

Abstract
Objective
To determine if there was a genetic contribution to our patient’s unusual clinical presentation of nephrolithiasis and nonhealing stress fracture.
Methods
We describe a 31-year-old man who had rickets as a child and developed a femur insufficiency fracture and recurrent nephrolithiasis as an adult after moving to the United States from India. The patient’s clinical course and results from radiographic and biochemical analyses are described. Analysis of the SLC34A3 gene was performed using genomic DNA samples from the patient and his family members.
Results
Prior to referral to Yale Bone Center, the patient was treated with calcitriol, ergocalciferol and phosphate. Changing therapy to phosphate alone led to clinical improvement. Genetic analysis revealed that the patient is a compound heterozygote for mutations in the SLC34A3 gene. On one allele he has a previously identified missense mutation in exon 7 c.575C>T (p.S192L). The other allele carries a novel nonsense mutation in exon 3 c.145C>T (p.Q49*). One unaffected sibling is a carrier of the missense mutation and 1 sister with a history of flank pain is a carrier of the novel mutation.
Conclusion
The patient is a compound heterozygote for mutations in the SLC34A3 gene including a novel nonsense mutation in exon 3. His atypical clinical presentation illustrates that both environmental and genetic factors potentially affect phenotypic expression of SLC34A3 mutation.
INTRODUCTION
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) is characterized by hypophosphatemia due to renal phosphate wasting and typically presents clinically with bone pain, muscle weakness, and rickets/osteomalacia (1,2). The appropriately elevated serum 1,25(OH)2D levels in response to hypophosphatemia distinguishes HHRH from other types of hypophosphatemic rickets. Elevated 1,25(OH)2D levels lead to enhanced intestinal absorption of calcium resulting in an increased renal filtered calcium load and hypercalciuria (1). Surprisingly, despite the hypercalciuria, nephrolithiasis and nephrocalcinosis have rarely been reported (3, 4).
Treatment of HHRH with phosphate supplementation alone can correct the clinical abnormalities associated with the disease even though the renal phosphate wasting persists (1).
It is important to distinguish HHRH from other Mendelian renal phosphate wasting disorders, such as X-linked hypophosphatemic rickets (XLH), autosomal dominant hypophosphatemic rickets (ADHR), and autosomal recessive hypophosphatemia (ARHP). While ADHR, XLH, and ARHP require both calcitriol and phosphate supplementation, treatment with calcitriol can worsen the hypercalciuria and increase the risk of nephrolithiasis in patients with HHRH (5).
HHRH was first described in a large consanguineous Bedouin kindred in 1985 and was thought to have an autosomal recessive mode of inheritance (1). The genetic basis for HHRH has been identified as a mutation in the SLC34A3 gene, which encodes the renal sodium phosphate co-transporter NaPi-IIc (3, 5).
We report a patient referred to us for a non-healing stress fracture and recurrent nephrolithiasis. He proved to be a compound heterozygote for a known missense mutation in exon 7 and a novel nonsense mutation in exon 3 of NaPi-IIc. His somewhat atypical clinical presentation highlights the fact that both environmental and genetic factors affect phenotypic expression of SLC34A3 mutations.
CASE REPORT
A 31-year-old Indian male was referred to the Yale Bone Center in July 2005 for evaluation of recurrent nephrolithiasis and a non-healing right femur insufficiency fracture. He had bilateral genu valgum as a child and underwent bilateral supracondylar osteotomies in India at 12 years of age. He had been treated as a child with vitamin D for presumed nutritional rickets. In his late teens, the patient discontinued Vitamin D supplementation. He had no other skeletal symptoms and no history of nephrolithiasis until he came to the United Sates.
In 2000, he moved to the United States. Two years later he passed a calcium oxalate stone. He passed a second calcium oxalate stone the following year and was noted to have an elevated blood alkaline phosphatase value of 270 IU/L (normal 20–125 IU/L), a normal serum calcium and a low serum phosphate of 1.5 mg/dL (normal 2.5–4.5 mg/dL). Calcitriol 0.5 µg/day was started for a presumptive diagnosis of XLH. One year later he complained of increasing right leg pain. Radiographs showed a right femur insufficiency fracture (Figure 1A). His treating physician increased the dose of calcitriol to 0.75 µg/day and sodium phosphate supplementation was begun. One month later he passed a third calcium oxalate stone and the calcitriol and sodium phosphate were discontinued. Urine studies revealed that his urine calcium was elevated at 4.5 mg/kg/bw/day (normal ≤4.0 mg/kg/bw/day), as was his urinary oxalate at 44.3 mg/day (normal 3.6–38.0 mg/day). His urinary citrate excretion was normal at 315 mg/day (normal 100–1300 mg/day). His urinary phosphate was 264 mg/day. A DXA scan revealed an L-spine bone mineral density (BMD) of 0.670 gm/cm2 (T-score: −3.83) and a left total hip BMD of 0.841 gm/cm2 (T-score: −1.27).
