

Abstract
Meckel syndrome (MKS) is a rare autosomal recessive disease causing perinatal lethality associated with a complex syndrome that includes occipital meningoencephalocele, hepatic biliary ductal plate malformation, postaxial polydactyly and polycystic kidneys. The gene mutated in type 1 MKS encodes a protein associated with the base of the cilium in vertebrates and nematodes. However, shRNA knockdown studies in cell culture have reported conflicting results on the role of Mks1 in ciliogenesis. Here we show that loss of function of mouse Mks1 results in an accurate model of human MKS, with structural abnormalities in the neural tube, biliary duct, limb patterning, bone development and the kidney that mirror the human syndrome. In contrast to cell culture studies, loss of Mks1 in vivo does not interfere with apical localization of epithelial basal bodies but rather leads to defective cilia formation in most, but not all, tissues. Analysis of patterning in the neural tube and the limb demonstrates altered Hedgehog (Hh) pathway signaling underlies some MKS defects, although both tissues show an expansion of the domain of response to Shh signaling, unlike the phenotypes seen in other mutants with cilia loss. Other defects in the skull, lung, rib cage and long bones are likely to be the result of the disruption of Hh signaling, and the basis of defects in the liver and kidney require further analysis. Thus the disruption of Hh signaling can explain many, but not all, of the defects caused by loss of Mks1.
INTRODUCTION
Meckel syndrome was first identified in 1822 (1). Since that time, six genomic loci in humans have been linked to MKS and, with the exception of Mks2, have been mapped to individual genes (2–7). Mks1and Mks6 (Cc2d2a) encode centrosomal proteins, whereas Mks3 (meckelin/Tmem67) is found in the plasma membrane and cilia (8,9). Mks4 (Cep-290/Nphp6) and Mks5 (Rpgrip1l) localize to both cilia and centrosomes (6,10,11). The localizations of these gene products suggest a commonality underlying the developmental defects in MKS individuals and suggest that MKS is one of a growing list of diseases (ciliopathies) that result from defects in a small cellular organelle, the cilium or its associated basal body, which is derived from the centrosome (reviewed in 12,13). Ciliopathies are a set of disorders with overlapping sets of clinical features and Meckel syndrome represents the most severe end of the phenotypic spectrum comprising defects in multiple organs and perinatal lethality.
Cilia are small microtubule-based extensions of the cell encapsulated by a membrane that is contiguous with the plasma membrane. Each cilium is nucleated by a centriole (basal body) within the centrosome, and a process called intraflagellar transport (IFT) is critical for ciliary outgrowth, signaling and resorption (14). The majority of cells in vertebrate embryos have a non-motile, primary cilium, while a subset of cells possess a single or multiple motile cilia (15,16), suggesting that this organelle has broad importance throughout the organism. Indeed, cilia have multiple roles in embryonic and postnatal development as well as disease and act in such diverse processes as signal transduction, mechanosensation and fluid flow (17,18).
The best-studied connection between cilia and signaling molecules involves the Hedgehog (Hh) pathway. The Hh receptor Patched (Ptch) localizes at the base of the cilium and acts to prevent movement of Smoothened (Smo) into the cilium (19). In the presence of Hh ligands, inhibition by Ptch is relieved and Smo can enter the cilium to activate Hh signaling. Additionally, the downstream Gli transcription factors localize to the tip of the cilium and require cilia for normal processing (20,21). In some tissues, defects in cilia or genes that are required for cilia formation or maintenance cause phenotypes that resemble alterations in Shh signaling. For example, cells in the limb with defective cilia fail to receive the Shh signal and display a loss of expression of downstream genes such as Gli1 (21,22). Further elucidation of the connections between ciliary proteins and the Hh signaling pathway is critical for our understanding of the role of this organelle in development and disease.
In a chemical mutagenesis screen, we identified a mouse mutant, kerouac (krc), based on its pre-axial polydactyly phenotype. Further examination revealed a suite of developmental defects in multiple organs. We mapped the mutation in this line to the Meckel Syndrome 1 (Mks1) gene, which is mutated in a subset of MKS patients (2). Mks1 is a cytosolic protein that is enriched at the centrosome in HEK293 and IMCD3 cells and localizes to the base of cilia in Caenorhabditis elegans (23,24). Mks1 encodes a protein with few functional motifs besides a B9 domain, however another B9 domain containing protein, stumpy, is required for cilia formation (25). To date, no mammalian model for type 1 MKS exists. Here we characterize the krc mutant phenotype with particular attention to ciliary defects and effects on Hh signaling. This mouse mutant is a faithful model for MKS and displays most of the core characters of this syndrome. Our phenotypic analysis reveals that Mks1 is required for cilia formation in multiple tissues and for normal Shh signaling in both the neural tube and the limb during development. Our data indicate that the level of Shh signaling is altered in krc mutants, which is likely to underlie many of the defects associated with Meckel syndrome.
RESULTS
Identification of the gene affected in krc mutants
We identified the recessive N-ethyl N-nitrosourea (ENU)-induced krc mutant based on a pronounced anterior bulge in the limbs of homozygous mutants at embryonic day (e) 12.5 (Fig. 1A and B). Analysis of embryonic patterning (see below) indicated that krc mutants shared a number of features seen when cilia formation is disrupted (21,26). We mapped the krc mutation to a 1 Mb interval on mouse Chromosome 11 corresponding to human Chromosome 17q23. The Mks1 gene was a candidate within this interval as siRNA knockdown experiments suggested that it was required for cilia formation in cultured cells (8). We identified a mutation in the splice donor site within intron 5 (c.515 + 6T > C) in krc mutants causing a 21-base insertion containing an inframe nonsense codon (E173stop) in the transcript. No wild-type splicing is detected in krc mutants by RT–PCR, suggesting that the krc allele (Mks1krc) does not produce any normal Mks1 transcripts (Supplementary Material, Fig. S1). Mks1 splicing defects are also found in human patients and, significantly, a mutation in the homologous splice donor site (c.515 + 1G > A) was reported in a human patient with classic MKS (27). Owing to the predicted early truncation in the protein and the strong phenotype of the similar human mutation, the Mks1krc allele is likely to be a null.
Figure 1.
