixture of control cells expressing CFP (cyan) and cells expressing the TGFβ receptor II lacking its kinase domain fused to GFP (TGFβRDN - green), collagen fibres in blue. White arrows indicate motile cells (see also Movie 15). B: TGFβRDN-GFP intravital primary tumour images from three different time points are shown overlaid in red, green and blue. Dashed yellow line highlights a region of cohesive cell movement (see also Movie11). C: Pie charts show the number of singly- and cohesively-moving cells is for control (EGFP-N1) and two different TGFβRDN expressing MTLn3E clones. Area is proportional to the number of motile cells – ≥4 mice analysed for each condition. D: Pie charts show the number of singly- and cohesively-moving cells is shown for control shRNA and two different Smad4 shRNA expressing MTLn3E clones. Area is proportional to the number of motile cells. E: Pie charts show the number of singly- and cohesively-moving cells is shown for control IRES-GFP and TGFβ IRES-GFP expressing MTLn3E clones. Area is proportional to the number of motile cells moving.
To test whether TGFβ-driven transcription is needed for this switch we generated clones stably depleted for Smad4 (Supplementary Figure8D-F). Intravital imaging confirmed that Smad4 is required for single cell motility (Figure7D). Our in vitro analysis suggested that TGFβ-driven transcription of various regulators of Rho/ROCK signalling is required for the switch to single cell movement. New analysis of ROCK inhibition in vivo28 revealed that there is a greater requirement for Rho/ROCK signalling for single cell movement as opposed to collective movement (Supplementary Figure9). These data support a role for TGFβ-dependent up-regulation of this pathway in single cell motility.
In a reciprocal approach we investigated the effect of activating TGFβ signalling. Cells stably over-expressing TGFβ1 were generated (characterised in Supplementary Figure8G&H) and their behaviour in vivo investigated. Figure7E shows that these cells exhibited greatly increased single cell motility in vivo although not all TGFβ1-expressing cells were motile (Movie17). These data demonstrate that TGFβ signalling is necessary for single cell motility in vivo and that its ectopic expression promotes single cell motility.
Mode of cell motility determines haematogenous versus lymphatic spread
The results presented so far have focused on cell motility but have not addressed how this relates to metastasis. To investigate this we generated mixed tumours containing control and experimentally manipulated cells and measured the relative efficiency of the cells to spread to the inguinal and axillary lymph nodes, enter the blood and form lung metastases. Both TGFβRDN expressing cells and Smad4shRNA cells showed reduced entry into the blood and lung colonisation (Figure8A&B). Interestingly, their spread to lymph nodes was similar to control cells suggesting that collective invasion may be used for lymphatic spread. To see if this was true we performed extensive imaging of tumour cells near lymphatic vessels. Figure8E shows a chain of MTLn3E cells extending into a lymphatic vessel. Timelapse analysis of this region shows that they are moving into the vessel (Movie18). We also observed numerous examples of groups of cancer cells within lymphatic vessels including TGFβRDN cells (Figure8E). These data demonstrate that collective invasion can mediate lymphatic dissemination.
Figure 8. TGFβ signalling is required for haematogenous but not lymphatic metastasis.

A: Representative images of primary tumours, lymph node metastases, intravasated cells, and lung metastases of ‘mixed’ tumours. Upper panels show mixed tumours containing control cells (red) and TGFβRDN cells (green). Middle panels show mixed tumours containing control cells (red) and Smad4shRNA cells (green). Lower panels mixed tumours containing control cells (red) and TGFβ1 over-expressing cells (green). Scale bar is 150μm. B: Quantification of metastasis of TGFβRDN and Smad4shRNA cells relative to control cells in the same mouse. Inguinal and axillary lymph nodes, circulating tumour cells and lung metastases are evaluated (average of 7, 4, 5 and 5 mice for TGFβRDN#1, TGFβRDN#2, shRNA#1 and shRNA#2, respectively). Quantification method is explained in Materials and Methods. ‘n.d’ indicates that no control or experimentally targeted cells were found in the blood. ‘0’ indicates that only control cells were detected. C: Quantification of TGFβ1 over-expressing MTLn3E metastasis relative to control cells in the same mouse. Inguinal and axillary lymph nodes, circulating tumour cells and lung metastases are evaluated (average of 8 mice – quantification performed as in part B). D: A 1:1 mixture of control cells expressing mCherry and control cells expressing GFP (columns 1&2) or control cells expressing mCherry and cells treated TGFβ1 2ng/ml for 24 hours expressing GFP (column 3) or a 1:1 mixture of control cells expressing mCherry and constitutively TGFβ1 IRES-GFP expressing cells (columns 4&5) were injected into the tail vein. 48hrs after injection the number of experimentally manipulated GFP expressing cells relative to control cells was assessed. Graph shows the ratio of GFP expressing cells to control mCherry expressing cells (the average and standard deviation from 4-6 different mice is shown). E: Left-hand image shows xy, xz, and yz sections of a ‘chain’ of MTLn3E cells (green) entering a lymphatic vessel. Middle panel shows motion analysis of the cells in the indicated region of the left-hand image. Right-hand panels show xy and yz sections of a group of cells within a lymphatic tumour draining from a mosaic tumour of control MTLn3E cells (red) and TGFβRDN MTLn3E cells (green).