Figure 1.

Radiographs of the study patient showing a right femur insufficiency fracture (Panel A) that was persistent 1 month after diagnosis (Panel B). After oral phosphate supplementation, the patient’s femoral stress fracture gradually healed (Panel C).
He continued to complain of right leg pain. Radiographs were repeated and demonstrated a persistent right femur insufficiency fracture (Figure 1B). He was restarted on a lower dose of calcitriol and ergocalciferol was added when his 25(OH)vitamin D level was found to be less than 5 ng/ml. He was seen in consultation by an orthopedic surgeon who referred the patient to the Yale Bone Center.
On presentation to the Yale Bone Center, the patient complained of right upper leg pain and difficulty bearing weight on that leg. He was 5 feet one-inch tall and weighed 114 pounds. He was ambulating with a walker. His hearing was normal. The patient’s dentition was good. He had mid-thoracic rotational scoliosis. There was genu valgum on the left and osteotomy scars over both distal femurs. There was no pretibial tenderness but he had compression tenderness of the metacarpals and metatarsals. He denied muscle weakness, dental, or hearing problems. He reported that he was a vegetarian and had a limited intake of protein and dairy products. He was working as a software consultant.
His family members live in India. His father was 5 feet 3 inches tall and was noted to have bowing of the lower extremities. He had died several years earlier from a myocardial infarction. His mother is 5 feet tall and has no skeletal complaints. He has four sisters with heights ranging from 4 feet 11 inches to 5 feet 2 inches. There is no history of skeletal complaints in his sisters, but one sister had complained of flank pain in the past. His maternal and paternal grandfathers lived to 90 years of age and had no medical problems. His maternal and paternal grandmothers both died at an early age and the details of their medical histories are unknown. He has three nephews, one of whom has a history of mild genu valgum, and four nieces all of whom are currently asymptomatic.
Materials and Methods
Biochemical studies
Biochemical studies were obtained two weeks after discontinuing calcitriol, ergocalciferol, and oral sodium phosphate supplementation. Serum and urine calcium were measured by atomic absorptiometry and phosphorus, creatinine and alkaline phosphatase by autoanalyzer as previously described (6). Serum levels of parathyroid hormone were measured using a sensitive mid-molecule PTH assay as previously reported (6). Serum 25(OH)vitamin D and 1,25(OH)2vitamin D concentrations were measured using commercially available radioimmunoassays (Diagnostics Systems Laboratories; Webster, TX). The renal phosphate threshold (TMP/GFR) was calculated as previously described by Walton and Bijvoet (7)
SLC34A3 mutational analyses
Nucleotide sequence analysis of the SLC34A3 gene was performed using genomic DNA samples from the patient and his family members as previously described (3). Written informed consent was obtained prior to sample collection and analysis.
Results
Biochemical Evaluation and Subsequent Clinical Course
At the Yale Bone Center, all vitamin D preparations were discontinued and biochemical studies obtained two weeks later. As shown in Table 1, at the time of initial evaluation, the patient had a normal serum calcium, a low serum phosphorus and a reduced renal phosphate threshold as evidenced by a low TMP/GFR of 1.9 mg/100 ml GF. His serum alkaline phosphatase was three-fold elevated above the upper limit of normal with a concomitantly normal serum 5′ nucleotidase, consistent with the alkaline phosphatase being of bone origin. The serum creatinine was 1.0 mg/dl and the GFR was 127 ml/min. He had a normal serum 25(OH)vitamin D level but a markedly elevated 1,25(OH)2vitamin D level of 222 pg/ml. On a weight-adjusted basis he was excreting 6.3 mg of calcium/kg/bw/d (normal, ≤4.0 mg/kg/bw/d). Based on these biochemical findings a presumptive diagnosis of HHRH was made.
Table 1.