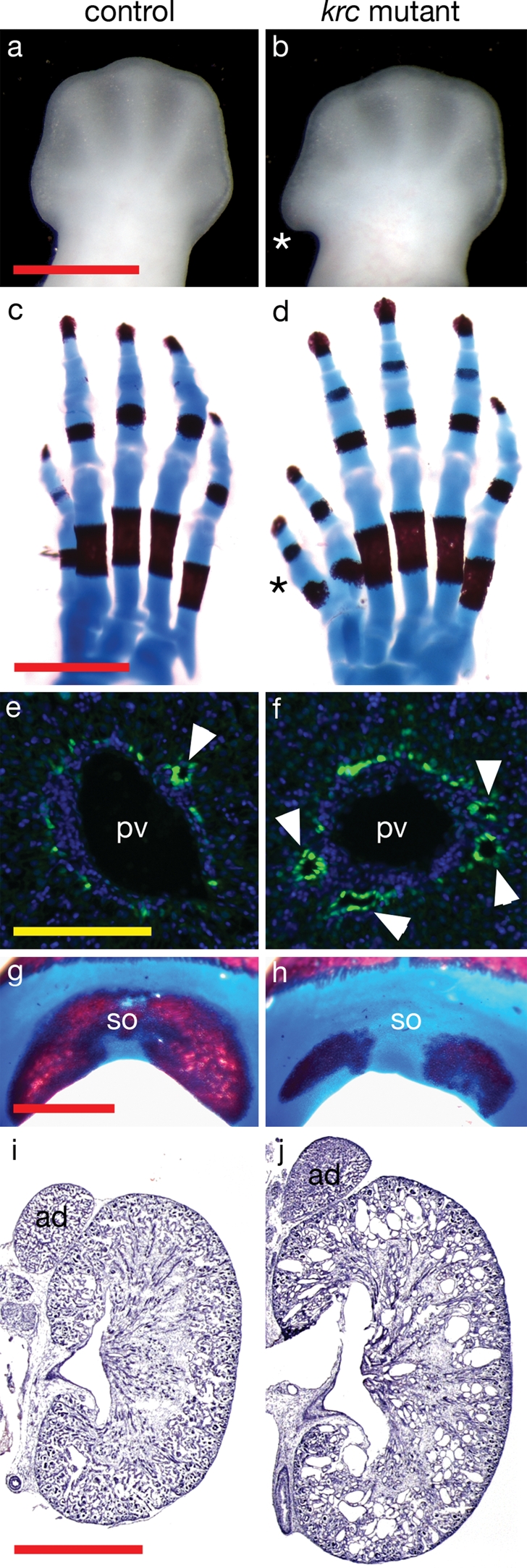
Mks1krc mutants develop defects similar to the Meckel syndrome tetrad. Dorsal views of (A and B) e12.5 hindlimbs and (C and D) e18.5 hindlimb skeletons showing polydactyly in Mks1krc mutants (asterisks). (E and F) Liver sections at e18.5 showing the formation of bile ducts (arrowheads) in the ductal plate flanking the portal veins [green, marked by Sox9; blue marks nuclei (DAPI)]. The ductal plate persists in mutants and multiple bile ducts form. (G and H) Posterior view of e18.5 skulls showing the mineralization of the supraoccipital bone. In 12/15 mutants, the supraoccipital bones showed reduced ossification compared with controls. (I and J) Sections through e18.5 kidneys and adrenal glands. The mutant kidney is larger than wild-type and multiple cysts are visible. In (C, D, G, H), the red staining marks bone and the blue is cartilage. ad, adrenal glands; pv, portal vein; so, supraoccipital bone. Controls and mutants are shown at the same magnification. Red scale bars = 1 mm, yellow scale bar = 0.1 mm.
krc mutants display typical Meckel syndrome characters
To test whether Mks1krc mutants represent a good mouse model for MKS, we examined whether krc mutants recapitulated the primary MKS characteristics. The four distinguishing hallmarks of Meckel syndrome are polydactyly, cystic kidneys, occipital encephalocoele and malformation of the biliary ductal plate. In contrast to the early lethality (e13.5–e14.5) on a C3H background, Mks1krc mutants survived as late as e18.5 on a mixed C3H/CD1 background, allowing assessment of later development. The anterior bulge in early Mks1krc mutant limbs resolves into pre-axial polydactyly, typically comprising a single extra anterior digit (Fig. 1C and D). There is variability in the penetrance and expressivity of this character, with ∼66% (n = 29/44) of e18.5 mutant fetuses showing some degree of polydactyly in at least one limb.
Dysplasia of the hepatic duct is also common in MKS. Disruptions in the ductal plate and bile duct formation in Mks1krc mutant livers are apparent at late stages. At e18.5, portal veins are flanked by 1–2 bile ducts that form in the ductal plate region during embryonic development (28). In Mks1krc mutants, we observed the formation of multiple bile ducts and persistence of the ductal plate, consistent with biliary defects reported in MKS human patients (Fig. 1E and F), although we did not detect biliary cysts, a frequent feature of MKS cases.
Occipital meningoencephalocoele, the protrusion of the brain through the occipital bone, is seen in ∼85% of MKS cases (29). Two types of related defects were seen in Mks1krc mutants. Exencephaly (failure of initial neural tube closure) was present in 28% (n = 32/113) of Mks1krc mutants in the C3H background (data not shown), although the penetrance was lower in the mixed C3H/CD1 background that allowed survival to birth. We also observed hypomineralization and/or splitting of the supraoccipital bone in late-term Mks1krc mutants (Fig. 1G and H) with 80% penetrance, which likely corresponds to the occipital defects in human patients.
Mks1krc mutants develop larger kidneys than wild-type, and those kidneys contain multiple cysts at e18.5 (Fig. 1I and J). The largest cysts are found in glomeruli and proximal tubules (Fig. 2A–D). Additional regions of mutant nephrons including collecting ducts and medullary thick ascending limbs show dilations of the epithelial tubules indicating that Mks1 is required throughout the kidney to regulate epithelial tubule diameter (Fig. 2E–H).
Figure 2.

Multiple segments of Mks1krc nephrons dilate or undergo cystogenesis. (A and B) Hematoxylin and eosin-stained sections of e18.5 kidneys show dilation of the Bowman's capsule (asterisk) surrounding the glomerulus in a mutant kidney. (C and D) Large cysts (asterisks) form in the proximal tubule region (green, marked by Lotus tetragonolobus lectin, LTL) of Mks1krc nephrons. (E and F) Collecting ducts (green, marked by Dolichos biflorus lectin, DBA) and (G, H) medullary thick ascending limbs (green, marked by Tamm–Horsfall protein, THP) are dilated in mutants (asterisks). In (C–H), blue marks nuclei (DAPI). gl, glomerulus. Controls and mutants are shown at the same magnification. Scale bars = 0.1 mm.