TGFβ1 over-expressing cells had an increased ability to enter the blood but this was not reflected in the overall numbers of lung metastases (Figure8C). The reduced lung metastases may reflect the growth inhibitory effects of TGFβ observed when measuring growth in soft agar (Figure2). To test this we directly measured lung colonisation following tail vein injection of a 1:1 mixture of control and constitutively over-expressing TGFβ1 cells. Two weeks after injection TGFβ1 cells were significantly under-represented in lung metastases (Figure8D). This was not due to a defect in the initial stages of lung colonisation because 24 hours after injection these cells were over-represented in the lungs. Cells pulsed with TGFβ1 ligand to induce a transient burst of signalling are favoured at both the early and late stages of lung colonisation (Figure 8D 29, 30). These data demonstrate that prolonged TGFβ signalling does not favour lung colonisation and that the ability to down-regulate signalling is critical for the growth of lung metastases. Thus optimal lung metastasis depends on both the ability to initially turn TGFβ signalling on and subsequently to turn it off.
Discussion
Many studies have proposed localised and transient changes in signalling as cells metastasize. Tumour imaging has shown that only a small proportion of cancer cells are motile even around the margins of metastatic breast cancer models17. This suggests that the signalling pathways that promote cancer cell motility may be heterogeneously active in tumours. We use intravital imaging of breast cancer cells engineered to express fluorescent reporters of TGFβ signalling to demonstrate localised and transient activation of signalling. Two distinct modes of motility are observed in vivo, collective and single, that differ in their speed and presence of cell-cell contacts. These probably reflect the differing patterns of invasion observed by pathologists3. TGFβ signalling is active in singly-moving cells but not in those moving cohesively; furthermore blockade of TGFβ signalling switches cells to cohesive motility. However, TGFβ signalling is not sufficient to drive cancer cell motility. We propose that other factors besides TGFβ determine whether cancer cells become motile. If TGFβ is also active, then single cell dissemination will occur whereas if it is not active then cohesive invasion occurs (Supplementary Figure10). It is likely that EGF is one such additional factor that may be heterogeneously distributed in tumours23, 31.
The switch to single cell motility requires regulation of a transcriptional program by TGFβ and Smad4. Different target genes are involved in modulating different aspects of cell behaviour required for single cell motility. Increased EGFR levels could promote sensing of chemotactic cues, while Nedd9 promotes actin polymerisation. RhoC, MPRIP and Farp1 combine to increase acto-myosin contractility which is important for destabilising cell-cell junctions and tail retraction in singly-moving cells.
Transient activation of TGFβ signalling could also explain why the growth suppressing effects of TGFβ do not lead to slow tumour growth. TGFβ signalling is low in the bulk of the primary tumour and metastases thereby allowing growth and is only active as cells disseminate. Strikingly, although forced and prolonged activation of TGFβ signalling promoted single cell motility in vivo it failed to promote lung metastasis. This was due to a failure of cells with high levels of TGFβ signalling to proliferate in the lungs. These observations highlight the importance of being able to down-regulate TGFβ signalling at certain stages of the metastatic process. It is tempting to speculate that singly-moving cells become more mesenchymal, however the available data provides only modest support for this hypothesis. Expression of Snail, Slug and Twist is not altered by TGFβ treatment (data not shown). Although vimentin expression is modulated by TGFβ signalling, it is observed in a significant proportion of non-motile cells and collectively-moving cells. Thus changes in vimentin expression may merely indicate increased TGFβ signalling, not that expression of mesenchymal markers drives the switch to single cell motility.