Biochemical studies at initial presentation to the Yale Bone Center and following treatment with oral phosphate supplementation.
| serum Ca (mg/dl) (9.0–10.5) |
Serum Pi (mg/dl) (2.5–4.5) |
PTH (nleq/ml) (10–25) |
25- OHD (ng/ml) (20–45) |
1,25-OHD (ng/ml) (25–66) |
Alk Phos (U/L) (30–130) |
24hr urine Ca (mg) (100–300) |
TMP/GFR (mg/100ml GF) (2.5–4.2) |
|
|---|---|---|---|---|---|---|---|---|
| Before treatment |
9.6 | 1.5 | 7 | 39 | 222 | 426 | 328 | 1.95 |
| 2 months after treatment |
9.4 | 2.5 | 10 | 21 | 155 | 416 | ||
| 4 months after treatment |
9.4 | 9 | 47 | 212 | 389 | |||
| 15 months after treatment |
9.4 | 15 | 23 | 82 | 144 |
Following his initial evaluation, he was prescribed oral phosphate supplementation alone. His femoral insufficiency fracture gradually healed (Figure 1C) and his biochemical abnormalities improved (Table 1). He was reseen in August of 2006 one year after therapy. At that point he was asymptomatic. A repeat bone density showed normalization of his spinal bone mass with a T-score of −0.6. His total hip BMD had improved to a T-score of −0.8. In November of 2006 he was seen for the last time. He remained completely asymptomatic. However, his 1,25(OH)2vitamin D level, although markedly improved, remained elevated and his alkaline phosphatase level remained minimally elevated as well. He continues to take an oral sodium phosphorus supplement.
Genetic Analysis
Sequence analysis of the patient's SLC34A3 gene identified him as a compound heterozygote. On one allele the patient had a previously identified cytidine to thymidine missense mutation in exon 7 (c.575C>T) (3). On his other allele he had a novel cytidine to thymidine nonsense mutation in exon 3 (c.145C>T). The missense mutation predicts a change in the amino acid sequence from serine to leucine at amino acid 192 (S192L). The nonsense mutation results in a truncated protein after amino acid 49 (Q49*), eliminating most of the membrane-spanning region and the intracellular c-terminal tail of NaPi-IIc.
Genetic screening of the patient’s family revealed that the missense mutation c.575C>T (p.S192L) was transmitted from his mother (Figure 2, I-2). Because the father (I-1) is deceased his haplotype was deduced. One unaffected sibling (II-5) is a carrier of the missense mutation c.575C>T (p.S192L). One sister with a history of flank pain (II-3) is a carrier of the novel mutation c.145C>T(p.Q49*). C.145C>T (p.Q49*) was not found in 136 control alleles.
Figure 2.
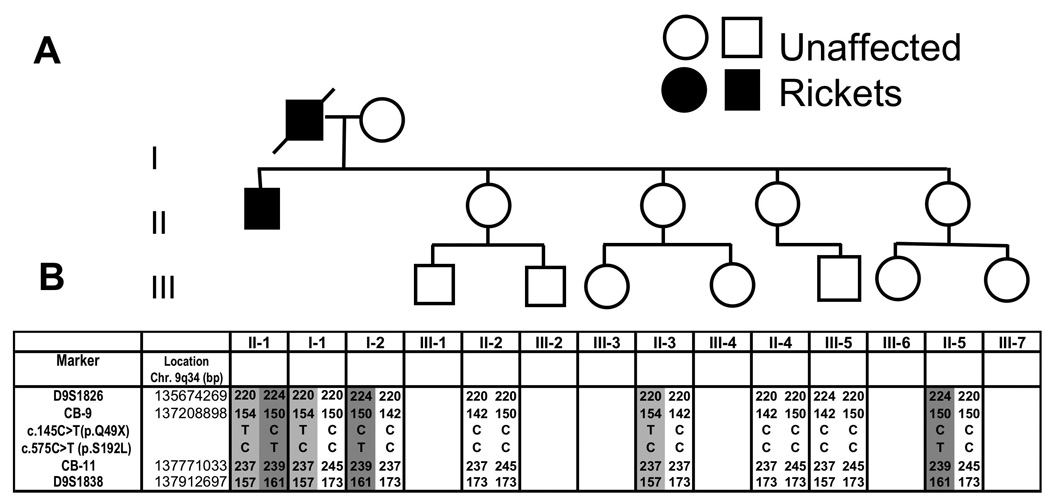
Family pedigree. Panel A, Pedigree of the patient’s family. Squares denote males; circles denote females. The arrow identifies the proband. Symbol with a diagonal dashed line denotes a deceased individual. Panel B, Haplotypes for chromosome region 9q34 between markers D951826 and D951838. Haplotypes associated with HHRH are depicted by numbers on a grey background. The haplotype of I-1 was deduced from genetic information from other family members.