In addition to the aforementioned clinical features, MKS patients variably display additional skeletal and soft tissue abnormalities (29). Mks1krc mutants exhibit comparable defects including left–right asymmetry disruption, campomelia (bowing of the long bones), pulmonary hypoplasia, genitourinary defects and ribcage abnormalities (Fig. 3A–J). Additional skull defects in Mks1krc mutants included hydrocephaly accompanied by reduced ossification of the frontal and parietal bones and, in one case, we observed cleft palate (Fig. 3K, L, O, P). The similarities of the phenotypes in Mks1krc mutants and MKS patients indicate that the mouse mutant provides an appropriate model to define the developmental genetics and molecular mechanisms underlying Meckel syndrome. Additional abnormalities were seen in the Mks1krc mutants, such as abnormal ovarian development (Fig. 3G and H) and reduced presphenoid bone (Fig. 3M and N), suggesting that detailed analysis of this mouse mutant may define novel aspects of Meckel syndrome.
Figure 3.

Mks1krc mutants display additional Meckel syndrome characteristics. (A and B) Ventral view of e12.5 hearts. Heart looping is a marker of left–right asymmetry. When heart looping is reversed, the outflow tract is on the left and left and right ventricles are reversed (B). In Mks1krc mutants, 32% of embryos show reversed heart looping, 41% have normal looping, 26% show partial looping (n = 87). (C and D) e18.5 hindlimbs showing shortening and bowing of the femur and tibia (campomelia; asterisks) in mutants (n = 16/16). (E and F) Dorsal view of e18.5 lungs. Mks1krc mutants also show pulmonary hypoplasia, with variable fusions between the right lungs lobes (n = 12/12). (G and H) e18.5 female genitourinary tracts. Ovaries normally lie largely posterior to the kidneys (asterisks). In Mks1krc mutants, ovaries are sometimes displaced (black arrow) and show abnormal morphology (n = 6/20). (I and J) e18.5 sternum and ribs. Mks1krc mutants show a shortening of the sternum and fusions or forking of the ribs (n = 14/16). (K and L) Dorsal view of e18.5 skull vaults showing reduced mineralization of the frontal and parietal bones in Mks1krc mutants (n = 14/14). (M and N) Dorsal view of the palate after removal of the skull vault. Mks1krc mutants show a reduction in the size of the presephenoid bone (n = 15/15). (O and P) Ventral view of the palate after removal of the mandible. One Mks1krc mutant (P) showed an absence of the maxilla and palatine shelves of the palate (cleft palate) and absence of the more dorsal presphenoid bone. In (C, D, I–P), the red staining marks bone and blue is cartilage. oft, outflow tract; rv, right ventricle; lv, left ventricle; fe, femur; ti, tibia; le, left lobe; cr, cranial lobe; me, medial lobe; ca, caudal lobe; ki, kidney; bl, bladder; st, sternum fr, frontal bone; pa, parietal bone; ip, interparietal bone; ps, presphenoid; ma, maxilla shelf; pa, palatine shelf. Controls and mutants are shown at the same magnification. Scale bars = 0.1 mm.
Mks1 is required for cilia formation in multiple tissues
siRNA experiments in IMCD3 cells indicated that the MKS1 gene product is required for proper cilia formation in cultured cells (8). We therefore examined primary cilia in several tissues from Mks1krc mutant embryos. We observed a dramatic reduction in the number of primary cilia that formed in most mesenchymal and epithelial tissues (Fig. 4A–F and Table 1). The left–right asymmetry defects in Mks1krc mutants (Fig. 3A and B) suggested a defect in the formation or function of motile nodal cilia, which are critical for proper left–right patterning of multiple organs (30). Nodal cilia are not detected at e7.5 in Mks1krc mutants (data not shown), but by e8.5 sparse nodal cilia of variable length and morphology are present (Fig. 4G and H). These defects in node cilia formation likely underlie the later defects in left–right patterning. Interestingly, some tissues do not display obvious reductions in cilia number including multi-ciliated lung cells and the bile ducts in the liver (Fig. 4M–P). The presence of cilia in Mks1krc mutant bile ducts is consistent with findings from Mks1 human liver tissue (31). Thus Mks1 is required for normal cilia formation in many, but not all, tissues.
Figure 4.

Defective cilia formation in Mks1krc mutants. Sections through (A and B) e11.5 hindlimb mesenchyme, (C and D) e18.5 cortical kidney regions and (E and F) e18.5 brains demonstrate a dramatic reduction in the number of cilia (green) in Mks1krc mutants. In limbs (A and B), cilia are associated with basal bodies (red) in controls but this is reduced in mutants. Cilia are located on the apical surface of kidney tubules (C) and lateral brain ventricles (E) but the number and length of cilia is reduced in mutants (D, section through a cyst). (G and H) Scanning electron micrograph of e8.25 mouse nodes show a reduction in the number and abnormal morphology of Mks1krc mutant nodal cilia. Insets show that the morphology of the node is normal in mutants. A few cilia are found that are nearly normal in length, but most cilia are reduced in size and some have bulges at their distal tips. In contrast to MKS1 siRNA knockdown (8), we do not see a noticeable difference in microvilli formation. (I–L) Basal bodies (red) localize to the apical side of polarized epithelia. (I and J) e12.5 lung epithelium. At this stage, the lung epithelial cells are monociliated. The white line indicates the basal side of the cells, and the apical side is to the right. (K and L) e11.5 neural tubes. The apical side of the cell faces the lumen (center, dashed line). In both of these tissue types, basal bodies are apically localized but there is a reduction in the number of cilia in mutants. (M and N) The lumens of e18.5 bile ducts (asterisks) show similar numbers of cilia in Mks1krc mutants compared with wild-type despite the persistence of the ductal plate and increased number of bile ducts. (O and P) e18.5 multiciliated lung airway cells. Mks1krc mutant cells form multiple, long cilia similar to controls. In all panels except (G and H) green marks cilia (Arl13b, A–F, I–L; or acetylated α-tubulin, M–P). In (A, B, I–L), red marks basal bodies (γ-tubulin). Purple (C–F, O, P) marks F-actin (phalloidin). In (M and N), red marks bile duct cells (Sox9). Controls and mutants are shown at the same magnification. White scale bars = 0.1 mm, red scale bars = 2 μm. (A–D, I–L, O, P) These are confocal X–Z projections.
