

Abstract
Histone modifications are implicated in epigenetic inheritance, and are of central importance in regulating chromatin structure and gene expression. A prototype example is the tri-methylation (Me3) of lysine 9 on histone 3 (H3), which is readout by an aromatic cage of the chromodomain of heterochromatin-associated protein 1 (HP1) thereby leading to transcriptional repression and heterochromatin formation. Considering that the lysine methylation does not change the charge state of the histone tail and such aromatic-cage mediated recognition of the quaternary ammonium moiety is emerging as the most striking mechanistic commonality for the state-specific recognition of histone lysine methylation, it is of particular interest and importance to understand the physical origin regarding how the aromatic cage distinguishes between the H3K9Me3 mark and its un-methylated counterpart. Here we have simulated relative binding free energies among HP1 chromodomain - H3 tail complexes differing at position 9 of H3 tail. Our simulated results further confirm the essential role of cation-π interactions for the binding of methylated H3 tail by HP1 chromodomain, but indicate that the effect from the electrostatic origin is not dominant in distinguishing between the H3K9Me3 mark and its un-methylated counterpart. Meanwhile, our calculated free energy difference between H3- tert-butyl norleucine 9 and H3- methylnorleucine 9 in their binding to the HP1 clearly manifests the importance of the charge independent interactions for the state-specific readout of histone lysine tri-methylation marks.
Introduction
In the nuclei of eukaryotic cells, DNA is tightly wrapped around histone proteins and is compacted into a dense structure known as chromatin. Histone tails, which protrude out of the surface of the chromatin assemblies, are subject to a variety of reversible covalent modifications, including acetylation, methylation, ubiquitination and phosphorylation 1, 2. These histone modifications can play critical roles in regulating chromatin structure and the access of the genomic DNA thereby leading to various important cellular functions, such as DNA replication, transcription, repair, etc 1-3. One prototype example is the histone 3 (H3) tri-methylation at lysine 9, which is directly linked to the recognition of the H3 tail by the chromodomain of heterochromatin-associated protein 1 (HP1) and thereby leading to transcriptional repression and heterochromatin formation 4-6. On the other hand, H3 with unmethylated lysine has almost no detectable binding to HP1 and would results in an opposite biological function. H3K9 tri-methylation has been implicated to be an epigenetic mark, and the mechanism for its transmission onto the chromatin of the replicating DNA has been proposed. Meanwhile, the cross-talk between H3K9Me3 and other histone modifications, including the phosphorylation of H3 serine 10 7, has been established, which leads to more complicated regulation in cellular function 7, 8.
Since the lysine methylation does not change the charge state of the histone tail, a critical question is how H3K9 tri-methylation would promote its recognition by HP1. The key structural feature for HP1 binding of the methylated lysine-9 was found to be an aromatic cage formed by three aromatic residues 9, 10 (see Fig. 1). Subsequently, such aromatic-cage mediated methyl-lysine recognition has been found to be the most striking mechanistic commonality for the state-specific readout of histone lysine tri-methylation marks 11, including the recognition of H3K4Me3 by the plant homeodomain (PhD) finger 12 and the readout of H3K27Me3 by Polycomb 13.
Figure 1.
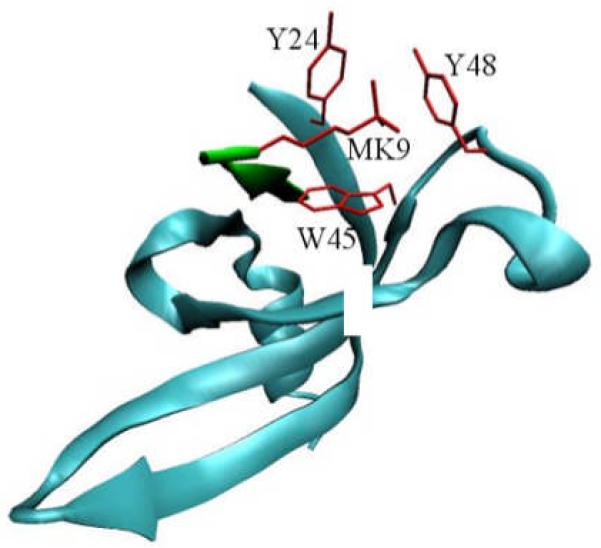
Illustration of the binding of HP1 chromodomain (blue) with histone H3 tail (green). Residues in red form the binding pocket for trimethylated K9.