A feature of this work is heterogeneous activity of TGFβ signalling in tumours. Immuno-histochemical analysis suggests that TGFβ signalling is active in <10% of cells throughout the tumour. Even in the tumour margins which we analyse by live imaging the proportion of cells with active signalling is only 30-50% (Figures 3&4). Analysis of human breast cancer samples shows considerable heterogeneity in TGFβ signalling suggesting that observations from our model systems are highly relevant (Supplementary Figure2). This raises the questions as to the source of this variation. We observe in experimental tumours that TGFβ signalling is greatest around tumour margins and blood vessels. These areas also have the greatest numbers of host cells. Species-specific RT-PCR for TGFβ ligands reveals that they are expressed by both murine host cells and rat MTLn3E cells (not shown). We believe that much of the heterogeneity results from uneven distribution of TGFβ producing non-tumour cells or possibly leaky vasculature. The reduction in TGFβ signalling at secondary sites may simply be caused by the cells being displaced from the TGFβ-rich microenvironments that activated signalling in the primary tumour.
Previous work had suggested rather stable changes in TGFβ signalling during cancer progression, often associated with a loss of anti-proliferative responses12. This was thought to occur either globally or for metastasis to specific sites32, 33. We provide a new framework for changes in TGFβ signalling in metastatic cancer. Loss of anti-proliferative responses is not necessary because TGFβ signalling is low in the majority of cancer cells in primary locations and metastases. TGFβ signalling is only transiently active in a small population of cells; activation of TGFβ signalling in these cells drives the expression of a range of genes that promote single cell motility. Cells that do not up-regulate TGFβ signalling are still observed moving collectively and can enter lymphatic vessels. Therefore, TGFβ signalling and subsequently the mode of cancer cell motility influence whether cancers metastasize via lymphatic or haematogenous routes. The activation of TGFβ signalling that promotes intravasation needs to be reduced for efficient growth of metastases in the lungs. Thus reversible changes in TGFβ signalling are necessary for blood-borne metastasis of breast cancer models.
Experimental procedures
Plasmids
To generate the CAGA12::ECFP the luciferase gene from CAGA12::luc24 was replaced with ECFP (Clontech). Expression of the GFP-Smad2 fusion protein was driven by a fusion of the EF-1alpha promoter and beta-globin 5'UTR; the GFP-Smad2 fusion is described in34. To generate dominant negative TGF Beta receptor II-GFP fusion, the EGFP coding sequence was cloned C-terminal of aminoacid 191 of human DN-TGFβ receptor II. The myr-EGFP and myr-Cherry constructs were a gift from Frank Gertler. mRFP-actin was a gift from Michael Way. Vimentin promoter – GFP was a gift from Christine Gilles35. The Smad4 shRNA vector was made by cloning GCAGGTGGCTGGTCGGAAAttcaagagaTTTCCGACCAGCCACCTGCttttt into pRetroSuper (equivalent to Smad4 siRNA#3). TGFβ1 IRES-GFP was made by cloning mouse TGFβ1 into pI-EGFP2.
Cell Lines
MTLn3E cells were grown in αMEM (GIBCO) containing 5% fetal calf serum (FCS). GFP-Smad2 Orange Fluorescent Protein-NLS (hereafter called Orange-NLS) cells lines were generated by co-transfection of the GFP-Smad2 plasmid together with pBabe-Puro, followed by selection with puromycin (2.5mg/ml). Following Fluorescent Activated Cell Sorting (FACS), a polyclonal cell line was obtained expressing GFP-Smad2. These cells were infected with lentivirus containing a CMV Orange -NLS cassette. Double positive cells were sorted by FACS to give raise to one polyclonal cells line and different monoclonal cell lines. Stable MTLn3E myr-cherry CAGA12::ECFP cell lines were generated by infection of MTLn3E with lentivirus containing a CMV myr-Cherry cassette. After FACS sorting a polyclonal cell line, these cells were further transfected with the CAGA12::ECFP construct in combination with pBabe-Puro. Following selection with puromycin, cells were induced with TGFβ1 for 6h and cells positive for CFP and mCherry were FACS sorted as single cells. Stable TGFβRDN MTLn3E cell lines were selected using G418 following plasmid transfection and single cell clones were chosen for further analysis. Stable Smad4 shRNA MTLn3E cell lines were selected following retroviral infection and single cell clones were chosen for further analysis. A polyclonal pool of cells expressing TGFβ1 IRES-GFP were selected using puromycin followed by FACS sorting.