Table 1.
krc mutants show reduced numbers of cilia compared with controls
| Tissue | Control | Krc Mutant |
|---|---|---|
| e8.25 Node | 25.9 ± 4.88 | 5.25 ± 1.67 |
| e10.5 Forelimb mesenchyme | 18.8 ± 3.47 | 6.96 ± 2.76 |
| e12.5 Lung mesenchyme | 19.7 ± 5.27 | 7.17 ± 2.87 |
| e18.5 Lateral brain ventricles: lateral side | 13.4 ± 1.98 | 2.42 ± 1.31 |
| e18.5 Lateral brain ventricles: medial side | 10.6 ± 2.84 | 7.25 ± 1.54 |
Cilia were counted in several embryonic tissues. We observed a two- to five-fold reduction in the number of cilia in most tissues. For e8.25 nodal cilia, whole or partial cilia were counted in a 144 square micron area. Three mutants and four controls were used and cilia in 1–3 SEM images per embryo were counted. For e10.5 limbs, e12.5 lung mesenchyme and e18.5 lateral brain ventricles, cilia were marked using an antibody against Arl13b on frozen sections. For lungs and limbs, we counted cilia in a 1.6 mm2 area and for brains, cilia were counted along a 40 μm length of the ventricle lumen. Three mutant and three control embryos were assayed for cilia formation in each of these tissues, with four (brains), six (lungs) or eight (limbs) images analyzed per embryo.
Mks1 is not required for basal body localization
MKS1 localizes to the basal body in cell culture, and MKS1 knockdown disrupts apical localization of the basal body in IMCD-3 cells (8). This led to the hypothesis that MKS1 is required for apical docking of the basal body and that the basal body abnormality was responsible for the defects in cilia formation. To test this in vivo, we examined the position of basal bodies in polarized epithelia, which form cilia on the apical side of the cell. Basal bodies showed normal apical localization in lung, neural tube and peritoneal epithelial cells in Mks1krc mutants, despite abnormal cilia formation in these cells (Fig. 4I–L and data not shown). These data indicate that Mks1 is required for the outgrowth of primary and motile cilia rather than basal body localization.
Mks1 is required for normal dorsal–ventral patterning in the neural tube
Cilia are important for signaling via the Hh pathway. Key pathway components localize to the cilium and mutations that affect cilia formation or maintenance cause phenotypes characteristic of disrupted Hh signaling (32). To test whether Hh signaling was affected in Mks1krc mutants, we examined specification of neuronal cell types in the neural tube, which is regulated by Shh signaling from the notochord (33). High levels of Shh are required for the most ventral cell fates, the floorplate and V3 interneurons, whereas lower levels specify motorneurons and additional interneuron fates. Mks1krc mutants look similar to wild-type at e10.5 (Fig. 5A, F) and their neural tubes specify dorsal and intermediate neuronal cell populations (Fig. 5B, C, G, H). However, HB9+ and Nkx6.1+ cells are found more dorsally than in wild-type (Fig. 5D, E, I, J) suggesting a broader domain of low-level Shh signaling. In addition, there is a reduction in the number of floor plate cells and V3 interneuron progenitors (Fig. 6C, D, G, H), accompanied by a ventral expansion of motor neurons at cervical, forelimb and hindlimb levels (Fig. 6B, F and data not shown). Loss of floor plate cells and the presence of V3 progenitors and motor neurons at the ventral midline are consistent with a reduction in high-level Shh signaling. The loss of cells requiring the highest level of Shh signaling accompanied by the expansion of cell types that require lower level Shh signaling is similar to, but milder than, the phenotype of hnn mutants, which have a mutation in Arl13b, a critical regulator of cilia architecture (34).
Figure 5.
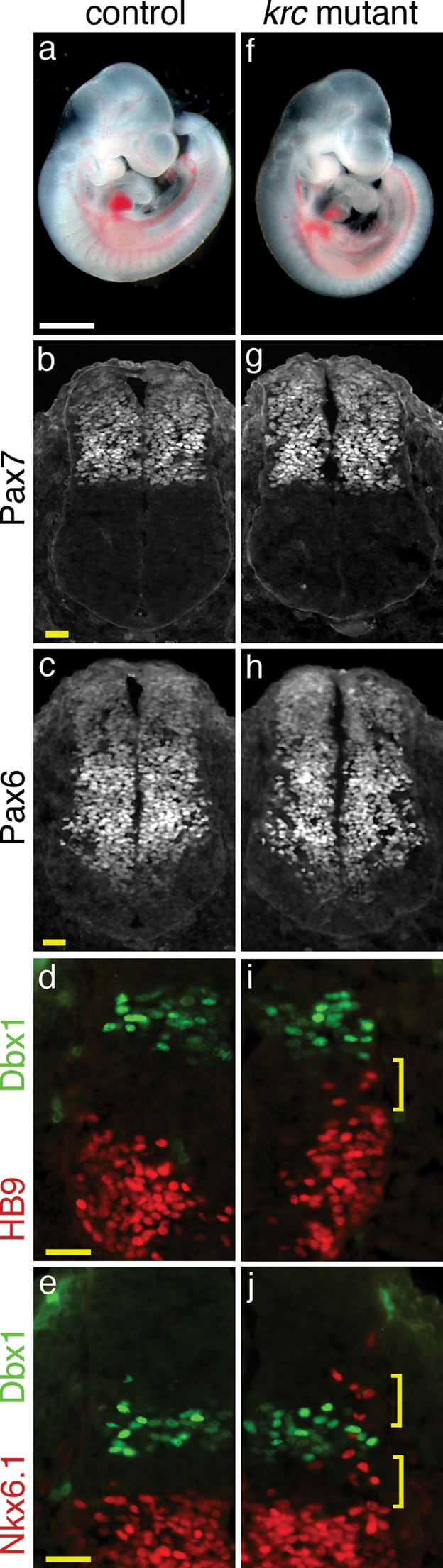
Mks1 is required for normal Shh-dependent patterning in the neural tube. (A and F) Lateral views of e10.5 embryos. Mks1krc mutants have a similar size and morphology as wild-type. (B–J) Immunofluorescent images of sections through e10.5 wild-type (B–E) and mutant (G–J) neural tubes at the level of the forelimb. HB9 marks motor neurons, NKX6.1 labels V2, V3 and motor neuron progenitors and DBX1 marks V0 progenitors. (B, C, G, H) PAX7 and PAX6, markers of dorsal and intermediate progenitor cells are expressed in similar domains in mutants and wild-type. In Mks1krc mutants, motor neurons (I) expand dorsally and approach the V0 progenitor domain (marked by Dbx1; bracket). Additionally, the dorsal boundary of Nkx6.1+ cells (J) is expanded dorsally (brackets). Controls and mutants are shown at the same magnification. White scale bar = 1 mm. Yellow scale bars = 0.05 mm.