As it has been demonstrated in numerous cases for other biological systems 14-17, the cation-π interaction originated from the π electrons of the aromatic residues and the positively charged methylammonium moiety is expected to play an important role in stabilizing the complex. Indeed a recent experimental study 18 showed that when the trimethyllysine (KMe3) is replaced by its neutral analogue, tert-butyl norleucine (tBuNle), the binding affinity between the histone 3A peptide and the choromodomain is reduced by 31 folds. So it has generally assumed that the cation-π interaction is mainly responsible for the readout of the trimethylysine mark while the charge independent interactions, including van der Waals and hydrophobic effect, play a less important role. However, the lysine methylation does not change the charge state, and it has been known that ammonium cation interacts more strongly with aromatic systems than tetramethylammonium cation does 19. To address this apparent dilemma, we have applied computer simulations to elucidate the physical origin regarding how the aromatic cage distinguishes between the H3K9Me3 mark and its un-methylated counterpart in the binding of H3 tail to the HP1 chromodomain. Since molecular recognition and ligand binding are of central importance to biology and medical chemistry, computational approach to studying the absolute/relative binding free energies has made significant progress in recent years and the results are in good agreement of experiments 20-24. Here we have employed long time molecular dynamics simulations and calculated the relative binding free energies by alchemically transforming the residue 9 of H3 tail from one to another (see Fig.2).
Figure 2.

Chemical structures of the side chains for KMe3, LYS, tBuNle and MeNle.
Computational Methods
I. MD simulations and free energy calculations
The starting models of the complex (HP1 chromodomain-histone H3 tail) were based on the crystal structure (PDB code 1KNE 9) and the histone tails were modeled with a short peptide corresponding to the residues 5 to 10 of histone H3, which participate directly in the binding to the chromodomain. Molecular dynamics simulations and thermodynamic integration were performed using GROMACS 3.3 25 and amber99 force field with a reparameterization of the backbone torsion terms (ff99SB) 26, 27.
For the non-standard residues (KMe3,tBuNle and MeNle), the atomic charges were determined with HF/6-31G* calculations and the restrained electrostatic potential (RESP) module in AMBER 9 28. The relative binding free energies were calculated by “alchemically” transforming KMe3 to tBuNle, tBuNle to MeNle and LYS to MeNle for H3 tails with the presence of HP1 chromodomain (complex) and in solution. When transforming tBuNle to MeNle, the process was decomposed into two steps with atomic charge transformation at the first step. At the second step, which involves the disappearance of the methyl groups, the method of soft-core potential was applied to ensure the stability of thermodynamic integration 29. Except for the box equilibration at the beginning, all the simulations were performed with Langevin dynamicis at NVT ensemble. The temperature was maintained at 300K and the MD integration time step is 2 fs. The bonds involving hydrogen atoms were constrained with the LINCS algorithm 30. The thermal dynamics integration was performed with 11 points or 16 point (if using soft-core potential). The simulation time at each point was 5 ns (the results of last 4 ns were used for data collection) in solution or 70 ns (the results of last 65 ns were used for data collection) in complex. To check the convergence of our simulations, two independent runs were performed for the transformations in complex. The relative binding free energies were obtained by taking the difference of the values calculated from the simulations in complex and in solution (see Scheme S1 for illustration). The detailed results from the simulations are shown in Table S1. We also performed 70 ns MD simulations of HP1 complexed with KMe3, tBuNle, MeMle and LYS H3 tails. The time dependant root mean square deviations (RMSDs) from the crystal structure are shown in Fig. S1.