siRNA
MTLn3E cells were transfected with 75nM siRNA using Dharmafect 2 (4μl/35mm well). The following rat siRNA reagents from Dharmacon were used: Smad1 J094977-05 & J094977-06 & J094977-07 & J094977-08, , Smad2 J-091698-07, Smad3 J-080100-06 & J-080100-07, Smad4 J-091337-10 (#2) & J-091337-11 (#3) & J-091337-12 (#4), Smad5 M-096022-00, RhoA M-095222-00, RhoC M-089673-00, MPRIP D-096225-02 & D-096225-03 & D-096225-04 , Farp-1 M-087852-00, CTGF M-080139-00, EGFR M-080049-00, Nedd9 M-094997-00, Fxyd5 L-100059-01, c-jun M-089158-00, junB M-087675-00, TC10 M-094213-00, PAI-1 M-096632-00.
410.4 cells were transfected with 75nM siRNA using Dharmafect 4 (4μl/35mm well). The following mouse siRNA reagents from Dharmacon were used: Smad3 M-040706-01, Smad4 D-040687-03, RhoA M-042634-01, RhoC M-064655-01, MPRIP M-058568-00, CTGF M-040018-01, EGFR M-040411-01, Nedd9 M-059282-01, Fxyd5 M-040425-00.
Tumour Imaging
Female nude mice (5-6 weeks old) were injected in the mammary fat under the fourth nipple with 106 cells for each cell line. When tumours reached 5-7 mm diameter (usually after 21-26 days) mice were anaesthetised and tumours exposed as described before 22. The Chameleon Coherent Ti-Sapphire laser was tuned to 870 for EGFP excitation and to 850 for ECFP excitation. For simultaneous imaging of EGFP or ECFP with either Orange, Cherry or mRFP, a single photon 543 laser was fired simultaneously to the Ti-Sapphire laser. Collagen was visualised through second harmonic generation. Typically four different areas were imaged for 20-30 minutes in each tumour. Cell speeds were determined for motile cells that remained visible in the same confocal section for several minutes using LSM image examiner. Cells that moved between confocal sections were not analysed for speed. Where appropriate, drift in the x-y plane was corrected using Imaris software prior to analysis. Lymphatic vessels were visualised by the injection of 10-20μl of high molecular weight TRITC-dextran into the tumour.
Smad2 localisation analysis
To analyse intravital movies of GFP-Smad2 localisation we categorised cells into having either cytoplasmic, nuclear and cytoplasmic, or nuclear Smad2. The nucleus was defined by Orange-NLS expression and we then measured the GFP-Smad2 pixel intensity in the nucleus and the cytoplasm. If these values were within 33% then we categorised the cell as having both nuclear and cytoplasmic Smad2 otherwise it was categorised as having either nuclear or cytoplasmic Smad2.
CAGA12-CFP and vimentin promoter-GFP analysis
Microscope settings were kept constant for each individual tumour analysed meaning that all images from a mouse are internally consistent. Slight differences between tumour position and the amount of surrounding tissue meant that it was not always possible to use identical settings between mice; nonetheless we tried to ensure that average pixel intensities were similar for all mice. Cells were simply divided into CFP or GFP negative (no signal above background noise) or positive (signal above background noise). We also tried to sub-divide positive cells depending on their amount of signal but this did not provide any further statistically significant insights (data not shown).
Analysis of cells in lymph nodes, blood and lungs
The numbers of disseminating cells were determined after sacrificing the mice. Inguinal and axillary lymph nodes were dissected and examined whole with a fluorescence microscope to determine the numbers of mCherry or mRFP and GFP positive cells. The lungs and heart were dissected together and the blood drained from the heart into a cell culture dish. Some PBS was added and the cells allowed to settle before the entire dish was scanned for mCherry or mRFP and GFP positive cells. The lungs were examined in a similar manner to the lymph nodes. The values shown in Figure 8B&C are the number of experimentally manipulated cells observed (green) divided by the number of control cells (red) in the lymph node, blood or lungs normalised to the ratio of experimentally manipulated cells to control cells observed in the primary tumour.