Figure 6.

Mks1 acts downstream of Ptch1 and Smo in neural tube patterning.(A–X) Immunofluorescent images of sections through e10.5 wild-type (A–D) Mks1krc (E–H), ptch1 (I–L), krc;ptch1 (M–P), smo (Q–T) and smo;krc (U–X) neural tubes at the level of the forelimb. FoxA2 marks floor plate cells, Nkx2.2 marks V3 interneuron progenitors, HB9 marks motor neurons, Nkx6.1 labels V2, V3 and motor neuron progenitors. In Mks1krc mutants, floor plate cells are reduced in number or absent (H) compared with wild-type (D). (G) V3 interneuron progenitors are found more ventrally in mutants than in controls (C) and (F) motor neurons expand ventrally to the midline. Additional ventral progenitors largely form normally in mutants (E). ptch1 Mutants fail to close the neural tube and show a gain of Shh signaling including an expansion of floorplate markers (L), V3 and other ventral progenitors (K, I). Note: ptch1 mutants are delayed compared with wild-type and have not formed significant numbers of motor neurons at this stage. Mks1krc;ptch1 double mutants look more similar to Mks1krc mutants than ptch1 mutants: only a few floor plate cells are specified (P), V3 progenitors are largely found at the ventral midline and motor neurons and other ventral progenitors are ventrally restricted (N, M). smo Mutants show a complete loss of Shh signaling in the neural tube and ventral cell types such are not specified (Q–T). smo;Mks1krc double mutants fail to specify the most ventral cell types (W, X) but some motor neurons (V) as well as Nkx6.1+ (U) and V0 progenitors (data not shown) are specified. Note: ptch1 (I–L) and smo (Q–T) mutant sections are shown at twice the magnification of the other sections. Scale bars = 0.1 mm. The scale bar in (A) is valid for all panels except (I–L; Q–T). See (I and Q) for appropriate scale bars.
Mks1 acts downstream of patched
To test whether Mks1 acts downstream of the Shh ligand during neuronal specification, we examined embryos doubly homozygous for Mks1krc and a mutation in the Shh receptor, Ptch1. Ptch1 is an inhibitor of the Hh pathway and ptch1 mutants show increased Shh signaling resulting in a loss of dorsal cell fates and dorsal expansion of ventral cell types throughout the neural tube (Fig. 6I–L) (35). Given the mild reduction in Shh signaling in Mks1krc mutants, it was striking that removal of Mks1 rescues many of the ptch1 defects. Mks1krc; ptch1 double mutants specify a small number of floor plate cells and V3 progenitors (Fig. 6O, P) and show a ventral expansion of motor neurons (Fig. 6N) similar to Mks1krc mutants. The dorsal exclusion of ventral progenitors in the double mutants (Fig. 6M–P contrasts with ptch1 mutants and resembles Mks1krc mutants, indicating that Mks1 lies downstream of Shh signal reception.
To assess whether the Mks1krc phenotype is solely due to decreased Shh signaling, we analyzed the effect of removing Mks1 function in smo and shh null backgrounds. Smo is a critical positive mediator of Hh signaling and smo mutants lack all ventral cell types in the neural tube (Fig. 6Q–T) (36). Given the reduced Shh signaling in Mks1krc neural tubes, it was surprising that loss of Mks1 rescues some smo defects indicating that the Mks1krc phenotype is not solely due to decreased Hh signaling. Although floor plate cells and V3 progenitors are not specified in smo; Mks1krc double mutants (Fig. 6W, X), motor neurons, V2 and V0 interneuron progenitors are present but ventrally localized and intermixed (Fig. 6U, V and data not shown). We observed similar results in shh; Mks1krc double mutants (data not shown). This suggests that Mks1 normally acts to inhibit motor neuron and V0–V2 interneuron fates, which is consistent with the observed dorsal expansion of Nkx6.1+ ventral progenitor cells and motor neurons in Mks1krc mutants.
The phenotype of krc is similar to that of tectonic (tctn1) (37) mutants: both krc and tctn1 have relatively mild effects on final neural patterning but can largely bypass the requirement for Shh and Ptch1, presumably by modifying the activity of the Gli transcription factors that mediate Shh signaling. The specification but spatial disorganization of ventral progenitors and motor neurons in smo; Mks1krc double mutants phenocopies smo; gli3 (36) and gli2; gli3 (38) double mutants, which supports the hypothesis that Mks1 acts through the Gli proteins to regulate the correct spatial patterning of ventral cell types in the neural tube.
Mks1 is required for normal anterior–posterior patterning in the limb
We initially identified the krc mutant based on its polydactylous phenotype, and as Shh has a well-defined role in limb patterning (39), we examined Shh expression in Mks1krc mutants and found that it appeared normal (Fig. 7A, B). The expression of Shh targets Ptch1 and Gli1 is dramatically reduced in some cilia mutants (21,22,40), however, these genes were expressed normally or in slightly larger posterior limb domains in Mks1krc mutants (Fig. 7E–H and data not shown). In a subset of Mks1krc mutant embryos (n = 2/12), we observed ectopic anterior expression of Gli1, indicating increased Shh signaling. We also observed anterior expansion of Gremlin1 (Grem1), an additional target of Shh signaling (41) (Fig. 7C, D). These effects on target gene expression parallel the findings in the Mks1krc neural tube: Grem1, Ptch1, Gli1 are broadly expressed in the central region of the limb, similar to the expression domain of HB9 and Nkx6.1 in the neural tube. In both Mks1krc mutant limbs and neural tubes, we see an expansion of both types of mid/low level targets of Shh, which suggests a common mechanism of action by Mks1 in these two tissues.
Figure 7.

Shh signaling is altered in Mks1krc mutant limbs. (A, C, E, G) Dorsal views of e11.5 wild-type forelimbs showing posteriorly restricted (A) Shh, (C) Grem1, (E) Gli1 and (G) Ptch1 expression. (B, D, F, H) Mks1krc mutant limbs show strong posterior expression of Shh and its target genes. (D) All Mks1krc mutant mutants assayed showed an anterior expansion of Grem1 (n = 12). (F) In a subset of mutants, Gli1 is ectopically expressed on the anterior side of the limb (n = 2/12) despite the lack of ectopic Shh. Arrowheads indicate ectopic expression in the anterior limb. Controls and mutants are shown at the same magnification. The scale bar in (A) is valid for all panels (0.5 mm).