II. Binding energy calculations for model systems
To validate the force field parameters for the non-standard residues, we calculated the binding energies between non-standard residues (side chains only) and three hydrophobic residues (Y24, W45 and Y48) present at the binding site for lysine-9 of H3 tail. The initial geometries were taken from the crystal structure and optimized at B3LYP/6–31G* level with side chain heavy atoms fixed. The binding energies were then calculated with molecular mechanics force field parameters and MP2/6-31G* level with basis set superposition error (BSSE) corrections. As shown in Table S2, the results between ab initio calculations and force field calculations are in quantitative agreement.
Results and Discussions
The key results of our current work, as listed in Table 1, are the relative binding free energies among HP1 chromodomain - H3 tail complexes differing at position 9 of H3 tail. It should be noted that a more negative binding free energy value indicates stronger binding. We can see that when KMe3 is transformed to its neutral analogue, tBuNle, the binding free energy is weakened by 3.1 kcal/mol, which is consistent with the experimental observation that cation-π interaction is essential for the binding of methylated H3 tail by HP1 chromodomain 18. We also found that after removing the methyl groups of tBuNle and mutating it to methylnorleucine (MeNle), the binding free energy is weakened by 1.9 kcal/mol, and changing LYS to its neutral analogue, MeNle, will destabilize the HP1 chromodomain - H3 tail complex by 2.1 kcal/mol. We note that the binding affinity between the MeNle H3 tail and HP1 chromodomain probably cannot be acquired via experiment because it is too weak to measure. We had attempted to directly calculate the relative binding free energy for the transformation from KMe3 to LYS, but were unable to obtain a converged value within affordable simulation time (in scale of 1000 ns in total). Therefore, as shown in Fig. 4, the relative binding free energy between H3K9Me3 and H3K9 in their bindings to HP1 is calculated via the completeness of thermodynamic cycle. The resulted value 2.9 kcal/mol is in agreement with the experimental one (> 2.7 kcal/mol) 18. Thus, our simulations have confirmed the experimental result that the tri-methylation of H3K9 promotes the binding of H3 tail to the HP1 chromodomain.
Table 1.
Relative binding free energies (in kcal/mol) between H3 tails and HP1 chromodomain. These values were calculated with the thermodynamic integration method by “alchemically” transforming the residue at position 9 of H3 tail from one to another, specifically, from KMe3 to tBuNle, tBuNle to MeNle, and MeNle to LYS. The transformations were performed for HP1 chromodomain – H3 tail complexes and H3 tails in solution and the relative binding free energies were obtained by taking the difference of the results from simulations in complex and in solution
| System | run 1 | run 2 | average |
|---|---|---|---|
| KMe3 -> tBuNle | 3.2 | 3.0 | 3.1±0.1 |
| tBuNle -> MeNle | 2.1 | 1.7 | 1.9±0.2 |
| LYS -> MeNle | 2.3 | 1.9 | 2.1±0.2 |
Figure 4.
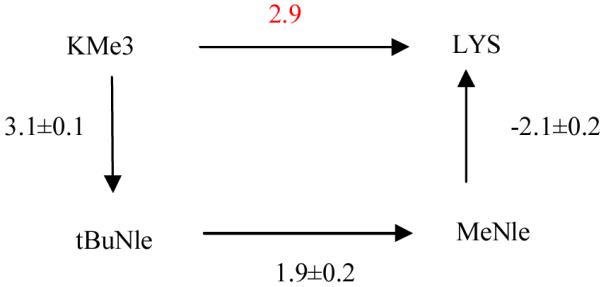
Thermodynamic cycle of relative binding free energies in kcal/mol. The relative binding free energy (shown in red) from H3-KMe3 to H3-LYS is obtained via the route KMe3 -> tBuNle -> MeNle -> LYS. The statistical errors were estimated by taking the differences of the results from two independent simulations.