Discussion
Our patient had lower limb bone deformities as a child in India and developed recurrent nephrolithiasis and a right femur stress fracture as an adult while living in the United States. Biochemical analyses revealed a low serum phosphate, a low TMP/GFR, and a high 1,25(OH)2vitamin D, findings consistent with HHRH. The appropriately elevated 1,25(OH)2vitamin D level made XLH, ADHR, ARHP and oncogenic osteomalacia all less likely diagnoses. The normal serum creatinine and the absence of aminoaciduria and glucosuria made Dent’s disease unlikely as well. An unremarkable urinalysis, normal serum bicarbonate, and normal electrolytes, also made a diagnosis of Fanconi’s syndrome remote.
Linkage studies have recently identified the genetic basis for HHRH as mutations in the SLC34A3 gene, which lead to loss of function of the NaPi-IIc protein (3,5). In addition to homozygous mutations in the SLC34A3 gene, compound heterozygous missense mutations or deletions were found in some of the affected individuals. FGF23 levels are low or normal in this disorder (4), which is consistent with the conclusion that HHRH is due to an intrinsic defect in renal phosphate reclamation.
Genotyping of our patient showed mutations affecting both alleles for the SLC34A3 gene, indicating that he is a compound heterozygote. In addition to having a known missense mutation in exon 7, he has a novel nonsense mutation in exon 3. Genotyping of family members revealed that his asymptomatic mother and one asymptomatic sister are both carriers of the missense mutation c.575C>T (p.S192L), a mutation which was previously identified in a kindred of mixed white and Aboriginal origin from Australia (3). The finding of the novel mutation in exon 3, adds to the list of more than 16 previously described disease-causing mutations (3,4,7). There is to date no evidence for genetic heterogeneity of HHRH or phenotypic variability between different NaPi-IIc mutations. The current kindred is not large enough to permit formal linkage analysis to determine whether p.Q49*, when present, alters the phenotype of HHRH. None the less it is interesting that both the father of the index case (who had a history of lower extremity bowing) and the sister (who had symptoms suggestive of nephrolithiasis) are both carriers of the novel mutation c.145C>T (p.Q49*), which may indicate that this mutation is a complete null allele leading to a more severe phenotype than p.S192L.
Clinical manifestations of HHRH typically begin in childhood (1). Despite marked elevation in serum 1,25(OH)2vitamin D levels and hypercalciuria, it is surprising that affected individuals rarely present with nephrolithiasis or nephrocalcinosis. While our patient had rickets as a child, it wasn’t until he moved to the United States that he developed symptoms as an adult. Our patient is a vegetarian, and a change in his diet while living in the United States may have contributed to the development of hypercalciuria and recurrent nephrolithiasis. Furthermore, the initiation of treatment with calcitriol, which is contraindicated in this disease, likely led to further increases in urinary calcium excretion contributing to stone formation. His stones were analyzed and found to be 90% calcium oxalate, which is what would be predicted in the setting of marked hypercalciuria and hyperoxaluria. His hyperoxaluria is likely due to the fact that calcium is hyperabsorbed in HHRH. Consequently, there is less gut calcium available to bind enteric oxalate leaving more free oxalate available for absorption in the colon (8). Although hyperoxaluria has not been associated with renal calcifications in appropriately treated patients with HHRH (9) the use of calcitriol and a change to a more lithogenic Western diet may have, together with the hypercalciuria, put him at greater risk for renal stone disease. The magnitude of our patient's hypercalciuria is consistent with reports in the literature. While in the range of that reported for affected members of the original Bedouin kindred (8.6±1.0 mg/kg/bw/day) (1), our patient’s urine calcium of 6.3 mg/kg/bw/day, is actually slightly lower than this average.
It is possible that nephrolithiasis and nephrocalcinosis have been under appreciated as clinical manifestations of HHRH. Bergwitz et al. reported nephrocalcinosis and nephrolithasis in two kindreds affected by HHRH (3) and Ichikawa et al. recently described recurrent nephrolithiasis in an HHRH kindred (7). A systematic radiologic evaluation for nephrolithiasis and nephrocalcinosis has not been reported in the majority of the cases of HHRH published to date, so an estimate of the prevalence of this complication cannot be ascertained from the literature.
The etiology of the patient’s femur insufficiency fracture is unclear. It is possible that a change in environment (more walking) could have contributed to his fracture. Additionally, less sun exposure while living on the East coast of the United States could have contributed to the development of vitamin D deficiency before being referred to us. Vitamin D-deficiency would have increased his susceptibility to fracture. Alternatively, since 1,25(OH)2vitamin D in high doses can stimulate bone resorption (10), when calcitriol therapy was added to his already high endogenous 1,25(OH)2vitamin D production it is possible that this led to increased bone resorption at a vulnerable skeletal site.