DISCUSSION
Our analysis of the krc allele of Mks1 provides a better understanding of the developmental defects of MKS. Our findings demonstrate that Mks1 is required in vivo for normal cilia formation in multiple tissues during development. Those defects in ciliogenesis are responsible for disruption of Hh signaling in a range of different cell types, which leads to a variety of developmental abnormalities.
In contrast to siRNA knockdown experiments in cell culture, Mks1 is not required for apical localization of basal bodies, but rather for cilia length and morphology. This role for Mks1 is consistent with findings for several other mouse mutants that disrupt basal body proteins, including Mks4–6, Oro-Facial-Digital syndrome 1 and most Bardet-Biedl syndrome proteins (9–11,42,43). The mechanisms by which proteins localized to the basal body affect cilia are not understood but may include regulation of trafficking to the cilium or transport within the cilium itself (43).
The cilia abnormalities in Mks1krc mutants cause developmental defects in a variety of tissues and organs. The neural tube and limb phenotypes can be explained as disruptions in cilia-dependent Shh signaling. However, the defects seen in these tissues do not correspond to the simple loss of Hh signaling seen in other mutants with reduced cilia number (21,22,26). In the Mks1krc neural tube, there is both a reduced response to high level Shh activity accompanied by an expansion of the domain of Shh signaling; a similar expansion of the domain of Shh activity is also seen in the limb. The abnormalities in the skull, lungs and ribcage in Mks1krc mutants are similar to phenotypes in mouse loss of function mutants in the Hh pathway (44,45), suggesting that these defects in Mks1krc mutant mice and in MKS patients may also be due to altered Hh signaling.
However, MKS phenotypes cannot be solely explained by altered Hh signaling. Reversal of left–right asymmetry, ductal plate malformations and cystic kidneys have not been reported in Hh pathway mutants, but do occur as a result of ciliary or basal body defects (13). While the left–right asymmetry defects are likely the result of disrupted motile cilia-dependent flow in the Mks1krc node, the cause of the kidney cysts and extra bile ducts requires further examination. Together, our findings suggest that the ciliogenesis defects in Mks1krc mutants likely underlie both the Hh-dependent and -independent phenotypes in MKS. Interestingly, a mutation in the C. elegans homolog of Mks1 (Xbx-7) does not affect cilia formation or morphology on its own but acts with nephrocystin genes to regulate cilia formation, suggesting that the role of Mks1 may have diverged between the nematode and vertebrate lineages (24,46). The differences between the cell culture, nematode and mouse studies highlight the importance of developing vertebrate models to better define the basis of some human disorders.
Five of six genomic loci that are linked to Meckel syndrome have been identified and studied to varying extents (2–7). Although no mouse models for MKS types 2 or 4 exist, Mks3 and Mks5 mutants have recently been reported, but the full spectrum of MKS characters has not been analyzed in these models. Similar to Mks1, Mks5 is also required for left–right asymmetry and Shh signaling in the limb and neural tube; however, the mutants have not been assayed for additional skeletal phenotypes or cyst formation (47). Mks3 mutant mice develop polycystic kidneys but lack other MKS characteristics, whereas Mks3 mutant rats (wpk) show cilia defects in kidneys, eyes and sperm (11), The milder phenotypes in Mks3 animals compared with Mks1 mutants may reflect the relatively milder defects in Mks3 patients compared with those with mutations in Mks1 (48,49). Our analysis reveals that Mks1krc mutants currently provide the most complete parallel to the human syndrome and identifies defects in both ciliogenesis and Hh signaling as key underlying defects in several tissues. Because Mks1krc mutants provide a faithful model of MKS, this mutant provides the means to further assess the molecular mechanisms underlying the broad spectrum of developmental defects in Meckel syndrome.
MATERIALS AND METHODS
Animals and mutagenesis
C57BL/6J and C3HeB/FeJ mice were obtained from The Jackson Laboratory. ENU mutagenesis was performed essentially as described (50); 10 mg/ml ENU in phosphate–citrate buffer was injected intraperitoneally into male mice 9–10 weeks old at the time of the first injection. C57BL/6J males received ENU in three doses of 100 mg/kg body weight administered once a week for 3 weeks. These males were used to establish lines by crossing to C3HeB/FeJ females (Supplementary Material, Fig. S2). Fertility was recovered starting ∼7 weeks after treatment.
Screening and recovery of mutations
F1 sons of mutagenized mice were crossed as shown in Supplementary Material, Figure S2 to produce lines of mice for screening. Each F1 male was mated to two C3HeB/FeJ females, to generate second-generation (G2) females. Four to ten G2 females per line were mated to the F1 male; lines with two or more litters containing embryos with similar abnormal morphology were considered potentially mutant. G2 males and females from these lines were produced and intercrossed, and litters were screened at 12.5 days post-coitum to confirm the heritability of the phenotype and to identify G2 males that carried the mutation. Lines were maintained initially by outcrossing the carrier males to C3HeB/FeJ, intercrossing the resulting progeny and examining embryos to identify new male carriers. After the mutation responsible for the phenotype was initially mapped, carriers of both sexes were identified by PCR as those carrying the C57BL/6J alleles of markers flanking the induced mutation. Out of 97 lines screened, we recovered 14 lines that appeared to have a heritable, recessive mutation. Of these lines only one had multiple segregating phenotypes, indicating that most mutants were monogenic.
Generation and mapping of krc
krc Mutants were initially identified based on pre-axial polydactyly. However, additional phenotypes were observed at e12.5–e14.5 in the krc line including micropthalmia, exencephaly, somite derivative defects and a kinked neural tube. Embryos were considered mutant if they contained two or more of these defects. We crossed eight G2 females to the F1 male and we obtained 12 morphological mutants out of 74 total implantation sites (16%) compared with an expectation of 9.25 mutants (12.5%). We recovered slightly higher than Mendelian frequency as five of eight G2 females were carriers. These data are consistent with a single gene recessive mutation.