In comparison with the available experimental data, one novel aspect of our current study is the determination of free energy difference with reference to the H3MeNle9 tail, which allows us to provide further physical insights into the role of lysine methylation on the recognition of the quaternary ammonium moiety by the aromatic cage. The lysine methylation can have its influence on the binding from two aspects: one can be attributed to the effect from electrostatic origin, including cation-π interactions and hydrogen bondings with water molecules; and the other can be the charge independent effect including the hydrophobicity and van der Waals interactions. With our calculated results shown in Fig. 4, these two aspects responsible for the state-specific readout of the trimethylation mark can be separated out. The relative binding free energy 1.9 kcal/mol between tBuNle and MeNle manifests the strength of the charge independent effect given by the methyl groups, while the difference of the values between KMe3->tNuNle and LYS->MeNle transformations, which is only about 1 kcal/mol, indicates the effect of lysine methylation on binding coming from the electrostatic origin. Therefore, our work suggests that the charge independent interactions are important for the aromatic cage to distinguish between the H3K9Me3 mark and its un-methylated counterpart in the binding of H3 tail to the HP1 chromodomain. Meanwhile, the effect from the electrostatic origin cannot be ignored but not dominant for readout of the trimethylation mark by HP1. Our results are consistent with previous experimental result that both charge-independent and electrostatic interactions are important in recognizing the quaternary ammonium moiety by the aromatic residues in the active site of acetylcholinesterase 15.
In order to further elucidate the structural effect of lysine methylation on its binding to the aromatic cage, we have also analyzed the trajectories of MD simulations of all four complex systems and calculated the populations of the distances between residue 9 of H3 tail and the aromatic residues forming the binding cage. As shown in Fig. 3, unlike KMe3 which resides well inside the binding cage and interacts with all three aromatic residues, the unmethylated lysine only interacts more strongly with TRP 48 by stacking with it in a closer distance. The other two distance distributions become much wider and especially the distance between LYS-9 and TYR48 is significantly longer, which indicates that Lys-9 has partially left the aromatic cage. On the other hand, for both MeNle and tBuNle at residue 9, their distance distributions are more similar to those of KMe3 and indicate that both of them still reside in the aromatic cage. These results further emphasize the importance of charge-independent effects in the read-out of the trimethylation mark by an aromatic cage with three aromatic residues.
Figure 3.

Distance population calculated from MD simulations. The distances are measured from the mass center of the side chain of residue 9 of H3 tail to the mass center of three aromatic residues (Y24, W45 and Y48) forming the aromatic cage. The vertical lines indicate the distances measured from the crystal structure (PDB code 1KNE).
Conclusions
In order to elucidate the physical origin regarding how the aromatic cage distinguishes between the H3K9Me3 mark and its un-methylated counterpart in the binding of H3 tail to the HP1 chromodomain, we have employed molecular dynamics simulations and alchemical free energy calculations to calculate the relative binding free energies among HP1 chromodomain - H3 tail complexes differing at position 9 of H3 tail. We found that when KMe3 is transformed to its neutral analogue, tBuNle, the binding free energy is weakened by 3.1 kcal/mol, which is consistent with the experimental observation that cation-π interaction plays an important role for the binding of methylated H3 tail with HP1 chromodomain. However, the change of Lys9 to its neutral analogue, MeNle, is also found to destabilize the HP1 chromodomain - H3 tail complex by 2.1 kcal/mol. This indicates that the difference between KMe3 and Lys in their complexation to the aromatic cage coming from the electrostatic origin is only about 1.0 kcal/mol, which cannot fully account for the methylation-state specific recognition. Meanwhile, the calculated free energy difference of 1.9 kcal/mol between H3-tBuNle9 and H3-MeNle9 in their binding to the HP1 manifests the importance of the charge independent effects for the state-specific readout of histone lysine trimethylation marks. We hope that our current study will inspire further experimental and theoretical work toward a better understanding of the histone lysine methylation effect.