Luciferase assays
105 MTLn3E cells were transfected, using Fugene 6 (Roche), with 2.0μg of CAGA12::luc in combination with 1μg EF-LacZ. 6 hours post transfection cells were treated for 18h with either TGFβ1 2ng/ml, SB431542 10μm, DMSO 1:1000 or combinations thereof. Using the luciferase assay kit (Promega), luminescence was assayed in Wallac 1420 plate reader. For each sample, luciferase activity was divided by β- galactosidase activity. Values are expressed as fold activation relative to the untreated control cells.
Soft agar assay
Cells are treated for 18h with either DMSO, 2ng/ml TGFβ1+DMSO or 10uM SB431542 before washing with PBS and preparation for the soft-agar assay. Base agar is prepared at 42°C by mixing 2x medium (αMEM + 5% FCS) with 1.2% agarose dissolved in water. 1.5 ml was added to 6 well plate and allowed to set for >30min at room temperature. Top agar was prepared by mixing 2x medium (αMEM + 5% FCS) with 0.75% agarose gel dissolved in water. 1.5 ml of mix is added to 5000 pelleted cells and the mixture is overlaid on the base agar. Plates are allowed to set for 30min at room temperature then overlaid with αMEM + 5% FCS, which was changed twice weekly. At day 21-24 plates were fixed for 10 minutes in ice cold 10% methanol and stained with Giemsa stain. Excess stain is removed by washing with water and visible colonies were then counted.
Western blotting
Whole cell extract were prepared using D0.4 buffer (20mM Hepes pH7.5, 10% glycerol, 0.4M NaCl, 0.4% Triton X-100, 10mM EGTA, 5mM EDTA 1M DTT, 1x protease inhibitors (Roche), 10mg/ml Aprotonin). For cytoplasmic/nuclear fractionation cells were treated as described in 36. Western blotting was performed using standard procedure. The following primary antibodies were used in this work: Smad2/3 (BD #610843), pSmad2 (Cell Signaling #3101), Smad3 (Zymed #511500), Smad 4 (Santa Cruz sc-7966), β-Tubulin (Sigma), PARP (Roche #1183528001), Grb2, vimentin (Santa Cruz sc-6260), Anti-GFP (Roche #11814460001).
Immunofluorescence
Cells were fixed using 4% PFA in PBS then permeabilized with 0.25% Triton X100. After blocking with 5% BSA, primary antibodies β-catenin antibody (Santa Cruz sc-7963) or pS63-c-jun (Santa Cruz sc-822) were diluted in 1% BSA in PBS and incubated with sample overnight at 4C. Secondary antibodies and/or phalloidin were diluted in 1% BSA in PBS and incubated with sample for 1hr at room temperature.
Tumour Staining
Frozen tumour samples, either MTLn3E or human breast carcinoma, were fixed using 4% paraformaldehyde in PBS following by 0.2%Triton X100 in PBS and blocked with 5%BSA in PBS. The following antibodies were used pSmad3 423/425 (Cell Signaling #9514), pSmad3 423/425 (Epitomics #1880), Smad2/3 (BD #610843), β-catenin (Santa Cruz #7963), CD31 (BD Pharmingen #553370) and TRITC-phalloidin and DAPI were used to visualize F-actin and DAPI, respectively.
Inhibitors and recombinant proteins
Recombinant TGFβ1 (Peprotech) was dissolved in 4mM HCl + 1mg/ml BSA at a stock concentration of 1μg/ml. The following inhibitors were used at 10μM working concentrations: SB431542 (ALK4,5,7 - Tocris), Y27632 (ROCK1&2 - Tocris), AG1478 (EGFR -Sigma), SP600125 (JNK - Calbiochem), UO126 (MEK).
Scattering assay
Cells were plated as single cells at low density, so that each single cell could give rise to a small cluster. 18-24h after plating cells were treated for 18-24h with TGFβ alone or in combination with inhibitors. For MTLn3E cells the extent of scattering was determined by measuring the area underlying a colony divided by the number of cells within that colony (scattering index). For 410.4 the number of single cells per 10x microscope field was counted in four fields for each condition. For time-lapse imaging of these assays cells were treated for 36-40 before imaging.