For initial mapping of the krc mutation, we examined two to five simple-sequence length polymorphism markers per chromosome in an attempt to establish linkage between the phenotype and C57BL/6J DNA polymorphisms. We began with Chromosome 1 and proceeded sequentially. For markers on Chromosomes 1 through 10, we observed recombination frequencies from 25 to 66% indicating minor or no linkage between the phenotype and these chromosomes. In contrast, on Chromosome 11, markers close to the 89 Mb region showed 0% recombination between the phenotype and C57BL/6J polymorphisms, indicating tight linkage of the phenotype to this segment of Chromosome 11. Following the initial mapping, we chose potential carrier animals based on carrying C57BL/6J DNA on Chromosome 11. From 15 l embryos, we recovered 32 phenotypic animals, all of which mapped to the Chromosome 11 region. We have continued to outcross krc carrier males to C3HeB/FeJ females and by using recombination mapping narrowed the krc interval to Chromosome 11 between markers D11Mit39 (87.61 Mb) and D11Mit212 (88.67 Mb). We are currently at the eleventh (N11) generation on the C3HeB/FeJ background, which is equivalent to removing more than 99.9% of the original mutagenized B6 background, supporting the idea that the krc phenotype is monogenic.
The identification of the splice donor site in intron 5 (c.515 + 6 T > C) resulted from sequencing genomic DNA from the Mks1 locus in krc homozygous mutants.
Expression analysis
In situ hybridizations and immunofluorescence analyses were performed using standard methods as previously described (51). Monoclonal antibodies raised against Pax6, Nkx6.1, Nkx2.2, HB9, FoxA2 and Shh were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by Department of Biological Sciences, The University of Iowa, Iowa City, IA 52242, USA. A polyclonal antibody that recognizes Dbx1 was kindly provided by Thomas Jessell. For cilia staining, we used antibodies against Arl13b (34) and centrosomes were marked with γ-tubulin (Sigma).
Scanning electron microscopy
For SEM, we collected embryos on e7 and e8 and processed them as previously described (26).
Skeletal staining
Skeletons were prepared and stained with Alcian blue and Alizarin red as previously described (52).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by the National Institutes of Health [U01 HD43478 to K.V.A., L.A.N.; R01 NS044385 to K.V.A., NIH R01 HD32427 to L.A.N.] and institutional startup funds from the Yale Genetics Department (S.D.W.).
ACKNOWLEDGEMENTS
We thank A. Fernandez for comments on the manuscript, W. Horton and H. Alcorn for technical assistance and N. Lampen for help with scanning electron microscopy.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Meckel J.F. Beschreibung zweier, durch sehr aehnliche Bildungsabweichungen entstellter Geschwister. Dtsch. Arch. Physiol. 1822;7:99–172. [Google Scholar]
- 2.Kyttala M., Tallila J., Salonen R., Kopra O., Kohlschmidt N., Paavola-Sakki P., Peltonen L., Kestila M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006;38:155–157. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- 3.Roume J., Genin E., Cormier-Daire V., Ma H.W., Mehaye B., Attie T., Razavi-Encha F., Fallet-Bianco C., Buenerd A., Clerget-Darpoux F., et al. A gene for Meckel syndrome maps to chromosome 11q13. Am. J. Hum. Genet. 1998;63:1095–1101. doi: 10.1086/302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith U.M., Consugar M., Tee L.J., McKee B.M., Maina E.N., Whelan S., Morgan N.V., Goranson E., Gissen P., Lilliquist S., et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel–Gruber syndrome and the wpk rat. Nat. Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- 5.Baala L., Audollent S., Martinovic J., Ozilou C., Babron M.C., Sivanandamoorthy S., Saunier S., Salomon R., Gonzales M., Rattenberry E., et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C., et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 7.Tallila J., Jakkula E., Peltonen L., Salonen R., Kestila M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am. J. Hum. Genet. 2008;82:1361–1367. doi: 10.1016/j.ajhg.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dawe H.R., Smith U.M., Cullinane A.R., Gerrelli D., Cox P., Badano J.L., Blair-Reid S., Sriram N., Katsanis N., Attie-Bitach T., et al. The Meckel–Gruber syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum. Mol. Genet. 2007;16:173–186. doi: 10.1093/hmg/ddl459. [DOI] [PubMed] [Google Scholar]
- 9.Gorden N.T., Arts H.H., Parisi M.A., Coene K.L., Letteboer S.J., van Beersum S.E., Mans D.A., Hikida A., Eckert M., Knutzen D., et al. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am. J. Hum. Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang B., Khanna H., Hawes N., Jimeno D., He S., Lillo C., Parapuram S.K., Cheng H., Scott A., Hurd R.E., et al. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 2006;15:1847–1857. doi: 10.1093/hmg/ddl107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vierkotten J., Dildrop R., Peters T., Wang B., Ruther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development. 2007;134:2569–2577. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- 12.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 13.Sharma N., Berbari N.F., Yoder B.K. Chapter 13 ciliary dysfunction in developmental abnormalities and diseases. Curr. Top. Dev. Biol. 2008;85:371–427. doi: 10.1016/S0070-2153(08)00813-2. [DOI] [PubMed] [Google Scholar]
- 14.Pedersen L.B., Rosenbaum J.L. Chapter two intraflagellar transport (IFT) Role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 15.Wheatley D.N. Incidence and significance of oligocilia in normal and pathologic tissues. Ultrastruct. Pathol. 1993;17:565–566. doi: 10.3109/01913129309041308. [DOI] [PubMed] [Google Scholar]
- 16.Wheatley D.N. Primary cilia in normal and pathological tissues. Pathobiology. 1995;63:222–238. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- 17.Michaud E.J., Yoder B.K. The primary cilium in cell signaling and cancer. Cancer Res. 2006;66:6463–6467. doi: 10.1158/0008-5472.CAN-06-0462. [DOI] [PubMed] [Google Scholar]
- 18.Eggenschwiler J.T., Anderson K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohatgi R., Milenkovic L., Scott M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 20.Haycraft C.J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E.J., Yoder B.K. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu A., Wang B., Niswander L.A. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- 22.Haycraft C.J., Zhang Q., Song B., Jackson W.S., Detloff P.J., Serra R., Yoder B.K. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134:307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- 23.Dawe H.R., Farr H., Gull K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J. Cell Sci. 2007;120:7–15. doi: 10.1242/jcs.03305. [DOI] [PubMed] [Google Scholar]