Supplementary Material
Acknowledgement
The work has been supported by National Institute of Health (R01-GM079223) and National Science Foundation (CHE-CAREER-0448156, TeraGrid resources provided by LONI). We also thank NYU-ITS for providing computational resources and support.
REFERENCES
- 1.Shilatifard A. Annual Review of Biochemistry. 2006;75:243. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T. Cell. 2007;128:693. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Barski A, Cuddapah S, Cui KR, Roh TY, Schones DE, Wang ZB, Wei G, Chepelev I, Zhao KJ. Cell. 2007;129:823. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Nature. 2001;410:120. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 5.Lachner M, O’Carroll N, Rea S, Mechtler K, Jenuwein T. Nature. 2001;410:116. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 6.Nakayam J, Rice JC, Strahl BD, Allis CD, Grewal SIS. Science. 2001;292:110. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 7.Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Nature. 2005;438:1116. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 8.Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim YC, Minor W, Rastinejad F, Khorasanizadeh S. Nature. 2005;438:1181. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 9.Jacobs SA, Khorasanizadeh S. Science. 2002;295:2080. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 10.Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Nature. 2002;416:103. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 11.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. Nature Structural & Molecular Biology. 2007;14:1025. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li HT, Ilin S, Wang WK, Duncan EM, Wysocka J, Allis CD, Patel DJ. Nature. 2006;442:91. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischle W, Wang YM, Jacobs SA, Kim YC, Allis CD, Khorasanizadeh S. Genes Dev. 2003;17:1870. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma JC, Dougherty DA. Chem. Rev. 1997;97:1303. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 15.Quinn DM, Feaster SR, Nair HK, Baker NA, Radic Z, Taylor P. J. Am. Chem. Soc. 2000;122:2975. [Google Scholar]
- 16.Shi ZS, Olson CA, Kallenbach NR. J. Am. Chem. Soc. 2002;124:3284. doi: 10.1021/ja0174938. [DOI] [PubMed] [Google Scholar]
- 17.Hughes RM, Waters ML. J. Am. Chem. Soc. 2005;127:6518. doi: 10.1021/ja0507259. [DOI] [PubMed] [Google Scholar]
- 18.Hughes RM, Wiggins KR, Khorasanizadeh S, Waters ML. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11184. doi: 10.1073/pnas.0610850104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim KS, Lee JY, Lee SJ, Ha TK, Kim DH. J. Am. Chem. Soc. 1994;116:7399. [Google Scholar]
- 20.Deng YQ, Roux B. J. Phys. Chem. B. 2009;113:2234. doi: 10.1021/jp807701h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Straatsma TP, McCammon JA. Annu. Rev. Phys. Chem. 1992;43:407. [Google Scholar]
- 22.Kollman P. Chem. Rev. 1993;93:2395. [Google Scholar]
- 23.Simonson T, Archontis G, Karplus M. Accounts Chem. Res. 2002;35:430. doi: 10.1021/ar010030m. [DOI] [PubMed] [Google Scholar]
- 24.Gilson MK, Zhou HX. Annu. Rev. Biophys. Biomolec. Struct. 2007;36:21. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 25.Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. Journal of Computational Chemistry. 2005;26:1701. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 26.Wang JM, Cieplak P, Kollman PA. Journal of Computational Chemistry. 2000;21:1049. [Google Scholar]
- 27.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins-Structure Function and Bioinformatics. 2006;65:712. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Case DA, Darden TA, T.E. Cheatham I, Simmerling CL, Wang J, R.E. Duke R. Amber 9. University of California; 2006. [Google Scholar]
- 29.Beutler TC, Mark AE, Vanschaik RC, Gerber PR, Vangunsteren WF. Chemical Physics Letters. 1994;222:529. [Google Scholar]
- 30.Hess B, Bekker H, Berendsen HJC, Fraaije J. Journal of Computational Chemistry. 1997;18:1463. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.