Lung colonization assay
5×105 TGFβ1-IRES GFP MTLn3E cells and 5×105 control MTLn3E cells (labeled with mCherry) were mixed in 100μl PBS and injected intravenously. For the ‘pulse’ experiment GFP MTLn3E cells were treated with 2ng/ml TGFβ for 18 hours before injection. Mice were then sacrificed after either 48hrs or two weeks, the lungs removed and micro-metastases visualized using a fluorescence microscope and the ratio of red to green cells determined.
Microarray procedures and analysis
5 mg of total RNA were labelled according to Affimetrix genechip microarray protocol. The labelled RNA was hybridised to Rat genome 230 2.0 arrays. Samples for three independent repeats of each experimental condition were sent for analysis. Five conditions were assessed, namely DMSO 1:1000 for 18h, SB431542 10μM for 18h, TGF B1 2ng/ml + DMSO for 18h, TGFβ1 2ng/ml + DMSO for 2h, TGF-β1 100pg/ml for 18h. Expression levels of genes in TGFβ1 or SB431542 treated cells were compared to that of uninduced cells. Fold changes greater than 2 fold with p<0.000001 were deemed TGFβ1 regulated. Expression analysis was carried out at the Bioinformatics department at the Cancer Research UK London Research Institute.
RNA extraction and qRT-PCR
Total RNA was prepared from 106 cells using RNeasy (Qiagen), according to manufacturer's instruction. RNA aliquots were frozen at −70°C for qRT-PCR and microarray experiments. cDNA was generated from 1mg total RNA using M-MLV H-point mutant reverse transcriptase (Promega) and random primers. qPCR was carried out on 5ng c-DNA per sample using Platinum SYBR Green q PCR (Invitrogen), according to manufacturers instruction. The housekeeping gene coding for glyceraldehyde-3-phospate dehydrogenase (GAPDH) was used as control. Specific primer pair sequences are given below.
| Gene | Forward | Reverse |
|---|---|---|
| Tmepai | CTGGAGCTGAACCGAGAGTC | CGGTAGTGACCAATGACCT |
| Fxyd5 | TCCCAGTTCCAGATCAAACC | GGTACCTTTCTCGCTGCTTG |
| EGFR | ACCGTGGAGAGAATCCCTTT | AGAGGATGGGGTTGTTGCTA |
| PAI-1 | TGGTGAACGCCCTCTATTTC | GAGGGGCACATCTTTTTCAA |
| GAPDH | AGACAGCCGCATCTTCTTGT | CTTGCCGTGGGTAGAGTCAT |
| CeaCAM | AAGGTGACAGGGTGACGTTC | GGTTGAGGTTTGACCCTTGA |
| CTGF | GAGTCGTCTCTGCATGGTCA | CCACAGAACTTAGCCCGCTA |
| BhlhB2 | GGATTTTGCCCACATGTACC | TCAATGCTTTCACGTGCTTC |
| CDH 17 | TTCTGAAGACACAGCCATCG | GCTCAGGGTTTTCTGCTTTG |
| RhoC | GCAAGAAGACTACGATCGCC | GCCAGCTCTCTCCTGGTATG |
| Nedd9 | CGAAGACACCAGAGCCTTTC | GAGGAGGTGGAAGATGTGGA |
| CTGF | AGAGTGGAGATCGCCAGGAGA | TTCACTTGCCACAAGCTGTC |
| c-Jun | CATTGAGAAGTAGCCCCCAA | ATGGCTCTCAACTCAAGCGT |
| MPRIP | CATTGAAAAGGCAGAGCACA | GCCACGGAATCCCTGTAGTA |
| Farp-1 | TTCAACAGCGCATGTTCTTC | TTTTTCCATCTCTGATCGGG |
Supplementary Material
Acknowledgements
We thank lab colleagues for advice and comments on this project. We thank members of the LRI FACS laboratory, Clare Watkins and members of the LRI Biological Resources Unit, the Paterson Institute for Cancer Research microarray facility, the LRI Bioinformatics team, Mike Howell of the LRI Developmental Signalling laboratory and Debbie Aubyn of the LRI microscopy laboratory for technical assistance. This work was funded by the Breast Cancer Campaign project grant 12May05 and CRUK.
Footnotes
We have no conflicting interests to report.