- 24.Williams C.L., Winkelbauer M.E., Schafer J.C., Michaud E.J., Yoder B.K. Functional redundancy of the B9 proteins and nephrocystins in Caenorhabditis elegans ciliogenesis. Mol. Biol. Cell. 2008;19:2154–2168. doi: 10.1091/mbc.E07-10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Town T., Breunig J.J., Sarkisian M.R., Spilianakis C., Ayoub A.E., Liu X., Ferrandino A.F., Gallagher A.R., Li M.O., Rakic P., et al. The stumpy gene is required for mammalian ciliogenesis. Proc. Natl Acad. Sci. USA. 2008;105:2853–2858. doi: 10.1073/pnas.0712385105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huangfu D., Anderson K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl Acad. Sci. USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frank V., Ortiz Bruchle N., Mager S., Frints S.G., Bohring A., du Bois G., Debatin I., Seidel H., Senderek J., Besbas N., et al. Aberrant splicing is a common mutational mechanism in MKS1, a key player in Meckel–Gruber syndrome. Hum. Mutat. 2007;28:638–639. doi: 10.1002/humu.9496. [DOI] [PubMed] [Google Scholar]
- 28.Lemaigre F.P. Development of the biliary tract. Mech. Dev. 2003;120:81–87. doi: 10.1016/s0925-4773(02)00334-9. [DOI] [PubMed] [Google Scholar]
- 29.Chen C.P. Meckel syndrome: genetics, perinatal findings, and differential diagnosis. Taiwan J. Obstet. Gynecol. 2007;46:9–14. doi: 10.1016/S1028-4559(08)60100-X. [DOI] [PubMed] [Google Scholar]
- 30.Basu B., Brueckner M. Chapter six cilia multifunctional organelles at the center of vertebrate left-right asymmetry. Curr. Top. Dev. Biol. 2008;85:151–174. doi: 10.1016/S0070-2153(08)00806-5. [DOI] [PubMed] [Google Scholar]
- 31.Tammachote R., Hommerding C.J., Sinders R.M., Miller C.A., Czarnecki P.G., Leightner A.C., Salisbury J.L., Ward C.J., Torres V.E., Gattone V.H., II, et al. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Hum. Mol. Genet. 2009;18:3311–3323. doi: 10.1093/hmg/ddp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong S.Y., Reiter J.F. The primary cilium at the crossroads of mammalian hedgehog signaling. Curr. Top. Dev. Biol. 2008;85:225–260. doi: 10.1016/S0070-2153(08)00809-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stamataki D., Ulloa F., Tsoni S.V., Mynett A., Briscoe J. A gradient of Gli activity mediates graded Sonic Hedgehog signaling in the neural tube. Genes Dev. 2005;19:626–641. doi: 10.1101/gad.325905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caspary T., Larkins C.E., Anderson K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Goodrich L.V., Milenkovic L., Higgins K.M., Scott M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 36.Wijgerde M., McMahon J.A., Rule M., McMahon A.P. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev. 2002;16:2849–2864. doi: 10.1101/gad.1025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiter J.F., Skarnes W.C. Tectonic, a novel regulator of the Hedgehog pathway required for both activation and inhibition. Genes Dev. 2006;20:22–27. doi: 10.1101/gad.1363606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bai C.B., Stephen D., Joyner A.L. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev. Cell. 2004;6:103–115. doi: 10.1016/s1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 39.McGlinn E., Tabin C.J. Mechanistic insight into how Shh patterns the vertebrate limb. Curr. Opin. Genet. Dev. 2006;16:426–432. doi: 10.1016/j.gde.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 40.May S.R., Ashique A.M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A.S. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 41.Vokes S.A., Ji H., Wong W.H., McMahon A.P. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 2008;22:2651–2663. doi: 10.1101/gad.1693008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romio L., Fry A.M., Winyard P.J., Malcolm S., Woolf A.S., Feather S.A. OFD1 is a centrosomal/basal body protein expressed during mesenchymal–epithelial transition in human nephrogenesis. J. Am. Soc. Nephrol. 2004;15:2556–2568. doi: 10.1097/01.ASN.0000140220.46477.5C. [DOI] [PubMed] [Google Scholar]
- 43.Zaghloul N.A., Katsanis N. Mechanistic insights into Bardet–Biedl syndrome, a model ciliopathy. J. Clin. Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeong J., Mao J., Tenzen T., Kottmann A.H., McMahon A.P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mo R., Freer A.M., Zinyk D.L., Crackower M.A., Michaud J., Heng H.H., Chik K.W., Shi X.M., Tsui L.C., Cheng S.H., et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–123. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- 46.Bialas N.J., Inglis P.N., Li C., Robinson J.F., Parker J.D., Healey M.P., Davis E.E., Inglis C.D., Toivonen T., Cottell D.C., et al. Functional interactions between the ciliopathy-associated Meckel syndrome 1 (MKS1) protein and two novel MKS1-related (MKSR) proteins. J. Cell Sci. 2009;122:611–624. doi: 10.1242/jcs.028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cook S.A., Collin G.B., Bronson R.T., Naggert J.K., Liu D.P., Akeson E.C., Davisson M.T. A mouse model for Meckel syndrome type 3. J. Am. Soc. Nephrol. 2009;20:753–764. doi: 10.1681/ASN.2008040412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Consugar M.B., Kubly V.J., Lager D.J., Hommerding C.J., Wong W.C., Bakker E., Gattone V.H., II, Torres V.E., Breuning M.H., Harris P.C. Molecular diagnostics of Meckel–Gruber syndrome highlights phenotypic differences between MKS1 and MKS3. Hum. Genet. 2007;121:591–599. doi: 10.1007/s00439-007-0341-3. [DOI] [PubMed] [Google Scholar]
- 49.Khaddour R., Smith U., Baala L., Martinovic J., Clavering D., Shaffiq R., Ozilou C., Cullinane A., Kyttala M., Shalev S., et al. Spectrum of MKS1 and MKS3 mutations in Meckel syndrome: a genotype–phenotype correlation. Mutation in brief #960. Online. Hum. Mutat. 2007;28:523–524. doi: 10.1002/humu.9489. [DOI] [PubMed] [Google Scholar]
- 50.Russell W.L., Hunsicker P.R., Raymer G.D., Steele M.H., Stelzner K.F., Thompson H.M. Dose–response curve for ethylnitrosourea-induced specific-locus mutations in mouse spermatogonia. Proc. Natl Acad. Sci. USA. 1982;79:3589–3591. doi: 10.1073/pnas.79.11.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eggenschwiler J.T., Anderson K.V. Dorsal and lateral fates in the mouse neural tube require the cell-autonomous activity of the open brain gene. Dev. Biol. 2000;227:648–660. doi: 10.1006/dbio.2000.9918. [DOI] [PubMed] [Google Scholar]
- 52.Weatherbee S.D., Anderson K.V., Niswander L.A. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.