In summary, mutations in the SLC34A3 gene leading to loss of function of the NaPi-IIc protein have been identified as the molecular cause for HHRH. While elevated serum 1,25(OH)2vitamin D levels, increased urinary phosphate excretion, and marked hypercalciuria are distinguishing biochemical features of HHRH, nephrolithiasis and nephrocalcinosis are less often reported in these patients. The affected patient in the present case is a compound heterozygote for mutations in the SLC34A3 gene with a novel nonsense mutation in exon 3. He presented with rickets in childhood and developed nephrolithiasis and a femoral insufficiency fracture in adulthood after immigrating from India to the United States. This case illustrates the complex interplay of nutritional, environmental and genetic factors in affecting the phenotypic expression of SLC34A3 mutations.
Acknowledgements
The authors gratefully acknowledge the critical editorial comments of Dr. Harald Juppner.
This work was supported by the Yale Bone Center, the Yale Core Center for Musculoskeletal Disorders (AR46032, a P30 award to KLI), Young Investigator Awards from the National Kidney Foundation and the American Association for Clinical Investigation to CB and in part by Grant Number UL1 RR024139 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. Information on Re-engineering the Clinical Research Enterprise can be obtained from http://nihroadmap.nih.gov/clinicalresearch/overview-translational.asp
References
- 1.Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D, Liberman UA. Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med. 1985;312:611–617. doi: 10.1056/NEJM198503073121003. [DOI] [PubMed] [Google Scholar]
- 2.Gazit D, Tieder M, Liberman UA, Passi-Even L, Bab IA. Osteomalacia in hereditary hypophosphatemic rickets with hypercalciuria: a correlative clinical-histomorphometric study. J Clin Endocrinol Metab. 1991;72:229–235. doi: 10.1210/jcem-72-1-229. [DOI] [PubMed] [Google Scholar]
- 3.Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, Frappier D, Burkett K, Carpenter TO, Anderson D, Garabedian M, Sermet I, Fujiwara TM, Morgan K, Tenenhouse HS, Juppner H. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–192. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ichikawa S, Sorenson AH, Imel EA, Friedman NE, Gertner JM, Econs MJ. Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab. 2006;91:4022–4027. doi: 10.1210/jc.2005-2840. [DOI] [PubMed] [Google Scholar]
- 5.Lorenz-Depiereux B, Benet-Pages A, Eckstein G, Tenenbaum-Rakover Y, Wagenstaller J, Tiosano D, Gershoni-Baruch R, Albers N, Lichtner P, Schnabel D, Hochberg Z, Strom TM. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78:193–201. doi: 10.1086/499410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerstetter JE, Caseria DM, Mitnick ME, Ellison AF, Gay LF, Liskov TA, Carpenter TO, Insogna KL. Increased circulating concentrations of parathyroid hormone in healthy, young women consuming a protein-restricted diet. Am J Clin Nutr. 1997;66:1188–1196. doi: 10.1093/ajcn/66.5.1188. [DOI] [PubMed] [Google Scholar]
- 7.Walton RJ, Bijvoet OL. Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310. doi: 10.1016/s0140-6736(75)92736-1. [DOI] [PubMed] [Google Scholar]
- 8.Erickson SB, Cooper K, Broadus AE, Smith LH, Werness PG, Binder HJ, Dobbins JW. Oxalate absorption and postprandial urine supersaturation in an experimental human model of absorptive hypercalciuria. Clin Sci (Lond) 1984;67:131–138. doi: 10.1042/cs0670131. [DOI] [PubMed] [Google Scholar]
- 9.Tieder M, Blonder J, Strauss S, Shaked U, Maor J, Gabizon D, Manor H, Sela BA. Hyperoxaluria is not a cause of nephrocalcinosis in phosphate-treated patients with hereditary hypophosphatemic rickets. Nephron. 1993;64:526–531. doi: 10.1159/000187395. [DOI] [PubMed] [Google Scholar]
- 10.Maierhofer WJ, Gray RW, Cheung HS, Lemann J., Jr Bone resorption stimulated by elevated serum 1,25-(OH)2-vitamin D concentrations in healthy men. Kidney Int. 1983;24:555–560. doi: 10.1038/ki.1983.193. [DOI] [PubMed] [Google Scholar]