References
- 1.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 2.Sahai E. Illuminating the metastatic process. Nat Rev Cancer. 2007;7:737–749. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 3.Friedl P. Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol. 2004;16:14–23. doi: 10.1016/j.ceb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Leivonen SK, Kahari VM. Transforming growth factor-beta signaling in cancer invasion and metastasis. Int J Cancer. 2007;121:2119–2124. doi: 10.1002/ijc.23113. [DOI] [PubMed] [Google Scholar]
- 5.Lesko E, Majka M. The biological role of HGF-MET axis in tumor growth and development of metastasis. Front Biosci. 2008;13:1271–1280. doi: 10.2741/2760. [DOI] [PubMed] [Google Scholar]
- 6.Wells A. Tumor invasion: role of growth factor-induced cell motility. Adv Cancer Res. 2000;78:31–101. doi: 10.1016/s0065-230x(08)61023-4. [DOI] [PubMed] [Google Scholar]
- 7.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 9.Fox SB, Harris AL. The epidermal growth factor receptor in breast cancer. J Mammary Gland Biol Neoplasia. 1997;2:131–141. doi: 10.1023/a:1026399613946. [DOI] [PubMed] [Google Scholar]
- 10.Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 11.Donovan J, Slingerland J. Transforming growth factor-beta and breast cancer: Cell cycle arrest by transforming growth factor-beta and its disruption in cancer. Breast Cancer Res. 2000;2:116–124. doi: 10.1186/bcr43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–29. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 13.Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–457. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 14.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 15.Vincent-Salomon A, Thiery JP. Host microenvironment in breast cancer development: epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res. 2003;5:101–106. doi: 10.1186/bcr578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kokkinos MI, et al. Vimentin and epithelial-mesenchymal transition in human breast cancer--observations in vitro and in vivo. Cells Tissues Organs. 2007;185:191–203. doi: 10.1159/000101320. [DOI] [PubMed] [Google Scholar]
- 17.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3:921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 18.Wyckoff JB, Jones JG, Condeelis JS, Segall JE. A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res. 2000;60:2504–2511. [PubMed] [Google Scholar]
- 19.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 20.Wyckoff JB, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 21.Philippar U, et al. A Mena invasion isoform potentiates EGF-induced carcinoma cell invasion and metastasis. Dev Cell. 2008;15:813–828. doi: 10.1016/j.devcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahai E, et al. Simultaneous imaging of GFP, CFP and collagen in tumors in vivo using multiphoton microscopy. BMC Biotechnol. 2005;5:14. doi: 10.1186/1472-6750-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue C, et al. Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res. 2006;66:192–197. doi: 10.1158/0008-5472.CAN-05-1242. [DOI] [PubMed] [Google Scholar]
- 24.Dennler S, et al. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmierer B, Hill CS. Kinetic analysis of Smad nucleocytoplasmic shuttling reveals a mechanism for transforming growth factor beta-dependent nuclear accumulation of Smads. Mol Cell Biol. 2005;25:9845–9858. doi: 10.1128/MCB.25.22.9845-9858.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 27.Mi Z, et al. Differential osteopontin expression in phenotypically distinct subclones of murine breast cancer cells mediates metastatic behavior. J Biol Chem. 2004;279:46659–46667. doi: 10.1074/jbc.M407952200. [DOI] [PubMed] [Google Scholar]
- 28.Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr Biol. 2006;16:1515–1523. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- 29.Padua D, et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welch DR, Fabra A, Nakajima M. Transforming growth factor beta stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc Natl Acad Sci U S A. 1990;87:7678–7682. doi: 10.1073/pnas.87.19.7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goswami S, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005;65:5278–5283. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 32.Gupta GP, et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007;446:765–770. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- 33.Kang Y, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A. 2005;102:13909–13914. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicolas FJ, De Bosscher K, Schmierer B, Hill CS. Analysis of Smad nucleocytoplasmic shuttling in living cells. J Cell Sci. 2004;117:4113–4125. doi: 10.1242/jcs.01289. [DOI] [PubMed] [Google Scholar]
- 35.Gilles C, et al. Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer Res. 2003;63:2658–2664. [PubMed] [Google Scholar]
- 36.Wong C, et al. Smad3-Smad4 and AP-1 complexes synergize in transcriptional activation of the c-Jun promoter by transforming growth factor beta. Mol Cell Biol. 1999;19:1821–1830. doi: 10.1128/mcb.19.3.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.